

Abstract
Introduction
Intestinal atresias have long been hypothesized to result from either failure of recanalization of the intestinal lumen or in utero vascular accidents. Recent work in animal models is now calling for a reassessment of these widely held paradigms.
Purpose
In this review, we will examine the data that led to the original hypotheses and then evaluate more recent work challenging these hypotheses. Furthermore, we will discuss how defining the mechanism of atresia formation in animal models may provide insight into early intestinal development and the mechanism of lengthwise intestinal growth.
Conclusion
Such insight will be critical in developing regenerative therapies for patients with intestinal failure.
Keywords: Intestinal atresia, Mechanism, Intestinal growth, Hypothesis, Intestinal development, Vascular hypothesis, Epithelial plug, Duodenum
Introduction
Intestinal atresia is a phrase used to describe a segmental defect of the intestine which disrupts the luminal continuity of the intestinal tube during development. This is frequently accompanied by a loss of the surrounding mesoderm and blood supply to the affected region. A simple way of thinking about an atresia is that if the developing intestine is a series of pipes connected to one another end-to-end, in atresia, one of these segments of pipe disappears. Intestinal atresias can occur anywhere throughout the intestine, from the duodenum to the colon, and are one of the most common causes of neonatal intestinal obstruction, with an incidence between 0.57 and 6.6 per 10,000 live births (Table 1).1
Table 1.
Incidence of intestinal atresias versus allelic incidence of factor V Leiden
| Region/country | Incidence of small intestine atresia | Allele frequency (%) of factor V Leiden | |
|---|---|---|---|
| 1 | Spain (2001–2005) | 0.57 | 3.33 |
| 2 | Ireland (1995–2005) | 0.86 | 6.95 |
| 3 | Norway (1974–2005) | 0.9 | 3.7 |
| 4 | Chile (2002–2005) | 0.95 | 2.5 |
| 5 | Hungary (2001–2005) | 1.03 | 4.9 |
| 6 | UK | 1.04 | 3.61 |
| 7 | Finland | 1.12 | 4.2 |
| 8 | USA–Utah (2001–2005) | 1.32 | 4.2 |
| 9 | Italy (ISMAC) (2001–2005) | 1.5 | 2 |
| 10 | N. Netherlands (1985–2005) | 1.62 | 2.9 |
| 11 | Wales | 1.84 | 4 |
| 12 | Slovak (1995–2005) | 2.02 | 4 |
| 13 | New Zealand (2001–2005) | 2.27 | 3.8 |
| 14 | Germany (Northeastern) | 2.29 | 7 |
| 15 | Sweden (2001–2005) | 2.71 | 7.8 |
| 16 | Czech Republic (1995–2002) | 2.81 | 5.1 |
| 17 | Saudi Arabia | 2.88 | 1.3 |
| 18 | Australia | 3.14 | 3.6 |
| 19 | South America (2001–2005) | 3.2 | 1.6 |
| 20 | Canada | 3.84 | 5 |
| 21 | Japan (2001–2005) | 6.63 | 0 |
ICBDSR International Clearinghouse for Birth Defects Surveillance and Research
An Early Hypothesis Based on an Interesting Observation
Intestinal atresias were first described in 1684.2 The etiology of these defects were ascribed to any number of causes, including psychiatric fits of the mother, a lack of bile secretion, peritonitis, improper axial rotation of the intestine, compression by the transverse mesocolon as well as obliterative embryonic events.3 In 1900, a Viennese anatomist, Julius Tandler, published a hypothesis on the origins of intestinal atresia based on his studies of normal duodenal development. Tandler observed that the endoderm of the duodenum proximal to the “umbilical loop” (or C-loop of the duodenum) undergoes, “A remarkable thickening of the epithelium… until finally the duodenum becomes a solid string, in which no lumen at all is to be found.” This occlusion occurs at day 42 of development. Thereafter, the duodenum recanalizes between days 44 to 46 by forming multiple, small channels in the epithelial string which later coalesce into a single lumen. Tandler postulated that:
“If one keeps in mind the fact that on one hand the epithelial occlusion of the duodenum represents a normal event, but on the other hand that it is exactly in this place that most pathologic occlusions of the intestine occur, the question does not appear unjustified to ask whether these processes relate to each other, that is, whether they are causally related. It would not be impossible that in rare cases the physiologic atresia remains and develops into a congenital atresia…. It is clear to me that the opinion represented here does not exceed the status of a new hypothesis, and it is not meant to exceed this.”3
In spite of a cautiously worded qualification, Tandler's hypothesis has become dogma in the pediatric surgical community; however, researchers have begun to challenge this hypothesis over the last two decades. In his original paper, Tandler reported that, like humans, the duodenum of rats undergoes a similar developmental phase of luminal obliteration as a result of proliferation. More recent histological studies in rats indicate that this species do not undergo this developmental event.4 It remains unknown whether mice also fail to form a proliferative endodermal solid core or plug during development; however, duodenal atresias can be generated in mice by mutating either the gene for fibroblast growth factor receptor 2IIIb (Fgfr2IIIb) or its ligand Fgf10.5,6 If mice, like rats, fail to form a proliferative endodermal plug, it would indicate that this developmental event is not required for duodenal atresia formation.
