

Abstract
In familial and sporadic amyotrophic lateral sclerosis (ALS) and in rodent models of the disease, alterations in the ubiquitin-proteasome system (UPS) may be responsible for the accumulation of potentially harmful ubiquitinated proteins, leading to motor neuron death. In the spinal cord of transgenic mice expressing the familial ALS superoxide dismutase 1 (SOD1) gene mutation G93A (SOD1G93A), we found a decrease in constitutive proteasome subunits during disease progression, as assessed by real-time PCR and immunohistochemistry. In parallel, an increased immunoproteasome expression was observed, which correlated with a local inflammatory response due to glial activation. These findings support the existence of proteasome modifications in ALS vulnerable tissues. To functionally investigate the UPS in ALS motor neurons in vivo, we crossed SOD1G93A mice with transgenic mice that express a fluorescently tagged reporter substrate of the UPS. In double-transgenic UbG76V-GFP /SOD1G93A mice an increase in UbG76V-GFP reporter, indicative of UPS impairment, was detectable in a few spinal motor neurons and not in reactive astrocytes or microglia, at symptomatic stage but not before symptoms onset. The levels of reporter transcript were unaltered, suggesting that the accumulation of UbG76V-GFP was due to deficient reporter degradation. In some motor neurons the increase of UbG76V-GFP was accompanied by the accumulation of ubiquitin and phosphorylated neurofilaments, both markers of ALS pathology. These data suggest that UPS impairment occurs in motor neurons of mutant SOD1-linked ALS mice and may play a role in the disease progression.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the loss of motor neurons localized in motor cortex, brainstem and spinal cord (1,2). In ∼5–10% of patients the disease is inherited and 20% of these are associated with mutations in the gene coding for Cu,Zn superoxide dismutase (SOD1) (3). Transgenic mice that overexpress human SOD1 with the Gly93Ala substitution (SOD1G93A mice) develop a motor neuron dysfunction that mimics the human disease (4).
The presence of proteinaceous inclusions rich in ubiquitin in motor neurons is a neuropathological feature of both ALS patients and animal models of the disease (5–9). It has been proposed that alterations in the functionality of the ubiquitin-proteasome system (UPS) might play a role in this phenomenon (10–15).
The UPS is the main intracellular proteolytic system, responsible for the maintenance of protein turnover and for the selective removal of damaged and unfolded proteins (16–19). The 26S proteasome, that degrades poly-ubiquitinated proteins, consists of two sub-complexes: the 19S regulatory particle and the 20S particle, which contains the three catalytic subunits β1, β2 and β5. In mammals, upon induction by IFNγ and/or TNFα, these constitutive catalytic subunits can be replaced by the corresponding homologous ‘inducible’ subunits iβ1/LMP2, iβ2/LMP10 and iβ5/LMP7, forming the immunoproteasome (17,20).
It is known that mutant SOD1 is degraded by the proteasome (21,22) and partial inhibition of proteasome activity provokes the formation of large SOD1-containing aggregates (13,23,24). In a recent study we found that the levels of 20S constitutive catalytic subunits were significantly reduced in the spinal cord of SOD1G93A mice at an advanced stage of the disease, whereas at the same time the levels of their inducible counterparts were significantly increased. The replacement of the constitutive subunits with inducible subunits did not result in detectable changes in the 20S proteasome proteolytic activity. Other studies have demonstrated opposite changes in the expression of constitutive and inducible proteasome subunits in SOD1 mutant mice (11,25,26). So far it has not been clarified which mechanisms underlie the shift from constitutive proteasome to immunoproteasome and whether this effect may be related to the pathogenesis and/or disease progression. Moreover, although all these studies suggested the possibility that UPS is disrupted in ALS models, none of them conclusively demonstrated that the UPS impairment may occur in vivo in motor neurons.
An innovative approach to measure UPS functionality in vivo at the cellular level has been recently developed, based on the use of reporter proteins such as UbG76V-GFP. Under physiological conditions UbG76V-GFP is constitutively degraded by the UPS as the N-terminal ubiquitin moiety is recognized as a degradation signal leading to poly-ubiquitination and degradation of the ubiquitin-fusion substrate (27). An alteration in any step of the UPS may therefore result in the accumulation of the UbG76V-GFP protein. Hence, mice expressing the UbG76V-GFP reporter (from now referred to as GFP mice) may be a valuable tool to assess the overall functionality of the UPS at the cellular level in vivo (28). This model has already been used to demonstrate a functional UPS alteration in prion-infected mice (29) as well as to demonstrate that proteasome impairment does not occur in a mouse model of polyglutamine disease (30).
Therefore, in the present study we aimed to: (i) investigate the mechanisms that underlie the shift from constitutive proteasome to immunoproteasome during the course of the disease in the SOD1G93A mouse model and clarify their functional significance; (ii) assess the functional status of the UPS at the cellular level in the spinal cord of double-transgenic mice (GFP/SOD1G93A) at different stages during disease progression.
RESULTS
Increased immunoproteasome expression correlates with glial activation and TNFα induction in SOD1G93A spinal cord
To obtain information about the level of expression of various components of the 26S proteasome in SOD1G93A mice, the following transcripts were measured by real-time PCR in the lumbar spinal cord homogenate: constitutive catalytic β5, β1 and β2 subunits of 20S particle and their inducible counterparts LMP7, LMP2 and LMP10; non-catalytic 20Sα5 subunit; non-ATPAse S1 subunit of 19S complex; proteasome maturation protein (POMP). Transcript levels in SOD1G93A mice were reported as percentage of the levels in non-transgenic (NTg) littermates.
A remarkable decrease in the mRNA levels of all the constitutive catalytic subunits and POMP was observed in the lumbar spinal cord of end-stage SOD1G93A mice compared with NTg littermates (Fig. 1A). No changes were detected in these subunits at earlier disease stages except for a slight but significant decrease of the 19S subunit already at the pre-symptomatic stage and for a significant decrease of the non-catalytic 20Sα5 subunit at the symptomatic stage.
Figure 1.

Real-time PCR in the lumbar spinal cord of pre-symptomatic, symptomatic and end-stage SOD1G93A mice (black column) and NTg littermates (white column) for the following mRNAs: (A) β5, β1, β2, α5 subunit of 20S, S1 subunit of 19S and POMP; (B) LMP7, LMP2 and LMP10; (C) TNFα, GFAP, CD68 and CD8 transcripts. (A) Significant decrease of 19S at pre-symptomatic stage and of α5 subunit at the symptomatic stage as compared with NTg littermates. A remarkable decrease is found in the mRNA levels of almost all subunits in SOD1G93A mice at the end-stage in respect to NTg littermates. (B) Progressive increase of LMP7 mRNA from the pre-symptomatic to the end-stage. A small but significant increase of LMP10 is found only at the symptomatic phase, while LMP2 never changes. (C) High significant increase of TNFα, CD68 and GFAP starting from pre-symptomatic to the final stage. All transcripts were normalized versus β-actin and the ratios expressed as percentage of the values from NTg littermates. Each histogram shows the mean ± SEM of at least four mice. Data were analyzed by Student’s t-test. (*P < 0.05; **P < 0.01; ***P < 0.001 compared with NTg).
Also the levels of different immunoproteasome subunits changed in the lumbar spinal cord of SOD1G93A mice during disease progression (Fig. 1B). The mRNA of LMP7 subunit progressively increased from the pre-symptomatic stage until the end stage of disease. Such trend was not observed for the other inducible subunits except for a small but significant increase in the LMP10 only at the symptomatic stage. None of the transcript subunits examined was modified in the hippocampus of SOD1G93A mice at the end stage of the disease as compared with NTg controls (data not shown).
To investigate whether the induction of immunoproteasome in the lumbar spinal cord correlated with alterations of the immuno-inflammatory system, we measured the transcript levels of the pro-inflammatory cytokine TNFα as well as those of GFAP (glial fibrillary acidic protein), CD68 and CD8 as markers of activation for astrocytes, phagocytic microglial cells and cytotoxic T lymphocytes, respectively. The mRNAs for TNFα, CD68 and GFAP were progressively up-regulated from the pre-symptomatic stage, whereas the CD8 transcript remained unchanged (Fig. 1C).
Immunohistochemical studies on lumbar spinal cord sections from SOD1G93A, SOD1wt and NTg mice at different ages revealed selective cellular alterations in the expression of constitutive and inducible proteasomal subunits during disease progression (Figs 2 and 3).
Figure 2.
