

Abstract
Highly efficient and specific biomolecular recognition requires both affinity and specificity. Previous quantitative descriptions of biomolecular recognition were mostly driven by improving the affinity prediction, but lack of quantification of specificity. We developed a novel method SPA (SPecificity and Affinity) based on our funneled energy landscape theory. The strategy is to simultaneously optimize the quantified specificity of the “native” protein-ligand complex discriminating against “non-native” binding modes and the affinity prediction. The benchmark testing of SPA shows the best performance against 16 other popular scoring functions in industry and academia on both prediction of binding affinity and “native” binding pose. For the target COX-2 of nonsteroidal anti-inflammatory drugs, SPA successfully discriminates the drugs from the diversity set, and the selective drugs from non-selective drugs. The remarkable performance demonstrates that SPA has significant potential applications in identifying lead compounds for drug discovery.
Biomolecular recognition is central to cellular processes mediated by the formation of complexes between biomolecular receptors and their ligands. Understanding of biomolecular recognition is one of the most important issues in modern molecular biology1,2 and has direct applications in drug discovery and design3,4. The fast and accurate prediction of a ligand specifically binding to a target protein is a crucial step for lead discovery. During the past two decades, considerable efforts have been devoted to the development of docking algorithms and scoring functions5. There are many docking algorithms available, however, imperfections of scoring functions continue to be a major limiting factor6,7.
Highly efficient and specific biomolecular recognition requires both affinity and specificity8,9,10,11. The stability of the complex is determined by the affinity while the specificity is controlled by either partner binding to other competitive biomolecules discriminatively. The current scoring functions of protein-ligand binding12, whether force-field based, empirical, or knowledge-based scoring functions, are mainly focused on improving the ability of predicting the known binding affinities observed in experiments as accurately as possible. The strategy of developing these scoring functions seeks to optimize the stability based on the combination of energetics and shape complementarity without explicit consideration of binding specificity.
However, high affinity does not guarantee high specificity which is critical for function, e.g. drug-target recognition. To design a drug, one has to design a lead compound binding to a specific target receptor rather than others indiscriminately for avoiding the possible side effects. According to the Boltzman distribution (P ∼ exp[−F/KT]), the equilibrium population is exponentially dependent on the binding free energy. A gap in binding free energy or affinity will lead to significant population discrimination between the specific complex and non-specific ones. Thus to develop a scoring function, the strategy should satisfy the requirement that the stability of the specific complex is maximized while the stability of competing complexes is minimized, which can guarantee both the stability and the specificity for the specific complex.
The reason that the specificity usually was not taken into account explicitly is that the description of binding specificity was challenging to quantify. The conventional definition (Fig. 1a) of specificity is the ability of a ligand to specifically bind to a protein against other proteins, namely the relative difference in affinity of one specific protein-ligand complex to others8,9,10,11. The quantification of conventional specificity is challenging since specificity requires comparison of the affinities of all the different receptors with the same ligand. The receptor universe is huge and the information is often incomplete on those proteins. We proposed an alternative way to quantitatively determine the ligand-receptor binding specificity13,14. The new view of specificity is that a specific and native binding site of a ligand to its receptor is preferred to other binding sites on the same receptor (Fig. 1b). This concept is based on the assumption that a ligand binding to many protein receptors is equivalent to its binding to them with N and C terminus of these protein receptors linked together, leading to binding to an effectively large protein. Therefore, if the target protein is large enough, one can think of it as composed of many different segments each mimicking a protein receptor. The conventional specificity as relative affinity against different receptors now becomes specificity of the native binding mode against the other binding modes for this large target protein (Fig. 1a,b).
Figure 1. Illustration of the equivalence of conventional specificity to intrinsic specificity.

(a) Different receptors (green) binding to the same ligand (red) with the corresponding energy spectrum. (b) Different binding states (modes) of a particular ligand to its receptor. (c) Different ligands binding to the same receptor.
Also, one receptor protein with different sites binding with a ligand is similar to the case of the whole universe of different ligands binding with the same receptor (Fig. 1c). If the protein target size is large enough, then the spatial contact interactions can be sampled enough to cover all the contact interactions appeared in the ligand binding to different receptors. A recent work on molecular dynamics of a ligand searching for binding to the receptor has validated this assumption. In that work, Shaw et al.15 carried out a relatively long molecular dynamics simulation in which the inhibitor PP1 was initially placed at a random location to search the docking sites on the protein Src kinase. Persistent and noteworthy intermediate conformations with the inhibitor located diversely on the surface of the kinase were observed in the binding process, indicating multiple competing binding sites do exist on the same receptor.
Similar to protein folding, the binding process of protein-ligand can be physically quantified and visualized as a funnel-like energy landscape towards the native binding state with local roughness along the binding paths13,14,16,17,18,19,20,21,22,23. According to the theory of energy landscape (Fig. 2), the native conformation of the binding complex is the conformation with the lowest binding energy and the energies of the non-native conformations follow a statistical Gaussian distribution. A dimensionless quantity 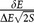 (termed as intrinsic specificity ratio (ISR), where δE is the energy gap between the native binding state and the average non-native binding states, ΔE is the energy variance of the non-native states and S is the configurational entropy) is defined to describe the magnitude of intrinsic specificity. A large ISR indicates a high level of discrimination of the native binding state from the non-native binding states, which means a high specificity. Therefore, ISR provides a quantitative measure of intrinsic specificity that can be determined without evaluating the conventional specificity by exploring the whole set of receptor or ligand universe.
