

Abstract
The feasibility of using technologies based on site-specific recombination in actinomycetes was shown several years ago. Despite their huge potential, these technologies mostly have been used for simple marker removal from a chromosome. In this paper, we present different site-specific recombination strategies for genome engineering in several actinomycetes belonging to the genera Streptomyces, Micromonospora, and Saccharothrix. Two different systems based on Cre/loxP and Dre/rox have been utilized for numerous applications. The activity of the Cre recombinase on the heterospecific loxLE and loxRE sites was similar to its activity on wild-type loxP sites. Moreover, an apramycin resistance marker flanked by the loxLERE sites was eliminated from the Streptomyces coelicolor M145 genome at a surprisingly high frequency (80%) compared to other bacteria. A synthetic gene encoding the Dre recombinase was constructed and successfully expressed in actinomycetes. We developed a marker-free expression method based on the combination of phage integration systems and site-specific recombinases. The Cre recombinase has been used in the deletion of huge genomic regions, including the phenalinolactone, monensin, and lipomycin biosynthetic gene clusters from Streptomyces sp. strain Tü6071, Streptomyces cinnamonensis A519, and Streptomyces aureofaciens Tü117, respectively. Finally, we also demonstrated the site-specific integration of plasmid and cosmid DNA into the chromosome of actinomycetes catalyzed by the Cre recombinase. We anticipate that the strategies presented here will be used extensively to study the genetics of actinomycetes.
INTRODUCTION
Actinomycetes, including the largest genus Streptomyces, are Gram-positive bacteria with a high GC content and a complex life cycle. They are the most productive bacteria with respect to the synthesis of bioactive metabolites used in pharmaceutical and agricultural applications (10). The spread of resistance to known antibiotics confronts scientists with the urgent need to find new active compounds (4, 24). Many natural products are too complex for trivial chemical synthesis. Therefore, metabolic engineering presents an alternative and attractive way to manufacture novel chemical entities. Metabolic engineering relies upon the modification of genes and genomes and thus requires a number of reliable genome modification methods (15). Additionally, the demand for functional gene characterization has increased greatly due to extensive whole-genome sequencing in actinomycetes. Our ability to sequence genomes greatly outpaces our ability to modify them. The genetic manipulation of actinomycetes still represents a serious challenge due to a lack of suitable tools and selection markers. Here, we apply well-established genetic tools from other bacteria for use in actinomycetes. Site-specific recombination (SSR) systems provide powerful and efficient instruments for high-throughput genetic analysis of bacteria in the postgenomic era (19). Site-specific genome recombination systems have been described for a number of bacteria (1, 3, 20, 28). Typical applications of site-specific recombinases include the construction of unmarked mutants (5), targeting of heterologous DNA into the chromosome (27), marker-free expression of foreign genes (12), transient and timed expression of genes (31), deletion of large genomic fragments (4), and in vivo cloning of genomic DNA regions among others (28). Many SSR applications are based on the Cre/loxP system from the P1 phage and the Flp/FRT system from yeast (3, 28). The Cre and Flp proteins are bidirectional tyrosine recombinases catalyzing reciprocal SSR of DNA at 34-bp target sites (loxP and FRT, respectively), which results in either excision or inversion depending on whether the target sites are located as direct or inverted repeats. Both recombinases are relatively context independent since they do not require any host cofactors or accessory proteins (33). Both Flp and Cre recombinases have been successfully expressed in actinomycetes. In 2006, successful expression of the Cre recombinase in Streptomyces coelicolor A3(2) was reported (16). The use of this system was limited, probably because the cre gene was introduced into streptomycetes on the native phage itself, which is not a widely used method of gene expression in streptomycetes. We have subsequently reported the successful expression of two synthetic genes encoding the Cre and Flp recombinases. These constructs were used to delete resistance markers in members of the Streptomyces and Saccharothrix genera (7, 8). The native Flp-encoding gene was also expressed in streptomycetes, though at lower efficiency (40).
In this study, we show that heterotypic lox sites in actinomycetes cannot be used for the construction of multiple mutations, since the Cre recombinase has a high affinity for the doubly mutated loxLERE site. Instead, we have efficiently expressed another recombinase called Dre, which recognizes a different target site called rox and thus provides an additional tool for marker removal. Dre recombinase was first described by Sauer and McDermott (27a) in the P1-like transducing bacteriophage D6 isolated from Salmonella enterica serovar Oranienburg. The genes encoding Dre and Cre recombinases share only 39% sequence similarity. Using transfection of mammalian CHO cells, Sauer and McDermott (27a) could show that Dre recombinase catalyzes site-specific DNA recombination recognizing rox sites, whereas Cre recombinase is not able to recognize rox sites, which are distinct from loxP sites. Using the Cre and Dre recombinases, we developed a marker-free expression system based on the VWB and φC31 integration-proficient vectors from which antibiotic resistance markers, integrase genes, and the remaining plasmid backbone can be evicted after integration of the desired sequences into the chromosome. Additionally, we describe a novel strategy for Cre/loxP-mediated deletion of large genomic fragments, which requires only two single crossovers. The system has been validated by the successful deletion of the three large gene clusters for phenalinolactone, lipomycin, and monensin biosynthesis. Finally, we were able to integrate foreign DNA into the streptomycete chromosome using the Cre recombinase. These methods for genome manipulation further expand the molecular genetics toolbox for actinomycetes and provide new opportunities for functional gene characterization.
MATERIALS AND METHODS
Strains and media.
Streptomyces lividans TK24, Streptomyces sp. strain Tü6071, S. coelicolor M145, Micromonospora sp. strain Tü6368, Streptomyces cinnamonensis A519, and Streptomyces aureofaciens Tü117 (13) were used as hosts for expression of the codon-optimized synthetic cre(a) and dre(a) genes encoding Cre and Dre recombinases, respectively. The Escherichia coli strain DH5α was used for cloning, and the strain ET12567/pUZ8002 was used to drive the conjugative transfer of nonmethylated DNA from E. coli to the actinomycete recipient as previously described (9, 22). Cultivation of the E. coli strains was performed as previously described (26). Streptomyces strains were grown on mannitol soy flour agar (MS agar) plates, yeast extract-agar plates, and ABB13 plates. ABB13 medium contained the following components (in grams per liter): Bacto Soytone, 5; soluble starch, 5; CaCO3, 3; MOPS (morpholinepropanesulfonic acid) buffer, 2.1, and Bacto Agar, 20. After autoclaving, 1.0 ml of a filter-sterilized 1.0% (wt/vol) stock solution of thiamine HCl and 1.0 ml of 1.2% (wt/vol) FeSO4 · 7H2O were added. Liquid growth was performed in tryptone soy broth (TSB) (17). Apramycin, spectinomycin, and hygromycin were added to the medium to final concentrations of 50 μg/ml, 100 μg/ml, and 150 μg/ml, respectively.
Plasmid construction.
