

Abstract
Although the identification of protein interactions by high-throughput (HTP) methods progresses at a fast pace, ‘interactome’ data sets still suffer from high rates of false positives and low coverage. To map the human protein interactome, we describe a new framework that uses experimental evidence on structural complexes, the atomic details of binding interfaces and evolutionary conservation. The structurally inferred interaction network is highly modular and more functionally coherent compared with experimental interaction networks derived from multiple literature citations. Moreover, structurally inferred and high-confidence HTP networks complement each other well, allowing us to construct a merged network to generate testable hypotheses and provide valuable experimental leads.
Keywords: protein interactions, protein structures, protein complexes, IBIS
Introduction
Proteins perform their functions through interactions with proteins and other biomolecules. The knowledge of entire sets of interactions combined with the locations and properties of protein-binding sites is essential for our understanding of cellular functions and the origin of many diseases. Recent advances in experimental high-throughput (HTP) approaches allowed the determination of protein interaction partners in various organisms on a large scale. Although detection of protein interactions through HTP methods progresses at a fast pace, current ‘interactome’ data sets still suffer from a high rate of false positives and low coverage. Comprehensive human interactome mapping is a daunting task with more than 80–90% of human protein–protein interactions remaining to be determined [1]. Given that the number of known structures of human protein complexes increases by thousands every year, low-throughput and high-resolution X-ray/NMR methods can be used to complement and verify interactions obtained from HTP screens. Ideally, complete structural coverage of protein complexes provides information on protein partnership combined with the atomic details of binding site locations and physicochemical properties of interaction interfaces.
Previously, structural data have been used to interrogate HTP yeast interactions using homologous structures [2], [3] or by comparing query proteins to known template protein–protein interfaces [4] (see supplementary information online). Here, we describe a framework that allows consistent inference of protein interactions and binding sites, using structural data on protein complexes. As interaction annotations transferred from one protein to another might result in incorrect assignment at larger evolutionary distances, our approach verifies interactions and binding interfaces by examining their evolutionary conservation, uses algorithms to evaluate correct biological forms of proteins in a cell and finally applies a rigorous scheme to rank binding sites with respect to their relevance to the query protein. Although our procedure can be applied to the interactome of any organism, we illustrate our method by rigorously determining a web of protein interactions in human. Our inferred protein interactions can be accessed at ftp://ftp.ncbi.nih.gov/pub/mmdb/humanIntNw/ and these data files also provide characteristics such as structural properties, evolutionary conservation and locations of binding sites on human protein sequences.
Results And Discussion
In Fig 1, we present a schematic outline of our procedure. Pooling all human gene sequences from the curated RefSeq database [5], we select the longest protein isoforms resulting in 20,846 protein sequences (step 1, Fig 1). For each protein sequence, we retrieve protein interaction partners and binding sites using our IBIS database [6, 7], which predicts and clusters protein interaction partners together with the locations of their binding sites on a query protein. IBIS provides experimentally ‘observed’ human interactions if a protein has at least five amino-acid residues ‘contacting’ another protein. Specifically, two residues are defined to be in contact if any of the heavy-atom inter-atomic distances is <6 Å, while the group of residues that have contacts with an interaction partner is called a ‘binding site’. On the basis of homology-based inference, we align query proteins to close homologues with known structural complexes (step 2, Fig 1), and transfer partner and binding site annotations from homologues to the query (step 3, Fig 1).
Figure 1.

Structural inference of human protein interactions. Using 20,846 human protein sequences (Step 1), we mapped each sequence to a homologous, experimentally determined structural complex (Step 2). Then we retrieved clusters of binding sites from homologous protein complexes allowing us to infer/predict protein interaction partners (Step 3). We also used structural information from organisms other than human and mapped interaction partners to their most similar homologous human proteins with more than 80% identity (Step 4). In total, we found 13,217 interactions between 3,614 human genes/proteins (Step 5).
