

Abstract
The direct interrogation of fleeting intermediates by rapid-mixing kinetic methods has significantly advanced our understanding of enzymes that utilize dioxygen. The gas’s modest aqueous solubility (< 2 mM at 1 atm) presents a technical challenge to this approach, because it limits the rate of formation and extent of accumulation of intermediates. This challenge can be overcome by use of the heme enzyme chlorite dismutase (Cld1) for the rapid, in situ generation of O2 at concentrations far exceeding 2 mM. This method was used to define the [O2] dependence of the reaction of the class Ic ribonucleotide reductase (RNR) from Chlamydia trachomatis, in which the enzyme’s MnIV/FeIII cofactor forms from a MnII/FeII complex and O2 via a MnIV/FeIV intermediate, at effective O2 concentrations as high as ~10 mM. With a more soluble receptor, myoglobin, an O2 adduct was accumulated to > 6 mM in < 15 ms. Finally, the C–H-bond-cleaving FeIV-oxo complex, J, in taurine:α-ketoglutarate dioxygenase and superoxo-Fe2III/III complex, G, in myo-inositol oxygenase, and the tyrosyl-radical-generating Fe2III/IV intermediate, X, in Escherichia coli RNR were all accumulated to yields more than twice those previously attained. This means of in situ O2 evolution permits a > 5 mM “pulse” of O2 to be generated in < 1 ms at the easily accessible [Cld] of 50 μM. It should therefore significantly extend the range of kinetic and spectroscopic experiments that can routinely be undertaken in the study of these enzymes and could also facilitate resolution of mechanistic pathways in cases of either sluggish or thermodynamically unfavorable O2-addition steps.
Aerobic organisms are replete with proteins and enzymes that react with O2 for such purposes as cellular and organismal respiration (1); oxidation reactions of primary and secondary metabolism (2-4); catabolism of drug and xenobiotic compounds (2); biosynthesis of enzyme cofactors (5-7), neurotransmitters (8) and natural products (4, 9); regulation of transcription (10-13); and uptake and storage of inorganic nutrients (14). Many of these enzymes employ reduced cofactors, made up of one or more reduced transition metal [typically FeII (2, 15-18) or CuI (19-21)] or a reduced flavin (22), that combine with O2 to form potently oxidizing intermediates that directly or indirectly transform their substrates. Rapid-mixing transient-kinetic studies have contributed greatly to our understanding of the mechanisms of these enzymes by permitting the direct detection, kinetic tracking, and spectroscopic characterization of fleeting intermediates in their catalytic cycles (15-18). The fleeting nature of intermediates formed (typical half-lives of < 100-101 s) has, with a few spectacular exceptions (23-25), precluded their three-dimensional structural characterization by X-ray crystallography. The alternative approach has been to (i) trap the intermediates at their maximum extents of accumulation by the freeze-quench method and (ii) subject them to a suite of spectroscopic methods. In combination with density functional theory (DFT1) calculations (26), these methods can afford local, high-resolution structural information on transient species, thereby providing ‘snap-shots’ along the reaction coordinate (27-29).
In the study of these enzymes, the physical properties of O2 impose certain challenges to the elucidation of reaction kinetics and the direct characterization of intermediates. Its gaseous nature makes systematic variation of its concentration more challenging and introduces greater uncertainty in concentration values than for non-volatile substrates. More importantly, the modest solubility of the gas imposes a very constraining upper limit of ~2 mM on the [O2] (30) that can be achieved without specialized apparatus. O2 has often been found to combine with the reduced enzyme cofactors with second-order rate constants of 104-107 M−1s−1, which, at the routinely accessible [O2] of 1 mM, are sufficient to give effective first-order rate constants of 101-104 s−1 (31-34). In many cases, these formation rate constants have proven to be comparable to or greater than the first-order rate constants for decay of the key intermediates. In these cases, accumulation of the intermediate states for detailed characterization has been possible (34-40). In other cases, isotopic or chemical modification of the substrate or mutagenesis of the protein has been used to slow decay of intermediates in order to permit their accumulation and characterization (41-43). In still other cases, however, the obstacle presented by the modest solubility of O2 has not been overcome, and many intriguing O2-dependent enzyme reactions have thus far proven resistant to this powerful approach to mechanistic dissection (44).
Several spectroscopic methods that can reveal important structural details for intermediates demand very concentrated samples that are highly enriched in the desired state. For example, application of extended X-ray absorption fine structure (EXAFS) spectroscopy, which in ideal cases can provide very precise metal-ligand and metal-metal distances for reactive intermediates, to dilute or heterogeneous freeze-quenched samples is notoriously problematic and in several notable cases has provided distances (45, 46) which are irreconcilable with those in DFT-derived structures (27, 28, 47) and inorganic model complexes (48, 49). Other methods, such as the developing technique of nuclear resonance vibrational spectroscopy (NRVS), which can reveal structural details of iron complexes, require that targets be present at concentrations exceeding the solubility of O2 (50). Long-lived complexes may be generated at such high concentrations by direct treatment of precursors with O2(g) (38, 39), but the sluggishness of transport across the gas-liquid interface makes such an approach impractical for complexes with half-lives of less than ~1 min. A rapid-mixing method permitting reaction with O2 at greater concentrations without the need for specialized equipment could, therefore, open doors to new experiments in this area of biochemistry.
The heme enzyme chlorite dismutase (Cld) rapidly converts chlorite (ClO2−) to chloride (Cl−) and O2, suggesting a simple approach to overcoming both the technical difficulties in the systematic variation of [O2] and its modest solubility (51). A number of proteobacteria have been shown to catalyze this reaction in conjunction with perchlorate (ClO4−) respiration. ClO4− is sequentially reduced to ClO3− and ClO2− by a membrane-bound molybdopterin-dependent perchlorate reductase, which couples the reductions to the generation of a proton gradient (52). The resulting ClO2− would accumulate and kill the organism without the detoxification reaction catalyzed by Cld. Accordingly, the reaction must be fast in order to serve its biological function: the homopentameric Cld from Dechloromonas aromatica (Da) is one of the fastest and most efficient Clds yet studied, with a kcat value of 2.0 (± 0.6) × 105 s−1 (per heme) at 4 °C and pH 5.2 (51, 53, 54). This rate constant suggests that Cld could support the generation of tens-of-millimolar O2 on the millisecond timescale. Importantly, the enzyme is not significantly inhibited by millimolar concentrations of either of its two products, Cl− and O2 (51). It is, moreover, capable of approximately 1.7 × 104 turnovers per heme before undergoing irreversible inactivation due to oxidative damage to the heme. We therefore reasoned that catalytic concentrations of Cld could be used to initiate the reaction of an O2-utilizing (metallo)enzyme by rapid mixing with the highly soluble, non-volatile ClO2− rather than with the sparingly soluble, gaseous O2. Here, we demonstrate that this approach can indeed simplify the experimental variation of [O2], expand the range of [O2] that can be interrogated with commonly available equipment, and permit preparation of O2-dependent intermediate states at concentrations and purities not accessible by conventional rapid mixing with O2-containing aqueous solutions.
EXPERIMENTAL PROCEDURES
Materials
Sodium chlorite (NaClO2), α-ketoglutaric acid, sodium ascorbate, and horse heart myoglobin (Mb) were purchased from Sigma-Aldrich. 1,1,2,2-[2H4]-2-aminoethane-1-sulfonic acid (d4-taurine) and 1,2,3,4,5,6-[2H6]-cyclohexan-(1,2,3,5/4,6)-hexa-ol (d6-myo-inositol or d6-MI) were purchased from C/D/N Isotopes.
