

Abstract
Non-technical summary
The orchestrated activity of an ensemble of voltage-gated ion channels determines the initiation, shape and duration of the action potential in excitable cells. In primary pain-sensing neurons, this ensemble includes a high voltage-activated potassium channel. However, its molecular identity, function and modulation were unknown. Here, we show that the rapidly inactivating Kv3.4 channel underlying the high voltage-activated potassium current is a major determinant of action potential repolarization. Furthermore, we found that physiological activation of protein kinase C dramatically slows Kv3.4 channel inactivation, which enhances the channel's ability to influence action potential repolarization. Based on these results and earlier work, we conclude that phosphorylation of the Kv3.4 channel inactivation gate is a mechanism by which pain-sensing neurons shape action potential repolarization. This modulation will influence Ca2+-dependent processes that play vital roles in nociception and might become deregulated in chronic pain.
Abstract
Fast inactivation of heterologously expressed Kv3.4 channels is dramatically slowed upon phosphorylation of the channel's N-terminal (N-type) inactivation gate by protein kinase C (PKC). However, the presence and physiological importance of this exquisite modulation in excitable tissues were unknown. Here, we employed minimally invasive cell-attached patch-clamping, single-cell qPCR and specific siRNAs to unambiguously demonstrate that fast-inactivating Kv3.4 channels underlie a robust high voltage-activated A-type K+ current (IAHV) in nociceptive dorsal root ganglion neurons from 7-day-old rats. We also show that PKC activation with phorbol 12,13-dibutyrate (PDBu) causes a 4-fold slowing of Kv3.4 channel inactivation and, consequently, accelerates the repolarization of the action potential (AP) by 22%, which shortens the AP duration by 14%. G-protein coupled receptor (GPCR) agonists eliminate IAHV fast inactivation in a membrane-delimited manner, suggesting a Kv3.4 channel signalling complex. Preincubation of the neurons with the PKC inhibitor bisindolylmaleimide II inhibits the effect of GPCR agonists and PDBu. Furthermore, activation of PKC via GPCR agonists recapitulates the effects of PDBu on the AP. Finally, transfection of the neurons with Kv3.4 siRNA prolongs the AP by 25% and abolishes the GPCR agonist-induced acceleration of the AP repolarization. These results show that Kv3.4 channels help shape the repolarization of the nociceptor AP, and that modulation of Kv3.4 channel N-type inactivation by PKC regulates AP repolarization and duration. We propose that the dramatic modulation of IAHV fast inactivation by PKC represents a novel mechanism of neural plasticity with potentially significant implications in the transition from acute to chronic pain.
Introduction
The mechanisms underlying the transition from acute to chronic pain are poorly understood (Costigan et al. 2009; Basbaum et al. 2009; Gold & Gebhart, 2010). It is, however, likely that the underlying maladaptive changes may affect the mechanisms of neural plasticity that regulate the intrinsic excitability of primary sensory neurons. The action potentials (APs) of nociceptors are typically long and display a characteristic shoulder in the repolarizing phase (Harper & Lawson, 1985; Traub & Mendell, 1988; Gold et al. 1996a), which is the result of tetrodotoxin (TTX)-resistant Na+ channel activity and a significant increase in Ca2+ conductance (Blair & Bean, 2002). The repolarization of these APs may also critically depend on the activity and modulation of a high voltage-activated K+ current (IAHV). Specifically, we hypothesize that Kv3.4 channels underlie the IAHV in nociceptors and that the modulation of Kv3.4 channel N-type inactivation by protein kinase C (PKC) dynamically regulates the rate of AP repolarization.
Kv3 channels are widely expressed in the mammalian nervous system, where they underlie TEA-hypersensitive high voltage-activated K+ currents (Rudy & McBain, 2001). Whereas, the Kv3.1 and Kv3.2 subunits generally generate fast delayed rectifier K+ current and the Kv3.3 subunit generates a fast-activating and slow-inactivating A-type K+ current, the Kv3.4 subunit underlies a fast-activating and fast-inactivating A-type K+ current. High-voltage activation and fast deactivation of Kv3.1, Kv3.2 and Kv3.3 channels allow them to hasten the repolarization of the AP and promote fast spiking (Rudy & McBain, 2001; Desai et al. 2008). The study of neuronal Kv3.4 channels has been hampered, however, because the corresponding fast A-type K+ currents are difficult to identify and isolate. The factors that contribute to this problem are their restricted regional and subcellular localization (Brooke et al. 2004), and the slowing of Kv3.4 channel inactivation by oxidation, phosphorylation and phospholipids, which obscures the distinct electrophysiological A-type signature of Kv3.4 channels (Ruppersberg et al. 1991; Covarrubias et al. 1994; Oliver et al. 2004). Nevertheless, several studies have also implicated the Kv3.4 channel in AP repolarization (Riazanski et al. 2001; Baranauskas et al. 2003; Martina et al. 2007; Alle et al. 2011).