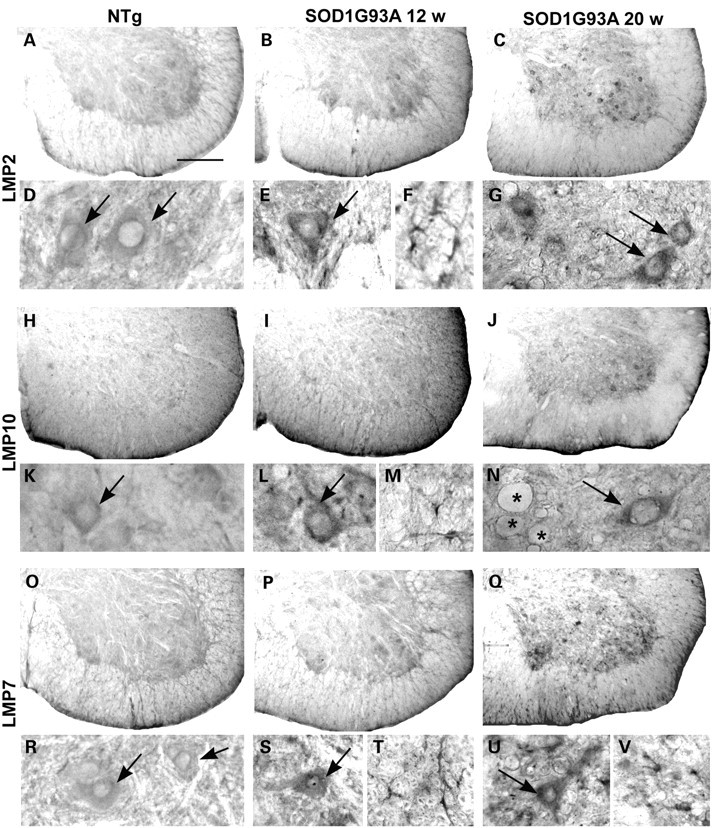
Immunoperoxidase detection of the proteasome inducible subunits LMP2, LMP10, LMP7 in lumbar spinal cord sections of NTg and SOD1G93A mice at pre-symptomatic (12 weeks) and late symptomatic (20 weeks) age. For each subunit the upper panels show low magnification images of the ventral horn, whereas the lower panels show details of motor neurons (arrows in D, E, G, K, L, N, R, S and U) and glial cells (F, M, T and V); asterisks = vacuoles in N. All the subunits are more intensely expressed in SOD1G93A mice than in NTg mice. Scale bar 10 µm in A–C, H–J, O–Q; 4 µm in D–G, K–N, R–U.
Figure 3.
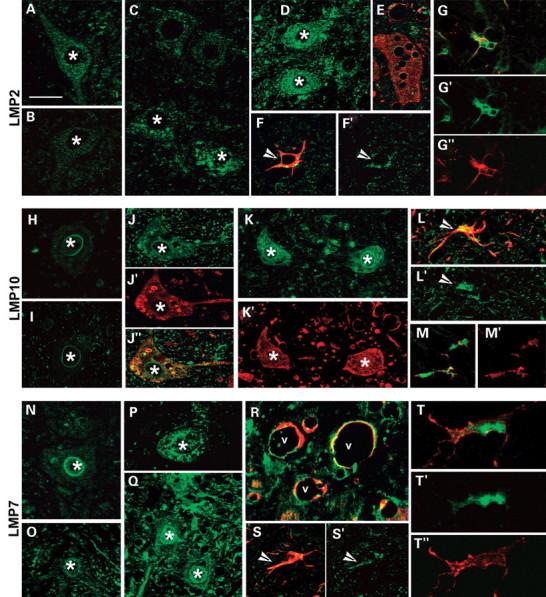
Confocal images of proteasome inducible subunits LMP2, LMP10, LMP7 (green) in lumbar spinal cord sections of NTg (A, H, and N), SOD1wt mice (B, I, and O) and SOD1G93A mice (C–G′; J–M′, P–T′) at different ages. In NTg (A, H and N) and SOD1wt (B, I and O) mice motor neurons (asterisks) have very low immunoreactivity (green) for the three subunits. Immunoproteasome labeling (green) is intense in motor neurons (asterisks) of pre-symptomatic (C, J and P) and symptomatic (D, E, K and Q) SOD1G93A mice; some of the motor neurons are vacuolated (E and J) and accumulate human SOD1 (red in E, J′ and K′); R shows vacuoles (v) in the neuropil rimmed by LMP7 (green) and human SOD1 (red) labeling. In SOD1G93A mice the immunoreativity for the three subunits (green) is also in astrocytes identified by GFAP (red, arrowheads in F, F′, L, L′, S and S′) and in microglial cells identified by CD11β (red in G, G′, M, M′, T and T′). Scale bar 4 µm in A–D, H–K, N–Q; 3.8 µm in E; 3 µm in F, L, M and S; 2.8 µm in G; 2.5 µm in T.
The constitutive structural 20S alpha subunits were decreased in some of the motor neurons of symptomatic SOD1G93A mice, compared with motor neurons of SOD1wt and NTg mice (not shown) confirming our previous results (10). The three inducible 20S beta subunits were very weakly expressed in the spinal cord of both NTg and SOD1wt mice at all ages (Figs 2 and 3), whereas their expression was increased in the ventral horn of SOD1G93A mice from pre-symptomatic stages (Figs 2 and 3). In particular, in SOD1G93A mice, an increased immunoreactivity for all the inducible beta subunits was found in activated astrocytes and microglia identified by specific markers (Fig. 3) and also in some motor neurons both with a normal appearance or vacuolated (Figs 2 and 3).
Proteasome inhibition induces the accumulation of UbG76V-GFP in cultured motor neurons from GFP mouse models
Both GFP1 and GFP2 mouse lines were analyzed to establish the basal levels of the reporter protein in the spinal cord. The expression of UbG76V-GFP was detected by immunohistochemistry with an anti-GFP antibody since native GFP fluorescence was under the detection threshold in the spinal cord of both lines (data not shown). A substantial difference was observed between the two transgenic lines: while in the GFP1 line basal UbG76V-GFP levels were clearly detectable by immunostaining in almost all the cell populations of both dorsal and ventral lumbar spinal cord (Supplementary Material, Fig. S1c and d), no immunostaining was observed in the GFP2 line (Supplementary Material, Fig. S1e and f) as in the NTg mice (Supplementary Material, Fig. S1a and b). No differences with age progression were found in either GFP1 or GFP2 lines (data not shown).
Both GFP1 and GFP2 lines were subsequently used for cross-breeding with SOD1G93A mice. However, since in GFP2 mice the basal reporter protein level was undetectable, this line was considered the most stringent model for detecting a specific and robust accumulation of UbG76V-GFP at the cellular level. Before cross-breeding, we verified whether GFP accumulated in the spinal cord motor neurons in response to UPS inhibition using in vitro spinal cord neuronal cultures (Fig. 4). Primary cultures obtained from the spinal cord of NTg or GFP2 embryos (14 days) were treated with the proteasome inhibitor MG132 and labeled for SMI-32 and GFP. In almost all cultured GFP2 neurons, the administration of 1.5 µm MG132 elicited a detectable increase in the levels of the reporter protein (Fig. 4D and F). The accumulation of GFP was particularly remarkable in neurons characterized by intense SMI-32 labeling, large cell body and prominent neuritic arborization, therefore identified as motor neurons (insets in Fig. 4F). The results were confirmed also with other doses of MG132 (0.5 and 4.5 µm, data not shown).
Figure 4.

Colocalization of GFP and SMI-32 immunoreactivity in spinal neurons cultures from GFP2 mouse embryos after proteasome inhibition. Cultures are treated with vehicle (A–C) or with the proteasome inhibitor MG132 1.5 μm (D–F) to induce an increase of GFP levels. An intense GFP immunostaining is observed in motor neurons characterized by an intense SMI-32 labeling after proteasome inhibition. Scale bar 50 µm, insets 20 µm.
On the basis of this evidence, the GFP2 mouse line was used for the majority of the experiments, although some of the key findings were confirmed also in GFP1 line.
Disease progression in SOD1G93A model is not modified by the presence of UbG76V-GFP
Since the presence of a transgene coding for a protein that is degraded by the UPS could represent an additional burden for a system already facing large amounts of unfolded proteins, we examined the disease progression of the GFP/SOD1G93A double-transgenic mice as compared with that of SOD1G93A mice. There were no indications for a more severe pathology in SOD1G93A mice expressing the reporter protein since the decline of the body weight, the progression of motor dysfunction and the length of survival were very similar in the two groups (Supplementary Material, Fig. S2). Hence, double-transgenic mice were analyzed at the same time points as SOD1G93A mice during disease progression.