To ensure the biological relevance of interaction partners and binding sites, we cluster similar binding sites of homologous protein complexes (‘conserved binding site clusters’). Specifically, similarity of binding sites is assessed by considering the sites’ sequence and structure conservation as well as physicochemical properties of protein assemblies. Interaction interfaces in complexes are additionally validated using the PISA algorithm. Using chemical thermodynamics, PISA computes a set of macromolecular assemblies that are expected to be stable in solution and presumed to represent the biological form of a protein in the cell [8]. We then map interaction partners from complexes of other organisms to their most similar human proteins that have more than 80% sequence identity and 80% protein sequence coverage (step 4a, Fig 1). We define a self-interaction (step 4b, Fig 1) if an interaction is inferred from a homooligomeric complex (complex of identical chains). We label each interaction with an ‘inference threshold’, defined as the average percentage of sequence identity between the query human protein and homologous structural complexes of the conserved binding site cluster.
As a result of our approach, we obtained 13,217 interactions (including 2,944 self-interactions) between 3,614 human genes, covering ∼18% of the human proteome (step 5, Fig 1) and ∼10% of the human interactome based on previous estimates of the size of the complete human interactome [1]. About 10% of all 13,217 interactions were ‘observed’ in actual protein structural complexes; 58% of all interactions were evolutionarily conserved among non-redundant homologous complexes (that is, part of ‘conserved binding site clusters’) as opposed to ‘singleton’ clusters formed by only one protein complex.
As a comparison benchmark, we used experimental protein interactions determined mostly from HTP screens. Pooling all interactions from Reactome [9], MINT [10] and HPRD [11] databases we assembled 61,240 unique interactions between 11,446 human proteins. In addition, we constructed a high-confidence (HC) set of 8,024 unique interactions between 3,168 proteins, demanding that each HC interaction was reported in at least two publications. We considered ‘structurally inferred’ (SI) interactions as those that were ‘observed’ or determined from a conserved binding site cluster. As a metric that allows the assessment of network similarity, we calculated the Jaccard distance, considering sets of edges of networks i and j. Using different inference thresholds, we compared SI networks with HTP and HC networks, respectively. Only considering interactions between proteins that appeared in both compared networks, we observed that Jaccard distances decreased with increasing inference threshold (Fig 2A). In particular, distances between SI and HC networks were not only smaller but also declined faster with increasing inference threshold compared with distances between SI and HTP networks. To estimate the significance of our results, we randomly distributed SI interactions, keeping the number of interactions per protein constant. Furthermore, we calculated distances of such randomized SI networks to HTP and HC interactions as previously described. Using a Z-test, we found that the observed distances at all inference thresholds were highly significant (P≪0.01). We also calculated Jaccard distances between proteins rather than edges, confirming our initial result (supplementary Fig S1 online).
Figure 2.

Comparisons of structurally inferred (SI) to high-throughput (HTP) and high-confidence (HC) interactions. In A, we determined Jaccard distances between SI, HC and HTP networks at different inference thresholds. In pairwise distance calculations, we only considered interactions between proteins that appeared in both networks. In B, we considered SI interactions that had an inference threshold of more than 50% identity. Considering interactions between proteins that appeared in all networks we calculated Jaccard distances between interacting proteins that were annotated with their corresponding GO-terms (inset) and pathways.
A different level of comparison is the assessment of network-dependent topological parameters. Using different inference thresholds, we compared SI with HTP and HC interactions between proteins that appeared in both networks. Consistent with a previous study [3] we found that the mean node degrees of proteins in the HTP network were generally much higher than for HC and SI networks (supplementary Fig S2a,b online), pointing to the possibility of false positives in the HTP network and possible physical constraints on the number of partners in SI networks. As for other topological measures, we observed similar mean clustering coefficients for HTP, HC and SI networks at different inference thresholds. Although mean shortest path lengths within connected components remained constant in HTP and HC networks, corresponding values of SI networks decreased with increasing inference threshold (supplementary Fig S4a,b online). As a different measure of centrality we also calculated betweenness centrality (supplementary Fig S5a,b online), indicating that centrality characteristics of SI networks appear closer to the HC than the HTP network.