Kv3.4 channels undergo fast N-type inactivation involving the first 28 amino acids of the channel's pore-forming subunit, which form a structured inactivation gate (Rettig et al. 1992; Antz et al. 1997). Earlier work from our laboratory showed that PKC-dependent phosphorylation of four serines within the Kv3.4 N-terminal inactivation domain (NTID) eliminates fast inactivation in a heterologous expression system (Covarrubias et al. 1994; Beck et al. 1998; Velasco et al. 1998). Also, we determined that cooperative conformational changes triggered by phosphorylation render the Kv3.4 NTID unstructured and incapable of causing fast N-type inactivation (Beck et al. 1998; Antz et al. 1999). Although it was then hypothesized that this modulation of N-type inactivation would impact the properties of the AP, this idea remained untested.
Here, we report that Kv3.4 channels are highly expressed in putative nociceptors. Moreover, we show that fast Kv3.4 channel inactivation is dramatically slowed by pharmacological and physiological activation of PKC, and that this modulation shapes the repolarization of the AP. We discuss possible implications of the results in terms of a mechanism of neural plasticity that may play a role in the transition from acute to chronic pain.
Methods
Isolation of DRG neurons
All animals were treated as approved by the IACUC of Thomas Jefferson University. Time-pregnant female Sprague–Dawley rats (Taconic Farms) were maintained in the Thomas Jefferson University Animal Facility for 1 week prior to the birth of the pups. For all experiments, 7-day-old pups were anaesthetized with isoflurane and killed by decapitation. Dorsal root ganglia (DRGs) were harvested from all accessible levels and placed into Hanks’ buffered saline solution (HBSS) with 10 mm Hepes. Ganglia were dissociated by treatment with 1.5 mg ml−1 collagenase in HBSS/Hepes solution for 30 min followed by a 15 min treatment with 1 mg ml−1 trypsin in HBSS/Hepes solution. DRG neurons were then transferred to L-15 Leibovitz medium supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 24 mm NaHCO3, 38 mm glucose, 2% penicillin–streptomycin, and 50 ng ml−1 nerve growth factor and mechanically dissociated with a fire polished Pasteur pipette. Neurons were plated onto poly l-ornithine coated coverslips and kept at 37°C for up to 48 h for all experiments.
Nucleofection of siRNA
Approximately 2 × 106 dissociated DRG neurons were transfected with 40 pmol of rat Kv3.4 (KCNC4) siRNA or control siRNA-A (Santa Cruz Biotechnology, Santa Cruz, CA, USA) using the Amaxa Nucleofector-II system (Lonza; programme G-013 and Rat Neuron Nucleofector Kit) or 4D-Nucleofector system (Lonza; programme DC-100 and P3 Primary Cell Kit). Immediately after transfection, neurons were allowed to recover for 10 min at 37°C before plating on cover slips.
Drugs and toxins
Unless indicated otherwise, all toxins and drugs were purchased from Sigma, stored as concentrated stocks and added to recording solutions immediately before recording. Phorbol 12,13-dibutyrate (PDBu, Enzo Life Sciences), phorbol-12-myristate-13-acetate (PMA), 4α-PDBu (Enzo Life Sciences, Farmingdale, NY, USA) and bisindolylmaleimide II (BIM, Calbiochem) stocks were made in DMSO and the final DMSO concentration in the recording solution was <0.001%. Serotonin was purchased from Acros Organics (Geel, Belgium).