Do Atresias Arise from Mechanical Events?
Fifty-five years after Tandler's work was published, surgeons J.H. Louw and Christiaan Barnard hypothesized that a “vascular accident” was the major etiology of intestinal atresias of the jejunum and ileum,7–9 an idea originally proposed by Spriggs in 1912.2 Their hypothesis was based on the clinical observation that thrombi were present in vascular arcades of the proximal intestine adjacent to the atretic region. They then tested whether segmental occlusion of the arterial blood supply to the intestine in utero would result in atresias. They successfully performed fetal surgery on two near-term canine fetuses, ligating arteries in the mesentery adjacent to the small intestine. Both pups developed atresias. Based on these observations, they concluded that interruption of the vasculature in utero (possibly from a thromboembolic event) was a major etiology of atresia formation.7–9
Louw and Barnard's work provided experimental evidence that mechanical compression of the arterial blood supply to the intestine can give rise to atresias, and there is clinical evidence that some intestinal atresias may arise from this mechanism. For example, gastroschisis and volvulus are two congenital defects that have a high association with atresia formation. In gastroschisis, a rupture occurs on the right side at the junction of the umbilical cord and the abdominal wall resulting in a ring-like abdominal wall defect. The intestine then herniates through the ring into the amniotic cavity. Seven percent of these patients will present with an atresia,10 which likely results from mechanical compression of the intestine by the ring-like defect. In volvulus (as a result of improper rotation and fixation or from a focal twist of the intestine around an adhesive band), intestinal blood flow is impaired resulting in involution of the affected segment of the intestine,8,9 loss of intestinal continuity, and atresia formation (Fig. 1). Not surprisingly, Louw and Barnard's hypothesis was based on observing an atresia arising from a focal volvulus around an adhesive band.7
Fig. 1.
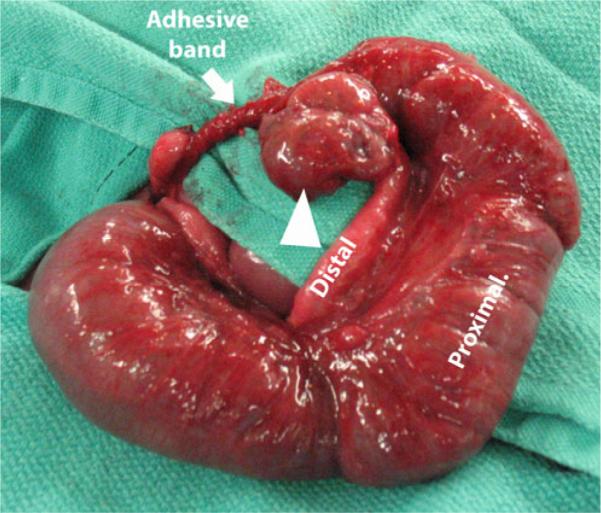
Atresia of the mid-ileum resulting from an adhesive band and subsequent volvulus. Proximal and distal limbs are indicated. Adhesive band is marked by a white arrow. The volvulized segment of the bowel is indicated by the white arrowhead
Cystic fibrosis (CF) has a high association with intestinal atresia formation but the mechanism is unknown. In fact, findings consistent with CF were associated with intestinal atresia long before CF was identified as a separate, distinct disease. Tandler credited the first description of this association to an investigator named Forer.3 Forer observed that atresia formation was associated with a lack of bile in the meconium, giving it the white or gray appearance commonly found in children with CF and intestinal atresias. Several studies have demonstrated a significantly higher incidence of CF in patients with jejunal–ileal atresias than would be expected in the general population.11,12 Cystic fibrosis with meconium ileus is reported to account for 11% of jejunoileal atresias and 5% of all intestinal atresias.13 If CF is included as a mechanical cause of atresia formation, together with gastroschisis and volvulus, these conditions account for approximately 50% of jejunoileal atresias and only 20% of all atresias.13 While the mechanism by which atresias occur in the setting of CF remains unclear, the majority of intestinal atresias (80%) do not appear to be associated with mechanical events.
Is There Epidemiological Evidence of In Utero Thromboembolic Events as a Cause of Intestinal Atresias?
Since the publication of Louw and Barnard's work, there has been a noticeable lack of genetic, molecular, or developmental evidence to support a “vascular accident” as an etiology of atresia formation. One paper was published that suggests a relationship between a predisposition to hypercoagulable states and atresia formation. That paper reported an associated increase in the allelic frequency of either factor V Leiden or the R353R mutation of the polymorphic region of factor VII in 28 patients with intestinal atresias.14 Whether any of the patients in this study were hypercoagulable at the time of their presentation was not determined. Factor V Leiden is very rarely associated with arterial thrombosis, the putative mechanism of the “Vascular hypothesis.” The R353R mutation results in increased levels of factor VII leading to coronary artery thrombosis in adults; however, gestational and perinatal levels of vitamin K-dependent clotting factors (including factor VII) are very low in infants due to a deficiency in vitamin K.15 Therefore, it is very unlikely that the R353R allele can result in atresia formation in utero. Finally, if either factor V Leiden or R353R were causative in atresia formation, then populations with high allelic frequencies of these mutations would have an increased incidence of intestinal atresias. This, however, is not the case. The Centers for Disease Control issues a report every year on the rate of congenital defects, including intestinal atresia, for a number of countries.1 Based on data from reporting countries, there is no statistical correlation between the incidence of intestinal atresia and the allelic incidence of factor V Lieden (correlation coefficient of −0.36) (Table 1; Fig. 2) or R353R (correlation coefficient of 0.24) (Table 2; Fig. 3).1,16–39,18,40–50 This analysis strongly suggests that neither mutation plays a role in, nor is associated with, intestinal atresia formation.