Preparation of Proteins
Methods for overexpression and purification of the β2 subunit of Chlamydia trachomatis (Ct) ribonucleotide reductase (RNR), Da Cld, Escherichia coli taurine:α-ketoglutarate(αKG) dioxygenase (TauD), Escherichia coli (Ec) RNR-β2, and Mus musculus myo-inositol oxygenase (MIOX) have been presented elsewhere (7, 35, 36, 51, 55). To prepare myoglobin (Mb) containing heme with natural abundance iron (56Fe-Mb), 400 mg of lyophilized horse-heart Mb (Sigma-Aldrich product no. M1882) was dissolved in 2 mL of 100 mM potassium phosphate buffer, pH 6.8. The protein was loaded onto a 50-mL anion exchange column (DE-52, Whatman) and eluted by gravity flow with the same buffer. Fractions with A409/A280 ratios (Rz) of at least 5 were pooled and concentrated to [heme] ~ 10 mM. For 57Fe-enriched heme (57Fe-Mb), metalloporphyrin synthesis was adapted from Adler, et al. (56) for 57Fe heme enrichment. Apo protein, generated using Teale’s method (57), was reconstituted at pH 7 (58). Excess heme was removed by anion exchange chromatography using Whatman DE-52 resin, as described above for 56Fe-Mb. Fractions with Rz > 5 were pooled and concentrated to [heme] ~ 10 mM.
Stopped-flow Absorption and Freeze-quench EPR and Mössbauer Experiments
Procedures for the stopped-flow and freeze-quench experiments and the spectrometers for the stopped-flow, EPR, and Mössbauer measurements have been described (7, 34, 36).
Analysis of the Stopped-flow Absorption Kinetic Data
A390-versus-time traces reflecting accumulation and decay of the MnIV/FeIV activation intermediate in Ct-RNR β2 were analyzed by nonlinear regression according to the equation,
| (1) |
which gives absorbance as a function of time (At) for two irreversible first-order reactions in terms of the rate constants (k1 and k2), the amplitudes associated with each reaction (ΔA1 and ΔA2) and the initial absorbance (A0) at time t = 0. The formation of the intermediate and its decay are sequential processes, but their well-resolved rate constants (k1[O2] >> k2) make the assumption of parallel reactions acceptable. Simulation of these traces was carried out by using KinTek Explorer (KinTek Corporation, USA). The kinetic mechanism for Cld and KD for the Cld•ClO2− Michaelis complex that were assumed in the simulations are given in Results. Kinetic constants for the formation and decay of the intermediate acquired from the regression fits in Fig. 1B were assumed in the simulations. Values of kcat of 30,000 s−1, 60,000 s−1, 120,000 s−1, and 200,000 s−1 (Fig. S2, panels A –D, respectively) were assumed for Cld.
Figure 1.

Activation of Ct β2 using Cld and ClO2−. (A) 390-nm absorbance-versus-time traces following rapid mixing at 5 °C of a solution containing 0.2 mM β2, 0.6 mM MnII, 0.2 mM FeII, and 0.01 mM Cld with an equal volume of either 20 mM ClO2− (black trace), O2-saturated 100 mM HEPES buffer, pH 7.6 (green trace), or O2-free buffer (orange trace). Traces from control reactions, from which either Cld or β2 was omitted, are shown in red and blue, respectively. (B) Delineation of the [O2] dependence of the Ct β2 activation reaction by variation of [ClO2−]. Reactions were carried out as for the black trace in A, but with the concentration of the ClO2− reactant solution varied to give the final values of [ClO2−] noted in the figure. Traces were analyzed by non-linear regression using the equation for two exponential phases (solid lines thru data; see SI for analysis) in order to extract observed first-order rate constants for formation of the MnIV/FeIV intermediate (kobs). (C) Plot of these observed first-order rate constants versus [ClO2−] or [O2]. The points with [ClO2−] ≤ 4 mM were fit by the equation for a line (solid line). Extrapolation of the kobs for the reaction with 16 mM ClO2− to the linear fit line (dashed lines) in this case gave an effective [O2] of 9 mM (arrow). The gray diamond points are values of kobs obtained after mixing with either O2-saturated buffer (as in A, green trace) or buffer prepared by diluting O2-saturated buffer 2- or 4-fold with O2-free buffer, as has been done in the past to define the [O2]-dependence of the reaction.
Analysis of EPR and Mössbauer Spectra to Quantify the MnIV/FeIV Intermediate in Ct-RNR β2
Double integration of first-derivative EPR signals was carried out using the graphing and analysis program KaleidaGraph (Synergy Software). Comparison to the corresponding double integral for the spectrum of a CuII(ClO4)2 standard with correction for the different g-values (59) permitted calculation of absolute spin concentration. The spectral contribution of mononuclear MnII was subtracted out by using the spectrum of a sample of apo β2 to which a known amount of MnII had been added. The contribution from the Fe2III/IV complex (X) was quantified by individually integrating the six peaks of the sextet signal of the MnIV/FeIV intermediate. The double integrals of five of the peaks (all except for the fourth, with which the spectrum of X overlaps) are identical within error. The difference between the area of the fourth peak and the average area of the other five peaks represents the contribution of X to the experimental spectrum, which corresponds to 6% of the total spin. In the freeze-quench method, the absolute spin concentration in the reaction solution depends also on the “packing factor,” which is the fraction of the packed material made up of the actual solution (the fraction not contributed by the frozen cryo-solvent). The [spin] determined from comparison of the double integral of samples to that of standard is divided by this packing factor to account for dilution of the frozen sample by the cryo-solvent. In our extensive experience using isopentane as the cryo-solvent, we have repeatedly measured packing factors of 0.52–0.60 (31, 34). Table S1 provides concentrations of the paramagnetic species determined over a range of packing factors of 0.50–0.60. The narrower range of 0.54–0.56, which agrees with the mean packing factor of 0.55 that we have determined over many years, gives ranges of 1.24–1.16 mM and 0.13–0.11 mM for the concentrations of the MnIV/FeIV complex and X, respectively. These ranges are in good agreement with the values determined by analysis of the Mössbauer spectra.
The multiple Fe-containing species present in the freeze-quench samples all contribute to the experimental Mössbauer spectra, requiring that the spectra be “deconvoluted” into their components to extract species concentrations. However, the field-orientation dependence of the predominant species, the MnIV/FeIV intermediate, provides an alternative means of accurate quantification. This analysis was carried out as previously described (34). The slightly different spin-Hamiltonian parameters used herein are provided in Table S2 and compared to the published values [given in parentheses (34)].
RESULTS
Activation of the β2 Subunit of Ct RNR by Mixing its MnII/FeII Complex with ClO2− in the Presence of Cld
We selected the activation reaction of the manganese- and iron-dependent class Ic Ct RNR as an ideal test case for the in situ generation of O2 by the Cld/ClO2− system (7, 60, 61). Previous studies showed that reaction of the MnII/FeII complex of the enzyme’s β2 subunit with O2 results in formation of the catalytically functional MnIV/FeIII cofactor (7, 62) via a novel MnIV/FeIV activation intermediate (34, 63). The reaction is a kinetically well-behaved, two-step sequence, in which the first step, formation of the MnIV/FeIV intermediate, is cleanly first-order in O2 (34). Both steps in the sequence are associated with absorbance changes, with the reactant complex being essentially transparent and the intermediate absorbing maximally at 390 nm with a molar absorptivity (ε390 ~ 4,500 M−1cm−1) approximately twice that of the MnIV/FeIII product (34). These characteristics permit convenient monitoring in stopped-flow absorption experiments.