The plasmids used in this work are displayed in Table 1. The plasmids were verified by restriction endonuclease mapping.
Table 1.
Plasmids used in this work
| Plasmid | Descriptiona | Reference or source |
|---|---|---|
| pAL1 | Replicative vector for actinomycetes; oriT, pSG5rep, oripUC18, tsr, hph, tipA | 7 |
| pALDRE | pAL1 derivative with the dre gene under a tipA promoter | This work |
| pALCRE | pAL1 derivative with the cre gene under a tipA promoter | This work |
| pUWLoriT | Replicative vector for actinomycetes; pIJ101 replicon, oriT, tsr, bla, ermE | 1 |
| pUWLDRE | pUWLoriT derivative with the dre gene under an ermE promoter | This work |
| pUWLCre | pUWLoriT derivative with the cre gene under an ermE promoter | 8 |
| pINTROX | Integrative vector for actinomycetes; oriT, int, and attP (φC31), aac(3′)IV is flanked by rox sites | This work |
| pIJ2925 | Cloning vector containing bla and lacZ | 14 |
| pIJrAmr | Derivative of pIJ2925 containing aac(3′)IV flanked by two rox sites | This work |
| pIJ773 | pBSK derivative containing aac(3′)IV flanked by two FRT sites | 11 |
| pUC19 | Cloning vector containing bla and lacZ | 38 |
| pMEnd-loxP | pUC19 derivative, oriT, aac(3′)IV, and loxP site | This work |
| pMStart2-loxP-hyg | pUC19 derivative, oriT and aac(3′)IV flanked by two loxP sites, hph | This work |
| pIJloxPbegin | pIJloxP derivative with a fragment homologous to the 5′ end of the α-phenalinolactone cluster | This work |
| pIJloxPend | pIJloxP derivative with a fragment homologous to the 3′ end of the α-lipomycin cluster | This work |
| cosmid 3-1O12 | pOJ436 derived cosmid containing the deleted part of the phenalinolactone gene cluster | 6 |
| pMOD3 | Customised Tn5 transposon construction vector | Epicentre, Madison, WI |
| pMOD3-acc | pMOD3 derivative containing aac(3′)IV | This work |
| pLERE | Cloning vector containing amp, two loxLE sites, and two loxRE sites | Geneart, Regensburg, Germany |
| pINT | pLERE derivative with aac(3′)IV flanked by two loxP sites | This work |
| pINTRE | pLERE derivative with aac(3′)IV flanked by two loxRE sites | This work |
| pINTLE | pLERE derivative with aac(3′)IV flanked by two loxLE sites | This work |
| pINTLERE | pLERE derivative with aac(3′)IV flanked by one loxLE site and one loxRE site | This work |
| pINTLERE2 | pLERE derivative with aac(3′)IV flanked by two loxLE sites and two loxRE sites | This work |
| pIJ2601 | pIJ2925 derivative containing aac(3′)IV and modified lacZ | 30 |
| pIJloxP | pIJ2601 derivative containing aac(3′)IV, modified lacZ and two loxP sites | This work |
| pIJloxP-lipbeg | pIJloxP derivative with a fragment homologous to the 5′ end of the α-lipomycin cluster | This work |
| pBeloBAC2601 | BAC cloning vector containing aac(3′)IV flanked by one loxP site | 30 |
| pBeloBAC2601-lipend | pBeloBAC2601 derivative with a fragment homologous to the 3′ end of the α-lipomycin cluster | This work |
| pSOK804 | Integrative vector for actinomycetes; oriT, int, and attP (VWB), aac(3′)IV | 29 |
| pTOS | pSOK804 derivative; attP flanked by rox sites | This work |
| pSET152 | Integrative vector for actinomycetes; oriT, int, and attP (φC31), aac(3′)IV | 1 |
| pTES | pSET152 derivative; attP flanked by loxP sites, ermE promoter flanked by tfd terminator sequences | This work |
aac(3′)IV, apramycin resistance-conferring gene; amp, ampicillin resistance-conferring gene; attP, attachment site on plasmid for phage integration; bla, carbenicillin/ampicillin resistance-conferring gene; cre, gene encoding Cre recombinase; dre, gene encoding Dre recombinase; ermE, constitutive promoter in streptomycetes; FRT, recognition site for FLP recombinase; hph, hygromycin resistance-conferring gene; lacZ, gene encoding β-galactosidase for blue/white selection; loxP, recognition site for Cre recombinase; int, phage integrase gene; oripUC18, origin of replication in E. coli; oriT, origin of transfer; pSG5rep, a temperature-sensitive replicon in streptomycetes; rox, recognition site for Dre recombinase; tfd, phage terminator sequence; tipA, thiostrepton-inducible promoter; tsr, thiostreptone resistance-conferring gene.
Comparison of the in vivo efficiency of Cre-mediated recombination of different lox pairs. (i) pLERE.
The synthetic vector pLERE contains two loxLE sites and two loxRE sites in addition to an ampicillin resistance gene and was ordered from Geneart (Regensburg, Germany). Different plasmids based on this vector were constructed to determine the recognition efficiency of mutated lox sites.
(ii) pINTRE.
An apramycin resistance gene was amplified from vector pMOD3-aac using EcoRI-containing (marked in italics) primers AG1, 5′-ACGTACCGAATTCGGTTCATGTGCAGCTCCATCAGC, and aac-r, 5′-ACGTACGAATTCATGAGCTCAGCCAATCGACTGG. Ligation of this fragment into MunI-digested pLERE resulted in an apramycin resistance gene flanked by two loxRE sites. The two redundant loxLE sites were removed by XbaI/NheI digestion and subsequent religation of the plasmid. A 3.4-kb NheI/XbaI restriction fragment of pSET152 containing oriT, the attP site, and the integrase gene was ligated into the SpeI-digested plasmid to produce pINTRE.
(iii) pINTLE and pINTLERE.
pINTLE and pINTLERE were constructed in a similar manner, but an apramycin resistance gene was flanked by loxLE (pINTLE) and loxLE plus loxRE (pINLERE).
(iv) pALCRE.
The synthetic gene cre(a) was subcloned from pUWLCRE into pAL1 using HindIII/BamHI digestion to produce pALCRE.
Dre recombinase plasmids. (i) pALDRE and pUWLDRE.
The synthetic gene dre(a) was synthesized by GenScript (Piscataway, NJ) and cloned via HindIII/BamHI digestion into pAL1 and pUWLoriT to produce pALDRE and pUWLDRE, respectively.
(ii) pINTROX.
The apramycin resistance cassette aac(3′)IV was amplified from pIJ773 using the primers roxF, 5′-AAGCTTTAACTTTAAATAATGCCAATTATTTAAAGTTATTTCATGAGCTCAGCCAATCGAC, and roxR, 5′-GAATCCTAACTTTAAATAATTGGCATTATTTAAAGTTAATTCCGGGGATCCGTCGACC (rox sites underlined), and cloned into pIJ2925 (1) to produce pIJrAmr. The phage φC31 integrase gene and an attP site were digested from pSET152 using PstI and SphI and cloned into pIJrAmr to produce pINTROX.