(termed as intrinsic specificity ratio (ISR), where δE is the energy gap between the native binding state and the average non-native binding states, ΔE is the energy variance of the non-native states and S is the configurational entropy) is defined to describe the magnitude of intrinsic specificity. A large ISR indicates a high level of discrimination of the native binding state from the non-native binding states, which means a high specificity. Therefore, ISR provides a quantitative measure of intrinsic specificity that can be determined without evaluating the conventional specificity by exploring the whole set of receptor or ligand universe.
Figure 2. Distribution of energy and funneled energy landscape of the biomolecular binding.

(a) Multiple docking complex conformations of COX-2 with a selective drug (SC-558), the native pose of the drug shown in red and sticks while other decoys shown in blue and lines. (b) The binding energy spectrum for different binding decoys. (c) The corresponding distribution of binding energies where the standard deviation of energy (ΔE), average energy of decoys (< ED >) and the energy gap (δE) are represented. (d) Energy landscape of binding with a funneled shape towards the native state.
Given the inherent limitations of current scoring functions and the demands for the practical way of evaluating specificity, we developed a novel scoring function by optimizing both intrinsic specificity and affinity in this study (named as SPA, SPA stands for SPecificity and Affinity. The development flowchart of SPA is shown in Supplementary Fig. 1). The optimizing strategy for SPA is to simultaneously reach the maximization of the performances on both the specificity and the affinity predictions of the training set. SPA was validated by testing the benchmark set and the performance was compared with 16 previous scoring functions. The test results showed that SPA outperformed all these previous scoring functions. SPA was also applied for a target protein cyclooxygenase-2 (COX-2) of nonsteroidal anti-inflammatory drugs (NSAIDs) for discrimination of the drugs against the diversity set, and selective drugs against non-selective drugs. The results suggested that more reliable lead compounds can be screened with SPA through two dimensional screening of intrinsic specificity ISR and affinity.
Results
Binding pose prediction
The process of a ligand binding to a protein can be thought as a conformational search guided by a scoring function, and the final destination is to look for the “native” binding pose with the best score. Whether the scoring function can select out the best-scored binding pose which resembles the one in the crystal structure closely determines the performance of the scoring function for identifying the “native” binding conformation. Normally, the root mean square deviation (RMSD) is taken as the measure of the structural closeness between the scored binding poses and the “native” binding conformation which is the ligand pose in the crystal structure here. If the RMSD value of the best-scored binding pose is less than a predefined cutoff, it is considered as a successful recognition of the “native” or “near-native” binding pose by the scoring function.
To make a comparison with other scoring functions, the success rate for the benchmark set was calculated by SPA and compared to 16 scoring functions implemented in the mainstream commercial softwares or available from academic research groups24 (Table 1). The success rates of SPA as well as the other 16 scoring functions under five different cutoffs (1.0, 1.5, 2.0, 2.5 and 3.0 Å) clearly shows that SPA performs the best among all the scoring functions no matter what criterion of RMSD cutoff is used, suggesting that SPA is very successful on the ability to identify “native” or “near-native” binding poses.
Table 1. Success rates of identifying the “native” or “near-native” conformations under different RMSD cutoffs.
| Scoring Functiona | 1.0Å | 1.5Å | 2.0Å | 2.5Å | 3.0Å |
|---|---|---|---|---|---|
| SPA | 78.5 | 83.1 | 84.7 | 87.6 | 93.2 |
| GOLD/ASP | 69.3 | 79.2 | 82.5 | 85.2 | 89.1 |
| DS/PLP1 | 65.0 | 72.1 | 75.4 | 78.7 | 84.2 |
| DrugScorePDB/PairSurf | 62.8 | 69.4 | 74.3 | 77.6 | 81.4 |
| GlideScore/SP | 54.6 | 64.5 | 73.2 | 76.0 | 84.7 |
| DS/LigScore2 | 54.1 | 62.8 | 71.6 | 75.4 | 80.3 |
| GOLD/ChemScore | 54.6 | 62.8 | 70.5 | 71.6 | 79.2 |
| GOLD/GoldScore | 51.9 | 61.2 | 68.9 | 72.1 | 80.9 |
| X-Score1.2/HMScore | 51.4 | 59.6 | 68.3 | 72.1 | 78.1 |
| SYBYL/F-Score | 54.6 | 61.7 | 64.5 | 68.3 | 73.8 |
| SYBYL/ChemScore | 40.4 | 49.7 | 60.1 | 65.6 | 71.6 |
| DS/Ludi2 | 41.5 | 48.6 | 57.4 | 61.7 | 67.2 |
| SYBYL/PMF-Score | 37.2 | 41.5 | 48.1 | 53.0 | 56.8 |
| DS/Jain | 25.7 | 36.1 | 44.8 | 54.6 | 64.5 |
| DS/PMF | 32.2 | 36.1 | 43.7 | 47.5 | 53.6 |
| SYBYL/G-Score | 25.1 | 35.5 | 41.5 | 48.6 | 56.3 |
| SYBYL/D-Score | 15.3 | 23.5 | 30.6 | 39.3 | 47.5 |
aThe results except SPA are obtained from the literature24.