Electrophysiology
Electrophysiological recordings were obtained from small-diameter DRG neurons (diameter 18 ± 0.13 μm, n = 194) at room temperature (21–24°C). Patch electrodes were made with thin-walled patch glass (Warner Instruments, Hamden, CT, USA) and pulled with a PIP5 micropipette puller (HEKA Instruments Inc., Bellmore, NY, USA). Electrodes were coated with Sylgard (Dow Corning) and fire polished to have tip resistances of 1–3 MΩ. Signals were amplified by a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA), low-pass filtered at 2 kHz (4-pole Bessel), digitized at 10 kHz (Digidata 1440, Molecular Devices), and stored in a computer using Clampex 10.2 (Molecular Devices). In tail current experiments, recordings were low-pass filtered at 5 kHz (4-pole Bessel) and digitized at 50 kHz. Liquid junction potentials in cell-attached macropatch recordings (0 mV) and current-clamp recordings (+15.5 mV) were calculated using Clampex 10.2 and corrected off-line. Bath and pipette solution for all cell-attached macropatch experiments consisted of (in mm): 130 choline-Cl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 Hepes, 50 sucrose, pH 7.4. Cell-attached macropatches were stable for 20–60 min without evidence of change in IAHV amplitude or kinetics. For AP recordings the whole-cell configuration of the patch-clamp method was used. The AP external bath solution contained (in mm): 130 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 Hepes, pH 7.4. The pipette solution for AP recordings contained (in mm): 130 K-Mes, 1 CaCl2, 1 EGTA, 10 Hepes, 2 Mg-ATP, 0.3 Tris-GTP, pH 7.3. To determine the voltage in cell-attached patches, the following equation was used: V =Vc+Vr; where V is the actual voltage seen by the membrane patch, Vc is the command voltage, and Vr is the resting membrane potential. The Vr in all recordings was assumed to be –60 mV based on highly consistent measurements (Table 2), which are in agreement with published data from small-diameter DRG neurons (Cardenas et al. 1995; Villière & McLachlan, 1996). Confirming the stability of the resting membrane potential between cells and patches, the biophysical parameters were highly reproducible (Fig. 1 and Table 1).
Table 2.
Passive and active properties of APs and the effect of PKC activation
| PDBu | Hyperpolarization | GPCR Agonists | PKC activation + TEA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Control | DTX-K and IbTX | Pre | Post | Pre | Post | Pre | Post | Pre | Post | |
| RMP (mV) | −61.4 ± 1.1 | −61.0 ± 1.9 | −63.6 ± 2.0 | −69.0 ± 2.4** | −60.2 ± 3.3 | −76.8 ± 2.1** | −67.9 ± 3.8 | −69.1 ± 3.2 | −69.6 ± 3.1 | −71.3 ± 4.6 |
| Input resistance (GΩ) | 1.1 ± 0.1 | 0.9 ± 0.2 | 0.6 ± 0.1 | 0.5 ± 0.1 | ND | ND | 0.4 ± 0.1 | 0.8 ± 0.3 | 0.5 ± 0.2 | 0.6 ± 0.2 |
| Capacitance (pF) | 13.3 ± 0.7 | 16.2 ± 2.0 | 14.9 ± 0.8 | — | 13.1 ± 0.7 | — | 13.3 ± 0.8 | — | 13.8 ± 1.7 | — |
| Diameter (μm) | 18.2 ± 0.2 | 19.3 ± 0.4 | 17.6 ± 0.3 | — | 18.1 ± 1.1 | — | 19.0 ± 0.6 | — | 18.4 ± 0.6 | — |
| Rheobase (pA) | 63 ± 13 | 55 ± 18 | ND | ND | ND | ND | 137 ± 47 | 131 ± 44 | 115 ± 89 | 124 ± 104 |
| Threshold (mV) | −36.8 ± 1.0 | −38.1 ± 1.5 | −33.8 ± 2.3 | −36.8 ± 2.3* | −32.0 ± 3.1 | −36.4 ± 3.1 | −36.3 ± 2.6 | −39.3 ± 2.3 | −41.0 ± 2.8 | −43.3 ± 2.5 |
| Overshoot (mV) | 34.7 ± 1.6 | 33.7 ± 2.9 | 31.7 ± 3.5 | 26.4 ± 3.0* | 32.9 ± 4.2 | 36.5 ± 3.2 | 30.1 ± 3.9 | 33.0 ± 4.1 | 23.6 ± 5.5 | 25.1 ± 4.9 |
| AHP (mV) | −72.9 ± 0.98 | −76.2 ± 1.6 | −73.4 ± 1.6 | −79.2 ± 1.8** | −73.3 ± 1.3 | −79.4 ± 2.8* | −78.3 ± 2.4 | −80.8 ± 2.9 | −80.3 ± 2.6 | −75.7 ± 5.0 |
| APD50 (ms) | 4.4 ± 0.2 | 4.1 ± 0.3 | 4.7 ± 0.6 | 4.0 ± 0.4* | 4.3 ± 0.3 | 3.6 ± 0.4 | 5.0 ± 0.3 | 3.5 ± 0.3** | 3.5 ± 0.5 | 5.1 ± 1.3 |