To further measure the quality of structurally inferred interactions, we calculated pathway and functional distances between interacting proteins. Here we considered SI interactions that had an inference threshold of more than 50% sequence identity. Using pathway data from the Molecular Signature Database [12], we annotated each human protein with all human specific pathways with which it participated. Calculating pathway-specific Jaccard distances between interacting proteins (see Methods), we observed that proteins in SI and HC interactions are involved in more similar pathways than proteins in the HTP network (Fig 2B). Such a shift to lower distances is especially pronounced for SI networks (Wilcoxon rank-sum test, P≪0.01). As for biological functions, we annotated each protein with its corresponding Gene Ontology (GO)-terms [13]. In accordance with the previous result, HC and SI interactions appear to be more functionally coherent than HTP interactions (inset, Fig 2B). As for qualitative functional characteristics, we find several tightly connected and functionally coherent clusters of proteins in the SI network ranging from signalling, regulatory, cytoskeletal regulation, protein degradation to immune response functions (supplementary Fig S6 online).
Wondering whether SI and HC networks complement each other, we find that only 24% of interactions in the HC network are observed in the SI network, whereas only ∼22% of SI interactions are covered by HC interactions (even considering interactions between proteins that appear in both networks). Fig 3A shows the ‘merged’ network representing a union of HC and SI interactions. Both types of interactions appear to merge well together, suggesting that structurally inferred interactions might complement reliable HTP interactions. To test such an observation on a quantitative level, we define a ‘network coverage’ (see Methods). Considering a set of proteins from a certain pathway or GO category, we define coverage as the corresponding fraction of these proteins that appear in a given interaction network. Calculating coverage values for all pathways, we indeed observed that the coverage distribution of the merged network is significantly shifted to higher values compared with distributions of the network coverage of SI and HC (Wilcoxon rank-sum test, P≪0.01; Fig 3B), implying that the SI and HC networks complement each other in terms of pathway coverage. In Fig 3C, we use GO categories, allowing us to obtain a similar result (P=0.05).
Figure 3.

Merged network of high-confidence (HC) and structurally inferred (SI) interactions. In A, we show the largest component of a network composed of HC and SI interactions with inference threshold of more than 50% identity. Specifically, we considered proteins that appeared in both networks excluding self-interactions. (B) Pathway-specific coverage for SI, HC and merged networks. (C) GO-specific coverage for SI, HC and merged networks. (D) In the degree distribution of the merged network, we recovered a strong power-law tail. In the inset, we observed a power-law dependence of the clustering coefficient on the node degrees indicating the merged networks inherent modularity.
We also expected that the merged network largely recovered known topological characteristics of protein interaction networks. Indeed, we clearly observed that degree distributions of the merged network decayed as a power law (Fig 3D), similar to the separate SI and HC networks (supplementary Fig S7a online). At the same time, modularity of the SI and HC networks is evident from the strong power-law decay of the clustering coefficients with increasing node degree [14] (supplementary Fig S7b online), a characteristic that prevails in the merged network as well (inset, Fig 3D). The merged network comprises 5,464 protein nodes and 17,199 edges (excluding self-interactions) and is available at ftp://ftp.ncbi.nih.gov/pub/mmdb/humanIntNw/.
Here we present a comprehensive attempt to use the growing data on protein structural complexes to map the human interactome. Our framework not only uses invaluable experimental evidence to infer interactions but also taps the atomic details of binding interfaces and their evolutionary conservation, allowing the assessment of their functional importance. Our functional analysis and comparison with HTP networks show that our structurally inferred interactions are more functionally and pathway coherent than interactions obtained by HTP screens. As a proof of concept, the merged network of HC and SI interactions recovers general topological characteristics of protein interaction networks. Furthermore, SI and HC interactions complement each other well, suggesting that our approach might generate testable hypotheses and provide valuable experimental leads.
Methods
Software. Our pipeline was implemented using BioRuby [15], the NCBI Toolkit and the Entrez Programming Utilities [16] to facilitate data manipulation and analyses. Cytoscape [17] was used for interaction network visualization.