In practice, besides the best-scored binding pose, multiple other binding poses with good scores can also be selected out as putative “native” binding poses. Namely, it is probable in molecular docking to select a few binding poses with top-ranked scores for further evaluation in hierarchical database screenings25. The success rates were compared for the binding poses with top five scores under the commonly used criterion of RMSD cutoff ( = 2.0 Å) (Supplementary Table 1). We can see that SPA yields more than 90% success rate to identify the “native” binding pose when the binding scores are considered from top two to five binding scores. It has comparable performance as other three top-ranked scoring function GOLD/ASP, DrugScorePDB/PairSurf and DS/PLP1. This result further validates the outstanding performance of SPA on the “native” or “near-native” binding pose identification.
A high success rate of identifying the “native” conformation implies that the binding poses structurally close to the “native” conformation have high binding scores in energetics. This structure-energy correlation is consistent with the concept of funnel-shaped energy landscape of protein-ligand binding13, where the “native” binding conformation with lowest energy locates at the global minimum of the energy landscape and the conformations with low energies are structurally similar to the “native” conformation. According to the energy landscape theory16,17, it is promising that SPA with the highest success rate to identify the “native” conformation can search the global minimum with a fast convergence if it is applied to guide the conformational sampling in molecular docking. The high success rate also implies the capability of SPA to discriminate the “native” binding mode against decoys. This leads to a better quantification and discrimination of intrinsic specificity and therefore the (conventional) specificity of biomolecular recognition.
Binding affinity prediction
In addition to the prediction of binding pose, the prediction of binding affinity is another important criterion to evaluate the performance of scoring function. In contrast to binding pose prediction which emphasizes on the discrimination of the “native” conformation from the “non-native” decoys for each protein-ligand complex, the prediction of binding affinity relates to the ability of reproducing the experimentally measured binding affinities for different types of protein-ligand complexes. It determines the accuracy of binding scores predicted by the scoring functions compared with the experimental measurements. Due to scaling, the scoring functions usually cannot reproduce the absolute values of experimental binding affinities, the correlation between the predicted and experimental measured binding affinities is widely used to evaluate the accuracy of binding affinity prediction. Pearson correlation coefficients (CP) and Spearman rank correlation coefficients (CS) between the binding scores and the known binding constants were computed.
It can be seen (Table 2) that SPA gives the best correlations of both CP and CS. Compared to other scoring functions, this performance of SPA is surprisingly good since no other scoring functions simultaneously rank on the tops for both the predictions of binding pose (Table 1) and binding affinity (Table 2). For example, X-Score/HMScore performs well on the prediction of binding affinity but moderately on the prediction of binding pose. GOLD/ASP is able to identify the correct binding pose with a second highest success rate whereas it is less successful to reproduce the binding affinities. It is worth noticing that only SPA and X-Score/HMScore achieve CP over 0.6 in predicting affinity which is much superior to other scoring functions (Supplementary Fig. 2).
Table 2. Correlations between the predicted binding affinity and experimentally measured binding affinity.
| Scoring Functiona | CP | CS |
|---|---|---|
| SPA | 0.668 | 0.733 |
| X-Score/HMScore | 0.644 | 0.705 |
| DrugScoreCSD/PairSurf | 0.569 | 0.627 |
| SYBYL/ChemScore | 0.555 | 0.585 |
| DS/PLP1 | 0.545 | 0.588 |
| GOLD/ASP | 0.534 | 0.577 |
| SYBYL/G-Score | 0.492 | 0.536 |
| DS/Ludi3 | 0.487 | 0.478 |
| DS/LigScore2 | 0.464 | 0.507 |
| GlideScore/XP | 0.457 | 0.432 |
| DS/PMF | 0.445 | 0.448 |
| GOLD/ChemScore | 0.441 | 0.452 |
| NHA | 0.431 | 0.517 |
| SYBYL/D-Score | 0.392 | 0.447 |
| DS/Jain | 0.316 | 0.346 |
| GOLD/GoldScore | 0.295 | 0.322 |
| SYBYL/PMF-Score | 0.268 | 0.273 |
| SYBYL/F-Score | 0.216 | 0.243 |
aThe results except SPA are obtained from the literature24.
The best performance of SPA on the prediction of both binding pose and binding affinity suggests that the optimization strategy calibrated to improve specificity and affinity for the development of SPA is the correct route to explore the protein-ligand recognition. SPA is not only capable of discriminating the specific “native” conformation out of a large number of decoys by their scores but also accurately predicting the binding affinities of different protein-ligand complexes. This result is encouraging and motivates us to apply SPA in the virtual screening to identify the lead compounds for drug discovery with both affinity and specificity.