Marker-free expression. (i) pTOS.
The first plasmid is based on the vector pSOK804 (29), which uses the phage VWB integration system. The original plasmid attachment site (attP) was replaced with a synthesized DNA fragment consisting of the same attP site now flanked by rox sites and six unique restriction sites via BamHI/SphI digestion to produce pTOS (Fig. 1).
Fig 1.
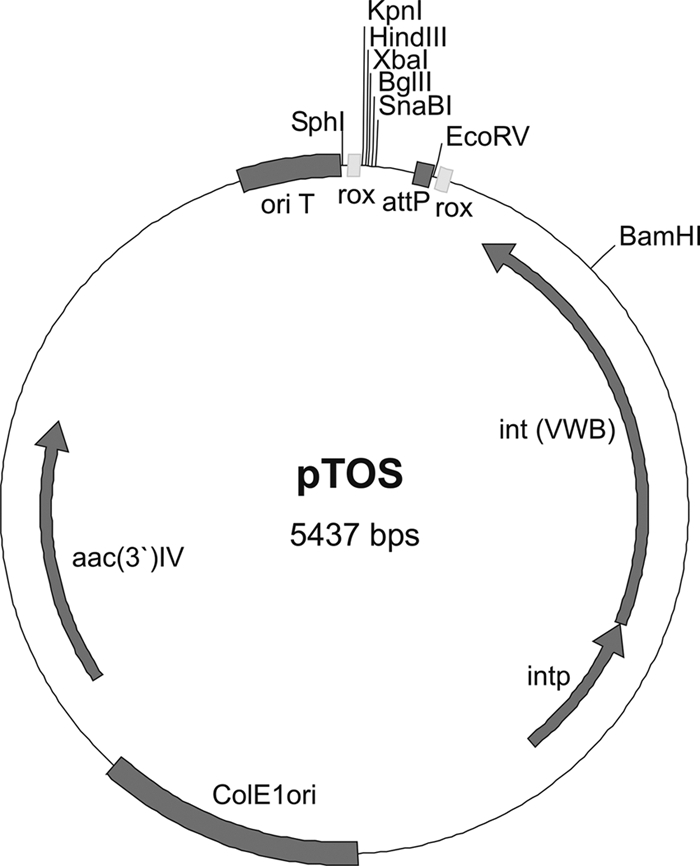
Plasmid map of pTOS. aac(3′)IV, apramycin resistance-conferring gene; attP, attachment site for integration; ColE1ori, origin of replication in E. coli; int (VWB), integrase of phage VWB; intp, integrase promoter; rox, recognition site for Dre recombinase; oriT, origin of plasmid transfer.
(ii) pTES.
The second plasmid is based on the vector pSET152 (1), which uses the phage φC31 integration system. The attP site of pSET152 overlaps the integrase promoter, which is not suitable for our application. Destruction of this attP site was accomplished by deleting two thymines in the core sequence through site-directed mutagenesis PCR, which leaves the integrase promoter functional and creates pSETdel2T. While previous work has shown that deletion of a single T in the TTT core sequence cannot promote recombination (32), we still obtained apramycin-resistant exconjugants. A synthesized DNA fragment consisting of an unmutated attP site, a tfd terminator, the strong promoter ermE, five unique restriction sites, and a second tfd terminator all bordered by two loxP sites was digested by SnaBI (order, SnaBI-loxP-attP-tfd-ermE-polylinker-tfd-loxP-SnaBI). This DNA fragment was blunt end ligated between the two PvuII restriction sites flanking the original polylinker of pSETdel2T to produce pTES (Fig. 2).
Fig 2.
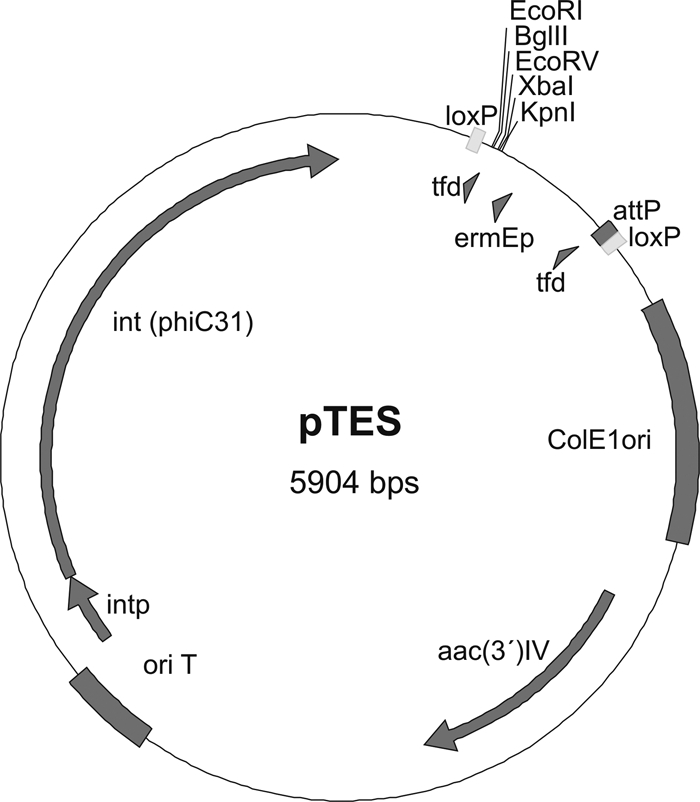
Plasmid map of pTES. aac(3′)IV, apramycin resistance-conferring gene; attP, attachment site for integration; ColE1ori, origin of replication in E. coli; ermEp, ermE promoter; int (phiC31), integrase of phage phiC31; intp, integrase promoter; loxP, recognition site for Cre recombinase; oriT, origin of plasmid transfer; tfd, phage fd terminator.
Plasmids for the lipomycin gene cluster deletion. (i) pIJloxP-lipbeg.
The polylinker of pUC18 was amplified by PCR using the primers laclox-f, 5′-CTGTCGAGATCTATAACTTCGTATAGCATACATTATACGAAGTTATCTGGCTTAACTATGCGGCAT, and laclox-r, 5′-CTGTCGAGATCTATAACTTCGTATAATGTATGCTATACGAAGTTATGGAAACAGCTATGACCATGA, containing loxP sites (underlined) and BglII restriction sites (marked in italics). This 428-bp PCR fragment was cloned into plasmid pIJ2601 (30) to produce pIJloxP. A 2,911-bp PCR fragment homologous to the 5′ end of the α-lipomycin cluster from S. aureofaciens Tü117 was amplified using the primers lipbegin-f, 5′-CTGTCGAAGCTTATTACCCTGTTATCCCTACGATCTGGACCCGGTTGTTGAG (HindIII restriction site marked in italics), and lipbegin-r, 5′-CTGTCGGAATTCTGGTCGGCCGTGTAGTAGTCCT (EcoRI restriction site marked in italics), and cloned via HindIII/EcoRI digestion into pIJloxP to give rise to pIJloxP-lipbeg.