Rapid mixing of a solution containing the Ct β2 protein (200 μM dimer), MnII (3 equiv relative to β2), FeII (1 equiv) and Cld (10 μM heme) with an equal volume of a 20 mM ClO2− solution (100 equiv relative to β2; 2000 equiv relative to Cld heme) results in a rapid increase in the absorbance at 390 nm (A390) followed by its slower decay to approximately half of the maximum value (Fig. 1A, black trace). The trace is qualitatively similar to that obtained by mixing the MnII/FeII-β2 complex with O2-saturated buffer (green trace). Analogous traces from control reactions from which either the MnII/FeII-β2 reactant (blue trace), the ClO2− reactant (orange trace), or the Cld catalyst (red trace) was omitted, do not show the characteristic behavior, suggesting that the transient behavior of the complete reaction reflects formation and decay of the MnIV/FeIV intermediate specifically as a result of the evolution of O2 from ClO2− by Cld.
To verify that the complete reaction containing MnII/FeII-β2, Cld, and ClO2− produces the expected MnIV/FeIV activation intermediate, freeze-quench EPR and Mössbauer samples were prepared from a concentrated MnII/57FeII-β2 reactant solution (giving a final concentration of 1.88 mM β2 with 1 equiv 57FeII and 2 equiv MnII). The Mössbauer and X-band EPR spectra of identical samples that were allowed to react for 1 s (near the time of maximum A390 in the black trace in Fig. 1A) are shown in Fig. 2, and quantitative analysis of these spectra is summarized in Tables S1 and S2. The spectra are dominated by the features of the MnIV/FeIV intermediate (34), confirming that it is formed in high yield [1.2 ± 0.3 mM by EPR and 1.2 ± 0.2 mM (63 ± 8% of total Fe) by Mössbauer]. A small fraction of the Fe2III/IV intermediate, X, resulting from reaction of O2 with Fe2II/II centers formed in competition with the desired MnII/FeII-β2 reactant complex, is also detected [0.2 ± 0.1 mM by EPR and 0.2 ± 0.1 mM (13 ± 5% of total Fe) by Mössbauer]. The spectroscopic results thus establish that the Cld/ClO2− system does indeed support formation of the expected intermediate.
Figure 2.

4.2-K/53-mT Mössbauer and EPR spectra of Ct β2 samples enriched in the MnIV/FeIV activation intermediate of Ct β2. Preparation of the samples is described in the text. (A) Experimental Mössbauer spectra were acquired with the magnetic field oriented parallel (top) or perpendicular (middle) to the γ beam, and the difference spectrum (bottom) was obtained mathematically. The solid blue and red lines are theoretical spectra of the MnIV/FeIV and Fe2III/IV intermediates, plotted at 63% and 13% of the total intensity. The theoretical spectrum of the Fe2III/IV intermediate was generated with published parameters (72). The parameters used to generate the theoretical spectrum of the MnIV/FeIV intermediate are slightly different from the previously published ones (see Table S2). The spectrum shown matches the experimental difference spectrum more precisely than that generated with the published parameters and therefore permits more precise quantification. We attribute the need for these slight adjustments to the facts that the previously published parameters were obtained by “global” simulation of multiple spectra, and the new spectra have significantly better signal-to-noise ratio because they were collected on a more concentrated sample. (B) EPR spectrum showing that the predominant EPR-active species is the MnIV/FeIV intermediate. The spectrum of the contaminating Fe2III/IV species (X) resulting from reaction of the Fe2II/II–β2 complex with O2 overlaps with the fourth line of the sextet and contributes 6% of the total spin (quantitative analysis of the spectrum is described in Experimental Procedures). Spectrometer conditions were: 9.5 GHz microwave frequency, 20 W microwave power, 14 ± 0.2 K temperature, 100 kHz modulation frequency, 10 G modulation amplitude, 167 s scan time, and 167 ms time constant.
The stopped-flow absorption kinetic traces of Fig. 1A suggest that the MnIV/FeIV intermediate forms much faster in the Cld/ClO2− reaction than in the O2-saturated-buffer reaction (compare black and green traces), consistent with a greater [O2] in the former case. To evaluate the effective [O2] more quantitatively, experiments were carried out with varying [ClO2−] (Fig. 1B). Effective first-order rate constants (kobs) for intermediate formation extracted by regression analysis of the A390 kinetic traces are linearly dependent on [ClO2−] at [ClO2− ] ≤ 4 mM (Figs. 1C and S2). The slope of the line, corresponding to the effective second-order rate constant, agrees precisely with that obtained by direct variation of [O2] by mixing with O2-containing buffer (gray diamonds in Fig. 1C). This result indicates that, at ≤ 4 mM ClO2−, the Cld completely converts the anion to O2 and Cl− sufficiently rapidly so as not to impose a lag phase on MnIV/FeIV intermediate formation (which would tend to diminish the extracted value of kobs). At greater [ClO2−], deviation from this strict first-order dependence is observed. Doubling the [Cld] from 5 μM to 10 μM had no significant effect on the values of kobs for [ClO2−] ≤ 4 mM but gave greater values (deviating less from the first-order dependence) at [ClO2−] > 4 mM (Fig. S1). This observation suggests that the deviation is not intrinsic to the Ct β2 reaction but rather reflects failure of the Cld reaction to rapidly reach completion at the higher [ClO2−]. This conclusion is consistent with the chlorite-dependent destruction of the heme previously shown to limit turnover in the steady state (53). Competition between Cld catalyst destruction and O2 formation begins to manifest at 8 mM [ClO2−] (1,600 equiv of chlorite per Cld heme, Fig. 1C). Extrapolation of the values of kobs obtained at the highest [ClO2−] tested (16 mM) to the fit line describing the first-order regime (dashed lines in Figs. 1C and S1) indicates that effective O2 concentrations of 7–11 mM were achieved in the stopped-flow experiments.
Simulation of the kinetic traces of Fig. 1C was undertaken to assess limitations of the Cld/ClO2− system for in situ generation of O2 and to extract an estimate of kcat for Cld under these reaction conditions (Fig. S2). A simple rapid-equilibrium-binding kinetic model with a value of KD equal to the published value of KM for ClO2− (215 μM) was assumed. The value of the rate-constant for the single-step conversion of the bound substrate to free Cl− and O2 (equivalent to kcat in this minimal kinetic scheme) was allowed to vary. Values of kcat of ≤ 60,000 s−1 gave simulated traces with excessively pronounced lag phases and insufficiently rapid rises in A390 compared to the experimental traces (panels A and B). Values of kcat ≥ 120,000 s–1 gave more acceptable agreement (panels C and D), and are consistent with those measured in the steady state at pH 7 and below. This extremely high turnover rate confirms that it should be possible to generate a > 5 mM “pulse” of O2 in < 1 ms at the easily accessible [Cld] of 50 μM.