Accumulation of UbG76V-GFP protein in spinal cord homogenate and motor neurons of double-transgenic GFP/SOD1G93A mice
The expression level of the reporter protein was examined by immunoblotting of homogenates and immunohistochemistry of lumbar spinal cord sections in symptomatic double-transgenic GFP/SOD1G93A mice compared with their GFP littermates. The levels of UbG76V-GFP in spinal cord homogenates of symptomatic GFP2 and GFP2/SOD1G93A mice were below the detection threshold in the immunoblot assay, whereas a specific band was detectable in the homogenate of the GFP1 mice, both single and double-transgenic (Fig. 5A). The quantification of the bands’ optical density (Fig. 5B) revealed a 1.2-fold increase in GFP1/SOD1G93A spinal cord compared with GFP1 mice, although the difference did not reach statistical significance (P = 0.058, Student’s t-test). To demonstrate that the accumulation of UbG76V-GFP protein was due to reduced protein degradation rather than to elevated protein production, the UbG76V-GFP transcript levels were also measured by real-time PCR in lumbar spinal cord of symptomatic GFP1/SOD1G93A mice compared with their GFP littermates. No differences were detected between the two groups (Fig. 5C). Similarly, no differences were observed in UbG76V-GFP transcript levels between GFP2/SOD1G93A mice and their GFP2 littermates (data not shown).
Figure 5.

(A) Representative immunoblot for UbG76V-GFP in the lumbar spinal cord of GFP1 and GFP1/SOD1G93A mice at the symptomatic stage. (B) Quantitative analysis of the immunoblot. The optical density was measured for each autoradiographic band and values for GFP1/SOD1G93A were compared with GFP1 after correction for total protein. Each histogram shows the mean ± SEM of at least four mice. Data were analyzed by Student’s t-test. P = 0.058 (C) Real-time PCR for UbG76V-GFP transcript in the lumbar spinal cord of GFP1/SOD1G93A mice and GFP1 littermates at the symptomatic stage of disease progression. Levels of GFP transcript were normalized to β-actin and the ratios from double-transgenic mice were expressed as percentage of the ratio value of the GFP1 mice. Each histogram shows the mean ± SEM of at least five mice. Data were analyzed by Student’s t-test.
To detect UbG76V-GFP at the cellular level, immunohistochemical analysis was performed on lumbar spinal cord sections of double-transgenic GFP1/SOD1G93A and GFP2/SOD1G93A mice at various stages of disease progression and in their GFP and SOD1G93A littermates. While in sections from GFP2 mice the reporter protein remained undetectable (Fig. 6C and D), a small number of cells with a clear accumulation of the reporter protein was found in the spinal grey matter of symptomatic GFP2/SOD1G93A mice. The percentage of GFP-positive cells over the total motor neurons per section in these mice was 11 ± 3% (mean ± SEM). It must be considered that the mean number of motor neurons counted in each lumbar section of symptomatic GFP2/SOD1G93A mice was 16 ± 3 (mean ± SEM, n = 10 sections/mouse, n = 4 mice), representing 57% of the motor neurons present in a lumbar spinal cord section of age-matched GFP2 mice (total motor neurons 28 ± 3, mean ± SEM, n = 10 sections/mouse, n = 4 mice). Moreover, the majority of the GFP-positive motor neurons showed morphological alterations such as vacuolization (Fig. 6G and H). This effect was observed also in GFP2/SOD1G93A mice at the end-stage of the disease, while no GFP-positive cells were detectable in the spinal cord of double-transgenic mice at the pre-symptomatic stage (data not shown).
Figure 6.
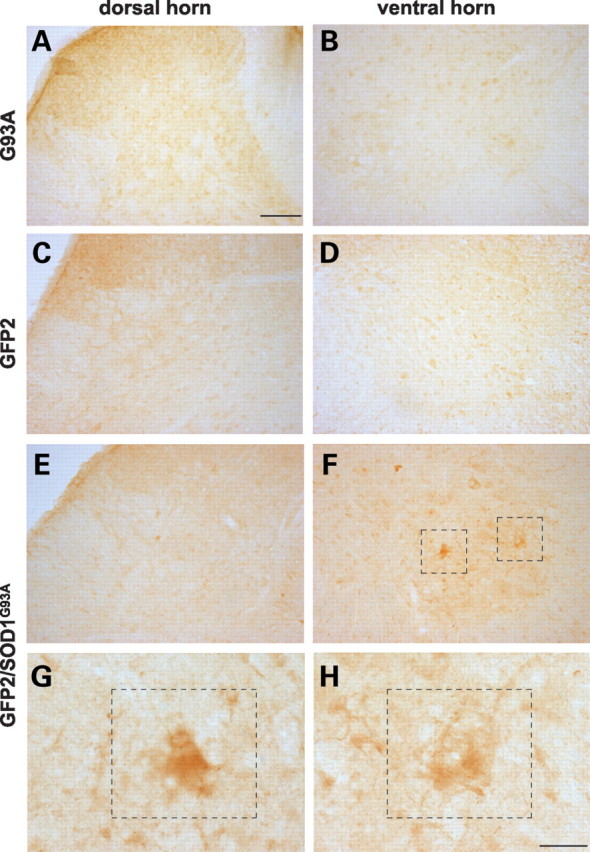
Immunohistochemistry of GFP in the dorsal and ventral horns of the lumbar spinal cord of symptomatic SOD1G93A (A and B), and GFP2/SOD1G93A (E and F) mice and in age matched GFP2 mice (C and D). While in sections from SOD1G93A and GFP2 mice the reporter protein remains undetectable (A–D), in the ventral horn of symptomatic GFP2/SOD1G93A mice a clear accumulation of the reporter protein occurs in motor neuronal-like cells; some of them show morphological alterations such as vacuolization (framed neurons in F, G and H). Scale bar 100 µm (A–F); 20 µm (G and H).
In the spinal cord of GFP1 mice, a diffuse basal GFP immunostaining was found in almost all cells of the ventral and dorsal regions, in line with higher constitutive reporter levels. In the spinal cord of double-transgenic GFP1/SOD1G93A mice, a few scattered motor neurons in the ventral horn showed a very intense GFP immunostaining, much stronger than the GFP1 control levels (data not shown), whereas no GFP immunostaining was found in the spinal cord sections of SOD1G93A mice (Fig. 6A and B).
No GFP immunostaining was found in sagittal brain sections of double-transgenic symptomatic and end-stage GFP2/SOD1G93A mice, with the exception of the brainstem where, at the end-stage of disease, an intense GFP labeling was detected in a few neurons and neurite-like structures present at the level of motor nuclei (facial, motor trigemini, Fig. 7).
Figure 7.

Representative microphotographs of GFP immunostaining in sagital sections from hindbrain and cerebellum of GFP2 (A and D) and GFP2/SOD1G93A mice at the end-stage (B, C, E and F). GFP signal is barely detectable in the hindbrain of GFP2 mice while it is remarkably intense in the same region of GFP2/SOD1G93A mice. Intense GFP-positive neuronal-like cells are found in nuclei trigemini (C) and facialis (F) of double-transgenic mice. Low GFP immunoreactivity is detectable in the cerebellum and no differences are found between the two mouse lines. Scale bar 500 µm (A, B, D and E); 20 µm (C and F).
Accumulation of UbG76V-GFP in spinal motor neurons colocalizes with markers of neurodegeneration
Double immunofluorescence confocal microscopy was performed on lumbar spinal cord sections of symptomatic GFP2/SOD1G93A mice to identify and characterize the cell types showing increased UbG76V-GFP levels. Double staining for GFP and the motor neuron marker choline acetyl transferase (ChAT) showed high quantities of reporter protein in some ChAT-positive motor neurons (Fig. 8D–F). Conversely, in the ventral horn of GFP2/SOD1G93A mice no colocalization was found between GFP and the GFAP, which specifically labels astrocytes or the anti-CD11β antibody, which labels reactive microglial cells (Fig. 8I and L).
Figure 8.