(ii) pBeloBAC2601-lipend.
To obtain the BAC vector pBeloBAC2601-lipend, a 2,064-bp PCR fragment homologous to the 3′ end of the α-lipomycin cluster from S. aureofaciens Tü117 was amplified using the primers lipend-f, 5′-CTGTCGGACTGCAGATCCGCATGCCGTACTGCGTG (SphI restriction site marked in italics), and lipend-r, 5′-CTGTCGAAGCTTGGACTCCACCGCGCAGAAACC (HindIII restriction site marked in italics), and cloned via HindIII/SphI digestion into pBeloBAC2601 (30).
Plasmids for the monensin gene cluster deletion. (i) pMStart2-loxP-hyg.
Homologous regions of the monensin biosynthetic gene cluster were amplified from cosmids Cos2 and Cos11 (24). A 1.6-kb PCR fragment was amplified from Cos2 using the primers MonStart-F, 5′-GCTCCGAATTCCCTCGACGGAG, and MonStart-R, 5′-GGCCAGAATTCTGCGGTGCACG, which introduced EcoRI sites at both ends of the fragment (marked in italics). This fragment contained the complete monAI gene (806 bp) and the beginning of the monAIX gene (460 bp) and was ligated into EcoRI-digested pUC19 (38) to produce pMStart. A second PCR fragment (1.4 kb) was amplified from pIJ774 using the primers 2loxPapra-F, 5′-TACTGCACGGCGAGCACCTCGACGCCGGGCGCGAGCAGCATTCCGGGGATCCGTCGACC, and 2loxPapra-R, 5′-CGAAGCAGCTCCAGCCTACACGCGGGCGGTTCGGCGAGTTACTACTTCGGGCTCTCCGG. It consisted of an apramycin resistance gene and one origin of transfer flanked by two loxP sites. The homologous regions of monAIX produced the ends of the fragment. This fragment was introduced into pMStart using the RedET recombination system. A hygromycin resistance gene was excised from pIJ10700 by XmnI/HindIII digestion. The ends were blunted using the Klenow fragment (New England BioLabs, Ipswich, MA), and the fragment was cloned into SpeI-digested pMStart to produce pMStart-2loxP-hyg.
(ii) pMEnd-loxP.
A 1.6-kb PCR fragment containing monD was amplified from Cos11 using the primers MonEnd-F, 5′-CGGTGACAAGCTTGAGGTATTCC (HindIII site marked in italics), and MonEnd-R, 5′-CGCACATGTGCAGAGGGAAG (PciI site marked in italics), and cloned into pUC19 via HindIII/PciI digestion. A second PCR fragment was amplified from pIJ774 using the primers loxPapra-F, 5′-ATTGTAAGCTTGAGCTGCTTC, and loxPapra-R, 5′-TATGAGGATCCCGCC, which introduced a HindIII site and a BamHI site (marked in italics), respectively. This fragment contained an apramycin resistance gene, the origin of transfer, and one loxP site and was ligated into the intermediate plasmid to produce the final plasmid, pMEnd-loxP.
Plasmids for the phenalinolactone gene cluster deletion. (i) pIJloxPbegin.
The homologous region was PCR amplified from the cosmid 3-1O12 using PfuI polymerase and the primers BG1 (5′-GCCAAGCTTGAGCCGGGCAACCC-3′) and BG2 (5′-TACGAATTCACGAATCGCTGCTCGACGC-3′), which introduced a HindIII site and an EcoRI site (marked in italics), respectively, at the ends of the fragment. This 2.2-kb fragment was ligated between two loxP sites of the digested vector pIJloxP.
(ii) pIJloxPend.
The homologous region for the second plasmid was also amplified by PfuI polymerase from the cosmid 3-1O12 template. The primers EN1 (5′-GCCAAGCTTCGCCGCGTTCTGGAACG-3′) and EN2 (5′-ACGCTGCAGTCAGACGAACTTGTGCC-3′) introduced a HindIII site and a PstI site (marked in italics), respectively, at the ends of the fragment. After HindIII/PstI digestion, the 2.4-kb fragment was ligated into HindIII/PstI-digested pIJloxP vector to produce pIJloxPend.
Cluster deletion procedure.
The relevant actinomycete strain contained two loxP sites as direct repeats flanking the region of interest. The plasmid pALCRE was introduced into the respective mutant strain by conjugation, and successful exconjugants were obtained by their ability to grow on hygromycin-supplemented MS agar plates. Exconjugants were grown in liquid culture supplemented with hygromycin but without apramycin until stationary phase. At this point, expression of the artificial Cre recombinase gene was induced by adding 5.0 μg/ml thiostrepton to the liquid culture and subsequent incubation for 10 h. This procedure of growth to the stationary phase followed by induction was repeated six times to ensure that the recombination event took place. Subsequently, the culture was cultivated twice at 37°C to counterselect for the temperature-sensitive replicon of pALCRE and thereby promote loss of the plasmid. The culture was diluted onto yeast extract-agar plates to obtain single colonies. Those colonies were replica-patched onto yeast extract-agar plates without antibiotics and yeast extract-agar plates with apramycin. The growth of colonies only in broth without apramycin indicated that the region of interest had been excised from the chromosome to leave a single loxP site. One of these colonies was randomly chosen for the following experiments.
Cluster complementation.
The φC31-based cosmid 3-1O12 was conjugated into the deletion mutant Streptomyces sp. strain Tü6071/Δpla-loxP and integrated into the attB site of the mutant chromosome. In the case of the two further deletion mutants S. cinnamonensis A519/Δmon-loxP and S. aureofaciens Tü117loxP2Δlip, no complementing cosmids were available to restore the production of the respective metabolite.
RESULTS
Introducing a novel Dre recombinase for actinomycetes.
Cre recombinase has low affinity for the double mutant loxLERE site, which is generated by Cre-mediated recombination between loxLE and loxRE together with a wild-type loxP site (21). We compared the in vivo Cre-mediated recombination efficiencies of different lox pairs in S. coelicolor M145. The Cre-mediated excision efficiency of an apramycin resistance gene flanked by loxLE and loxRE mutant sites was 100%. Surprisingly, an apramycin resistance gene flanked by loxLERE doubly mutated sites was excised in 80% of the clones, which was unexpected considering the reported low affinity of the Cre recombinase for loxLERE sites. Thus, the loxLERE site remaining in the chromosome may have a significant effect on subsequent manipulations involving Cre recombinase in actinomycetes.