Fig. 2.
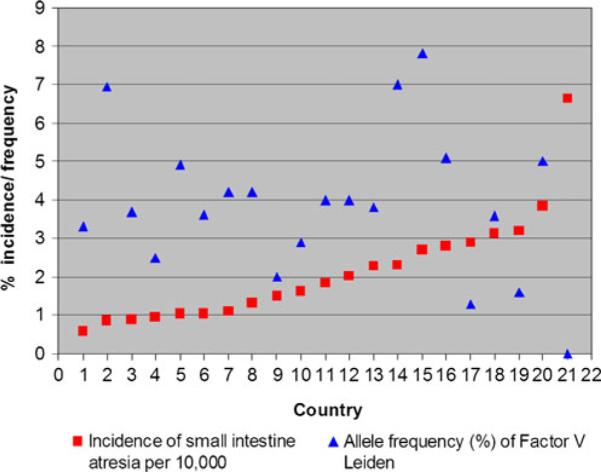
Graph of the incidence intestinal atresia versus allelic incidence of factor V Leiden
Table 2.
Incidence of Intestinal atresia versus allelic incidence of R353R mutation for factor VII
| Region/country | Incidence of small intestine atresia | Allele frequency (%) of factor V Leiden | |
|---|---|---|---|
| 1 | Spain (2001–2005) | 0.57 | 3.33 |
| 2 | Norway (1974–2005) | 0.9 | 3.7 |
| 3 | Chile (2002–2005) | 0.95 | 2.5 |
| 4 | UK | 1.04 | 4.4 |
| 5 | USA–Utah (2001–2005) | 1.32 | 4.2 |
| 6 | Italy (ISMAC) (2001–2005) | 1.5 | 2 |
| 7 | N. Netherlands (1985–2005) | 1.62 | 2.9 |
| 8 | Wales | 1.84 | 4 |
| 9 | Slovak (1995–2005) | 2.02 | 4 |
| 10 | New Zealand (2001–2005) | 2.27 | 3.8 |
| 11 | Germany (Saxony) | 2.29 | 8 |
| 12 | Sweden (2001–2005) | 2.71 | 7.8 |
| 13 | Saudi Arabia | 2.88 | 2.5 |
| 14 | Australia | 3.14 | 3.6 |
| 15 | South America (2001–2005) | 3.2 | 1.6 |
| 16 | Canada | 3.84 | 5 |
ICBDSR International Clearinghouse for Birth Defects Surveillance and Research
Fig. 3.
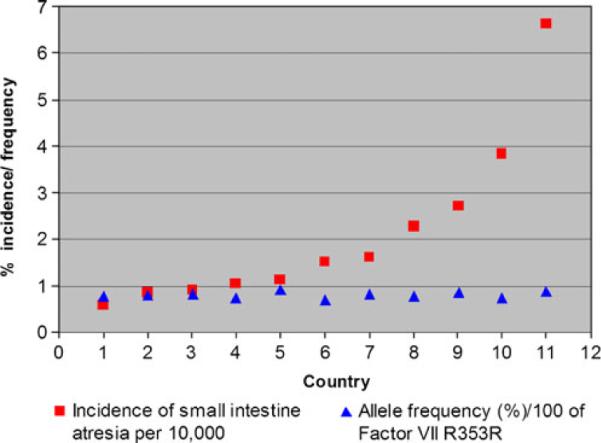
Graph of the incidence intestinal atresia versus factor allelic incidence of R353R mutation for factor VII
What Does the Clinical Presentation of Intestinal Atresias Reveal About Their Etiology?
Intestinal atresias present over a range of severity. The current categorization scheme reflects this range with types I, II, IIIa, and IIIb, organized by the amount of tissue absent and type IV reflecting the presence of multiple defects (Fig. 4). These five types of atresia occur with a relatively equal incidence and distribution throughout the intestinal tube. These characteristics are similar to other developmental defects that arise from genetic mutations that disrupt a common developmental process (cleft palate, omphalocele, and limb defects).51–55 Following this rationale, a type IIIb atresia would reflect an early disruption in this common developmental process in the embryonic gut, whereas less severe defects would reflect a later disruption in the same developmental process. Additionally, atresias primarily affect a limited area of intestine and are rarely hereditary.56 These clinical characteristics argue that atresias arise from a somatic mutation that disrupts a common developmental process in the embryonic gut.