Virtual screening test
With the excellent performance of SPA on the benchmark test, we want to evaluate the ability of SPA in real virtual screening test. The enzyme cyclooxygenase-2 (COX-2) was chosen as our target protein model which is the target of nonsteroidal anti-inflammatory drugs (NSAIDs) for reducing fever and inflammation, such as the commonly-taken drugs aspirin, motrin, telenoid, and advil. The virtual screening for COX-2 is challenging. Besides the importance to discriminate the drugs from the diversity set, it is more important to distinguish selective and nonselective drugs since selective drugs is more potent to inhibit COX-2 than non-selective drugs. Whether the differences can be evaluated according to the affinities and specificities determines the performance of SPA in the applications of virtual screening.
As seen from the enrichment curves (Fig. 3a), the selective drugs are obviously separated from the diversity set, while the non-selective drugs are weakly separated from the diversity set. This indicates that SPA has the capacity to discriminate the drugs from the diversity set, especially the selective drugs from the diversity set. The weak discrimination of non-selective drugs from the diversity set may result from the fact that the non-selective drugs are not specific for COX-2 and have much lower potency than the selective drugs. Clearly, the statistics of the top-ranked compounds of all the ligands (Supplementary Table 2), often taken as interest compounds in the virtual screening, shows that compared to affinity, the specificity is a more efficient criterion to select the drugs out of the top-ranked compounds. It is worth noticing that the performance of linear combination of the affinity and specificity is better than the single parameter to discriminate the selective drugs from the diversity set.
Figure 3. Statistics for the drugs and two dimensional contour map of drug screening.

(a) Enrichment for both selective and non-selective drugs with E, ISR and the linear combination of them (E & ISR) through logistic regression (b) KS statistic for the discrimination of selective drugs and non-selective drugs by the difference of cumulative fraction based on the parameters. (c) Two dimensional density contour map of binding affinity and ISR for 650 small molecules binding with COX-2, the selective drugs shown in red and non-selective drugs shown in green.
To further quantify the discrimination of selective drugs from non-selective drugs, the statistical discrimination KS test was calculated (Fig. 3b). The relative high values of KS statistic (higher than 40%) suggest significant differences between the selective drugs and the non-selective drugs in both affinity and specificity, and more obvious in terms of the combination of them. The KS statistic results demonstrate that SPA is capable to discriminate selective drugs against non-selective drugs, which is important for selecting drug candidates with specificity against targets such as COX-2.
Based on the performance of SPA on the screening, a two-dimensional projection of specificity and affinity is plotted (Fig. 3c) for COX-2 with the diversity set of 650 selected compounds as well as its 37 selective and 20 non-selective drugs. The basin center with the highest density locates in the area with small ISR and low affinity, indicating that random compounds which have weak thermodynamic stability also generally do not have high specificity. Whereas, most of the selective drugs have large ISR and high affinity, and most of the non-selective market drugs tend to have relatively smaller ISR and lower affinity. It is worth noticing that a few drugs have values near the basin center in one parameter (ISR or affinity), but have larger values in another parameter, which suggests that specificity and affinity can be complementary in searching some specific drug candidates in virtual screening. These results validate that specificity is an important property of drug-target system.
These results validate that specificity is an important property of drug-target system. Previously, both experimentally and computationally screening techniques mostly concentrated on the affinity selection for the lead compounds. The virtual screening test of SPA here demonstrates that the ISR is an appropriate indicator for the lead compounds with specificity selection. Experimentally, it is challenging to determine the binding specificity for a given target. Computationally, it is practical to employ the scoring functions to carry out two dimensional virtual screening using both affinity and ISR in drug discovery. SPA is a good choice based on its excellent performance.
Discussion
In this work, we developed a novel and quantitative descriptions of biomolecular interactions through the scoring function called SPA which takes into account of both specificity and affinity of protein-ligand binding. It represents a significant advance over the previous investigations on protein-ligand binding interactions and scoring functions that only focused on affinity for development. Two another important innovations are incorporated into SPA. Firstly, SPA provides an effective way to circumvent the calculation of the reference state which is an issue in the development of knowledge-based scoring functions26,27,28. Secondly, lacking of a large and high-quality set of protein-ligand complexes with experimentally determined binding affinities and three dimensional structures was a bottleneck for developing accurate and general scoring functions. SPA takes the largest data so far of high-quality set of protein-ligand complexes with experimentally determined binding affinities and 3D structures29. It gives SPA with the chance to be less training-set dependent and more general for applications, which is superior than previous empirical scoring functions.
The excellent performance of SPA was validated by the test on a benchmark set. Compared to the other 16 existing popular scoring functions, SPA achieves the highest success rate in identifying correct binding pose, yields the highest correlation coefficient in the prediction of the experimentally measured binding affinity. These significant improvements of performances over previous scoring functions are very encouraging and motivate us to apply SPA in identifying the lead compounds in drug discovery. The virtual screening test of SPA on a drug target COX-2 shows that it can successfully distinguish not only the drugs from the diversity set according to the binding affinity as well as the specificity, but also the selective drugs from non-selective drugs, the later discrimination is more demanding in the discovery of lead compounds for drug targets. Thus, more reliable lead compounds with both stability and specificity can be searched with SPA scoring. The success of SPA proves that the specificity is critical to the biomolecular recognition and necessary to be incorporated to the scoring function. In the computational design of the protein-protein interactions9,10, both stability and specificity can be considered to design the interactions for discriminating natural binding partners from many other possible ones with similar sequences and structures. In natural systems, both parameters may be subject to evolutionary optimization, giving the rationality of the optimization on affinity and specificity.