Considering the high activity of Cre on the doubly mutated site loxLERE, we have expressed a new Dre recombinase that recognizes completely different target sequences called rox sites. The synthetic dre(a) gene was optimized for expression in GC-rich organisms and was cloned into pUWLoriT and pAL1 to yield pUWLDRE and pALDRE, respectively. To test the activity of Dre recombinase, the plasmid pINTROX carrying the integration system of the phage φC31 and an apramycin resistance gene flanked by rox sites was constructed and integrated into the genome of S. coelicolor M145. After the transformation of pUWLHDRE and pALDRE into S. coelicolor exconjugants containing pINTROX, the strains were subjected to the same procedure used to test Cre activity. All 300 clones tested for resistance to apramycin showed sensitivity to the antibiotic. This experiment demonstrated the successful expression of the synthetic dre(a) gene and offers another efficient tool for resistance marker removal in actinomycetes. The Dre/rox system was proven to be effective with the same high efficiency of 100% in Micromonospora sp. strain Tü6368, Saccharothrix espanaensis, S. coelicolor T3456-20, and S. lividans TK24.
Marker-free expression system in actinomycetes.
Integrative plasmids have been successfully used in the genetic engineering of streptomycetes. Using components of phage integration systems such as attachment sites and integrases, genes of interest can be easily integrated into the corresponding attachment site in a streptomycete genome. However, integration of the entire plasmid, including a resistance marker and an integrase gene with its strong promoter, might lead to polar effects on neighboring genes in the chromosome. Additionally, there are only a limited number of selection markers available in streptomycetes. We developed a method by combining phage integration systems and recombinases where only the genes of interest remain in the genome, as shown in Fig. 3. Through the integration of target sites (loxP sites in the case of the Cre recombinase, rox sites in the case of the Dre recombinase) flanking the gene of interest and the phage attachment site (attP), we are able to delete the plasmid backbone, which is redundant after integration, by expressing the required site-specific recombinase. For this purpose, we constructed two plasmids using different phage integration systems and target sites, which allow marker-free integration of the genes of interest into two different sites in the streptomycetes chromosome.
Fig 3.

Schematic diagram depicting the mechanism of marker-free integration of a gene of interest (goi) into the streptomycete genome. (A) The integrase gene (int) on the plasmid is under the control of the native phage promoter, and its product catalyzes the integration of the plasmid between the attachment site on the plasmid (attP) and the attachment site on the bacterial chromosome (attB). (B) Selection for the integration of the plasmid into the genome is provided by the apramycin resistance-conferring gene (aac(3′)IV). (C) Expression of Dre recombinase will result in the deletion of the plasmid sequence between the rox sites, thus removing the marker and leaving behind only the gene of interest and one rox site in the genome. (D) Dre and Cre recombinase recognition sequences rox site and loxP site, respectively.
VWB-based plasmid pTOS.
The plasmid pTOS (Fig. 1) uses the VWB phage integration system with an attP site flanked by rox sites. It was inserted into S. lividans TK24 and S. coelicolor M145 by intergeneric conjugation with a frequency of ∼10−4. The S. lividans TK24 strain containing pTOS was again conjugated with the plasmid pUWLDre encoding the Dre recombinase. The resulting exconjugants were grown in antibiotic-free TSB medium to saturation and diluted on TSB plates with and without apramycin. One hundred percent of the cells were apramycin sensitive, which demonstrated that the Dre recombinase efficiently removed the backbone of the plasmid, including the apramycin resistance-conferring gene between the rox sites. Using this system, we integrated two genes of interest (goi1 and goi2) into the chromosome of S. lividans TK24 and proved the successful excision of the plasmid backbone by Dre recombinase by PCR and following sequence analysis of the PCR fragment.
φC31-based plasmid pTES.
The plasmid pTES (Fig. 2) uses the φC31 phage integration system with an attP site flanked by loxP sites. After the conjugation of the S. lividans TK24 strain containing pTES with the plasmid pALCre encoding the Cre recombinase, 99% of the 3,000 tested resulting exconjugants were immediately apramycin sensitive.
Generation of large-scale deletions.
The overall strategy for the construction of large deletions relies on a combination of homologous and site-specific recombination, with the latter mediated by the Cre/loxP system (Fig. 4). First, a homologous region (1.0 to 2.5 kb) flanking the 5′ end of the targeted regions is cloned between two loxP sites on a suicide vector and inserted via homologous recombination by a single crossover into the recipient chromosome to form a cointegrate. The relative orientation of the loxP sites with respect to the homologous fragment was chosen in a way that it would form direct repeats flanking the entire suicide vector, allowing subsequent excision of the vector sequences, including the resistance marker. Only one loxP site was left at the 5′ end of the targeted region as a result of Cre expression. The second homologous region flanking the 3′ end of the targeted regions was cloned adjacent to one loxP site on a suicide vector in such a way that the entire target region, including the suicide vector, was flanked by two loxP sites after insertion. After Cre expression, the region between the two loxP sites was excised.
Fig 4.

Schematic diagram depicting the steps for deleting large gene clusters from the streptomycete chromosome. Initially, a suicide plasmid carrying two recognition sites (rs) flanking a region homologous to the 5′ end of the cluster is introduced into the chromosome by a single crossover. Mutants are identified by selecting for antibiotic resistance that is conferred by a gene (R) on the newly integrated plasmid. Expression of the site-specific recombinase leads to excision of the plasmid backbone, including the resistance marker, and leaves one rs at the 5′ end of the cluster. A second suicide plasmid carrying one rs and a region homologous to the 3′ end of the cluster is integrated into the chromosome by means of single crossover. Mutants can again be selected by the resistance marker (R). Expression of the site-specific recombinase finally leads to excision of the entire cluster and loss of the resistance marker, leaving in its place only one rs “scar” in the chromosome.
Deletion of the phenalinolactone biosynthetic gene cluster from Streptomyces sp. strain Tü6071.
The biosynthetic gene cluster of the antibiotic diterpenoid phenalinolactone consists of 42 kb, which encode 35 open reading frames. To delete 38 kb of the phenalinolactone biosynthetic gene cluster in the genome, the above-mentioned strategy was used. The suicide plasmid pIJloxPbegin was integrated at the 5′ end of the phenalinolactone cluster by a single crossover to obtain Streptomyces sp. strain Tü6071/2loxPapra. The two inserted loxP sites flanking the plasmid backbone were used as target sites for Cre recombinase. The deletion of the plasmid core was accomplished as described in the Materials and Methods section. The frequency of the plasmid excision via Cre was approximately 50-fold higher in comparison to the plasmid removal via homologous recombination. The resulting mutant was named Streptomyces sp. strain Tü6071/loxP. The second vector pIJloxPend was introduced into Streptomyces sp. strain Tü6071/loxP by conjugation. By means of a single crossover, the plasmid integrated into the 3′end of the phenalinolactone cluster to give Streptomyces sp. strain Tü6071/2loxP. Positive exconjugants were identified by their resistance to apramycin. One of those exconjugants was randomly chosen and used for further experiments. The second excision step was performed as described in the Materials and Methods section. The resulting mutant strain was named Streptomyces sp. strain Tü6071/Δpla-loxP. Excision of the biosynthetic gene cluster was confirmed by PCR and following sequence analysis of the PCR fragment. The production of phenalinolactone could be restored by φC31-based integration of the cosmid 3-1O12. However, a phenalinolactone yield of only 4% relative to the wild-type level was obtained (Fig. 5).