Verification of High O2 Yield from the ClO2−/Cld System by Monitoring Conversion of FeII-myoglobin to Oxy-myoglobin
To demonstrate the potential of using Cld to generate O2 adducts at high concentration, we selected myoglobin (Mb) as a very soluble and efficient O2 receptor for which we could quantify the extent of reaction by Mössbauer spectroscopy (64). Horse heart Mb was enriched to ~25% with 57Fe. FeII-Mb was prepared by titration of the FeIII-Mb with stoichiometric sodium dithionite. In order to preclude prior redox equilibration of the concentrated FeII-Mb reactant with the dilute oxidized (FeIII) Cld catalyst (which could inactivate the Cld), the reaction was carried out by a sequential-mixing protocol. The FeII-Mb and Cld solutions were mixed first. After passage through a short connecting hose, this solution was mixed with the ClO2− solution, and the complete reaction was then freeze-quenched after ~15 ms. Comparison of the Mössbauer spectra of the FeII-Mb reactant solution (Fig. 3, top) and the freeze-quenched reaction sample (Fig. 3, bottom) reveals essentially quantitative (> 98 %) conversion of the 6.7 mM FeII-Mb to the oxy-form (the arrows indicate the small contribution from the remaining reactant). The spectrum of a control sample in which the FeII-Mb was mixed directly with the ClO2− solution (i.e., from which the Cld was omitted) reflects conversion of only (7 ± 3) % of the FeII-Mb to oxy-Mb (Fig. S3), establishing that the conversion observed in the complete reaction results from Cld-catalyzed evolution of O2. This ~7% conversion in the control sample could reflect either contact with atmospheric O2 during the mixing and freeze-quenching procedures or the accumulation of O2 in the ClO2− reactant solution as a result of a slow spontaneous breakdown process (65). Regardless, the spectrum of the complete reaction sample indicates that the 25 μM Cld catalyst produced a minimum of 6.5 mM O2 in ~ 15 ms. Thus, the Cld/ClO2− system appears to be capable of effectively eliminating O2 solubility as an obstacle for the preparation of O2-dependent reactive intermediates at high concentrations and purities.
Figure 3.
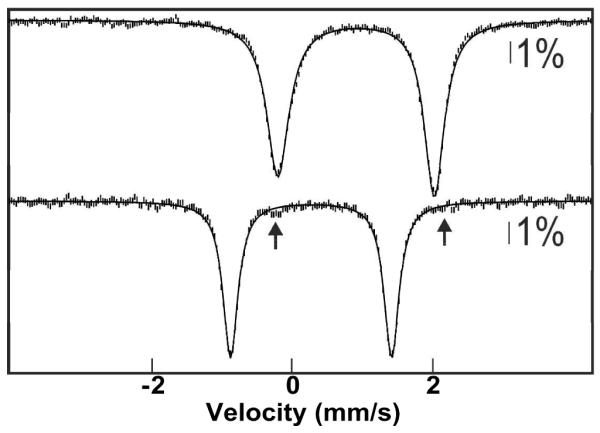
4.2-K/53-mT (parallel field) Mössbauer spectra demonstrating conversion of ferrous Mb to oxy-Mb by the Cld/ClO2− system. A solution of 10 mM Mb (2.5 mM 57Fe-Mb, 7.5 mM 56Fe-Mb) was reduced with stoichiometric sodium dithionite (top spectrum). The FeII-Mb reactant was mixed with 0.25 equivalent volumes of 0.125 mM Cld, this solution was mixed with 0.2 equivalent volumes of 100 mM ClO2−, and the complete reaction was freeze-quenched after 15 ms (bottom spectrum). The solid lines are quadrupole doublet simulations with parameters nearly identical to those previously published (64): δ = 0.91 mm/s; ΔEQ = 2.23 mm/s (top) and δ = 0.27 mm/s; ΔEQ = 2.29 mm/s (bottom).
Preparation of the Ferryl Intermediate, J, in Ec TauD at Unprecedented Concentration
As an additional demonstration of the utility of the approach for preparing O2-dependent reactive intermediates, we targeted the high-spin FeIV-oxo (ferryl) intermediate, J, which accumulates during O2 activation by Ec TauD. J cleaves the C1-H bond of the substrate, taurine, with a rate constant of 13 s−1 at 5 °C and is stabilized significantly (kdecay = 0.35 s−1) by inclusion of the deuterium-containing substrate, owing to a large deuterium kinetic isotope effect (33, 66). Even with this increased half-life of ~ 2 s, the complex is still sufficiently short-lived that it must be prepared by rapid-mixing methods. Thus, although J has been prepared in high purity (~80%) and interrogated by several spectroscopic methods (29, 67, 68), the maximum concentration of ~0.95 mM that has been obtained has precluded application of methods that require very high purity and concentration (e.g. NRVS). The ability to make more concentrated samples would make application of these methods feasible and afford the opportunity for further insight into the structure of J.
The Mössbauer spectrum of a sample prepared by mixing a solution containing the Ec TauD•FeII•αKG•d4-taurine complex (6.0 mM TauD, 4.8 mM 57FeII, 10 mM αKG, and 10 mM d4-taurine) with 0.25 equivalent volumes of 120 μM Cld, mixing the resultant solution with 0.2 volumes of a solution of 100 mM ClO2−, and then freeze-quenching the complete reaction after 0.03 s (Fig. 4, vertical bars) is dominated by the sharp quadrupole doublet of J (blue line plotted above the data, accounting for 77% of the total absorption area of the experimental spectrum). The contribution of the spectrum of J corresponds to a concentration of 2.5 mM. Thus, the Cld/ClO2− system supports preparation of the intermediate at more than twice the maximum concentration achieved in previous studies and at comparable purity (29).
Figure 4.

4.2-K/53-mT (parallel field) Mössbauer spectrum of a freeze-quenched sample from the reaction of the TauD•FeII•αKG•d4-taurine complex with the Cld/ClO2− system (see text for details). The red line is a simulation of the unreacted ferrous component (23%), the blue line is a simulation of the quadrupole doublet spectrum of J (parameters: δ = 0.29 mm/s and ∣ΔEQ∣ = 0.90 mm/s) accounting for 77% of the total intensity of the spectrum, and the solid black line is the summed contribution of both.
Preparation of the Fe2III/IV Intermediate, X, of Ec RNR at Unprecedented Concentration
As an additional demonstration of the utility of the approach for preparing O2-dependent reactive intermediates, we targeted the Fe2III/IV complex, X, that accumulates during activation of class Ia RNRs, including the most extensively studied ortholog from Ec (27, 28, 35, 45, 69-72). X oxidizes a conserved tyrosine residue by one electron to a tyrosyl radical [which is essential for the activity of these RNRs (73)] as the diiron cluster is reduced to the μ-oxo-Fe2III/III product (5, 35, 71). Previous studies established that the complex has a half-life of ~ 1 s at 5 °C and can be stabilized by ~ 5-fold by substitution of the tyrosine that it oxidizes (Y122) with a redox inert phenylalanine (35, 71). Even with this increased lifetime in the Y122F variant, the complex is still sufficiently short-lived that it must be prepared by rapid-mixing methods. As a result, the best samples yet reported have had ≤ 0.77 mM at a purity of ≤ 68% (45). Characterization of these optimized samples by EXAFS spectroscopy resulted in the report of an Fe–Fe distance of 2.5 Å (45), much shorter than for currently favored structural models (27, 28, 69). The availability of more concentrated or purer (or both) samples would motivate re-examination of this crucial structural metric either to confirm it with renewed confidence or to revise it upward to a distance more compatible with structural models.
The Mössbauer spectrum of a sample prepared by mixing a solution containing Ec β2-Y122F (2.6 mM dimer), 7.41 mM 57FeII, 10 mM ascorbate and 12.5 μM Cld with 0.25 equivalent volumes of 80 mM ClO2− and freeze-quenching after 0.30 s (Fig. 5, vertical bars) is dominated by the magnetic features of X. The solid line plotted over the data is the theoretical spectrum of X [generated with published parameters (72)] plotted at 70% of the total absorption area of the experimental spectrum. This contribution corresponds to [X] = 2.0 mM. Thus, the Cld/ClO2− system supports preparation of the intermediate at more than twice the concentration achieved in previous studies and at comparable purity.
Figure 5.