Fig. 4.
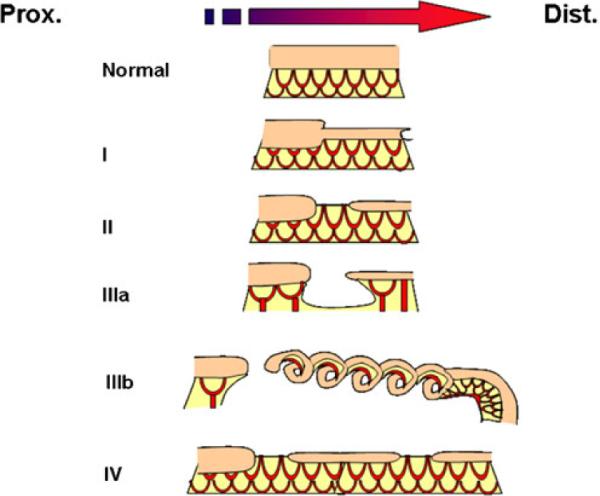
Atresias range in severity from a segmental narrowing or stenosis of the intestine to much more severe defects where there is an absence of a segment of the intestine and its blood supply. They are categorized based on increasing severity of the defect determined by three factors: (1) loss on intestinal tissue, (2) disruption of the accompanying intestinal blood supply, and (3) number of defects present. Type I atresias are a defect in which the intestinal mesoderm remains in continuity, but the lumen is obstructed by a diaphragm of tissue. In some cases, there is a small central opening, which allows for the restricted passage of contents. This variant includes the defect known as intestinal stenosis. Type II atresias are a defect wherein a small solid core of tissue connects the proximal and distal portions of the intestine; the core of tissue entirely lacks a lumen. In type IIIa atresias, the affected segment of intestine disappears and a V-shaped defect is visible in the adjacent mesentery or blood supply to the intestine. Type IIIb defects have an extensive gap in the blood supply between the proximal and distal limbs in intestine and they are frequently referred to as “apple peel defects” or “Christmas tree defects” because the distal limb of the bowel coils around its own mesentery. Type IV atresias have multiple points of interruption in the intestine, but the intestinal mesentery remains largely intact
In What Tissue Would a Disruption in a Common Developmental Process Have to Occur for an Atresia to Develop?
Further examination of the clinical presentation of atresias points to a defect in endoderm development as a leading event in atresia formation. This is most evident in duodenal atresias. Duodenal atresias, in addition to having a significant association with Down syndrome, also present with a high incidence of anomalies of other midline structures, including the esophagus, pancreatic duct, bile duct, heart, and rectum.56,57 Like the duodenum, these other structures initiate development in the midline of the embryo, and with the exception of the heart, are all derived in part from endoderm. The heart is not composed of endoderm, but cardiac myoblasts come into direct contact with the foregut endoderm during their migration out of the heart fields prior to forming the heart tube.58 It appears that this developmental interaction with the endoderm is critical in programming these cells in the proper formation of the heart.58 The common thread running through all these defects is that the endoderm plays a central role in their development. Thus, disruptions in endoderm development would appear to be central to the etiology of these defects.
What Do Animal Models Tell Us About Atresias?
Recent data from animal models contests the view that the leading event in atresia formation is vascular. These data, instead, suggest that a disruption in endoderm development results in atresia formation. For example, mutation of Fgfr2IIIb or the gene encoding its ligand Fgf10 results in both colonic and duodenal atresias. This suggests that the mechanism of formation of both atresias is the same.5,6,59–61 The Fgfr2IIIb gene encodes for a membrane-bound tyrosine kinase receptor that is thought to be expressed in the endoderm, but not the mesoderm, of the developing intestine.62 Mutation of Fgfr2IIIb or Fgf10 results in the loss of receptor signaling function leading to atresia formation. The leading cellular events in this model are increased in cell death and decreased proliferation specifically, and exclusively, in the endoderm.61 These endodermal cellular events precede any changes in the vasculature by at least a full day in the mouse (the equivalent of 4–6 days in a human). Second, disruption of Hedgehog signaling in the results in atresia formation in mice.64 Hedgehogs are signaling proteins that are generated by the endoderm and act on targets in the mesoderm. Mouse embryos that are homozygous for mutation in Shh develop a stenosis of the duodenum and a variant of imperforate anus, both of these defects fall within the spectrum of intestinal atresias.63 Finally, it has been shown that mutation of a gene encoding a transcription factor expressed exclusively in the colonic endoderm (Cdx-2) after embryonic day 12 in mice also results in atresia formation.64 Taken together, these data appear to refute the vascular hypothesis and instead point to a disruption in endoderm development or endoderm to mesoderm signaling as the leading events in atresia formation.
A New Round of Questions
The accumulating evidence favoring a disruption in endoderm development as the leading event in atresia formation raises a number of important questions. First, when do atresias occur? Second, what genes are involved? Third, what are the downstream morphogenetic events that result in the improper development of the affected intestinal segment? Finally, what do these defects reveal about the normal processes of intestinal growth and development?