Fig 5.

Deletion and reconstruction of phenalinolactone A (PL A) and phenalinolactone D (PL D) production. (A) Analysis of PL production of the wild-type strain Streptomyces sp. strain Tü6071 and the deletion mutant Streptomyces sp. strain Tü6071/Δpla-loxP by LC. (B) Characteristic UV spectrum of PL D measured in the complementation mutant Streptomyces sp. strain Tü6071/Δpla-loxP + Cos 3-O12 and the wild type. (C) The extracted mass of m/z = 714 u/e [M-H]− and m/z = 698 u/e [M-H]− represents the negative ion of PL A and PL D detected during the MS-chromatography of the complementation mutant. These masses were not present in the deletion mutant.
Deletion of the lipomycin biosynthetic gene cluster from S. aureofaciens Tü117.
The identical strategy was used to remove 67 kb of the α-lipomycin biosynthetic gene cluster from the chromosome using plasmids pIJloxP-lipbeg and pBeloBAC2601-lipend. The acyclic polyene antibiotic α-lipomycin is produced by S. aureofaciens Tü117. Its biosynthetic gene cluster (lip gene cluster) is encoded on 74 kb, divided in 28 open reading frames (2). Excision of the biosynthetic gene cluster was confirmed by PCR and high-pressure liquid chromatography (HPLC) analysis.
The frequencies of the deletion of both gene clusters (67 kb in the case of lipomycin, 38 kb in the case of phenalinolactone) as well as a resistance gene [aac(3′)IV, 800 bp] were all the same, showing that the length of DNA between two loxP sites is irrelevant for the Cre recombinase.
Deletion of the monensin biosynthetic gene cluster from S. cinnamonensis A519.
The deletion of the monensin biosynthetic gene cluster was our first effort of directed cluster deletion. Although this procedure works quite well, it is more labor intensive than the strategy which was used for the deletions of the other clusters and is depicted in Fig. 4. The biosynthetic gene cluster of the polyether antibiotic monensin comprises 97 kb. To delete 83 kb of the cluster from the genome, a slightly different approach was used. Instead of two different inactivation constructs, two plasmids based on the same vector were used. This resulted in the need for one double crossover and one single crossover instead of two single crossovers to avoid the integration of the second plasmid into the first integrated plasmid. Therefore, the plasmid pMStart2-loxP-hyg was placed at the 5′end of the monensin cluster by a double crossover, integrating an apramycin resistance cassette flanked by two loxP sites. To remove the apramycin resistance gene, the cre(a) expression plasmid pALCRE was introduced into S. cinnamonensis A519/2loxPapra by conjugation. The following steps are described in the Materials and Methods section. The resulting mutant was named S. cinnamonensis A519/loxP. The second vector, pMEnd-loxP, was introduced into S. cinnamonensis A519/loxP by conjugation. The plasmid integrated into the 3′ end of the monensin gene cluster via a single crossover to give S. cinnamonensis A519/2loxP. The procedure for monensin biosynthetic gene cluster removal from the chromosome is described in the Materials and Methods section. The resulting mutant strain was named S. cinnamonensis A519/Δmon-loxP. Excision of the biosynthetic gene cluster was confirmed by PCR, sequencing analysis, and the loss of antibiotic production.
Insertion of a plasmid into the streptomycete chromosome using the Cre/loxP system.
Heterologous expression is commonly based on phage integration systems. We have performed here a Cre-mediated integration to insert a single copy of foreign DNA into a predetermined locus within a genome. The Cre/loxP system can be used to remove the biosynthetic gene cluster of a natural product from the chromosome as well as to integrate desired DNA sequences into the same position. This system allows the expression of a mutated gene cluster responsible for the natural product biosynthesis under the same conditions as the native one. To prove the integration activity of Cre recombinase, a simple plasmid of 5 kb (pMEnd-loxP) and the deletion mutant Streptomyces sp. strain Tü6071/Δpla-loxP, both containing one loxP site, were used. The cre(a) expression plasmid pALCRE was introduced into Streptomyces sp. strain Tü6071/Δpla-loxP by conjugation. The resulting exconjugants were picked from the plate and grown in TSB medium containing hygromycin. The expression of the artificial recombinase gene by the tipA promoter was induced by adding 5 μg/μl thiostrepton to the liquid culture. After 6 h, the plasmid pMEnd-loxP was introduced by conjugation. The exconjugants were grown in TSB medium containing apramycin and plated onto ABB13 plates also containing apramycin to obtain single colonies. Growth of colonies in broth with apramycin demonstrated the successful insertion of plasmid pMEnd-loxP in the chromosome. The integration was confirmed by PCR and sequence analysis.
DISCUSSION
The use of site-specific recombinases for genomic engineering is no longer novel. They have been employed in many different eukaryotic and prokaryotic systems. Although the site-specific recombinase systems are extremely efficient and easy to use, their true potential in streptomycete genetics has not been fully exploited.
Construction of unmarked mutants.
Gene replacement strategies most often result in the introduction of a selectable marker into the genome, which remains in the chromosome permanently. The number of consecutive deletions of additional genes in the same strain may be limited by the number of available resistance genes (17). Additionally, the presence of the resistance gene may result in polar effects on the expression of genes located upstream and downstream. The expression of selectable markers may also influence bacterial fitness. Such obstacles could be overcome if the resistance markers are eliminated after mutant selection, which can be achieved via SSR. Therefore, removal of a selectable marker from the genome using SSR has been frequently used in plants, mouse cell lines, and yeast and, recently, in actinomycetes (3, 8, 23, 28). In a typical application, Cre and Flp are employed to delete a resistance marker, which was previously inserted in the chromosome after targeted gene inactivation. Flp- and Cre-mediated excision of the antibiotic resistance cassette leaves a “scar” in the chromosome and generates a single target FRT or loxP site. The length of this scar is usually less than 50 bp. Upon excision of the antibiotic resistance cassette from the chromosome, the FRT and loxP sites left behind in the chromosome do not exert polar transcriptional effects. Marker rescue and reuse offer a simple and efficient way to introduce multiple gene deletions. However, consecutive deletions of additional genes in the same strain may be limited because of undesired DNA rearrangements generated by the multiple target sites left after each excision. To minimize genetic instability, different heterotypic lox sites containing mutations within the inverted repeats (loxLE and loxRE) have been used for plants, chicken cell lines, and bacteria (3, 21). Recombination of loxLE and loxRE results in a double mutant loxLERE site, which is a poor substrate for Cre. Generation of this site allows for repeated gene deletions in a single genetic background. Surprisingly, Cre could very efficiently remove an apramycin resistance cassette flanked by loxLERE from the chromosomes of actinomycetes. Thus, the risk of undesired chromosomal rearrangements in actinomycetes is still very high if Cre is used in combination with loxLE and loxRE. The successful functional expression of a Dre recombinase encoding gene reported in this study increases our ability to construct marker-free multimutant strains. Dre recognizes rox sites, which differ notably from the previously reported FRT and loxP sites in actinomycetes. This characteristic makes Dre an ideal partner for Flp and Cre in performing multiple mutations in a single genetic background. Additionally, the xis and int genes from the pSAM2 plasmid could be used to remove antibiotic resistance markers from the chromosomes of streptomycetes (25). Therefore, at least four consecutive resistance marker deletions can be performed in actinomycetes in a single genomic background using Cre, Flp, Dre, and xis and int from pSAM2.