| APD90 (ms) | 1.20 ± 0.05 | 1.16 ± 0.09 | 1.22 ± 0.14 | 0.98 ± 0.08* | 1.10 ± 0.05 | 0.97 ± 0.05* | 1.05 ± 0.05 | 0.82 ± 0.05** | 0.90 ± 0.13 | 1.28 ± 0.30 |
| Max depolarization rate (mV ms−1) | 66.6 ± 4.0 | 64.6 ± 7.6 | 65.6 ± 8.8 | 68.7 ± 8.9 | 59.3 ± 9.4 | 82.1 ± 13.0* | 62.7 ± 11.3 | 76.2 ± 6.0 | 76.0 ± 18.2 | 81.9 ± 20.5 |
| Max repolarization rate (mV ms−1) | 31.8 ± 1.5 | 32.4 ± 3.1 | 33.7 ± 4.4 | 39.7 ± 4.5** | 31.9 ± 2.3 | 37.4 ± 2.0* | 31.7 ± 2.4 | 40.2 ± 3.1** | 44.0 ± 8.0 | 28.8 ± 7.0* |
| n | 54 | 15 | 13 | 9 | 6 | 6 | ||||
AP properties were measured as described in Methods and Student's paired t test used for comparisons of paired pre and post data. Values for DTX-K and IbTX are APs recorded with 100 nm DTX-K and IbTX in the bath and compared to controls using Student's independent t test; values for PKC activation + TEA are data collected after PKC activation, pre = PKC activation only and post = PKC activation + 100 μm TEA
P≤ 0.05
P≤ 0.01
RMP = resting membrane potential; ND = not determined.
Figure 1. Biophysical properties of IAHV in small-diameter DRG neurons.
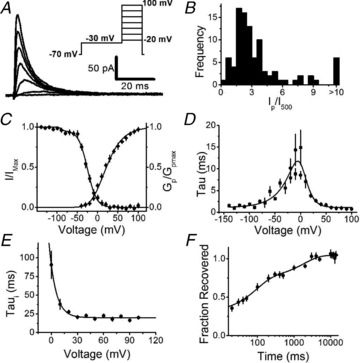
A, representative family of currents elicited in a cell-attached macropatch by the protocol shown in the inset. The durations of the conditioning and test pulses were 1 s and 500 ms, respectively. B, frequency histogram of the peak current over the current at 500 ms (Ip/I500) at +100 mV. C, normalized peak conductance–voltage relation (Gp/Gpmax) and steady-state inactivation curve (I/Imax). The continuous black lines represent the best-fit Boltzmann functions as described in Methods (Table 1 summarizes the best-fit parameters). For steady-state inactivation, the non-inactivating component of the curve comprised 24% of the total current and was manually subtracted. This fraction accounted for a slow-activating delayed-rectifier current seen in a majority of patches. The best-fit parameters of the small (4.3%) hyperpolarized component of the steady-state inactivation curve were V1/2=–90 mV and k = 3 mV. D, voltage dependence of the time constants of activation (•) and deactivation (▪). These time constants were obtained separately from exponential fits to the rising phase of outward currents and tail currents, respectively. The continuous black line represents the best-fit sum of two exponentials. The derived estimates of the associated apparent charges (z) are summarized in Table 1. E, voltage dependence of the time constant of inactivation (n = 76). The continuous black line represents the best-fit to an exponential function. The derived estimate of the associated apparent charge (z) is summarized in Table 1. F, recovery from inactivation at –100 mV. The continuous black line represents the best-fit sum of two exponentials. The fast time constant associated with low-voltage A-type currents was 91 ± 16 ms and represents 44% of the total current and the slow time constant associated with IAHV (Table 1) represents 30% of the total current. Some error bars are occluded by the symbols.
Table 1.
IAHV biophysical properties
| Activation | n | |
|---|---|---|
| V1/2 | +21.6 ± 2.0 mV | 79 |
| Vs | –20.9 ± 1.5 mV | 79 |
| zGp | 1.00 ± 0.04 e0 | 79 |
| zτa | 2.32 ± 0.71 e0 | 69 |
| zτd | 0.95 ± 0.25 e0 | 12 |
| Inactivation | ||
|---|---|---|
| V1/2 | −25.7 ± 0.1 mV | 39 |
| zssi | 2.30 ± 0.02 e0 | 39 |
| τr | 1.8 ± 0.4 s | 15 |
| zτi | 3.31 ± 0.31 e0 | 76 |
All parameters were calculated as described in Methods. For z, the subscripts indicate the parameter from which the z was calculated: Gp, peak chord conductance relation; ssi, steady state inactivation curve. τa, time constant of activation; τd, time constant of deactivation; τi, time constant of inactivation; τr, time constant of recovery from inactivation at –100 mV.