4.2-K/53-mT (parallel field) Mössbauer spectrum showing accumulation of the Fe2III/IV activation intermediate, X, in Ec RNR-β2-Y122F mediated by the Cld/ClO2− system. A solution containing 2.6 mM Ec β2-Y122F dimer, 7.41 mM 57FeII, 10 mM ascorbate and 12.5 μM Cld was mixed with 0.25 equivalent volumes of 50 mM ClO2−, and the reaction was freeze-quenched after 0.30 s. The solid line is the theoretical spectrum of X generated with published parameters (72) and plotted at 70% (2.0 mM) of the total absorption area of the experimental spectrum. The remaining contributions from the Fe2II/II (red) and Fe2III/III (blue) complexes represent 15% (each) of the total absorption area.
Use of the Cld/ClO2− System to Drive the Reversible O2-Addition Step Generating the Superoxo-Fe2III/III Complex, G, in MIOX
A growing number of O2-utilizing non-heme metalloenzymes are thought to employ mid-valent metal-superoxo complexes, formed by the one-electron oxidative addition of O2 to the reduced cofactors, to cleave C–H or C–C bonds (or both) (18, 20, 21, 44, 74-76). For only two such cases have the postulated superoxo complexes been directly detected (43, 77). For the unusual di-iron enzyme, myo-inositol (MI) oxygenase (MIOX) (55, 75, 77-79), it was shown that the reversible addition of O2 to the Fe2II/III–MIOX•MI complex results in formation of a putatively superoxo-Fe2III/III complex, G, that cleaves the C1–H bond of MI to initiate its C–C-bond-cleaving four-electron oxidation to D-glucuronate (77). It was further shown that, even with the C–H cleavage step slowed by use of d6-MI, the two pathways for decay of G (forward by C1–2H cleavage at ~ 48 s−1 and backward by reductive elimination of O2 at ~ 40 s−1) conspire to make the net rate constant for its breakdown comparable to the effective first-order rate constant for its formation at the maximum accessible [O2] of ~ 1 mM (k ~ 95 mM−1s−1 giving kobs ~ 95 s−1), thereby limiting its maximum accumulation to ~ 40% of the initial concentration of the reactant complex (77). The inherent limitations on the concentration and purity of G imposed by the facile C–2H-cleavage and O2-dissociation steps are largely responsible for the fact that its characterization has not progressed beyond the observation and analysis of its characteristic rhombic g = (2.06, 1.98, 1.92) EPR spectrum and 57Fe nuclear hyperfine coupling thereupon. Moreover, it is likely that, for other enzyme reactions involving mid-valent metal-superoxide complexes, high O2-dissociation rates (perhaps in combination with efficient forward conversion steps) are responsible for having prevented the complexes from accumulating even to detectable levels.
To test whether the approach can be used to overcome a reversible O2-addition step for the specific case of MIOX, thereby allowing greater accumulation of G, a solution containing the Fe2II/III–MIOX•MI complex was reacted with the Cld/ClO2− (20 μM/16 mM) system for the minimum accessible reaction time (transit time 3 ms, total reaction time ~ 10 ms) before being freeze-quenched. Comparison of the EPR spectrum of this sample (Fig. 6, red spectrum) to that of a control sample prepared by mixing the complex with two equivalent volumes O2-saturated buffer (giving [O2] of 1 mM) and freeze-quenching at the same reaction time (Fig. 6, green spectrum) shows that the Cld/ClO2− system does permit G to be accumulated to a greater extent: the resolved g = 2.06 and 1.92 features (green and black arrows) are ~ 2-fold more intense in the spectrum of this sample than in that of the control. Moreover, the hallmark of reversible and disfavored O2 addition, a high level of residual Fe2II/III–MIOX•MI complex (~ 50% of the initial) remaining after accumulation of G to its maximum extent, is quite evident in the spectrum of the control sample (features at g = 1.95 and 1.81; see blue spectrum for their positions), but the associated signal is greatly diminished (by 15-fold) in the spectrum of the Cld/ClO2− sample. Both observations are consistent with the expected increase in [O2] from ~ 2.5 times to ~ 25 times the KD in G (0.4 mM) and the associated enhanced kinetic resolution of the formation and decay of the intermediate. Interestingly, a prominent g = 2.0 signal corresponding to ~ 80 μM total spin is also observed in the spectrum of the Cld/ClO2− sample (red arrow). Although much less intense, this signal is, nevertheless, still evident in the spectrum of the (−ClO2−/+O2−) control sample and in that of an additional control prepared with omission also of Cld. The g = 2.0 signal is thus associated with the MIOX reaction rather than the Cld/ClO2− reaction and could reflect accumulation of a substrate-based radical. Whether this previously undetected radical is an on-pathway intermediate that accumulates to a greater extent as a consequence of the greater accumulation of G or is an off-pathway species that forms in a side reaction that is favored by very high [O2] will require more extensive kinetic studies to correlate the changes observed by EPR with formation of MI-derived intermediates and products. Irrespective of the answer, the greater accumulation of G and diminution of residual Fe2II/III–MIOX•MI reactant should facilitate further characterization of G. More generally, the results illustrate the capacity of the Cld/ClO2− system to overcome the obstacle presented by an O2-addition equilibrium that is unfavorable at the [O2] accessible by conventional methods and equipment.
Figure 6.
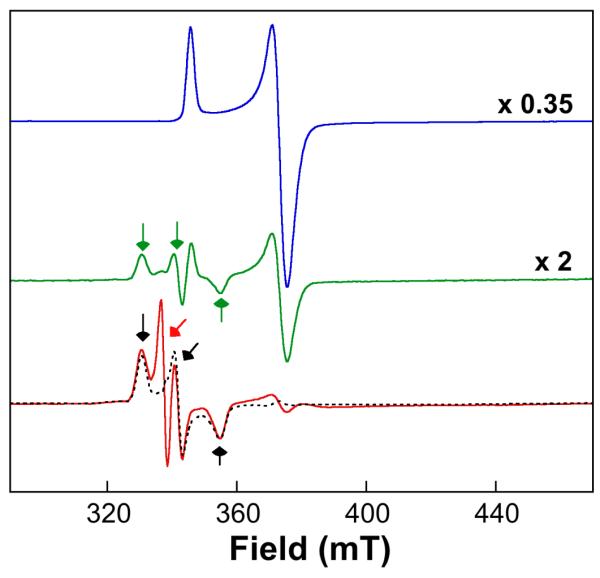
EPR spectra showing accumulation of the C1-H-abstracting superoxo-Fe2III/III intermediate, G, in the MIOX reaction with d6-MI initiated by the Cld/ClO2− system. A solution containing 1.13 mM Fe2II/III-MIOX (3 mM total MIOX protein), 60 mM d6-MI (blue spectrum), and 30 μM Cld was mixed either with two equivalent volumes of O2-saturated 50 mM Bis-Tris chloride (pH 6.0) buffer (green spectrum) or with 0.5 equivalent volumes of 48 mM ClO2− solution (red spectrum), and the reaction was freeze-quenched at a reaction time of ~ 10 ms. The spectrometer conditions were: 9.5 GHz microwave frequency; 100 KHz modulation frequency; 10 G modulation amplitude; 10 ± 0.2 K temperature; 100 W power; 0.167 s time constant; 10 scans per spectrum. The spectra were scaled as indicated to the right to account for dilution and “packing factor” (the fraction of the sample not contributed by the cryosolvent). The black, dashed line overlaid with the red spectrum is the published spectrum of G, acquired under similar spectrometer conditions. The green and black arrows indicate the g-values of G (2.05, 1.98, 1.91), whereas the red arrow indicates the organic radical signal present in the spectra of both reaction samples but featured more prominently in the Cld/ClO2− sample.