With the availability of rapidly increasing number of protein structures and the advent of high-performance computing system, computational virtual screening offers an effective and practical route to discovering new drug molecules, a complementary or even an alternative way of experimental high-throughput screening30. In the processes of virtual screening, the performance of the scoring function has a major impact on the quality of molecular docking predictions. The outstanding performance of SPA shown in this work makes it practical to be implemented in the docking software and widely applied in virtual screening for identifying the lead compounds.
Methods
Derivation of distance-dependent potentials
The initial atom-pair potential to be optimized is directly derived from the Boltzmann relation widely used in the knowledge-based statistical potentials27,28,31, which is
where gij(r) is the observed pair distribution function which can be calculated by
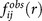 is the observed number density of atom pair ij within a spherical shell between r and r+δr and the
is the observed number density of atom pair ij within a spherical shell between r and r+δr and the 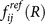 is the expected number density within the sphere of the reference state where there were no interactions between atoms. The former can be directly extracted from the database of protein-ligand complexes, while the later is obtained based on the approximation that the atom pair ij is uniformly distributed in the sphere of the reference state32. Respectively, they are calculated as
is the expected number density within the sphere of the reference state where there were no interactions between atoms. The former can be directly extracted from the database of protein-ligand complexes, while the later is obtained based on the approximation that the atom pair ij is uniformly distributed in the sphere of the reference state32. Respectively, they are calculated as
where M is the number of protein-ligand complexes, 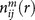 and
and 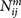 are the numbers of atom pair ij within the spherical shell and the reference sphere for a given protein-ligand complex m, where
are the numbers of atom pair ij within the spherical shell and the reference sphere for a given protein-ligand complex m, where 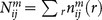 .
. 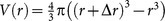 and
and 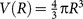 are the volumes of the spherical shell and the reference sphere, where Δr is the bin size and R is the radius of sphere. In this work, Δr and R are set as 0.3Å and 7.0Å, respectively. In total, there are 16 spherical shells with bin size 0.3 Å from the shortest radius 2.2Å which is the value to exclude the protein-ligand complexes with the steric atom clashes in PDBbind database29.
are the volumes of the spherical shell and the reference sphere, where Δr is the bin size and R is the radius of sphere. In this work, Δr and R are set as 0.3Å and 7.0Å, respectively. In total, there are 16 spherical shells with bin size 0.3 Å from the shortest radius 2.2Å which is the value to exclude the protein-ligand complexes with the steric atom clashes in PDBbind database29.
In fact, the initial potential can be extracted from any database of protein-ligand complex structures, while a good set of initial potentials can make optimizing search more efficient. The database of protein-ligand complexes used here was taken from the refined set of 2011 version in PDBbind database29,33. This database provides a comprehensive and highquality collection of the experimentally determined biomolecular complexes with measured binding affinities which were filtered from the Protein Data Bank (PDB) by applying a series of criteria. Due to infrequent occurrence of metal atoms in the protein-ligand complexes, the complexes containing metal atoms are excluded and only the potentials between nonmetal atoms are considered. 2316 protein-ligand complexes are remaining as our database to extract the initial potentials. Based on the definition of atom type by SYBYL34, 22 atom types are used to cover protein-ligand interactions (Supplementary Table. 3), these atom types were converted from PDB files by BABEL35. A cutoff ( = 600) of Nij was employed to ignore the contribution from the atom pairs with statistically insufficient occurrences. This leads to 101 effective types of atom pairs in our calculation. In addition, if the atom pair has no occurrence in a particular spherical shell, the corresponding pair potential was set as the van der Waals interaction within this shell.
Generation of docking decoys
To calculate the ISR for the optimization of SPA, enough conformations need to be sampled for each ligand docking to its specific protein receptor to explore the underlying binding energy landscape13,14. For each protein-ligand complex, except the “native” protein-ligand conformation obtained from the PDB structure, all the conformational decoys of protein-ligand complexes were generated by the molecular docking with software AutoDock4.236 in this work. Given that enough sampling of binding decoys to generate binding energy landscape is dependent on the docking space that the ligand can explore, the grid box for ligand docking should be sufficiently large to cover the active site as well as significant portions of the surrounding surface. The edges of the grid box were set as five times as the radius of gyration of the naive conformation of ligand to guarantee the enough exploration of the active site, and the grid box was centered on the geometric center of the native pose with a grid spacing of 0.375. Within the grid box, Autodock4.2 stochastically generates a population of conformational, rotational, and translational isomers from the starting structure of the ligand and docks them with the conformational search method of Lamarckian genetic algorithm. During the search, the ligand was considered conformationally flexible with its torsional bonds defined by AutoDock4.2 according to their chemical features. For each protein-ligand complex, 500 separate docking runs were performed which resulted in a database of 500 decoys for each complex. Other parameters were set as the default values of AutoDock4.2.