Single cell quantitative PCR
Individual small-diameter DRG neurons (diameter <25 μm) were harvested by applying negative pressure to a large bore pipette. The neuron was deposited into a sterile PCR tube and rapidly frozen after adding 10 μl of RNase-free water. Single-cell mRNA was reverse transcribed (RT) in a reaction containing 5 μl sample, 65 ng random primers (Invitrogen), 8 units μl−1 MMLV reverse transcriptase (Fischer Scientific), 50 mm Tris-HCl (pH 8.3), 75 mm KCl, 3 mm MgCl2, 10 mm dithiothreitol, 0.5 mm dNTPs and 1 unit μl−1 RNase inhibitor (Promega, Madison, WI, USA). A 5 μl aliquot of the cell lysates was identically treated except water was substituted for reverse transcriptase in the reaction mixture (RT-). Quantitative real-time single cell PCR was conducted using an Agilent Mx3005P real-time PCR machine and Brilliant SYBR Green reaction mix (Agilent Technologies, Santa Clara, CA, USA). The reaction was conducted in a 25 μl sample volume containing 12.5 μl of 2× SYBR Green master mix (including 30 nm ROX reference dye), 1–3 μl of sample cDNA, 1 μl of primer set (final concentration 100 nm), and nuclease-free water. The thermal profile for real time qPCR includes 10 min at 95°C, and 40 cycles at 95°C for 30 s, 60°C for 1 min, and 72°C for 1 min. Ct values were determined using the MxPro software. The housekeeping gene β−actin was used to normalize expression. The Ct value from each sample was read from a standard curve and corrected for handling. Melting curve analysis was performed on each sample. For construction of cDNA standards for each target sequence, the mRNA of whole DRG was reverse transcribed using a standard MMLV reverse transcriptase reaction. Standard PCR was performed in a 25 μl reaction volume containing 0.2 mm dNTPs, 0.4 μm primers, 1 μl of cDNA sample, and 0.2 μl of Taq DNA polymerase. The PCR products were analysed on an ethidium bromide stained 2% agarose gel. PCR controls, without cDNA or primer sets were analysed in parallel. A single band with the predicted size was then excised from the gel and purified using QIAEXII (Qiagen, Valencia, CA, USA). The cDNA concentration was determined using a Bio-mini DNA/RNA analyzer (Shimadzu, Kyoto, Japan). The copy numbers of the standards were calculated assuming the average weight of a base pair of 660 Da. Standard cDNAs were serially diluted to concentrations between 2 and 106 copies μl−1. Primer sets for each channel were designed to cross multiple exon–intron borders in order to reduce the contamination by genomic DNA. Outer primers were used to generate cDNA fragments for standards and inner primers used for real-time qPCR detections. Forward (f) and reverse (r) primers used were as follows.
Outer primers:
Kv3.1 (515 bp): (f) GCCACGCATCTGGGCACTGT, (r) ACTCGTTGGTGCTGGCACGG
Kv3.2 (627 bp): (f) ACGCTGAGCGAGTAGGGGCT, (r) ATCTGGCCTCGGCTTGCGTG
Kv3.3 (557 bp): (f) CGGGCGTCTTCGCCTACGTG, (r) AGAGGCCTGCGTCACCGTCT
Kv3.4 (577 bp): (f) CCACACCGACTTCAAGAACA, (r) GCCTTCTTTCTGGACACTGC
Nav1.9 (628 bp): (f) TCTCCTTCCTCATCGTGGTC, (r) AAGCTGTGAGGCAGTGAGGT
β-actin (502 bp): (f) GCTATGTTGCCCTAGACTTC, (r) AACGCAGCTCAGTAACAGTCC
Inner primers:
Kv3.1 (172 bp): (f) ATCCCGCTATGCGCGGTACG, (r)CTCCGTCTCCGCTTCCCGGT
Kv3.2 (168 bp): (f) ACCTCCGGAAAGGCTCCCCA (r) GCCTCGGCTTGCGTGTAACCA
Kv3.3 (194 bp): (f) TGACCTACCGCCAGCACCGT (r) TCCGCCCGCGTCCTGAAAAC
Kv3.4 (177 bp): (f) CAGAAGCTTCCCAAGAAACG, (r) TAGCGTCACCATTCTGCTTG
Nav1.9 (236 bp): (f) TGCATGGATGTTCTCTTTGC, (r) GACCTGTCCTTCAGCCTCAG