DISCUSSION
The Cld/ClO2− system can be used for rapid generation of concentrations of O2 exceeding the normally achievable 2 mM to drive accumulation of metalloenzyme intermediates and surmount reversible and disfavored equilibria in the O2-addition steps that initiate the reactions of some enzymes in this class. In addition to allowing for preparation of intermediate complexes that have already been identified, such as the MnIV/FeIV activation intermediate in Ct RNR and the H•-abstracting ferryl and superoxo-Fe2III/III complexes in TauD and MIOX, respectively, at concentrations and purities required for well-established and developing approaches to structural characterization (e.g., EXAFS and NRVS), this method could permit identification of previously undetected precursors to known complexes, thus further resolving the complex reaction pathways of these enzymes. For example, the reactions of the α-KG-dependent oxygenases and the pterin-dependent aromatic amino acid hydroxylases are known to proceed through ferryl complexes that form rapidly without demonstrated accumulation of precursor complexes (17). In the former enzymes, ferryl formation involves addition of O2, cleavage of both the O–O bond of O2 and the C1–C2 bond of αKG, and formation of a new C2–O bond in the succinate co-product. Similarly, in the pterin-dependent enzymes, ferryl formation requires O2 addition, O–O-bond cleavage, and formation of a new C4a–O bond to the pterin. The kinetic masking of precursors leaves the pathways to ferryl formation experimentally unresolved. With [O2] greater by as much as ten-fold, addition of O2 should occur ten times more rapidly, perhaps permitting accumulation and identification of ferryl precursors. Alternatively, the failure of precursors to accumulate might reflect the reversible addition of O2 to produce adducts with relatively high dissociation constants (>> 1 mM). This situation is also likely in cases for which the initial adducts are proposed to effect difficult H•-abstraction steps, as in MIOX. Here again, the ability to access ~ 10 mM O2 could permit this obstacle to be overcome. In many characterized reactions, including those of the α-ketoglutarate(αKG)-dependent oxygenases, acceleration of the initial step would necessarily move the reaction times at which precursors accumulate to << 10 ms, a regime that is inaccessible by conventional cryosolvent-based freeze-quenching. In these cases, the recently developed “microsecond freeze-hyperquenching” technique (80) should permit the reactions to be terminated at these shorter times.
Two crucial requirements for the successful application of the system are the extraordinarily high efficiency of the Cld catalyst and the modest reactivities of target enzymes to ClO2−. These characteristics combine to ensure that the vastly predominant pathway is evolution of O2 by the Cld catalyst and then reaction of O2 with the target enzyme. High efficiency of the Cld also requires that it not be strongly inhibited by components of the target enzyme reaction, such as by reduction or coordination to its FeIII-heme cofactor (53). The potential for this complication is minimized by a sequential mixing protocol, which ensures that the Cld catalyst is exposed to the components of the target enzyme reaction for only a few ms before the Cld is exposed to its substrate. In cases for which the target enzyme is more reactive to ClO2− or components of the reaction inhibit Cld, it may be important to increase [Cld] to maintain the homogeneity of the reaction pathway. Additionally, the catalyst concentration must be elevated in the presence of very high [ClO2−] to keep the ClO2−/heme ratio < ~1,500 in order to avoid deleterious competition between chlorite-mediated degradation of the Cld heme and generation of O2. This is likely to be an issue primarily when delivery of exact quantities of O2 is desirable (for example, in kinetic studies). The Da Cld used herein is soluble to concentrations of at least 500 μM, and so, even with use of high target-enzyme/Cld volume ratios (e.g., 4:1 in the Mb experiment) to minimize dilution of the target enzyme, Cld concentrations of > 100 μM are readily accessible. Given estimates of the kcat of Cld (> 100,000 s−1) and the reasonably low KM for ClO2− of 215 μM, this [Cld] is theoretically capable of generating a 10 mM pulse of O2 in 1 ms. With these impressive parameters, it seems likely that the system will be robust and widely applicable.
Supplementary Material
ACKNOWLEDGMENTS
This paper is dedicated to our friend Vincent Huynh, with whom we have often agonized over the low solubility of O2.
Footnotes
SUPPORTING INFORMATION AVAILABLE Figure showing kobs for formation of the Ct RNR-β2 MnIV/FeIV intermediate as a function of [ClO2−] in three different experiments at two different Cld concentrations; simulations of the stopped-flow kinetic traces reflecting formation and decay of the MnIV/FeIV intermediate; tables summarizing analysis of Mössbauer and EPR spectra for quantification of the MnIV/FeIV intermediate in freeze-quenched samples; Mössbauer spectrum of a control freeze-quenched sample prepared by mixing FeII-Mb with ClO2− in the absence of Cld. This material is available free of charge at the journal website http://pubs.acs.org.
This work was supported by the National Institutes of Health (GM-55365 to JMB, CK, and MTG, DK-74641 to JMB and CK, and GM-090260 to JLD) and the Alfred P. Sloan Foundation Minority PhD Scholarship Program (to LMKD).
ABBREVIATIONS: RNR, ribonucleotide reductase; Ct, Chlamydia trachomatis; Ec, Escherichia coli; Da, Dechloromonas aromatica; TauD, taurine: α-ketoglutarate dioxygenase; α-KG, α-ketoglutarate; MIOX, myo-inositol oxygenase; DFT, density functional theory; EXAFS, extended X-ray absorption fine structure; NRVS, nuclear resonance vibrational spectroscopy; Cld, chlorite dismutase; EPR, electron paramagnetic resonance; d4-taurine, 1,1,2,2-[2H4]-2-aminoethane-1-sulfonic acid; MI, myo-inositol or cyclohexan-(1,2,3,5/4,6)-hexa-ol; d6-MI, 1,2,3,4,5,6-[2H6]-cyclohexan-(1,2,3,5/4,6)-hexa-ol.