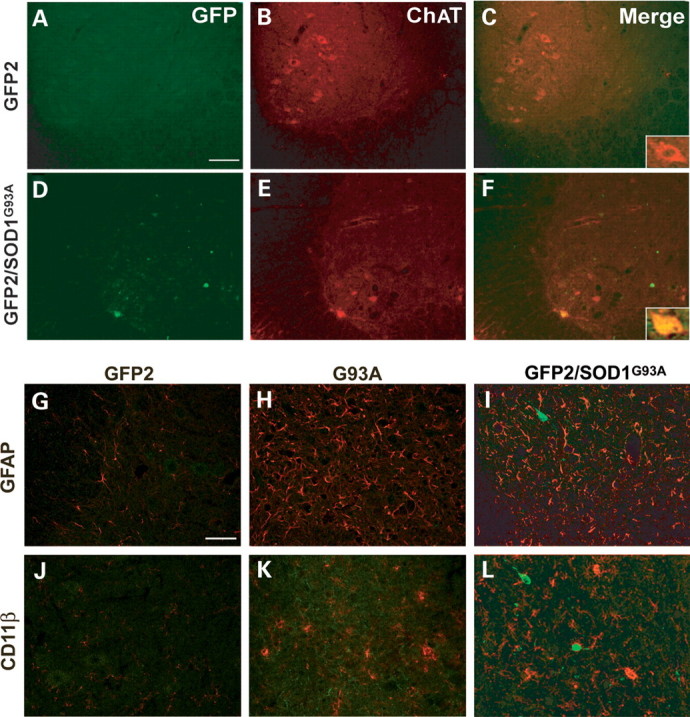
(A) Representative laser scanning confocal microphotographs of colocalization of GFP and ChAT immunostaining in the ventral horn of the lumbar spinal cord from GFP2 (A–C) or GFP2/SOD1G93A mice at the symptomatic stage (D–F). As shown in merge (C and F), intense GFP immunostaining is observed in some ChAT motor neurons (inset in F) of GFP2/SOD1G93A mice. Scale bar 100 µm (inset 1500×). (B) Representative laser scanning confocal microphotographs of GFP and GFAP or CD11β immunostaining in the ventral horn of the lumbar spinal cord of GFP2 (G and J), SOD1G93A (H and K) and symptomatic GFP2/SOD1G93A (I and L) mice. No colocalization is found with GFAP and CD11β. Scale bar 50 µm.
Since the accumulation of phosphorylated neurofilaments in the neuron perikarya as well as the increased levels of ubiquitin are considered markers of neuronal degeneration, we also examined the colocalization of GFP with SMI-31, an antibody specific for phosphorylated neurofilaments normally present only in axons (Fig. 9A–C), or with an anti-ubiquitin antibody (Fig. 9D–F), on serial sections from symptomatic GFP2/SOD1G93A lumbar spinal cord. About one-third of the GFP-positive cells in the ventral horns of GFP2/SOD1G93A mice also displayed an accumulation of SMI-31 immunoreactivity, while colocalization of GFP and ubiquitin was found in ∼50% of the UbG76V-GFP-positive cells (Fig. 9G).
Figure 9.
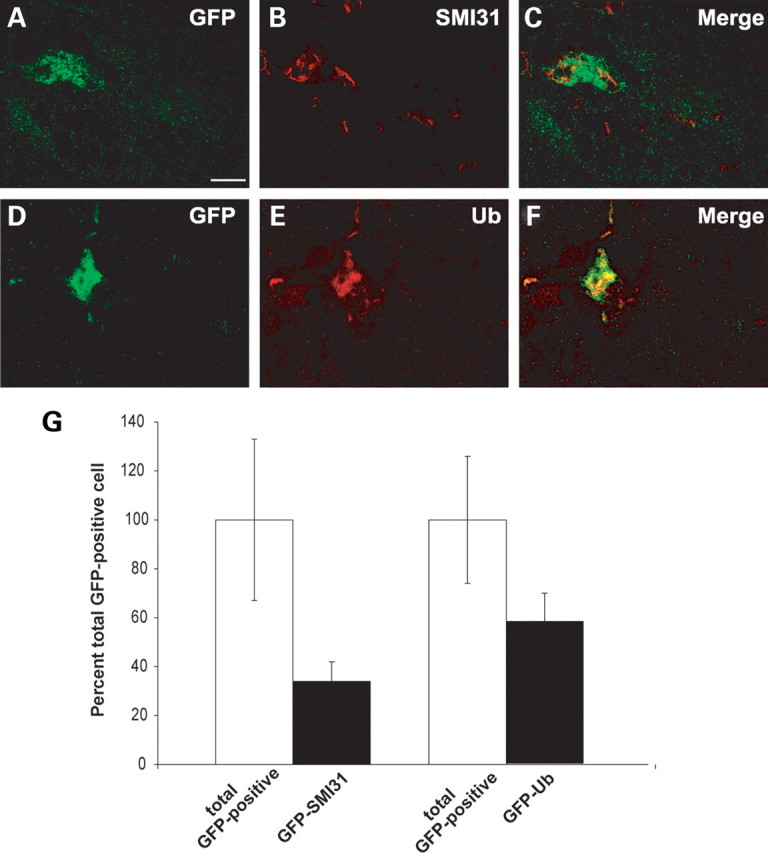
Representative laser scanning confocal microphotographs of GFP (A and D) and SMI-31 (B) or ubiquitin (E) immunostaining on ventral horn of the lumbar spinal cord of symptomatic GFP2/SOD1G93A mice. Colocalization is found between GFP and ubiquitin (C) or SMI-31 (F) labeling. Scale bar 20 µm. (G) Quantitative analysis of SMI-31 or ubiquitin-labeled neurons within GFP-positive population in the ventral horns of symptomatic GFP2/SOD1G93A mice. White histogram represents total GFP-positive cells, expressed in percentage (white bars, mean ± SEM of five mice), while black histogram represents the fraction that is also SMI-31 or ubiquitin-positive (black bars, mean ± SEM of five mice).
UbG76V-GFP transcript is up-regulated in small but not in large cells of double-transgenic mouse spinal cord
Although real-time PCR revealed no changes in UbG76V-GFP mRNA in spinal cord homogenates of symptomatic double-transgenic mice, the expression of this transcript was also evaluated at the cellular level by in situ hybridization as differences were found in the accumulation of UbG76V-GFP between motor neurons and other cells.
Figure 10 shows representative images of sections from the lumbar ventral horn of NTg (A), GFP2 (B) and GFP2/SOD1G93A (C) mice hybridized with a radiolabeled RNA probe for the UbG76V-GFP mRNA detected by autoradiographic emulsion. An intense silver grain density, higher than the background signal found in NTg mice, was visible in the cells from GFP2 and GFP2/SOD1G93A samples, indicating the specificity of the probe. The graph (Fig.10D) shows the levels of UbG76V-GFP transcript, quantified as grain densities, localized in small (100–250 µm2) and large cells, presumably motor neurons (>250 µm2) of GFP2/SOD1G93A mice compared with their GFP2 littermates. In ventral horns of symptomatic mice, a significant increase of the UbG76V-GFP mRNA was detected in small cells, but not in large cells, of GFP2/SOD1G93A mice compared with GFP2 mice. Although we do not have proof of the phenotype of these small cells, on the basis of their size we suggest that they are likely glial cells and/or small neurons but not motor neurons. Importantly, the elevation in transcript in these cells is not associated with an increase of UbG76V-GFP protein suggesting either a stabilization of the transcript and/or an efficient protein degradation in these cells.
Figure 10.

Representative autoradiographs of lumbar spinal cord sections from NTg (A), GFP2 (B) and symptomatic GFP2/SOD1G93A mice (C) hybridized with 35S-labelled anti-sense GFP-RNA probe. An intense silver grain density is observed in the cells of GFP2/SOD1G93A sections (B and C). Scale bar 100 µm. (D) Quantification of the grain density in cells from the ventral horn lumbar spinal cord of GFP2 and GFP2/SOD1G93A mice. Each histogram represents the mean ± SEM of five mice. Data were analyzed by Student’s t-test (*P < 0.05 compared with GFP). In symptomatic GFP2/SOD1G93A a significant increase of UbG76V-GFP mRNA is detected in small cells (<250 µm2), but not in large cells (>250 µm2) compared with GFP2 mice.
DISCUSSION
UPS dysfunction occurs in ALS motor neurons
The presence of inclusion bodies containing aggregated and ubiquitinated proteins in motor neurons is a typical feature both in ALS patients and in murine models of the disease. Although this finding has been linked to UPS alterations, so far this hypothesis has not been substantiated from in vivo studies. The present investigation, exploiting a mouse model which allows to monitor UPS activity in vivo (UbG76V-GFP mouse, 28), cross-bred with an established mouse model of ALS (SOD1G93A mouse, 4), demonstrates for the first time that a selective impairment of UPS function takes place in spinal motor neurons during disease progression. In fact, an increase in UbG76V-GFP reporter levels, indicating accumulation of the protein not degraded by the proteasome, was observed only in areas vulnerable to the pathology, such as the ventral spinal cord and the brainstem motor nuclei of double-transgenic GFP/SOD1G93A mice at symptomatic stages. This effect was related to a deficient degradation of the reporter protein and not to a transcriptional deregulation.
The demonstration that the UPS impairment was not observed prior to symptoms onset in GFP/SOD1G93A mice suggests that UPS dysfunction is unlikely to be a primary defect in the process of motor neuron degeneration, as it occurs later than other specific cellular abnormalities such as swelling of mitochondria, vacuolization, Golgi fragmentation or axonal transport impairment that are detectable since pre-symptomatic stages (31–34). The absence of detectable reporter accumulation at early stages of the disease process does not rule out the possibility that more subtle perturbations to UPS activity may be occurring in motor neurons earlier in the pathogenic process. Although insufficient to result in the detectable accumulation of reporter, subtle perturbations to UPS activity could contribute over time to the accumulation of misfolded proteins and hence play a role in the disease pathogenesis.