Based on data from mouse models, intestinal atresias begin forming very early in development between E10.5 and E12.5 (the equivalent of days 33–48 in humans); however, the likelihood of demonstrating similar timing of these events in humans either from existing embryo collections or with the current prenatal imaging technologies seems remote at best. The animal models implicate disruptions in several molecular pathways in atresia formation, including the Fgfs and the Hedgehogs. Encouragingly, work in humans has demonstrated that mutations in the Fgfr2 coding region are associated with duodenal stenosis.65 Work in mice has demonstrated that mutations in a number of members of the Hedgehog signaling pathway genes (Gli-1, Gli-2, Gli-3, Ihh, and Foxf1) do not result in intestinal atresias but in some cases can cause variations in imperforate anus.63,66 These results suggest that formation of atresias may require disruptions of other signaling cascades in addition to the Hedgehog pathway. Unraveling the cellular events downstream of the initial insult to the endoderm will be challenging because these events will involve cellular differentiation, movement, and organization into specific tissue layers. These processes in early intestinal development have not been well defined.
What atresias may reveal about intestinal development and lengthwise intestinal growth could be far more profound. In mice, atresias begin forming at E10.5, the beginning of the most rapid phase in linear intestinal growth which runs from E10.5 to E15.5 and is equivalent to weeks 4.5 to 12 in the human.61 At the beginning of this phase, the intestinal length is approximately one third that of the embryo. By the end of this phase, the intestine is five to six times the length of the embryo.67 During this period, the lengthwise growth of the intestine outpaces that of the embryo by a factor of nearly 15–1. The characterization of cellular events during atresia formation should provide critical insight into normal intestinal development during this most rapid phase of lengthwise growth. These insights will be essential if we are to develop much needed novel therapies to stimulate lengthwise intestinal growth for some 30,000 patients in the U.S.A. with intestinal failure.
Acknowledgements
The authors would like to thank Dr. Silke Niederhaus for her translation of J. Tandler's original publication and Anastasia Lopukhin for her assistance in editing this manuscript.
References
- 1.International Clearinghouse for Birth Defects Surveillance and Research. Annual report 2007 [Google Scholar]
- 2.O'Neill JA Jr, RM, Grosfeld JL, et al., editors. Pediatric Surgery. Vol. 2. Mosby; St Louis, Mo: 1998. Duodenal atresia and stenosis. [Google Scholar]
- 3.Tandler J. Zur Entwicklungsgeschichte des menschlichen Duodenum in fruhen Embryonalstadien. Morphol Jahrb. 1900;29:187. [Google Scholar]
- 4.Cheng W, Tam PK. Murine duodenum does not go through a “solid core” stage in its embryological development. Eur J Pediatr Surg. 1998;8(4):212–5. doi: 10.1055/s-2008-1071156. [DOI] [PubMed] [Google Scholar]
- 5.Fairbanks TJ, Kanard R, Del Moral PM, et al. Fibroblast growth factor receptor 2 IIIb invalidation–a potential cause of familial duodenal atresia. J Pediatr Surg. 2004;39(6):872–4. doi: 10.1016/j.jpedsurg.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 6.Kanard RC, Fairbanks TJ, De Langhe SP, et al. Fibroblast growth factor-10 serves a regulatory role in duodenal development. J Pediatr Surg. 2005;40(2):313–6. doi: 10.1016/j.jpedsurg.2004.10.057. [DOI] [PubMed] [Google Scholar]
- 7.Louw JH, Barnard CN. Congenital intestinal atresia; observations on its origin. Lancet. 1955;269(6899):1065–7. doi: 10.1016/s0140-6736(55)92852-x. [DOI] [PubMed] [Google Scholar]
- 8.Barnard CN. The genesis of intestinal atresia. Surg Forum. 1957;7:393–6. [PubMed] [Google Scholar]
- 9.Barnard CN, Louw JH. The genesis of intestinal atresia. Minn Med. 1956;39(11):745. passim. [PubMed] [Google Scholar]
- 10.Abdullah F, Arnold MA, Nabaweesi R, et al. Gastroschisis in the United States 1988–2003: analysis and risk categorization of 4344 patients. J Perinatol. 2007;27(1):50–5. doi: 10.1038/sj.jp.7211616. [DOI] [PubMed] [Google Scholar]
- 11.Stollman TH, Wijnen RM, Draaisma JM. Investigation for cystic fibrosis in infants with jejunoileal atresia in the Netherlands: a 35-year experience with 114 cases. Eur J Pediatr. 2007;166(9):989–90. doi: 10.1007/s00431-006-0342-6. [DOI] [PubMed] [Google Scholar]