REFERENCES
- 1.Miller S. Ferguson, Babcock GT. Heme/copper terminal oxidases. Chem. Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 3.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 4.Hausinger RP. Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 5.Atkin CL, Thelander L, Reichard P, Lang G. Iron and free radical in ribonucleotide reductase. Exchange of iron and Mössbauer spectroscopy of the protein B2 subunit of the Escherichia coli enzyme. J. Biol. Chem. 1973;248:7464–7472. [PubMed] [Google Scholar]
- 6.Klinman JP, Mu D. Quinoenzymes in biology. Annu. Rev. Biochem. 1994;63:299–344. doi: 10.1146/annurev.bi.63.070194.001503. [DOI] [PubMed] [Google Scholar]
- 7.Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak M-A, Krebs C, Bollinger JM., Jr A manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Science. 2007;316:1188–1191. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]
- 8.Fitzpatrick PF. Mechanism of aromatic amino acid hydroxylation. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kershaw NJ, Caines MEC, Sleeman MC, Schofield CJ. The enzymology of clavam and carbapenem biosynthesis. Chem. Comm. 2005:4251–4263. doi: 10.1039/b505964j. [DOI] [PubMed] [Google Scholar]
- 10.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 11.Jaakkola P, Mole DR, Tian Y-M, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIFα to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 12.Tsukada Y.-i., Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 13.Cloos PAC, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 14.Liu XF, Theil EC. Ferritins: Dynamic management of biological iron and oxygen chemistry. Acc. Chem. Res. 2005;38:167–175. doi: 10.1021/ar0302336. [DOI] [PubMed] [Google Scholar]
- 15.Baik M-H, Martin N, Friesner RA, Lippard SJ. Mechanistic studies on the hydroxylation of methane by methane monooxygenase. Chem. Rev. 2003;103:2385–2419. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- 16.Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 17.Krebs C, Fujimori D. Galonić, Walsh CT, Bollinger JM., Jr Non-heme Fe(IV)-oxo intermediates. Acc. Chem. Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Finding intermediates in the O2 activation pathways of non-heme iron oxygenases. Acc. Chem. Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Solomon EI, Sundaram UM, Machonkin TE. Multicopper oxidases and oxygenases. Chem. Rev. 1996;96:2563–2605. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 20.Chen P, Solomon EI. Oxygen activation by the noncoupled binuclear copper site in peptidylglycine α-hydroxylating monooxygenase. Reaction mechanism and role of the noncoupled nature of the active site. J. Am. Chem. Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 21.Klinman JP. The copper-enzyme family of dopamine beta-monooxygenase and peptidylglycine alpha-hydroxylating monooxygenase: resolving the chemical pathway for substrate hydroxylation. J. Biol. Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 22.Massey V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 23.Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovaleva EG, Lipscomb JD. Intermediate in the O-O bond cleavage reaction of an extradiol dioxygenase. Biochemistry. 2008;47:11168–11170. doi: 10.1021/bi801459q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Crystal Structure of Naphthalene Dioxygenase: Side-on Binding of Dioxygen to Iron. Science. 2003;299:1039–1042. doi: 10.1126/science.1078020. [DOI] [PubMed] [Google Scholar]
- 26.Neese F. A critical evaluation of DFT, including time-dependent DFT, applied to bioinorganic chemistry. J. Biol. Inorg. Chem. 2006;11:702–711. doi: 10.1007/s00775-006-0138-1. [DOI] [PubMed] [Google Scholar]
- 27.Han WG, Liu TQ, Lovell T, Noodleman L. Active site structure of class I ribonucleotide reductase intermediate X: A density functional theory analysis of structure, energetics, and spectroscopy. J. Am. Chem. Soc. 2005;127:15778–15790. doi: 10.1021/ja050904q. [DOI] [PubMed] [Google Scholar]
- 28.Mitić N, Clay MD, Saleh L, Bollinger JM, Jr., Solomon EI. Spectroscopic and electronic structure studies of intermediate X in ribonucleotide reductase R2 and two variants: a description of the FeIV-oxo bond in the FeIII-O-FeIV dimer. J. Am. Chem. Soc. 2007;129:9049–9065. doi: 10.1021/ja070909i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinnecker S, Svensen N, Barr EW, Ye S, Bollinger JM, Jr., Neese F, Krebs C. Spectroscopic and computational evaluation of the structure of the high-spin Fe(IV)-oxo intermediates in taurine:α-ketoglutarate dioxygenase from Escherichia coli and its His99Ala ligand variant. J. Am. Chem. Soc. 2007;129:6168–6179. doi: 10.1021/ja067899q. [DOI] [PubMed] [Google Scholar]
- 30.Hitchman ML. Measurement of Dissolved Oxygen. Vol. 49. Wiley; New York: 1978. [Google Scholar]
- 31.Baldwin J, Krebs C, Ley BA, Edmondson DE, Huynh BH, Bollinger JM., Jr Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 1. Evidence for a transient tryptophan radical. J. Am. Chem. Soc. 2000;122:12195–12206. [Google Scholar]
- 32.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM., Jr Kinetic dissection of the catalytic mechanism of taurine:α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2005;44:8138–8147. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 33.Bollinger JM, Jr., Krebs C. Stalking intermediates in oxygen activation by iron enzymes: Motivation and method. Journal of Inorganic Biochemistry. 2006;100:586–605. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 34.Jiang W, Hoffart LM, Krebs C, Bollinger JM., Jr A manganese(IV)/iron(IV) intermediate in assembly of the manganese(IV)/iron(III) cofactor of Chlamydia trachomatis ribonucleotide reductase. Biochemistry. 2007;46:8709–8716. doi: 10.1021/bi700906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bollinger JM, Jr., Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase. Science. 1991;253:292–298. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- 36.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr., Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 37.Galonić DP, Barr EW, Walsh CT, Bollinger JM, Jr., Krebs C. Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nature Chem. Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 38.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr., Krebs C. A long-lived, substrate-hydroxylating peroxodiiron(III/III) intermediate in the amine oxygenase, AurF, from Streptomyces thioluteus. J. Am. Chem. Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Substrate-triggered formation and remarkable stability of the C-H bond-cleaving chloroferryl intermediate in the aliphatic halogenase, SyrB2. Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rittle J, Green MT. Cytochrome P450 compound I: Capture, characterization, and C-H bond activation kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 41.Bollinger JM, Jr., Krebs C, Vicol A, Chen S, Ley BA, Edmondson DE, Huynh BH. Engineering the diiron site of Escherichia coli ribonucleotide reductase protein R2 to accumulate an intermediate similar to Hperoxo, the putative peroxodiiron(III) complex from the methane monooxygenase catalytic cycle. J. Am. Chem. Soc. 1998;120:1094–1095. [Google Scholar]
- 42.Saleh L, Krebs C, Ley BA, Naik S, Huynh BH, Bollinger JM., Jr Use of a chemical trigger for electron transfer to characterize a precursor to cluster X in assembly of the iron-radical cofactor of Escherichia coli ribonucleotide reductase. Biochemistry. 2004;43:5953–5964. doi: 10.1021/bi036099e. [DOI] [PubMed] [Google Scholar]
- 43.Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, Lipscomb JD. Trapping and spectroscopic characterization of an Fe(III)-superoxo intermediate from a nonheme mononuclear iron-containing enzyme. Proc. Natl. Acad. Sci. U. S. A. 2010;107:16788–16793. doi: 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Donk WA, Krebs C, Bollinger JM. Substrate activation by iron superoxo intermediates. Curr. Opin. Struct. Biol. 2010;20:673–683. doi: 10.1016/j.sbi.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riggs-Gelasco PJ, Shu L, Chen S, Burdi D, Huynh BH, Que L, Jr., Stubbe J. EXAFS characterization of the intermediate X generated during the assembly of the Escherichia coli ribonucleotide reductase R2 diferric tyrosyl radical cofactor. J. Am. Chem. Soc. 1998;120:849–860. [Google Scholar]