The fraction of motor neurons displaying GFP accumulation in symptomatic GFP/SOD1G93A spinal cord was only 11%. However, since at this stage of the disease 43% of the spinal motor neurons are already lost and it is impossible to know which percentage of them was GFP-positive, this effect could be underestimated. Moreover, the observation of a relative small proportion of GFP-positive motor neurons may depend on the fact that (i) a transient inhibition of the UPS occurs in a non-synchronous way in the total motor neuron population and (ii) in each single sample we are analyzing only a snapshot in a sequence of events occurring at different times in a progressive disease such as ALS. In line with this, we also found that not all the ventral horn neurons showing reporter immunoreactivity also accumulated phosphorylated neurofilaments or ubiquitin, two markers of degeneration in ALS. This suggests that, in motor neuron perikarya, the UPS dysfunction visualized by GFP detection precedes the massive accumulation of phosphorylated filaments and ubiquitinated proteins that occurs only at later stages of disease.
The dysfunction of UPS may be related to decreased levels of the 26S proteasome components that need to be properly assembled to form a proteolytically efficient complex. We previously showed a marked decline of constitutive 20S and 19S subunits in motor neurons of symptomatic SOD1G93A mice (10), in line with the decreased catalytic activity of 20S proteasome reported in lumbar spinal cord homogenates of the same mouse model (12). A pre-symptomatic reduction of the 20S constitutive catalytic β3 and β5 subunits without changes in their mRNAs was also reported (11). In the present study, we also found no changes in 20S β5 mRNA levels in the lumbar spinal cord of pre-symptomatic and symptomatic SOD1G93A mice, whereas a moderate decrease in the mRNA levels of the constitutive 19S and 20S α5 occurred at these stages of the disease, suggesting a control at the transcriptional level of these subunits. In line with this, a recent paper showed that the selective loss of a 19S subunit in a conditional knock-out mouse caused neurodegeneration and formation of intraneuronal inclusions (35).
The marked decreases in the transcript levels of almost all constitutive proteasome subunits that we detected in end-stage SOD1G93A mice are likely due to the massive loss of motor neurons. However, since UbG76V-GFP needs to be poly-ubiquitinated and recognized by the 26S complex in order to be degraded, a failure in other steps in this process, such as the availability of free ubiquitin, the action of ubiquitinating enzymes and the delivery of the substrate to the proteasome, could also account for the accumulation of the reporter protein in motor neurons.
Involvement of non-neuronal cells
Small cells, presumably glial cells, in the ventral horn of symptomatic GFP/SOD1G93A mice did not display accumulation of UbG76V-GFP but showed an increase in the reporter mRNA levels. This can be due to an increase in the reporter mRNA stability or in the transcriptional activation occurring in reactive astrocytes and microglia, as demonstrated by the remarkable increase of the mRNA levels of their selective markers, GFAP and CD68, respectively, in spinal cord homogenates of symptomatic SOD1G93A mice. A significant increase of UbG76V-GFP mRNA levels was previously observed in retinal neurons of double-transgenic mice resulting from the cross-breeding of a murine model of polyglutamine disease (SCA7266Q) with UbG76V-GFP mice (30). In that case however, and in contrast to our observations, the higher transcript levels correlated with a significant increase in the reporter protein levels. Therefore, the absence of reporter protein in hypertrophic astrocytes or reactive microglia in the spinal cord of our GFP/SOD1G93A mice, despite the increased UbG76V-GFP transcript levels, suggests that the UPS degradation system remains functional and may even be overactive in glial cells. This is in line with the evidence that hypertrophic astrocytes and reactive microglia in symptomatic SOD1G93A mice do not accumulate mutant SOD1 or ubiquitin (36). The increased expression of immunoproteasome subunits that we observed in spinal cord glial cells of SOD1G93A mice, in line with previous data (25,26), may account for their enhanced protein degradation activity.
This is also consistent with the significant up-regulation of the mRNA of LMP7 and LMP10 immunoproteasome subunits that we found in lumbar spinal cord homogenates of SOD1G93A mice already at symptomatic ages and which became even more pronounced for the LMP7 subunit at the end-stage of disease when the glia is maximally activated. Inducible proteasome subunits are up-regulated in response to inflammatory cytokines (37) and TNFα treatment was shown to increase LMP7 immunoreactivity in spinal cord astrocytes and microglia of both NTg and SOD1G93A mice (26). In line with this, we found a remarkable and progressive increase of TNFα mRNA in the spinal cord of SOD1G93A mice compared with NTg littermates, thus confirming previous studies (38,39). In contrast with previous data (26), we found no changes in the mRNA levels of LMP2 subunit in the spinal cord of SOD1G93A mice at any stage of the disease analyzed. This discrepancy may be due to the different transgenic mouse lines studied, high- versus low-number of transgene copies of SOD1G93A.
Since immunoproteasome is required for the efficient generation of certain MHC class I molecules on the cell surface, and for the activation of CD8+ T cells, we analyzed the transcript levels of these cells in lumbar spinal cord homogenates of SOD1G93A mice. Even if we can not exclude the infiltration of rare CD8+ T cells, whose markers remain undetectable by real-time PCR in homogenates, the finding of similar levels of CD8 mRNA in SOD1G93A and NTg mice at any stage of disease suggests that CD8+ T lymphocytes do not play a relevant role in the immune-inflammatory reactions occurring in SOD1 mutant mice. Additionally, the influx of peripheral lymphocytes is a rare event also in ALS patients and is only associated with the end-stage of the disease (40–42).
Role of immunoproteasome in motor neurons
Immunoproteasome is normally poorly expressed in the central nervous system (43), but its neuronal expression can be induced under pathological conditions (44). A role of immunoproteasome in ALS has been suggested by the increase in inducible subunit mRNA and protein levels in spinal cord homogenates of rodent models of the pathology (10,11,25,26). At the cellular level, however, the increased expression of immunoproteasome LMP7 (the only subunit investigated) has been principally attributed to glial cells (25,26). At variance with reported data, in spinal cord sections from SOD1G93A mice, compared with NTg and SOD1wt mice, we found increased labeling for the three immunoproteasome subunits also in motor neurons and from pre-symptomatic stages. This discrepancy can be explained by (i) the use of different animal models or (ii) technical reasons such as the use of paraffin sections instead of free floating vibratome or cryostat sections or a different antiserum. In support of our findings, an early increase in LMP7 mRNA expression has been recently reported in lumbar motor neurons of SOD1G93A mice using laser capture microdissection (11). This is at variance with the reduced LMP7 protein and mRNA reported in surviving motor neurons of human sporadic ALS patients (45), however, it might depend on the post-mortem interval affecting protein detection or to a loss of inducible subunits occurring in motor neurons at terminal stages of disease.
As for the functional significance, it can be speculated that an increased immunoproteasome expression occurs in motor neurons as a compensatory mechanism to cope with the presence of aberrant proteins affecting the constitutive subunits. The proteolytic activity achieved in this way is however unable to rescue motor neurons from fatal disease progression, since we also found immunoproteasome labeling in some degenerating neurons that accumulate mutant SOD1.
Recently, it has been shown that blocking the induction of LMP7 in astrocytes and microglia exacerbated the pathology in SOD1G93A transgenic rats and reduced their survival (25), suggesting that glial immunoproteasomes may play a protective role on motor neurons. However, when SOD1G93A mice were cross-bred with mice lacking the LMP2 immunoproteasome subunit (46) no changes in the disease progression and survival was observed, suggesting that this subunit is not directly involved in the pathogenesis of mutant SOD1-induced disease.
Conclusions and perspectives
In the present study, we have used a novel approach to monitor the functional status of the UPS in vivo at the cellular level, and, for the first time, report the occurrence of UPS impairment in motor neurons of a mouse model of familial ALS. The reduced expression of some constitutive subunits of the 26S proteasome observed in SOD1G93A mice may contribute to this impairment. In agreement with other studies we have also confirmed that the immunoproteasome, in particular its LMP7 subunit, is up-regulated during disease progression in the SOD1G93A mouse model of familial ALS. Its prevalent up-regulation in astrocytes and microglia in the absence of down-regulation of the constitutive proteasome subunits, may explain the lack of aggregates and ubiquitin accumulation in these cells.
On the basis of this evidence, we suggest that interventions aimed at increasing the activity of the proteasome in motor neurons could have beneficial effects in protecting these cells by preventing the aberrant protein aggregates and in slowing the progression of pathology in ALS.
Although little is known about the mechanisms of proteasome regulation, recent reports have suggested that expression of UPS subunits can be modulated by chemicals in mouse (47–49). In particular, the catalytic subunits of the 26S proteasome are inducible in mouse brain by oral treatment with chemicals that also reduce levels of mutant human SOD1 protein in murine neuroblastoma cells (47). Thus, testing these chemicals on the progression of the disease in SOD1 mutant mice may be warranted.