Generation of the marker-free expression system.
The other important part of Streptomyces genome engineering strategies is the stable integration of desired genetic traits into the chromosome. The integration of regulatory elements, gene fusions, and complementation of gene deletions is generally achieved using plasmids carrying a phage integrase gene (int) and the corresponding phage attachment site (attP). Commonly used integration vectors based upon the Streptomyces phages φC31 and VWB integrate within a chromosome condensation protein-encoding gene and the arginine tRNA gene, respectively. Another system based on the phage φBT1 has gained popularity as well in recent years (9). However, it is cumbersome to create multiple, stable, unmarked chromosomal integrations in actinomycetes using currently available vectors. For example, these methods usually necessitate antibiotic selection and thus leave behind a selection marker as well as additional unnecessary delivery vehicle sequences, such as an int gene and an E. coli origin of replication. Permanent expression of the int gene might also lead to an unstable construct (30). Therefore, the φC31- and VWB-based vectors, which are able to integrate at a specific attB site into the chromosome, were engineered to remove unwanted delivery vehicle DNA, the phage integrase and the antibiotic selection marker. Removal of the φC31 integrase gene together with an apramycin resistance marker from the pTES vector is performed by Cre recombinase, while Dre recombinase deletes the VWB integrase gene together with the apramycin resistance marker from the pTOS plasmid. Successful removal is easily identified by the loss of the backbone reporter gene and was observed in almost 100% of the tested clones. These pTES and pTOS vectors enable the engineering of stable recombinant actinomycete strains devoid of any selection marker. Furthermore, pTES is already equipped with the strong promoter ermE for overexpression of target genes. Additionally, the polylinker of pTES is protected from transcriptional interference by two tfd terminators. The ability to create multiple, stable, unmarked integrations, to express several genes simultaneously, or to increase overall expression by increasing copy number will be beneficial in many applications.
Generation of large-scale deletions.
The third important strategy for genome engineering described here is the generation of large-scale deletions by the Cre/loxP system in streptomycetes. It has been applied in several studies to create large-scale deletions in Corynebacterium glutamicum (34, 35), Bacillus subtilis (37), Streptomyces avermitilis (18), and E. coli (39). Our method has been specifically designed for actinomycetes by the modification of a pair of conjugative suicide vectors (pKC1132 and pBeloBAC11) for the introduction of loxP sites. The specific advantage of our strategy is that it works with single crossovers, which are usually easier to obtain than double-crossover events. In the examples shown, we observed single insertion frequencies between 2.0 × 10−4 and 1.0 × 10−7. The subsequent Cre-mediated deletion of the target fragment could usually be detected immediately in exconjugants, even without induction of the Cre-encoding recombinase gene. Precise excision of the target fragments was obtained in almost 100% of the tested clones. The total time requirement to generate a large-scale deletion using this method depended on the Streptomyces strain but could be achieved within 2 to 3 months in our case. This is generally faster and more efficient than using conventional allelic replacement techniques since the physical distance between recognition sites does not appear to limit the capacity of the recombinase. We have demonstrated the usefulness of this method in the construction of three deletions spanning the α-lipomycin, phenalinolactone, and monensin biosynthetic gene clusters in S. aureofaciens Tü117, Streptomyces sp. strain Tü6071, and S. cinnamonensis A519, respectively. As expected, all three deletion mutants cannot produce the corresponding antibiotic. The successful complementation of phenalinolactone production by introduction of the corresponding gene cluster presents opportunities to modify a cosmid directly in E. coli and perform gene deletion using mutated cosmids for complementation. However, the yield of phenalinolactone after complementation of the mutant with the cosmid must be improved. In addition to a systematic functional analysis of biosynthetic gene clusters, this method could be utilized for targeted deletion of genomic regions that are not required for growth and contain a large number of repeats and transposons that cause instability of the chromosomal DNA in actinomycetes. This would result in genetically stable host strains for the synthesis of natural products.
Cre-mediated DNA integration.
In genomic engineering, the ability to insert a gene of interest into a certain location is very desirable. Various factors appear to affect the expression and stability of heterologous genes. The most prominent of these factors is the genomic location of gene integration (positional effect). Surrounding genomic elements may cause the heterologous expression of a gene to be increased, decreased, or misregulated (36). The site-specific integration of vectors into actinomycete chromosomes does not provide much flexibility, since only a few different integrative systems are available. Thus, the integration can occur only at fixed positions that are not always optimal for target gene expression. Here, we were able to perform a Cre-mediated integration to insert a single copy of foreign DNA into predetermined loci within a genome. In summary, we have used the Dre and Cre recombinases in the construction of unmarked multiple mutations, marker-free expression of target genes, large-scale deletions, and chromosomal integration of biosynthetic gene clusters in different genera of actinomycetes. We expect that these techniques will find a broad application in actinomycete genetics and will facilitate functional studies of the large number of newly sequenced genes.
ACKNOWLEDGMENTS
This work was partly funded by a BMBF grant (GenomikPlus) and a grant from DFG (Lu1524/2-1) (both to A.L.).