- 12.Roberts HE, Cragan JD, Cono J, et al. Increased frequency of cystic fibrosis among infants with jejunoileal atresia. Am J Med Genet. 1998;78(5):446–9. doi: 10.1002/(sici)1096-8628(19980806)78:5<446::aid-ajmg9>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 13.Dalla Vecchia LK, Grosfeld JL, West KW, et al. Intestinal atresia and stenosis: a 25-year experience with 277 cases. Arch Surg. 1998;133(5):490–6. doi: 10.1001/archsurg.133.5.490. discussion 496–7. [DOI] [PubMed] [Google Scholar]
- 14.Johnson SM, Meyers RL. Inherited thrombophilia: a possible cause of in utero vascular thrombosis in children with intestinal atresia. J Pediatr Surg. 2001;36(8):1146–9. doi: 10.1053/jpsu.2001.25733. [DOI] [PubMed] [Google Scholar]
- 15.Greer FR. Vitamin K the basics—What's new? Early Hum Dev. 2010;86:43–47. doi: 10.1016/j.earlhumdev.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 16.Bowen DJ, Bowley S, John M, Collins PW. Factor V Leiden (G1691A), the prothrombin 3–-untranslated region variant (G20210A) and thermolabile methylenetetrahydrofolate reductase (C677T): a single genetic test genotypes all three loci–determination of frequencies in the S. Wales population of the UK. Thromb Haemost. 1998;79(5):949–54. [PubMed] [Google Scholar]
- 17.Arruda VR, Annichino-Bizzacchi JM, Costa FF, Reitsma PH. Factor V Leiden (FVQ 506) is common in a Brazilian population. Am J Hematol. 1995;49(3):242–3. doi: 10.1002/ajh.2830490312. [DOI] [PubMed] [Google Scholar]
- 18.Asindi AA, Al-Daama SA, Zayed MS, Fatinni YA. Congenital malformation of the gastrointestinal tract in Aseer region, Saudi Arabia. Saudi Med J. 2002;23(9):1078–82. [PubMed] [Google Scholar]
- 19.Frances F, Portoles O, Gabriel F, et al. Factor V Leiden (G1691A) and prothrombin-G20210A alleles among patients with deep venous thrombosis and in the general population from Spain. Rev Med Chil. 2006;134(1):13–20. doi: 10.4067/s0034-98872006000100002. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Gala JM, Alvarez V, Pinto CR, et al. Factor V Leiden (R506Q) and risk of venous thromboembolism: a case-control study based on the Spanish population. Clin Genet. 1997;52(4):206–10. doi: 10.1111/j.1399-0004.1997.tb02548.x. [DOI] [PubMed] [Google Scholar]
- 21.Gibson CS, MacLennan AH, Rudzki Z, et al. The prevalence of inherited thrombophilias in a Caucasian Australian population. Pathology. 2005;37(2):160–3. doi: 10.1080/00313020500058250. [DOI] [PubMed] [Google Scholar]
- 22.Hepner M, Roldan A, Pieroni G, et al. Factor V Leiden mutation in the Argentinian population. Thromb Haemost. 1999;81(6):989. [PubMed] [Google Scholar]
- 23.Herrmann FH, Koesling M, Schroder W, et al. Prevalence of factor V Leiden mutation in various populations. Genet Epidemiol. 1997;14(4):403–11. doi: 10.1002/(SICI)1098-2272(1997)14:4<403::AID-GEPI5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 24.Hudecek J, Dobrotova M, Hybenova J, et al. Factor V Leiden and the Slovak population. Vnitr Lek. 2003;49(11):845–50. [PubMed] [Google Scholar]
- 25.McCowan LM, Craigie S, Taylor RS, et al. Inherited thrombophilias are not increased in “idiopathic” small-for-gestational-age pregnancies. Am J Obstet Gynecol. 2003;188(4):981–5. doi: 10.1067/mob.2003.218. [DOI] [PubMed] [Google Scholar]
- 26.Nurk E, Tell GS, Refsum H, et al. Factor V Leiden, pregnancy complications and adverse outcomes: the Hordaland Homocysteine Study. QJM. 2006;99(5):289–98. doi: 10.1093/qjmed/hcl040. [DOI] [PubMed] [Google Scholar]
- 27.Palomo I, Pereira J, Alarcon M, et al. Factor V Leiden and prothrombin G20210A among Chilean patients with venous and arterial thrombosis. Rev Med Chil. 2005;133(12):1425–33. doi: 10.4067/s0034-98872005001200003. [DOI] [PubMed] [Google Scholar]
- 28.Pecheniuk NM, Marsh NA, Walsh TP. Multiple analysis of three common genetic alterations associated with thrombophilia. Blood Coagul Fibrinolysis. 2000;11(2):183–9. [PubMed] [Google Scholar]
- 29.Zoller B, Norlund L, Leksell H, et al. High prevalence of the FVR506Q mutation causing APC resistance in a region of southern Sweden with a high incidence of venous thrombosis. Thromb Res. 1996;83(6):475–7. doi: 10.1016/0049-3848(96)00157-0. [DOI] [PubMed] [Google Scholar]
- 30.Balogh I, Poka R, Pfliegler G, et al. High prevalence of factor V Leiden mutation and 20210A prothrombin variant in Hungary. Thromb Haemost. 1999;81(4):660–1. [PubMed] [Google Scholar]