MATERIALS AND METHODS
Mouse models
Mice were maintained at a temperature of 21 ± 1°C with a relative humidity 55 ± 10% and 12 h of light. Food (standard pellets) and water were supplied ad libitum.
Procedures involving animals and their care were conducted according to the institutional guidelines, that are in compliance with national (D.L. no. 116, G.U. suppl. 40, Feb. 18, 1992, Circular No.8, G.U., 14 luglio 1994) and international laws and policies (EEC Council Directive 86/609, OJ L 358, 1 Dec.12, 1987; NIH Guide for the Care and use of Laboratory Animals, U.S. National Research Council, 1996).
Transgenic SOD1G93A mice expressing about 20 copies of mutant human SOD1 with a Gly93Ala substitution (B6SJL-TgNSOD-1-SOD1G93A-1Gur) or wild-type human SOD1 (SOD1wt) were originally obtained from Jackson Laboratories and maintained on a C57BL/6 genetic background at Harlan Italy S.R.L., Bresso (MI), Italy.
UbG76V-GFP mice express the transgene from a chicken β-actin promoter with a cytomegalovirus immediate early enhancer (28). Both UbG76V-GFP mouse strains, named UbG76V-GFP1 (GFP1) and UbG76V-GFP2 (GFP2), were used in this study. For the maintenance of these lines, GFP1 or GFP2 males were bred with C57BL/6 NTg females.
Since SOD1G93A females are sterile, double-transgenic GFP/SOD1G93A mice were obtained by cross-breeding SOD1G93A males with GFP1 or GFP2 females. Both SOD1G93A and GFP mice are derived from the same C57BL/6 strain, thus minimizing the confounding effects of different genetic backgrounds. The genotyping of the litters was conducted by polymerase chain reaction (Primer sequences: SOD1 F′: CAT CAG CCC TAA TCC ATC TGA, R′: CGC GAC TAA CAA TCA AAG TGA; UbG76V-GFP F′: ACC ACA TGA AGC AGC ACG ACT, R′: CTT GTA CAG CTC GTC CAT GC) on DNA extracted from tail biopsies. Mice were killed at 12, 16 and 22–23 weeks of age, corresponding, respectively, to pre-symptomatic, early symptomatic and end stage of progression of the motor dysfunction.
Cell cultures
Primary spinal neurons were prepared from spinal cord of 14-day-old GFP2 or NTg mouse embryos. Cells were plated into wells previously coated with a layer of confluent NTg astrocytes, cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2 and used after 5–6 days in vitro. The growth of UbG76V-GFP glial cells was minimized by adding to the culture medium the anti-mitotic drug Ara-c (10 µm).
The proteasome inhibitor MG132 (0.5–4.5 µm from Sigma) was added in the culture medium. After 6 h of treatment, the cells were rinsed in PBS, fixed with 4% paraformaldehyde in PBS for 30 min at RT and processed for immunohistochemistry as described below.
Immunohistochemistry
Anaesthetized mice were perfused with 4% paraformaldehyde and brain and spinal cord were removed, post-fixed and either frozen at −80°C after cryoprotection or sectioned with a vibratome. Light microscopic immunohistochemical analyses were done on lumbar spinal cord and brain sections (40 µm thick vibratome sections and 30 µm thick cryosections) and on primary spinal neurons.
The following mice were analyzed: at least three NTg and three SOD1G93A mice for each age and three 22-week-old SOD1wt mice; at least four GFP1, GFP2, GFP1/SOD1G93A and GFP2/SOD1G93A mice for each age examined.
The following primary antibodies were used: (i) monoclonal anti-20Sα (PW8195); polyclonal anti-19S regulator ATPase subunit Rpt3 (PW8175); polyclonal anti-LMP2 (PW8205); polyclonal anti-LMP10 (PW8150; polyclonal anti-LMP7 (PW8200) to detect proteasome subunits (all from Biomol,1:1000); (ii) monoclonal anti-human SOD1 (Stressgen, 1:1000); (iii) polyclonal anti-ubiquitin (DAKO, 1:200); (iv) monoclonal SMI-32 and SMI-31 (Sternberger Inc, 1:200) to visualize non-phosphorylated and phosphorylated epitopes of neurofilaments, respectively; (v) monoclonal anti-ChAT (Calbiochem, 1:200) to identify motor neurons; (vi) monoclonal anti-GFAP to detect astrocytes (Chemicon, 1:2500); (vii) monoclonal anti-CD11b to detect microglia (The Chemical Society, 1:1000); (viii) polyclonal anti-GFP (Molecular Probes, 1:3500).
For the immunoperoxidase procedure, tissue sections were treated with 1% hydrogen peroxide in PBS to inhibit endogenous peroxidases, blocked in 1% BSA in PBS containing 0.2% Triton X-100 for 30 min and then incubated overnight with the primary antibodies diluted in PBS containing 0.1% BSA. Immune reactions were revealed by 75 min incubation in the appropriate secondary biotinylated antiserum (goat anti-rabbit for polyclonals, horse anti-mouse or goat anti-rat for monoclonals, both from Vector Laboratories, 1:200), followed by 75 min incubation in the avidin-biotin-peroxidase Complex (Vector) and using diaminobenzidine as chromogen. Control sections processed with omission of the primary antiserum and developed under the same conditions gave no immunostaining.
For double-label immunofluorescence, the samples were incubated overnight in a mixture of polyclonal and monoclonal primary antibodies revealed by the appropriate secondary antibodies conjugated to different fluorochromes (Invitrogen, 1:200).
For the immunofluorescence detection of GFP on tissue sections and fixed cells, the anti-GFP antibody (1:10 000) was used and revealed with the TSA amplification kit (Cy5, Perkin Elmer), as already described (50).
In each immunohistochemical experiment, some of the sections were processed without the primary antibody, in order to verify the specificity of the staining. For the colocalization between GFP and ubiquitin, it was verified that no direct reaction was detectable between the secondary fluorescent antibody and the primary antibody amplified with tyramide procedure.
The number of motor neurons, of GFP-positive cells and of cells showing colocalization of GFP and SMI-31 or of GFP and ubiquitin was determined on serial sections (one every 10th) of lumbar spinal cord segment L3. A total of ten sections were acquired with a camera, using AnaliSYS software (Soft Imaging Systems, ver. 3.2); only the neurons showing intense GFP immunostaining were considered for the quantitative analysis. In these sections Nissl’s substance was visualized with NeuroTrace 500/525 Nissl stain (1:500 Invitrogen) in order to count the number of motor neurons in the ventral horn. Motor neurons were defined as large polymorphic cells with an area larger than 250 µm2. Data were expressed as mean number of neurons per section.
For the colocalization analyses, fluorescence-labeled samples were analyzed under an Olympus Fluoview laser scanning confocal microscope (Olympus BX61 light microscope) or a TCS NT confocal laser scanning microscope (Leica Lasertecknik GmbH, Heidelberg, Germany) equipped with a 75 mW Kripton/Argon mixed gas laser.
Western blot and immunoblotting
Mice were killed according to ethical procedures by decapitation. The spinal cord was flushed from the vertebral column and sectioned into cervical, thoracic and lumbar segments. The samples were immediately frozen on dry-ice and stored at −80°C.
Frozen lumbar spinal cords were homogenized by sonication in ice-cold homogenization buffer (20 mm HEPES pH 7.4, 100 mm NaCl, 10 mm NaF, 1% Triton X-100, 1 mm orthovanadate, 10 mm EDTA, 20 mm NEM and protease inhibitor cocktail Roche, Basel, Switzerland) and centrifuged at 10 000g for 10 min at 4°C. Protein concentration was determined by BCA Protein Assay (Pierce Biotechnology, Rockford, IL, USA) against BSA standards and homogenates were adjusted to equal protein concentration in homogenization buffer. Equal amounts of total protein (30 µg) were separated on 12% Tris–glycine polyacrylamide gels and electroblotted onto nitrocellulose membranes (Protran, Scheicher and Schuell). To check for even protein loading and transfer, membranes were briefly immersed in Ponceau-S stain (0.2% Ponceau-S in 3% trichloro-acetic acid) and rinsed in water. Membranes were then placed between plastic sheets and scanned for later analysis.
For detection of GFP reporter protein, membranes were immunoblotted with mixed-mouse monoclonal anti-GFP antibody (1:2000 from Roche), followed by HRP-conjugated anti-mouse secondary antibody (1:4000 from GE Helathcare, UK) and visualized by enhanced chemiluminescent. Films were scanned on a CanoScan 5200F scanner (Canon) and band intensities measured using Image J (public domain software).