Footnotes
Published ahead of print 13 January 2012
REFERENCES
- 1. Bierman M, et al. 1992. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49 [DOI] [PubMed] [Google Scholar]
- 2. Bihlmaier C, et al. 2006. Biosynthetic gene cluster for the polyenoyltetramic acid alpha-lipomycin. Antimicrob. Agents Chemother. 50:2113–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Branda CS, Dymecki SM. 2004. Talking about a revolution: the impact of site-specific recombinases on genetic analyses in mice. Dev. Cell 6:7–28 [DOI] [PubMed] [Google Scholar]
- 4. Campo N, Daveran-Mingot ML, Leenhouts K, Ritzenthaler P, Le Bourgeois P. 2002. Cre-loxP recombination system for large genome rearrangements in Lactococcus lactis. Appl. Environ. Microbiol. 68:2359–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 6. Dürr C, et al. 2006. Biosynthesis of the terpene phenalinolactone in Streptomyces sp. Tu6071: analysis of the gene cluster and generation of derivatives. Chem. Biol. 13:365–377 [DOI] [PubMed] [Google Scholar]
- 7. Fedoryshyn M, Petzke L, Welle E, Bechthold A, Luzhetskyy A. 2008. Marker removal from actinomycetes genome using Flp recombinase. Gene 419:43–47 [DOI] [PubMed] [Google Scholar]
- 8. Fedoryshyn M, Welle E, Bechthold A, Luzhetskyy A. 2008. Functional expression of the Cre recombinase in actinomycetes. Appl. Microbiol. Biotechnol. 78:1065–1070 [DOI] [PubMed] [Google Scholar]
- 9. Flett F, Mersinias V, Smith CP. 1997. High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes. FEMS Microbiol. Lett. 155:223–229 [DOI] [PubMed] [Google Scholar]
- 10. Ganesan A. 2008. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 12:306–317 [DOI] [PubMed] [Google Scholar]
- 11. Gust B, Challis GL, Fowler K, Kieser T, Chater KF. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U. S. A. 100:1541–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72 [DOI] [PubMed] [Google Scholar]
- 13. Hütter R. 1967. Systematik der Streptomyceten unter besonderer Berücksichtigung der von ihnen gebildeten Antibíotica. S. Karger AG, Basel, Switzerland: [PubMed] [Google Scholar]
- 14. Janssen GR, Bibb MJ. 1993. Derivatives of pUC18 that have BglII sites flanking a modified multiple cloning site and that retain the ability to identify recombinant clones by visual screening of Escherichia coli colonies. Gene 124:133–134 [DOI] [PubMed] [Google Scholar]
- 15. Keasling JD. 2010. Manufacturing molecules through metabolic engineering. Science 330:1355–1358 [DOI] [PubMed] [Google Scholar]
- 16. Khodakaramian G, et al. 2006. Expression of Cre recombinase during transient phage infection permits efficient marker removal in Streptomyces. Nucleic Acids Res. 34:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. John Innes Foundation, Norwich, United Kingdom [Google Scholar]
- 18. Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H. 2010. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc. Natl. Acad. Sci. U. S. A. 107:2646–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kühn R, Torres RM. 2002. Cre/loxP recombination system and gene targeting. Methods Mol. Biol. 180:175–204 [DOI] [PubMed] [Google Scholar]
- 20. Kuhstoss S, Richardson MA, Rao RN. 1991. Plasmid cloning vectors that integrate site-specifically in Streptomyces spp. Gene 97:143–146 [DOI] [PubMed] [Google Scholar]
- 21. Leibig M, et al. 2008. Marker removal in staphylococci via Cre recombinase and different lox sites. Appl. Environ. Microbiol. 74:1316–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luzhetskyy A, et al. 2006. IncP plasmids are most effective in mediating conjugation between Escherichia coli and streptomycetes. Genetika 42:595–601 [PubMed] [Google Scholar]
- 23. Marx CJ, Lidstrom ME. 2002. Broad-host-range Cre-lox system for antibiotic marker recycling in Gram-negative bacteria. Biotechniques 33:1062–1067 [DOI] [PubMed] [Google Scholar]
- 24. Oliynyk M, et al. 2003. Analysis of the biosynthetic gene cluster for the polyether antibiotic monensin in Streptomyces cinnamonensis and evidence for the role of monB and monC genes in oxidative cyclization. Mol. Microbiol. 49:1179–1190 [DOI] [PubMed] [Google Scholar]
- 25. Raynal A, Karray F, Tuphile K, Darbon-Rongère E, Pernodet JL. 2006. Excisable cassettes: new tools for functional analysis of Streptomyces genomes. Appl. Environ. Microbiol. 72:4839–4844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 27. Sauer B, Henderson N. 1990. Targeted insertion of exogenous DNA into the eukaryotic genome by the Cre recombinase. New Biol. 2:441–449 [PubMed] [Google Scholar]
- 27a. Sauer B, McDermott J. 2004. DNA recombination with a heterospecific Cre homolog identified from comparison of the pac-c1 regions of P1-related phages. Nucleic Acids Res. 32:6086–6095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schweizer HP. 2003. Applications of the Saccharomyces cerevisiae Flp-FRT system in bacterial genetics. J. Mol. Microbiol. Biotechnol. 5:67–77 [DOI] [PubMed] [Google Scholar]
- 29. Sekurova ON, et al. 2004. In vivo analysis of the regulatory genes in the nystatin biosynthetic gene cluster of Streptomyces noursei ATCC 11455 reveals their differential control over antibiotic biosynthesis. J. Bacteriol. 186:1345–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Siegl T, Petzke L, Welle E, Luzhetskyy A. 2010. I-SceI endonuclease: a new tool for DNA repair studies and genetic manipulations in streptomycetes. Appl. Microbiol. Biotechnol. 87:1525–1532 [DOI] [PubMed] [Google Scholar]
- 31. Slauch JM, Camilli A. 2000. IVET and RIVET: use of gene fusions to identify bacterial virulence factors specifically induced in host tissues. Methods Enzymol. 326:73–96 [DOI] [PubMed] [Google Scholar]
- 32. Smith MC, et al. 2004. Synapsis and DNA cleavage in phiC31 integrase-mediated site-specific recombination. Nucleic Acids Res. 32:2607–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stark WM, Boocock MR, Sherratt DJ. 1992. Catalysis by site-specific recombinases. Trends Genet. 8:432–439 [PubMed] [Google Scholar]
- 34. Suzuki N, Nonaka H, Tsuge Y, Inui M, Yukawa H. 2005. New multiple-deletion method for the Corynebacterium glutamicum genome, using a mutant lox sequence. Appl. Environ. Microbiol. 71:8472–8480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Suzuki N, et al. 2005. Multiple large segment deletion method for Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 69:151–161 [DOI] [PubMed] [Google Scholar]
- 36. Wilson C, Bellen HJ, Gehring WJ. 1990. Position effects on eukaryotic gene expression. Annu. Rev. Cell Biol. 6:679–714 [DOI] [PubMed] [Google Scholar]
- 37. Yan X, Yu HJ, Hong Q, Li SP. 2008. Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl. Environ. Microbiol. 74:5556–5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119 [DOI] [PubMed] [Google Scholar]
- 39. Yu BJ, et al. 2002. Minimization of the Escherichia coli genome using a Tn5-targeted Cre/loxP excision system. Nat. Biotechnol. 20:1018–1023 [DOI] [PubMed] [Google Scholar]
- 40. Zelyas N, Tahlan K, Jensen SE. 2009. Use of the native flp gene to generate in-frame unmarked mutations in Streptomyces spp. Gene 443:48–54 [DOI] [PubMed] [Google Scholar]