- 31.Clark P, Walker ID, Govan L, et al. The GOAL study: a prospective examination of the impact of factor V Leiden and ABO(H) blood groups on haemorrhagic and thrombotic pregnancy outcomes. Br J Haematol. 2008;140(2):236–40. doi: 10.1111/j.1365-2141.2007.06902.x. [DOI] [PubMed] [Google Scholar]
- 32.Dizon-Townson DS, Meline L, Nelson LM, et al. Fetal carriers of the factor V Leiden mutation are prone to miscarriage and placental infarction. Am J Obstet Gynecol. 1997;177(2):402–5. doi: 10.1016/s0002-9378(97)70205-9. [DOI] [PubMed] [Google Scholar]
- 33.Jaaskelainen E, Toivonen S, Romppanen EL, et al. M385T polymorphism in the factor V gene, but not Leiden mutation, is associated with placental abruption in Finnish women. Placenta. 2004;25(8–9):730–4. doi: 10.1016/j.placenta.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 34.Kobashi G, Kato EH, Morikawa M, et al. MTHFR C677T Polymorphism and factor V Leiden mutation are not associated with recurrent spontaneous abortion of unexplained etiology in Japanese women. Semin Thromb Hemost. 2005;31(3):266–71. doi: 10.1055/s-2005-872430. [DOI] [PubMed] [Google Scholar]
- 35.Lee DH, Henderson PA, Blajchman MA. Prevalence of factor V Leiden in a Canadian blood donor population. CMAJ. 1996;155(3):285–9. [PMC free article] [PubMed] [Google Scholar]
- 36.Prochazka M, Lubusky M, Slavik L, et al. Frequency of selected thrombophilias in women with placental abruption. Aust N Z J Obstet Gynaecol. 2007;47(4):297–301. doi: 10.1111/j.1479-828X.2007.00741.x. [DOI] [PubMed] [Google Scholar]
- 37.Saour JN, Shoukri MM, Mammo LA. The Saudi Thrombosis and Familial Thrombophilia Registry. Design, rational, and preliminary results. Saudi Med J. 2009;30(10):1286–90. [PubMed] [Google Scholar]
- 38.Sottilotta G, Mammi C, Furlo G, et al. High incidence of factor V Leiden and prothrombin G20210A in healthy southern Italians. Clin Appl Thromb Hemost. 2009;15(3):356–9. doi: 10.1177/1076029607310218. [DOI] [PubMed] [Google Scholar]
- 39.Vaughan J, Power C, Nolan C, et al. The incidence of factor V Leiden in a normal Irish population and its relationship to the laboratory diagnosis of APC resistance. Thromb Haemost. 1999;81(4):661–3. [PubMed] [Google Scholar]
- 40.Batalla A, Alvarez R, Reguero JR, et al. Synergistic effect between apolipoprotein E and angiotensinogen gene polymorphisms in the risk for early myocardial infarction. Clin Chem. 2000;46(12):1910–5. [PubMed] [Google Scholar]
- 41.Berthier MT, Houde A, Bergeron J, et al. Effect of the factor VII R353Q missense mutation on plasma apolipoprotein B levels: impact of visceral obesity. J Hum Genet. 2003;48(7):367–73. doi: 10.1007/s10038-003-0039-x. [DOI] [PubMed] [Google Scholar]
- 42.Bowman R, Joosen AM, Welch AA, et al. Factor VII, blood lipids and fat intake: gene-nutrient interaction and risk of coronary heart disease with the factor VII R353Q polymorphism. Eur J Clin Nutr. 2009;63(6):771–7. doi: 10.1038/ejcn.2008.28. [DOI] [PubMed] [Google Scholar]
- 43.Di Castelnuovo A, D'Orazio A, Amore C, et al. Genetic modulation of coagulation factor VII plasma levels: contribution of different polymorphisms and gender-related effects. Thromb Haemost. 1998;80(4):592–7. [PubMed] [Google Scholar]
- 44.Eriksson-Berg M, Deguchi H, Hawe E, et al. Influence of factor VII gene polymorphisms and environmental factors on plasma coagulation factor VII concentrations in middle-aged women with and without manifest coronary heart disease. Thromb Haemost. 2005;93(2):351–8. doi: 10.1160/TH04-09-0616. [DOI] [PubMed] [Google Scholar]
- 45.Lindman AS, Pedersen JI, Arnesen H, et al. Coagulation factor VII, R353Q polymorphism, and serum choline-containing phospholipids in males at high risk for coronary heart disease. Thromb Res. 2004;113(1):57–65. doi: 10.1016/j.thromres.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Kakko S, Elo T, Tapanainen JM, et al. Polymorphisms of genes affecting thrombosis and risk of myocardial infarction. Eur J Clin Invest. 2002;32(9):643–8. doi: 10.1046/j.1365-2362.2002.01047.x. [DOI] [PubMed] [Google Scholar]
- 47.Doggen CJ, Manger Cats V, Bertina RM, et al. A genetic propensity to high factor VII is not associated with the risk of myocardial infarction in men. Thromb Haemost. 1998;80(2):281–5. [PubMed] [Google Scholar]
- 48.Heywood DM, Carter AM, Catto AJ, et al. Polymorphisms of the factor VII gene and circulating FVII:C levels in relation to acute cerebrovascular disease and poststroke mortality. Stroke. 1997;28(4):816–21. doi: 10.1161/01.str.28.4.816. [DOI] [PubMed] [Google Scholar]