β-actin (160 bp): (f) CCGATGCCCCGAGGCTCTCT, (r) GCCTGGGTACATGGTGGTGCC
Data analysis and statistics
Data analysis was conducted in Clampfit 10.2 (Molecular Devices) and Origin Pro 8.0 (OriginLab Corp., Northampton, MA, USA). The criterion for the presence of IAHV at +100 mV was based on a ratio of peak current over current at 75 ms (Ip/I75) >1.5. To normalize the peak IAHV, the current at 75 ms was chosen as a reference because activation of a slowly activating delayed rectifier current becomes apparent at this time. All patches with Ip/I75 >1.5 had a clear inactivation phase that was readily described by an exponential function. To compare IAHV to the total delayed-rectifier current present in macropatches, the current at 500 ms (end of the voltage pulse; I500) was used and changes in the amplitude of IAHV were estimated as the ratio of the peak current over the I500 (Ip/I500) or its reciprocal (I500/Ip). Peak chord conductance (Gp) was calculated using the following:
where Ip is the peak current, V is the actual voltage seen by the patch (see Electrophysiology above) and Vrev is the observed reversal potential (–72.5 ± 3.5 mV, n = 12). Conductance was normalized to the maximum conductance (Gpmax) calculated from the best fit fourth-order Boltzmann function:
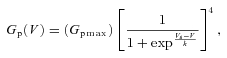 |
Where k is the slope factor and Vs is the activation mid-point voltage of a single subunit. V1/2, the mid-point voltage of peak conductance curve, was calculated from the equation:
Steady-state inactivation was described assuming a double Boltzmann function. Slope factors were converted into equivalent charge (z) using the following:
where R and F are known physical constants and T is absolute temperature. Kinetics of inactivation, activation and deactivation were described assuming exponential trajectories. The z associated with each process was determined by the equation above except that k is the slope derived from the exponential fit.
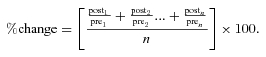
For AP analysis, AP overshoot (peak depolarization) and afterhyperpolarization (AHP, peak hyperpolarization) were determined and the difference between them yielded the spike height. AP duration was measured as the time it took to cross the voltage at 50% (APD50) and 90% (APD90) of the spike height. Rheobase is the magnitude of a 100 ms current injection pulse sufficient to elicit an AP, and threshold was defined as the voltage at which the dV/dt deviated from 0 mV ms−1 as determined from the phase-plane plot. Percentage changes in properties from paired samples were determined by taking the average of the percentage change for each pair:
 |
For control vs. siRNA experiments, where the data is not paired, the percentage changes and propagated errors were calculated as follows:
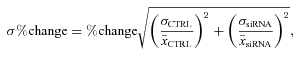 |
where 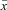 is the mean and σ is the standard error of the mean (SEM) of controls (CTRL) and Kv3.4 siRNA (siRNA) transfected cells.
is the mean and σ is the standard error of the mean (SEM) of controls (CTRL) and Kv3.4 siRNA (siRNA) transfected cells.
All results are expressed as mean ± SEM and data points that were >2 standard deviations (SD) from the mean were considered outliers. One-way ANOVA (with post hoc Bonferroni corrections) and Student's t test were used to evaluate differences between samples. Student's paired t test was used in experiments where pre- and post-data are available.