Quantitative description of specificity and affinity
To get a potential energy function which can maximize the binding specificity and the consistence between predicted and experimental affinity, we rewrote the initial energy function by introducing a set of adjustable parameters as the coefficients ck for the potentials of atom pair, that is
where E is the total intermolecular energy of a protein-ligand complex. k stands for the type of atom pair interaction, there are 1616 types by multiplying the number of effective atom pairs ( = 101) and the number of shells ( = 16). fk represents the occurrences of the interaction type k between the protein and ligand, and uk is an alternative representation of uij(r) in equation 1.
The intrinsic specific ratio (ISR) for a given protein-ligand complex m is calculated as
where α is a scaling factor which accounts for the contribution of the entropy to the specificity13. Here, it approximately depends on the number of torsional bonds of the ligands 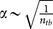 . δE is the energy gap between the energy of native conformation EN and the average energy of ensemble of decoys < ED >, and ΔE is the energy fluctuation or the width of the energy distribution of the decoys (Fig. 2), namely
. δE is the energy gap between the energy of native conformation EN and the average energy of ensemble of decoys < ED >, and ΔE is the energy fluctuation or the width of the energy distribution of the decoys (Fig. 2), namely
<> means the average over the ensemble of decoys. Combined equations 5–8 together, the λm can be represented as
where k,l are the indices of the interaction types. Once fk is computed for each interaction type in the decoys, one can easily compute the value of λm for a given set of ck.
The λm above is defined for a single protein-ligand complex, while we seek the potential energy function that simultaneously makes λm values large enough for the whole proteinlingand complexes in the training set. Therefore we need a single objective function that reflects the λm values for all the protein-ligand complexes in the training set. We chose the Bolzmann-like weighted average of λm as the objective function which is
where βλ is a constant value for weighting which is set as −0.1. The Bolzmann-like weighted average has the advantage over the normally used algebraic average since the protein-ligand complex with the smallest absolute value of λm contribute most to the objective function λ. If we optimize the smallest value of λm from the distribution pool among different protein-ligand binding complexes, we will be sure that even the smallest λm will be large enough for discrimination of separating the “native” from non-native decoys. Therefore Bolzmann-like weighted average is an appropriate combination of individual λm to match our purpose that all the resulting protein-ligand complexes of the training set will be optimized with large λm value. This average approach has similar function as some other weighted average approaches used for optimizing energy function of protein folding37,38,39.
The quantitative measurements of the correlation between predicted and experimental affinity is depicted with Pearson's correlation coefficient by
 |
The predicted binding affinity 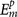 for the protein-ligand complex is represented by the binding scores calculated from our scoring function with a given set of ck. The experimentally measured affinity
for the protein-ligand complex is represented by the binding scores calculated from our scoring function with a given set of ck. The experimentally measured affinity 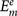 is expressed in log Kd or log Ki units, where Kd and Ki are experimentally determined dissociation constant and inhibition constant respectively for the protein-ligand complex m.
is expressed in log Kd or log Ki units, where Kd and Ki are experimentally determined dissociation constant and inhibition constant respectively for the protein-ligand complex m.
Optimization of potential energy function
After getting the initial potential energy and decoy ensembles, the energy function can be readily optimized. The aim of optimization here is to maximize the value of λ for specificity and the value of γ for affinity, a combination parameter (ρ = λγ) which couples specificity and affinity is constructed to evaluate the performance of scoring function during the optimization. The optimization is performed by Monte Carlo (MC) search with simulated annealing in the space of adjustable coefficients ck. The initial values of coefficients are set as 1.0 here, which means optimization of the scoring function starts from the energy function obtained through equation 1-4. A constraint is applied to ck by restricting it varied within [ , 5] times of its initial value, otherwise some coefficients could become very large or small due to infrequent occurrences of the interactions. At each MC step one of the coefficients is chosen at random and added with 0.2 or −0.2. The resulting change in E (E is defined as E = −ρ, minimizing E is equivalent of maximizing ρ) is accepted with the probability
, 5] times of its initial value, otherwise some coefficients could become very large or small due to infrequent occurrences of the interactions. At each MC step one of the coefficients is chosen at random and added with 0.2 or −0.2. The resulting change in E (E is defined as E = −ρ, minimizing E is equivalent of maximizing ρ) is accepted with the probability
where 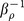 is the optimization temperature for ρ. This guarantees the chosen MC steps statistically prefer the low E and high ρ. The temperature
is the optimization temperature for ρ. This guarantees the chosen MC steps statistically prefer the low E and high ρ. The temperature 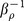 decreases exponentially during the search and the starting temperature is 0.5. The search converges well within 300,000 MC steps (Supplementary Fig. 2a), which suggests that a set of ck are found maximally optimized for ρ. The convergence of both λ and γ (Supplementary Fig. 2b) indicates the optimized scoring function reaches the maximal performance of simultaneously quantifying the specificity and affinity. For each MC search, 1500 protein-ligand complexes are randomly selected as the training set from the refined set of PDBbind database except the complexes in the test set. 5 independent MC simulations were performed, and the average of correlation between the coefficients from different MC simulations is 0.80. This high correlation indicates our optimization is successful and robust on the training set.
decreases exponentially during the search and the starting temperature is 0.5. The search converges well within 300,000 MC steps (Supplementary Fig. 2a), which suggests that a set of ck are found maximally optimized for ρ. The convergence of both λ and γ (Supplementary Fig. 2b) indicates the optimized scoring function reaches the maximal performance of simultaneously quantifying the specificity and affinity. For each MC search, 1500 protein-ligand complexes are randomly selected as the training set from the refined set of PDBbind database except the complexes in the test set. 5 independent MC simulations were performed, and the average of correlation between the coefficients from different MC simulations is 0.80. This high correlation indicates our optimization is successful and robust on the training set.