- 46.Shu L, Nesheim JC, Kauffmann KE, Münck E, Lipscomb JD, Que L., Jr An Fe2IVO2 diamond core structure for the key intermediate Q of methane monooxygenase. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 47.Gherman BF, Baik M-H, Lippard SJ, Friesner RA. Dioxygen activation in methane monooxygenase: A theoretical study. J. Am. Chem. Soc. 2004;126:2978–2990. doi: 10.1021/ja036506+. [DOI] [PubMed] [Google Scholar]
- 48.Hsu HF, Dong YH, Shu LJ, Young VG, Que L. Crystal structure of a synthetic high-valent complex with an Fe2(μ-O)2 diamond core. Implications for the core structures of methane monooxygenase intermediate Q and ribonucleotide reductase intermediate X. J. Am. Chem. Soc. 1999;121:5230–5237. [Google Scholar]
- 49.Xue GQ, Wang D, De Hont R, Fiedler AT, Shan XP, Munckt E, Que L. A synthetic precedent for the [Fe2(IV)( μ-O)2] diamond core proposed for methane monooxygenase intermediate Q. Proc. Natl. Acad. Sci. U. S. A. 2007;104:20713–20718. doi: 10.1073/pnas.0708516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bell CB, Wong SD, Xiao YM, Klinker EJ, Tenderholt AL, Smith MC, Rohde JU, Que L, Cramer SP, Solomon EI. A combined NRVS and DFT study of Fe-IV=O model complexes: A diagnostic method for the elucidation of non-heme iron enzyme intermediates. Angew. Chem.-Int. Edit. 2008;47:9071–9074. doi: 10.1002/anie.200803740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Streit BR, DuBois JL. Chemical and steady-state kinetic analyses of a heterologously expressed heme dependent chlorite dismutase. Biochemistry. 2008;47:5271–5280. doi: 10.1021/bi800163x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coates JD, Achenbach LA. Microbial perchlorate reduction: Rocket-fuelled metabolism. Nat. Rev. Microbiol. 2004;2:569–580. doi: 10.1038/nrmicro926. [DOI] [PubMed] [Google Scholar]
- 53.Streit BR, Blanc B, Lukat-Rodgers GS, Rodgers KR, DuBois JL. How Active-Site Protonation State Influences the Reactivity and Ligation of the Heme in Chlorite Dismutase. J. Am. Chem. Soc. 2010;132:5711–5724. doi: 10.1021/ja9082182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goblirsch B, Kurker RC, Streit BR, Wilmot CM, DuBois JL. Chlorite Dismutases, DyPs, and EfeB: 3 Microbial Heme Enzyme Families Comprise the CDE Structural Superfamily. J. Mol. Biol. 2011;408:379–398. doi: 10.1016/j.jmb.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xing G, Barr EW, Diao Y, Hoffart LM, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr Oxygen activation by a mixed-valent, diiron(II/III) cluster in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. Biochemistry. 2006;45:5402–5412. doi: 10.1021/bi0526276. [DOI] [PubMed] [Google Scholar]
- 56.Adler AD, Longo FR, Kampas F, Kim J. On the preparation of metalloporphyrins. J. Inorg. Nucleic Chem. 1970;32:2443–2445. [Google Scholar]
- 57.Teal FW. Cleavage of Haem-Protein Link by Acid Methylketone. Biochem. Biphys. Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]
- 58.Wagner G, Perez M, Toscano WA, Gunsalus I. Apoprotein Formation and Heme Reconstitution of Cytochrome P450cam. J. Biol. Chem. 1981;256:6262–6265. [PubMed] [Google Scholar]
- 59.Aasa R, Vänngård T. EPR signal intensity and powder shapes: A reexamination. Journal of Magnetic Resonance. 1975;19:308–315. [Google Scholar]
- 60.Jiang W, Xie J, Nørgaard H, Bollinger JM, Jr., Krebs C. Rapid and quantitative activation of Chlamydia trachomatis ribonucleotide reductase by hydrogen peroxide. Biochemistry. 2008;47:4477–4483. doi: 10.1021/bi702085z. [DOI] [PubMed] [Google Scholar]
- 61.Bollinger JM, Jr., Jiang W, Green MT, Krebs C. The manganese(IV)/iron(III) cofactor of Chlamydia trachomatis ribonucleotide reductase: structure, assembly, radical initiation, and evolution. Curr. Opin. Struct. Biol. 2008;18:650–657. doi: 10.1016/j.sbi.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 62.Jiang W, Bollinger JM, Jr., Krebs C. The active form of Chlamydia trachomatis ribonucleotide reductase R2 protein contains a heterodinuclear Mn(IV)/Fe(III) cluster with S = 1 ground state. J. Am. Chem. Soc. 2007;129:7504–7505. doi: 10.1021/ja072528a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang W, Saleh L, Barr EW, Xie J, Gardner MM, Krebs C, Bollinger JM., Jr Branched activation- and catalysis-specific pathways for electron relay to the manganese/iron cofactor in ribonucleotide reductase from Chlamydia trachomatis. Biochemistry. 2008:8477–8484. doi: 10.1021/bi800881m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Debrunner PG. Mössbauer spectroscopy of iron porphyrins. (Series 4).Physical Bioinorganic Chemistry. 1989:137–234. [Google Scholar]
- 65.Greenwood NN, Earnshaw A. Chemistry of the elements. 2nd ed Butterworth Heinemann; Oxford, U.K.: 1997. [Google Scholar]
- 66.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr Evidence for hydrogen abstraction from C1 of taurine by the high-spin Fe(IV) intermediate detected during oxygen activation by taurine:α-ketoglutarate dioxygenase (TauD) J. Am. Chem. Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 67.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. Direct detection of oxygen intermediates in the non-heme Fe enzyme taurine/α-ketoglutarate dioxygenase. J. Am. Chem. Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 68.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Jr., Krebs C. EXAFS spectroscopic evidence for an Fe=O unit in the Fe(IV) intermediate observed during oxygen activation by taurine:α-ketoglutarate dioxygenase. J. Am. Chem. Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 69.Shanmugam M, Doan PE, Lees NS, Stubbe J, Hoffman BM. Indentification of Protonated Oxygenic Ligands of Ribonucleotide Reductase Intermediate X. J. Am. Chem. Soc. 2009;131:3370–3376. doi: 10.1021/ja809223s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stubbe J. Di-iron-tyrosyl radical ribonucleotide reductases. Curr. Opin. Chem. Biol. 2003;7:183–188. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 71.Ravi N, Bollinger JM, Jr., Huynh BH, Edmondson DE, Stubbe J. Mechanism of assembly of the tyrosyl radical-diiron(III) cofactor of E.coli ribonucleotide reductase. 1. Mössbauer characterization of the diferric radical precursor. J. Am. Chem. Soc. 1994;116:8007–8014. [Google Scholar]
- 72.Sturgeon BE, Burdi D, Chen S, Huynh BH, Edmondson DE, Stubbe J, Hoffman BM. Reconsideration of X, the diiron intermediate formed during cofactor assembly in E. coli ribonucleotide reductase. J. Am. Chem. Soc. 1996;118:7551–7557. [Google Scholar]
- 73.Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 74.Bollinger JM, Jr., Krebs C. Enzymatic C-H activation by metal-superoxo intermediates. Curr. Opin. Chem. Biol. 2007;11:151–158. doi: 10.1016/j.cbpa.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 75.Bollinger JM, Jr., Diao Y, Matthews ML, Xing G, Krebs C. myo-Inositol oxygenase: A radical new pathway for O2 and C-H activation at a nonheme diiron cluster. Dalton Trans. 2009;6:905–914. doi: 10.1039/b811885j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cicchillo RM, Zhang HJ, Blodgett JAV, Whitteck JT, Li GY, Nair SK, van der Donk WA, Metcalf WW. An unusual carbon-carbon bond cleavage reaction during phosphinothricin biosynthesis. Nature. 2009;459:871–U810. doi: 10.1038/nature07972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr Evidence for C-H cleavage by an iron-superoxide complex in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. Proc. Natl. Acad. Sci., U.S.A. 2006;103:6130–6135. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Charalampous FC, Lyras C. Biochemical studies on inositol. IV. Conversion of inositol to glucuronic acid by rat kidney extracts. J. Biol. Chem. 1957;228:1–13. [PubMed] [Google Scholar]
- 79.Xing G, Hoffart LM, Diao Y, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr A coupled dinuclear iron cluster that is perturbed by substrate binding in myo-inositol oxygenase. Biochemistry. 2006;45:5393–5401. doi: 10.1021/bi0519607. [DOI] [PubMed] [Google Scholar]
- 80.Cherepanov AV, de Vries S. Microsecond freeze-hyperquenching: development of a new ultrafast micro-mixing and sampling technology and application to enzyme catalysis. Biochim. Biophys. Acta-Bioenerg. 2004;1656:1–31. doi: 10.1016/j.bbabio.2004.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.