Results
Kv3.4 channels underlie IAHV in nociceptors
To isolate IAHV under minimally invasive conditions in acutely dissociated small-diameter DRG neurons, we used the cell-attached macropatch configuration (Methods). In this configuration, a 1 s conditioning pulse to –30 mV from a holding voltage of –70 mV was sufficient to isolate a robust IAHV evoked by a family of step depolarizations (Fig. 1A). The pre-pulse to –30 mV eliminates the low voltage-activated A-type K+ current typically mediated by Kv4.1 and Kv4.3 channels in these neurons (Phuket & Covarrubias, 2009). IAHV was present in 80 out of 95 patches, was on average 3.6 ± 0.4 times larger than the current at 500 ms (I500) (Fig. 1B), and was similarly expressed in isolectin B4 negative and positive (IB4- and IB4+) neurons (see on-line Supplemental Material, Fig. S1). Voltage-dependent activation of IAHV requires depolarizations that are more positive than the AP threshold in these neurons (Fig. 1C, Table 1). Accordingly, fast activation and deactivation kinetics also exhibit high-voltage dependence (Fig. 1D). The voltage dependence of steady-state inactivation induced by 5 s conditioning pulses (from a holding voltage of –70 mV) to various voltages displays two components: a small hyperpolarized component, consistent with the presence of a residual Kv4 current at –70 mV, and a dominant high-voltage component (Fig. 1C). The time constant of inactivation (τi), determined from the best exponential fits to the decay phase of the currents, was not voltage dependent at positive voltages >+20 mV (Fig. 1E). The recovery from inactivation of the A-type K+ currents at –100 mV was biphasic. The fast and slow time constants are consistent with low and high voltage-activated A-type K+ channels, respectively (Fig. 1F). Low concentrations of TEA in the patch electrode blocked IAHV almost completely. Indicating the loss of IAHV, Ip/I75 decreased by 3-fold (P = 0.01, Student's t test; Fig. 2D). Overall, the biophysical (Table 1) and pharmacological properties of IAHV in small-diameter DRG neurons agree closely with the hallmarks of heterologously expressed Kv3.4 channels (Beck et al. 1998; Gutman et al. 2005).
Figure 2. Single-cell qPCR and pharmacology indicate the presence of Kv3.4 channels in small-diameter DRG neurons.
A, Kv3.4 mRNA qPCR amplification plots of standard curve (open diamonds) and 1 μl of cDNA from 49 Nav1.9 positive small-diameter neurons (filled diamonds). Solid horizontal line indicates critical threshold for relative fluorescence (RF). The flat curve corresponds to no template control (arrow). B, standard curve of data in A. To determine copies per cell from 1 μl of cDNA reaction, data were multiplied by 50 to correct for using 1/50 of the original 10 μl (Methods). C, mRNA copy numbers for Kv3 channel transcripts in Nav1.9 positive small-diameter DRG neurons. Ratios above the bar graph indicate the fractions of neurons where the corresponding transcripts were found. D, representative traces at +100 mV with either control solution (CTRL) or 300 μm TEA in the patch pipette. The peak current over the current at 75 ms (Ip/I75) decreased from 3.29 ± 0.21 (n = 90, two outliers with ratio >20 were excluded from analysis) in control patches to 1.18 ± 0.07 (n = 6) in the presence of TEA. These results pooled observations at 0.3 and 2 mm TEA (n = 4 and 2, respectively). A ratio ≤1.5 indicates that IAHV is absent. The asterisks indicate P < 0.05.
To investigate the molecular identity of the IAHV, we screened for Kv3 mRNAs using single-cell qPCR in small-diameter DRG neurons (15–25μm). All neurons screened expressed the Nav1.9 transcript, which is specifically found in nociceptors (Dib-Hajj et al. 1998; Amaya et al. 2000; Ho & O'Leary, 2011). Whereas mRNA encoding Kv3.4 was detected in 94% of neurons (Fig. 2), transcripts encoding Kv3.1 and Kv3.2 were rarely found (0% and 20% of neurons, respectively). In contrast, 88% of the neurons expressed Kv3.3 mRNA; however, compared to all other Kv3 mRNAs, the copy numbers for the Kv3.4 mRNA are substantially higher (P < 0.0001 for individual comparisons, t test with Bonferroni correction; Fig. 2C).
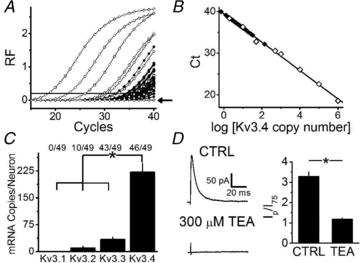
Finally, to confirm that Kv3.4 channels underlie IAHV in DRG neurons, we silenced the Kv3.4 mRNA by transfecting the cells with specific siRNAs (Methods). As a result, the Ip/I75 is reduced 2-fold (P = 0.0005, ANOVA; Fig. 3). This effect is also apparent when examining the raw peak currents (Fig. S2). Showing that the transfection per se was not responsible for the inhibition of the IAHV, three separate controls (non-transfected, mock-transfected and control siRNA-transfected neurons) exhibited indistinguishable Ip/I75 values (Fig. 3B). In addition, single-cell qPCR 24 h post-transfection detected Kv3.4 mRNA in 93% of control siRNA-transfected neurons and 60% of Kv3.4 siRNA-transfected neurons. The Kv3.4 siRNA actually decreases expression from 220 ± 60 transcripts/cell in neurons transfected with control siRNA to 15 ± 7 transcripts/cell in neurons transfected with Kv3.4 siRNA (n = 15 and 25, respectively; P < 0.001, Bonferroni-corrected t test; Fig. 3C). Interestingly, neurons kept in culture for 24 h after transfection with the control siRNA have Kv3.4 mRNA levels identical to control neurons harvested immediately after dissociation (Fig. 3C). Altogether, the biophysical, pharmacological and molecular results constitute compelling evidence for a dominant contribution of Kv3.4 channels to the IAHV in small-diameter DRG nociceptors.