Validation of SPA
To validate SPA, two kinds of tests were taken. First, SPA was tested on a benchmark of protein-ligand complexes which is a high-quality set of 195 protein-ligand complexes selected out from the refined set of 2007 version of the PDBbind database24. This benchmark was taken as testing set to compare the performance for a large collection of 16 scoring functions implemented in main-stream commercial softwares or available from academic research groups, which offers a reference for the performance of SPA. Each protein-ligand complex of the benchmark set was docked with the same parameter as the training set above to generate a binding energy landscape with decoy ensemble. Second, SPA was applied on a target protein cyclooxygenase-2 (COX-2) for the virtual screening test. COX-2 is the inhibition target of nonsteroidal anti-inflammatory drugs (NSAIDs) for reducing fever and inflammation. A diverse set of 650 small molecules were selected from the NCI-Diversity database40 having molecular weights similar to that of the reference compound SC-558, with which the crystal structure of the COX-2 complex is available (PDB code 1CX2)41,42. 37 COX-2 selective and 20 nonselective drugs (Supplementary Table. 4) of NSAIDs are taken for the test of discrimination of drugs from the diversity set, as well as the discrimination of selective from non-selective drugs. COX-2 selective inhibitors are specific to inhibit only COX-2, while COX-2 non-selective drugs inhibit both the COX-2 and its isoenzyme COX-1. Also, each ligand was docked to COX-2 to generate a binding energy landscape with 2000 decoy conformations, and the box size of docking was set as 100 * 100 * 100 grids centered at the compound SC-558. Since the crystal structures of COX-2 with the ligands in the diversity set are not available, by clustering the decoys the same as implemented in AutoDock software, the decoy with the lowest energy in the largest population cluster was considered as the “native” pose43. The evaluation methods44 including the enrichment test and the Kolmogorov-Smirnov test (KS test) were employed to describe the performance of SPA for COX-2 system.
Author Contributions
JW designed the project. ZY performed the research. All authors wrote and reviewed the manuscript.
Supplementary Material
Specificity quantification of biomolecular recognition and its implication for drug discovery
Acknowledgments
This work was supported by National Science Foundation.
References
- Koshland Jr D. E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. 44, 98–104 (1958). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCammon J. A. Theory of biomolecular recognition. Curr. Opin. Struct. Biol. 8, 245–249 (1998). [DOI] [PubMed] [Google Scholar]
- Cohen N. C. Guidebook on molecular modeling in drug design. Academic Press pages 1–361 (1996). [Google Scholar]
- Warren G. L. et al. Computational and Structural Approaches to Drug Discovery: Ligand-Protein Interactions. RSC Publishing (2008). [Google Scholar]
- Gregory L. et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 49, 5912–5931 (2006). [DOI] [PubMed] [Google Scholar]
- Kim R. & Skolnick J. Assessment of programs for ligand binding affinity prediction. J. Comput. Chem. 29, 1316–31. (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen D. B., Decornez H., Furr J. R. & Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applicaitons. Nat. Rev. Drug Discov. 3, 935–949 (2004). [DOI] [PubMed] [Google Scholar]
- Janin J. Principles of protein-protein recognition from structure to thermodynamics. Biochimie 77, 497–505 (1995). [DOI] [PubMed] [Google Scholar]
- Havranek J. J. & Harbury P. B. Automated design of specificity in molecular recognition. Nat. Struct. Biol. 10, 45–52 (2003). [DOI] [PubMed] [Google Scholar]
- Kortemme T. et al. Computational redesign of protein-protein interaction specificity. Nat. Struct. Biol. 11, 371–379 (2004). [DOI] [PubMed] [Google Scholar]
- Bolon D. N., Grant R. A., Baker T. A. & Sauer R. T. Specificity versus stability in computational protein design. Proc. Natl. Acad. Sci. USA 102, 12724–12729 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. Y., Grinter S. Z. & Zou X. Q. Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys. Chem. Chem. Phys. 12, 12899–12908 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. & Verkhivker G. M. Energy landscape theory, funnels, specificity, and optimal criterion of biomolecular binding. Phys. Rev. Lett. 90, 188101–188104 (2003). [DOI] [PubMed] [Google Scholar]
- Wang J. et al. Quantifying intrinsic specificity: A potential complement to affinity in drug screening. Phys. Rev. Lett. 99, 198101–198104 (2007). [DOI] [PubMed] [Google Scholar]
- Shan Y. et al. How does a drug molecule find its target binding site? J. Am. Chem. Soc. 133, 9181–9183 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryngelson J. D. & Wolynes P. G. Spin-glasses and the statistical-mechanics of protein folding. Proc. Natl. Acad. Sci. USA 84, 7524–7528 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryngelson J. D., Onuchic J. N., Socci N. D. & Wolynes P. G. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins: Struct. Funct. Bioinf. 21, 167–195 (1995). [DOI] [PubMed] [Google Scholar]