- 49.Mrozikiewicz PM, Cascorbi I, Ziemer S, et al. Reduced procedural risk for coronary catheter interventions in carriers of the coagulation factor VII-Gln353 gene. J Am Coll Cardiol. 2000;36(5):1520–5. doi: 10.1016/s0735-1097(00)00925-6. [DOI] [PubMed] [Google Scholar]
- 50.Nishiuma S, Kario K, Nakanishi K, et al. Factor VII R353Q polymorphism and lacunar stroke in Japanese hypertensive patients and normotensive controls. Blood Coagul Fibrinolysis. 1997;8(8):525–30. doi: 10.1097/00001721-199711000-00007. [DOI] [PubMed] [Google Scholar]
- 51.Perveen R, Lloyd IC, Clayton-Smith J, et al. Phenotypic variability and asymmetry of Rieger syndrome associated with PITX2 mutations. Invest Ophthalmol Vis Sci. 2000;41(9):2456–60. [PubMed] [Google Scholar]
- 52.Eggenschwiler J, Ludwig T, Fisher P, et al. Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith-Wiedemann and Simpson-Golabi-Behmel syndromes. Genes Dev. 1997;11(23):3128–42. doi: 10.1101/gad.11.23.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rice R, Spencer-Dene B, Connor EC, et al. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113(12):1692–700. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Riley BM, Mansilla MA, Ma J, et al. Impaired FGF signaling contributes to cleft lip and palate. Proc Natl Acad Sci U S A. 2007;104(11):4512–7. doi: 10.1073/pnas.0607956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riley BM, Schultz RE, Cooper ME, et al. A genome-wide linkage scan for cleft lip and cleft palate identifies a novel locus on 8p11–23. Am J Med Genet A. 2007;143A(8):846–52. doi: 10.1002/ajmg.a.31673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conrad C, Freitas A, Clifton MS, Durham MM. Hereditary multiple intestinal atresias: 2 new cases and review of the literature. J Pediatr Surg. 2010;45(4):E21–4. doi: 10.1016/j.jpedsurg.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 57.Pameijer CR, Hubbard AM, Coleman B, Flake AW. Combined pure esophageal atresia, duodenal atresia, biliary atresia, and pancreatic ductal atresia: prenatal diagnostic features and review of the literature. J Pediatr Surg. 2000;35(5):745–7. doi: 10.1053/jpsu.2000.6049. [DOI] [PubMed] [Google Scholar]
- 58.Nijmeijer RM, Leeuwis JW, DeLisio A, et al. Visceral endoderm induces specification of cardiomyocytes in mice. Stem Cell Res. 2009;3(2–3):170–8. doi: 10.1016/j.scr.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 59.Fairbanks TJ, Kanard RC, De Langhe SP, et al. A genetic mechanism for cecal atresia: the role of the Fgf10 signaling pathway. J Surg Res. 2004;120(2):201–9. doi: 10.1016/j.jss.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 60.Fairbanks TJ, Kanard RC, Del Moral PM, et al. Colonic atresia without mesenteric vascular occlusion. The role of the fibroblast growth factor 10 signaling pathway. J Pediatr Surg. 2005;40(2):390–6. doi: 10.1016/j.jpedsurg.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 61.Fairbanks TJ, Sala FG, Kanard R, et al. The fibroblast growth factor pathway serves a regulatory role in proliferation and apoptosis in the pathogenesis of intestinal atresia. J Pediatr Surg. 2006;41(1):132–6. doi: 10.1016/j.jpedsurg.2005.10.054. discussion 132-6. [DOI] [PubMed] [Google Scholar]
- 62.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2(3):1–9. doi: 10.1186/gb-2001-2-3-reviews3005. 3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramalho-Santos M, Melton DA, McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development. 2000;127(12):2763–72. doi: 10.1242/dev.127.12.2763. [DOI] [PubMed] [Google Scholar]
- 64.Gao N, White P, Kaestner KH. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev Cell. 2009;16(4):588–99. doi: 10.1016/j.devcel.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martinovic-Bouriel J, Bernabe-Dupont C, Golzio C, et al. Matthew-Wood syndrome: report of two new cases supporting autosomal recessive inheritance and exclusion of FGF10 and FGFR2. Am J Med Genet A. 2007;143(3):219–28. doi: 10.1002/ajmg.a.31599. [DOI] [PubMed] [Google Scholar]
- 66.Mo R, Kim JH, Zhang J, et al. Anorectal malformations caused by defects in sonic hedgehog signaling. Am J Pathol. 2001;159(2):765–74. doi: 10.1016/S0002-9440(10)61747-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.FitzSimmons J, Chinn A, Shepard TH. Normal length of the human fetal gastrointestinal tract. Pediatr Pathol. 1988;8(6):633–41. doi: 10.3109/15513818809022320. [DOI] [PubMed] [Google Scholar]