The obtained values were corrected for Ponceau staining of total protein and expressed as a percentage of the basal value detected in the single transgenic UbG76V-GFP control tissues. Statistical comparisons were performed using Student’s t-test.
Real-time PCR
Mice were killed in accordance with ethical procedures (Equithesin: 1% Phenobarbital, 4% chloral hydrate) by decapitation. The brain was removed and the hippocampus was isolated. The spinal cord was flushed out from the vertebral column and sectioned into cervical, thoracic and lumbar segments. The samples were immediately frozen on dry-ice and stored at −80°C. Total RNA was extracted using the Trizol method (Invitrogen), purified according to the manufacturers’ recommendation and resuspended in sterile water. RNA samples were treated with DNase I (1× DNase I reaction buffer and 2 U of DNAse I from Invitrogen).
cDNA used for the Taq Man technology was obtained by reverse transcription of DNase-treated RNA using the High Capacity reverse transcription kit (1× RT Buffer, Multiscribe MuLV reverse transcriptase 1.25 U/μl, RNAsi inhibitor 1 U/μl, Random Primers 1× and 4 mm deoxyNTP mix from Applied Biosystems).
cDNA used for real-time PCR experiments with the SYBR Green chemistry was obtained by reverse transcription of total RNA using Gene Amplification RNA PCR-core kit (1× PCR Buffer, MuLV reverse transcriptase 2.5 U/μl, RNAsi inhibitor 1 U/μl, oligo dT 16 2.5 µm and deoxyNTP 1 mm each, Applied Biosystems).
Real-time PCR quantification for β-actin, GFAP, CD68 and TNFα was performed on cDNA specimens in triplicate, using 1× Universal PCR master mix and 1× mix containing specific probes (Supplementary Material, Table S1) (Taq Man Gene expression assays, Applied Biosystems). All the constitutive and inducible proteasome subunits, the CD8 and the GFP, were quantified on SYBR Green chemistry-derived cDNA specimens (in triplicate) using 1× SYBR Green PCR master mix and specific primers (0.3 µm each, Supplementary Material, Table S2).
The levels of all transcripts were normalized to β-actin mRNA levels and the ratio values in transgenic mice were expressed as percentage of the ratio estimated from NTg mice. Levels of GFP transcript in double-transgenic mice (GFP1/SOD1G93A) were expressed as percentage of the levels measured in the single GFP1 littermates. Mean values of the triplicate for each animal (four mice per group) were used as individual data for statistical analysis using Student’s t-test.
In situ hybridization
Mice were killed in accordance with ethical procedures, by decapitation. The spinal cord was flushed out from the vertebral column and sectioned into cervical, thoracic and lumbar segments. The samples were frozen in 2-methylbutane at −45°C and conserved at −80° until use. Lumbar spinal cords sections (14 µm) were cut using a cryostat, mounted on poly-l-lysine-coated microscope slides and fixed in 4% paraformaldehyde, acetylated (0.1 m triethanolamine and 0.25% acetic anhydride in 0.9% NaCl), dehydrated through a graded series of ethanol, delipidated in chloroform, air dried and stored frozen at −80°C.
35S-labeled RNA probes were obtained by in vitro transcription (51) and in situ hybridization was performed as previously described (52). Briefly, fixed spinal cord sections (14 µm, L2–L4 levels) of GFP/SOD1G93A and GFP mice (four mice in each group) were hybridized with 35S-labelled RNA probes complementary to GFP mRNAs, which were prepared through in vitro transcription of cDNA fragments. GFP insert (514 bp in length) was cloned in pCDNA3 plasmid, and antisense and sense transcripts were obtained by using SP6 and T7 RNA polymerase enzymes, respectively.
The templates were hydrolyzed in mild alkaline buffer at 60°C to obtain fragments of about 200 bp in length. The mixtures were neutralized and the 35S-labelled riboprobes were purified by G-50 Sephadex Quick Spin Columns (Roche). Probes were diluted to 4000 cpm/µl with hybridization buffer and slides were incubated with the 35S-labelled RNA probes overnight at 55°C in sealed humidified chambers. After hybridization, sections were washed, treated with RNase A and dehydrated through a graded series of ethanol as previously described (52).
The slides were exposed to Beta-max film (Amersham Biosciences) for 7 days, dipped in photographic emulsion (Ilford K5 diluted 1:1 with 0.1% Tween 20) and exposed for 15 days at 4°C. Sections were then developed, counterstained with cresyl violet and examined by light and dark field microscope before the quantitative analysis of the grain density. The specificity of the in situ hybridization for the transcript was verified by the absence of the signal using sense radiolabeled probe.
Grain density in single cell was quantitatively evaluated using AnaliSYS software (Soft Imaging Systems, ver. 3.2). Cresyl violet stained cells were used to define the circular frame outlining the cell and the grain density over a single cell was expressed as number of grains/μm2 cell area. At least two sections from each animal were analyzed and the cells were subdivided on the basis of their size. Mean values of grain density for each size category in each animal were used for statistical analysis by Student’s t-test.
SUPPLEMENTARY MATERIAL
FUNDING
This study was supported by grants from Telethon (GGP06063 to C.B. and S.D.B.). C.J.M. and N.P.D. were supported by the Swedish Research Council and the Nordic Centre of Excellence Neurodegeneration.
Conflict of Interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Rowland L.P., Shneider N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 2.Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 3.Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O’Regan J.P., Deng H.X., et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 4.Gurney M.E., Pu H., Chiu A.Y., Dal Canto M.C., Polchow C.Y., Alexander D.D., Caliendo J., Hentati A., Kwon Y.W., Deng H.X., et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 5.Bendotti C., Carri M.T. Lessons from models of SOD1-linked familial ALS. Trends Mol. Med. 2004;10:393–400. doi: 10.1016/j.molmed.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 6.Bruijn L.I., Becher M.W., Lee M.K., Anderson K.L., Jenkins N.A., Copeland N.G., Sisodia S.S., Rothstein J.D., Borchelt D.R., Price D.L., et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 7.Leigh P.N., Whitwell H., Garofalo O., Buller J., Swash M., Martin J.E., Gallo J.M., Weller R.O., Anderton B.H. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain. 1991;114:775–788. doi: 10.1093/brain/114.2.775. [DOI] [PubMed] [Google Scholar]
- 8.Migheli A., Autilio-Gambetti L., Gambetti P., Mocellini C., Vigliani M.C., Schiffer D. Ubiquitinated filamentous inclusions in spinal cord of patients with motor neuron disease. Neurosci. Lett. 1990;114:5–10. doi: 10.1016/0304-3940(90)90419-a. [DOI] [PubMed] [Google Scholar]
- 9.Morrison B.M., Janssen W.G., Gordon J.W., Morrison J.H. Time course of neuropathology in the spinal cord of G86R superoxide dismutase transgenic mice. J. Comp. Neurol. 1998;391:64–77. doi: 10.1002/(sici)1096-9861(19980202)391:1<64::aid-cne6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 10.Cheroni C., Peviani M., Cascio P., Debiasi S., Monti C., Bendotti C. Accumulation of human SOD1 and ubiquitinated deposits in the spinal cord of SOD1G93A mice during motor neuron disease progression correlates with a decrease of proteasome. Neurobiol. Dis. 2005;18:509–522. doi: 10.1016/j.nbd.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Kabashi E., Agar J.N., Hong Y., Taylor D.M., Minotti S., Figlewicz D.A., Durham H.D. Proteasomes remain intact, but show early focal alteration in their composition in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2008;105:2353–2366. doi: 10.1111/j.1471-4159.2008.05317.x. [DOI] [PubMed] [Google Scholar]
- 12.Kabashi E., Agar J.N., Taylor D.M., Minotti S., Durham H.D. Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2004;89:1325–1335. doi: 10.1111/j.1471-4159.2004.02453.x. [DOI] [PubMed] [Google Scholar]
- 13.Puttaparthi K., Wojcik C., Rajendran B., DeMartino G.N., Elliott J.L. Aggregate formation in the spinal cord of mutant SOD1 transgenic mice is reversible and mediated by proteasomes. J. Neurochem. 2003;87:851–860. doi: 10.1046/j.1471-4159.2003.02028.x. [DOI] [PubMed] [Google Scholar]
- 14.Urushitani M., Kurisu J., Tateno M., Hatakeyama S., Nakayama K., Kato S., Takahashi R. CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J. Neurochem. 2004;90:231–244. doi: 10.1111/j.1471-4159.2004.02486.x. [DOI] [PubMed] [Google Scholar]