- Janin J. Quantifying biological specificity: the statistical mechanics of molecular recognition. Proteins: Struct. Funct. Bioinf. 25, 438–445 (1996). [DOI] [PubMed] [Google Scholar]
- Rejto P. A. & Verkhivker G. M. Unraveling principles of lead discovery: From unfrustrated energy landscapes to novel molecular anchors. Proc. Natl. Acad. Sci. USA 93, 8945–8950 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C. J., Kumar S., Ma B. & Nussinov. R. Folding funnels, binding funnels, and protein function. Protein Sci. 8, 1181–1190 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy B. N. & Shakhnovich E. I. Native atom types for knowledge-based potential: Applications to binding energy prediction. J. Med. Chem. 47, 4838–4558 (2004). [DOI] [PubMed] [Google Scholar]
- Liu Z., Dominy B. N. & Shakhnovich E. I. Structural mining: Self-consistent design on flexible protein-peptide docking and transferable binding affinity potential. J. Am. Chem. Soc. 126, 8515–8528 (2004). [DOI] [PubMed] [Google Scholar]
- Levy Y., Wolynes P. G. & Onuchic P. G. Protein topology determines binding mechanism. Proc. Natl. Acad. Sci. USA 101, 511–516 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T. J., Li X., Li Y., Liu Z. H. & Wang R. X. Comparative assessment of scoring functions on a diverse test set. J. Chem. Inf. Model. 49, 1079–1093 (2009). [DOI] [PubMed] [Google Scholar]
- Wang J., Kang K., Kuntz I. D. & Kollman P. A. Hierarchical database screenings for HIV-1 reverse transcriptase using a pharmacophore model, rigid docking, solvation docking, and MM-PB/SA. J. Med. Chem. 48, 2432–2444 (2005). [DOI] [PubMed] [Google Scholar]
- Thomas P. D. & Dill K. A. An iterative method for extracting energy-like quantities from protein structures. Proc. Natl. Acad. Sci. USA 93, 11628–11633 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. & Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 11, 2714–2726 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. Y. & Zou X. An Iterative knowledge-based scoring function to predict proteinligand interactions: I. Derivation of interaction potentials. J. Comput. Chem. 27, 1866–1875 (2006). [DOI] [PubMed] [Google Scholar]
- Wang R., Fang X., Lu S. & Wang Y. The pdbbind database: Collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 47, 2977–2980 (2004). [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. Virtual screening of chemical libraries. Nature 432, 862–865 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Lu M., Chen M. Li J. & Ma J. OPUS-Ca: A knowledge-based potential function requiring study only Cα positions. Protein Sci. 16, 1449–1463 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippl M. J. Calculation of conformational ensembles from potentials of mean force. an approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 213, 859–883 (1990). [DOI] [PubMed] [Google Scholar]
- PDBbind Database website: http://www.pdbbind-cn.org/
- Clark M., Cramer III R. D. & Opdenbosch N. V. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 10, 982–1012 (1989). [Google Scholar]
- Guha T. et al. The blue obelisk – interoperability in chemical informatics. J. Chem. Inf. Model 46, 991–998 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M. et al. Automated docking using a lamarckian genetic algorithm and and empirical binding free energy function. J. Comput. Chem. 19, 1639–1662 (1998). [Google Scholar]
- Leonid A. M. & Shakhnovich E. I. How to derive a protein folding potential? A new approach to an old problem. J. Mol. Biol. 264, 1164–1179 (1996). [DOI] [PubMed] [Google Scholar]
- Koretke K. K., Luthey-Schulten Z. & Wolynes P. G. Self-consistently optimized energy functions for protein structure prediction by molecular dynamics. Proc. Natl. Acad. Sci. USA 95, 2932–2937 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitsuka Y., Takada S., Luthey-Schulten Z. A. & Wolynes P. G. Optimizing physical energy functions for protein folding. Proteins: Struct. Funct. Bioinf. 54, 88–103 (2004). [DOI] [PubMed] [Google Scholar]
- NCI Website: http://dtp.nci.nih.gov/branches/dscb/diversity_explanation.html
- Kurumbail R. G. et al.Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 384, 644–648 (1996). [DOI] [PubMed] [Google Scholar]
- Dewitt D. L. Cox-2-selective inhibitors: The new super aspirins. Mol. Pharmacol. 55, 625–631 (1999). [PubMed] [Google Scholar]
- Wei D., Zheng H., Su N., Deng M. & Lai L. Binding energy landscape analysis helps to discriminate true hits from high-scoring decoys in virtual screening. J. Chem. Inf. Model. 50, 1855–1864 (2010). [DOI] [PubMed] [Google Scholar]
- Kirchmair J., Markt P., Distinto S., Wolber G. & Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection - what can we learn from earlier mistakes. J. Comput. Aided. Mol. Des. 22, 213–228 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Specificity quantification of biomolecular recognition and its implication for drug discovery