Inferring protein–protein interactions. To infer interactions based on homology, we first collected template proteins with known structures that are similar to a given query protein and have at least 80% sequence identity and more than 80% of the query sequence aligned using cBlast [18]. For each template protein, we retrieved all homologous (ensuring more than 30% identity to a query) and structurally similar proteins with known structural complexes from the Protein Data Bank [19]. Template and homologous structural complexes were structurally aligned using the VAST algorithm [20]. Subsequently, homologous complexes were grouped based on their binding site similarity with the concept that a binding site is more likely to be functionally important and not lineage specific if it is evolutionarily conserved among non-redundant homologues. A binding site cluster represents a collection of structures that are related to the query protein where all members of the cluster contain similar binding sites. We measured similarity between binding sites in terms of sequence similarity, and assigned an additional weight to structurally aligned positions (see supplementary information online). Such a two-step process of mapping a query protein to homologous structural complexes was necessary to ensure high quality alignments through structure–structure comparisons. Sequence similarity between the query and homologous structural complexes in the conserved binding site cluster was calculated and defined as the ‘inference threshold’. Binding sites were additionally verified using the PISA algorithm [8].
HTP protein–protein interactions. For a representative set of human protein–protein interactions that have been determined mostly by experimental HTP methods (although some low-throughput interactions were also included), we pooled all interactions from Reactome [9], MINT [10] and HPRD[11] to construct a network of 61,240 unique interactions between 11,446 human proteins. In addition, we accounted for interactions that have been reported in at least two different publications by these databases, allowing us to compile a HC network of 8,024 unique interactions between 3,168 human proteins.
Jaccard distance. Representing each protein i by a list of attributes, Γi (GO or pathway annotations), we defined the Jaccard distance between interacting proteins i and j as
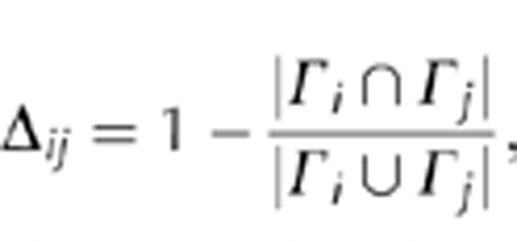 |
where 0⩽Δij⩽1. We also calculated Jaccard distance between two networks i and j by considering the corresponding sets of edges/interactions Γi and Γj between proteins that appeared in both networks.
Functional and pathway coverage of interactions. As a measure of a network's G=(V,E) functional or pathway coverage, we considered sets of proteins that appeared in a certain pathway or represented a GO function P=(p1, p2, …,
pn) We constructed a corresponding graph GP=(Vp, Ep) by accounting for all interactions in the given network G between proteins that appeared in a given set P, 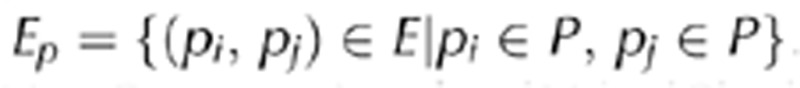 . Therefore, Vp is a subset of P, Vp⊆P, suggesting that |Vp |⩽ |P|, allowing us to define the set-specific coverage of a network regarding set P as
. Therefore, Vp is a subset of P, Vp⊆P, suggesting that |Vp |⩽ |P|, allowing us to define the set-specific coverage of a network regarding set P as
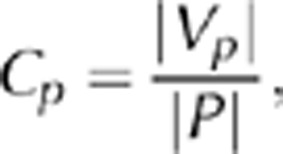 |
where 0⩽Cp⩽0. Using all pathways or GO categories, we obtained corresponding frequency distributions of Cp values.
Pathway information. As a resource of pathway information, we used data from the Molecular Signatures Database [12] that compiles pathway information from KEGG [21], Biocarta and Reactome [9]. We discarded redundant pathways that shared more than 95% of their proteins with other pathways and ended up with 480 non-redundant pathways.