- 15.Urushitani M., Kurisu J., Tsukita K., Takahashi R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- 16.Ciechanover A., Schwartz A.L. The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proc. Natl Acad. Sci. USA. 1998;95:2727–2730. doi: 10.1073/pnas.95.6.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeMartino G.N., Slaughter C.A. The proteasome, a novel protease regulated by multiple mechanisms. J. Biol. Chem. 1999;274:22123–22126. doi: 10.1074/jbc.274.32.22123. [DOI] [PubMed] [Google Scholar]
- 18.Glickman M.H., Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 19.Voges D., Zwickl P., Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 20.Goldberg A.L., Cascio P., Saric T., Rock K.L. The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol. Immunol. 2002;39:147–164. doi: 10.1016/s0161-5890(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 21.Di Noto L., Whitson L.J., Cao X., Hart P.J., Levine R.L. Proteasomal degradation of mutant superoxide dismutases linked to amyotrophic lateral sclerosis. J. Biol. Chem. 2005;280:39907–39913. doi: 10.1074/jbc.M506247200. [DOI] [PubMed] [Google Scholar]
- 22.Hoffman E.K., Wilcox H.M., Scott R.W., Siman R. Proteasome inhibition enhances the stability of mouse Cu/Zn superoxide dismutase with mutations linked to familial amyotrophic lateral sclerosis. J. Neurol. Sci. 1996;139:15–20. [PubMed] [Google Scholar]
- 23.Hyun D.H., Lee M., Halliwell B., Jenner P. Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins. J. Neurochem. 2003;86:363–373. doi: 10.1046/j.1471-4159.2003.01841.x. [DOI] [PubMed] [Google Scholar]
- 24.Johnston J.A., Dalton M.J., Gurney M.E., Kopito R.R. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl Acad. Sci. USA. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahtoniemi T., Goldsteins G., Keksa-Goldsteine V., Malm T., Kanninen K., Salminen A., Koistinaho J. Pyrrolidine dithiocarbamate inhibits induction of immunoproteasome and decreases survival in a rat model of amyotrophic lateral sclerosis. Mol. Pharmacol. 2007;71:30–37. doi: 10.1124/mol.106.028415. [DOI] [PubMed] [Google Scholar]
- 26.Puttaparthi K., Elliott J.L. Non-neuronal induction of immunoproteasome subunits in an ALS model: possible mediation by cytokines. Exp. Neurol. 2005;196:441–451. doi: 10.1016/j.expneurol.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 27.Neefjes J., Dantuma N.P. Fluorescent probes for proteolysis: tools for drug discovery. Nat. Rev. 2004;3:58–69. doi: 10.1038/nrd1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindsten K., Menendez-Benito V., Masucci M.G., Dantuma N.P. A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol. 2003;21:897–902. doi: 10.1038/nbt851. [DOI] [PubMed] [Google Scholar]
- 29.Kristiansen M., Deriziotis P., Dimcheff D.E., Jackson G.S., Ovaa H., Naumann H., Clarke A.R., van Leeuwen F.W., Menendez-Benito V., Dantuma N.P., et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell. 2007;26:175–188. doi: 10.1016/j.molcel.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Bowman A.B., Yoo S.Y., Dantuma N.P., Zoghbi H.Y. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum. Mol. Genet. 2005;14:679–691. doi: 10.1093/hmg/ddi064. [DOI] [PubMed] [Google Scholar]
- 31.Bendotti C., Calvaresi N., Chiveri L., Prelle A., Moggio M., Braga M., Silani V., De Biasi S. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J. Neurol. Sci. 2001;191:25–33. doi: 10.1016/s0022-510x(01)00627-x. [DOI] [PubMed] [Google Scholar]
- 32.Mourelatos Z., Gonatas N.K., Stieber A., Gurney M.E., Dal Canto M.C. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc. Natl Acad. Sci. USA. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williamson T.L., Cleveland D.W. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 34.Wong P.C., Pardo C.A., Borchelt D.R., Lee M.K., Copeland N.G., Jenkins N.A., Sisodia S.S., Cleveland D.W., Price D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 35.Bedford L., Hay D., Devoy A., Paine S., Powe D.G., Seth R., Gray T., Topham I., Fone K., Rezvani N., et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 2008;28:8189–8198. doi: 10.1523/JNEUROSCI.2218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basso M., Massignan T., Samengo G., Cheroni C., De Biasi S., Salmona M., Bendotti C., Bonetto V. Insoluble mutant SOD1 is partly oligoubiquitinated in amyotrophic lateral sclerosis mice. J. Biol. Chem. 2006;281:33325–33335. doi: 10.1074/jbc.M603489200. [DOI] [PubMed] [Google Scholar]
- 37.Bochtler M., Ditzel L., Groll M., Hartmann C., Huber R. The proteasome. Annu. Rev. Bioph. Biom. 1999;28:295–317. doi: 10.1146/annurev.biophys.28.1.295. [DOI] [PubMed] [Google Scholar]
- 38.Elliott J.L. Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Brain Res. Mol. Brain Res. 2001;95:172–178. doi: 10.1016/s0169-328x(01)00242-x. [DOI] [PubMed] [Google Scholar]
- 39.Ishigaki S., Niwa J., Ando Y., Yoshihara T., Sawada K., Doyu M., Yamamoto M., Kato K., Yotsumoto Y., Sobue G. Differentially expressed genes in sporadic amyotrophic lateral sclerosis spinal cords–screening by molecular indexing and subsequent cDNA microarray analysis. FEBS Lett. 2002;531:354–358. doi: 10.1016/s0014-5793(02)03546-9. [DOI] [PubMed] [Google Scholar]
- 40.Graves M.C., Fiala M., Dinglasan L.A., Liu N.Q., Sayre J., Chiappelli F., van Kooten C., Vinters H.V. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph. Lateral Scler. 2004;5:213–219. doi: 10.1080/14660820410020286. [DOI] [PubMed] [Google Scholar]
- 41.Kawamata T., Akiyama H., Yamada T., McGeer P.L. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992;140:691–707. [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang R., Gascon R., Miller R.G., Gelinas D.F., Mass J., Hadlock K., Jin X., Reis J., Narvaez A., McGrath M.S. Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS) J. Neuroimmunol. 2005;159:215–224. doi: 10.1016/j.jneuroim.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 43.Noda C., Tanahashi N., Shimbara N., Hendil K.B., Tanaka K. Tissue distribution of constitutive proteasomes, immunoproteasomes, and PA28 in rats. Biochem. Biophy. Res. Commun. 2000;277:348–354. doi: 10.1006/bbrc.2000.3676. [DOI] [PubMed] [Google Scholar]
- 44.Diaz-Hernandez M., Hernandez F., Martin-Aparicio E., Gomez-Ramos P., Moran M.A., Castano J.G., Ferrer I., Avila J., Lucas J.J. Neuronal induction of the immunoproteasome in Huntington’s disease. J. Neurosci. 2003;23:11653–11161. doi: 10.1523/JNEUROSCI.23-37-11653.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen S., Heath P.R., Kirby J., Wharton S.B., Cookson M.R., Menzies F.M., Banks R.E., Shaw P.J. Analysis of the cytosolic proteome in a cell culture model of familial amyotrophic lateral sclerosis reveals alterations to the proteasome, antioxidant defenses, and nitric oxide synthetic pathways. J. Biol. Chem. 2003;278:6371–6383. doi: 10.1074/jbc.M209915200. [DOI] [PubMed] [Google Scholar]
- 46.Puttaparthi K., Van Kaer L., Elliott J.L. Assessing the role of immuno-proteasomes in a mouse model of familial ALS. Exp. Neurol. 2007;206:53–58. doi: 10.1016/j.expneurol.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwak M.K., Cho J.M., Huang B., Shin S., Kensler T.W. Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 2007;43:809–817. doi: 10.1016/j.freeradbiomed.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 48.Kwak M.K., Wakabayashi N., Greenlaw J.L., Yamamoto M., Kensler T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell Biol. 2003;23:8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwak M.K., Wakabayashi N., Itoh K., Motohashi H., Yamamoto M., Kensler T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 50.Tortarolo M., Veglianese P., Calvaresi N., Botturi A., Rossi C., Giorgini A., Migheli A., Bendotti C. Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol. Cell Neurosci. 2003;23:180–192. doi: 10.1016/s1044-7431(03)00022-8. [DOI] [PubMed] [Google Scholar]
- 51.Bendotti C., Hohmann C., Forloni G., Reeves R., Coyle J.T., Oster-Granite M.L. Developmental expression of somatostatin in mouse brain. II. In situ hybridization. Brain Res. Dev. Brain Res. 1990;53:26–39. doi: 10.1016/0165-3806(90)90121-e. [DOI] [PubMed] [Google Scholar]
- 52.Bendotti C., Tortarolo M., Suchak S.K., Calvaresi N., Carvelli L., Bastone A., Rizzi M., Rattray M., Mennini T. Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J. Neurochem. 2001;79:737–746. doi: 10.1046/j.1471-4159.2001.00572.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.