Network parameters. A local measure of protein centrality in the network is its degree, defined as the number of interaction partners. We calculated mean shortest path length, defined as the shortest paths between all protein pairs in a given connected component and then averaged over connected components. As a global measure of protein centrality, we calculated betweenness centrality, reflecting a protein's appearance in shortest paths through the whole network. In particular, we defined betweenness centrality of a node ν as
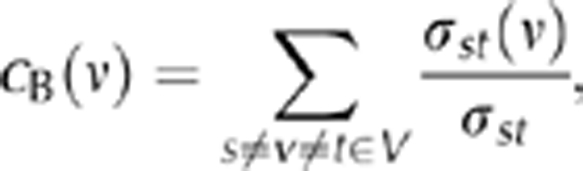 |
where σst was the number of shortest paths between proteins s and t, while σst(ν) was the number of shortest paths running through ν [22]. The clustering coefficient, Ci, was defined as the fraction of observed interactions Ei, among all possible interactions between Ni neighbours of a protein i,
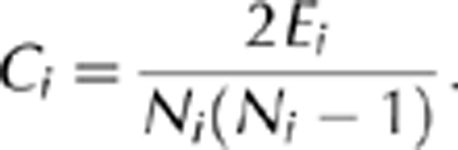 |
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health/Department of Health and Human Service (Intramural Research program of the National Library of Medicine).
Author contributions: A.R.P. designed the study; M.T. and K.H. implemented the approach; B.A.S. and S.W. analysed results; A.R.P. and S.W. wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Venkatesan K et al. (2009) An empirical framework for binary interactome mapping. Nat Methods 6: 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloy P, Russell RB (2002) Interrogating protein interaction networks through structural biology. Proc Natl Acad Sci USA 99: 5896–5901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PM, Lu LJ, Xia Y, Gerstein MB (2006) Relating three-dimensional structures to protein networks provides evolutionary insights. Science 314: 1938–1941 [DOI] [PubMed] [Google Scholar]
- Tuncbag N, Gursoy A, Nussinov R, Keskin O (2011) Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat Protoc 6: 1341–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Klimke W, Maglott DR (2009) NCBI reference sequences: current status, policy and new initiatives. Nucleic Acids Res 37: D32–D36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker BA, Zhang D, Thangudu RR, Tyagi M, Fong JH, Marchler-Bauer A, Bryant SH, Madej T, Panchenko AR (2010) Inferred Biomolecular Interaction Server – a web server to analyze and predict protein interacting partners and binding sites. Nucleic Acids Res 38: D518–D524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker BA, Zhang D, Tyagi M, Thangudu RR, Fong JH, Marchler-Bauer A, Bryant SH, Madej T, Panchenko AR (2012) IBIS (Inferred Biomolecular Interaction Server) reports, predicts and integrates multiple types of conserved interactions for proteins. Nucleic Acids Res 40: D834–D840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Croft D et al. (2011) Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 39: D691–D697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol A, Chatr Aryamontri A, Licata L, Peluso D, Briganti L, Perfetto L, Castagnoli L, Cesareni G (2010) MINT, the molecular interaction database: 2009 update. Nucleic Acids Res 38: D532–D539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava Prasad TS et al. (2009) Human Protein Reference Database—2009 update. Nucleic Acids Res 37: D767–D772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GO (2008) The gene ontology project in 2008. Nucleic Acids Res 36: D440–D444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi AL, Oltvai ZN (2004) Network biology: understanding the cell's functional organization. Nat Rev Genet 5: 101–113 [DOI] [PubMed] [Google Scholar]
- Goto N, Prins P, Nakao M, Bonnal R, Aerts J, Katayama T (2010) BioRuby: bioinformatics software for the Ruby programming language. Bioinformatics 26: 2617–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW et al. (2011) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 39: D38–D51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bryant S, Tatusov R, Tatusova T (2000) Links from genome proteins to known 3-D structures. Genome Res 10: 1643–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat JF, Madej T, Bryant SH (1996) Surprising similarities in structure comparison. Curr Opin Struct Biol 6: 377–385 [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32: D277–D280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh KI, Oh E, Jeong H, Kahng B, Kim D (2002) Classification of scale-free networks. Proc Natl Acad Sci USA 99: 12583–12588 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.