Figure 3. Kv3.4 siRNA specifically reduces IAHV.
A, representative families of macropatch currents from mock-transfected, control siRNA-transfected (CTRL siRNA) and Kv3.4 siRNA-transfected neurons. Currents were elicited as described in Fig. 1 legend. B, the peak current over the current at 75 ms (Ip/I75) at +100 mV was 3.3 ± 0.2 in control, non-transfected neurons (n = 90), 2.9 ± 0.4 in mock-transfected neurons (n = 24), 3.0 ± 0.4 in control siRNA transfected neurons (n = 23), and 1.5 ± 0.1 (n = 24) in patches from Kv3.4 siRNA transfected neurons. Dashed line = 1.5 and indicates the cut off for IAHV in the recording (Methods). Four outliers (1 in mock-transfected, 2 in control siRNA-transfected, and 1 in Kv3.4 siRNA-transfected) were excluded from analysis with Ip/I75 > 2 SD from their respective means; however comparisons including these outliers still indicate significant reduction in Ip/I75 in Kv3.4 siRNA transfected neurons as compared to all other groups. C, average Kv3.4 mRNA copies/neuron in the indicated conditions. Ratios quantify the fraction of neurons that express Kv3.4 transcripts. The asterisks indicate P≤ 0.05.
PKC activation slows fast Kv3.4 channel inactivation in small-diameter DRG neurons
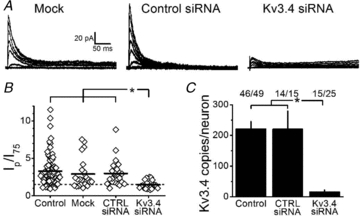
To test whether the previously reported modulation of Kv3.4 channels by PKC occurs in small-diameter DRG nociceptors, we obtained cell-attached macropatches and perfused the bath with phorbol esters and PKC inhibitors while recording the IAHV in the patch (Fig. 4). Upon application of 500 nm PDBu, the τi increases from 15 ± 5 ms to 60 ± 19 ms (P = 0.03, Student's paired t test), and the (I500/Ip) also increases from 0.15 ± 0.07 to 0.52 ± 0.13 (P = 0.01, Student's paired t test). Similarly, 100 nm PMA increases I500/Ip 2.5-fold (P = 0.03, Student's paired t test; Fig. 4B). These observations show that fast inactivation of Kv3.4 channels is substantially slowed upon pharmacological activation of PKC. By contrast, the inactive phorbol ester 4α-PDBu does not affect Kv3.4 channel inactivation (Fig. 4). The slowing of fast Kv3.4 channel inactivation induced by PKC activation is inhibited upon bath application of 10 μm BIM for 5 min prior to the addition of PDBu and BIM (Fig 4). Given that the extracellular side of the membrane patch was not exposed to the reagents, these results clearly demonstrate that pharmacological activation of PKC in small-diameter DRG neurons modulates fast Kv3.4 channel inactivation in a non-membrane-delimited manner.
Figure 4. PKC activation slows Kv3.4 channel inactivation.
A, representative normalized currents before and after addition of the indicated pharmacological agents washed into the bath. From a holding of –70 mV, the currents were elicited by a depolarization to +100 mV after a 1 s condition pulse to –30 mV. Thick traces were recorded before drug addition and thin traces were recorded 5 min after addition of drug. BIM + PDBu example includes three traces: no drugs, BIM alone and BIM + PDBu. Scale bars are identical for all currents displayed. B, summary of τi and current at 500 ms over the peak current (I500/Ip) at +100 mV for all conditions. In PMA condition, τi increases from 18 ± 4 ms to 43 ± 18 ms and the I500/Ip increases from 0.12 ± 0.04 to 0.31 ± 0.08. Upon 4α-PDBu addition, τi remained unaltered at 25 ± 13 ms before and 23 ± 12 ms after as did I500/Ip (0.27 ± 0.06, before; and 0.20 ± 0.07, after). For BIM experiments, the τi also remained unaltered at 20 ± 8 ms with BIM alone and 23 ± 10 ms after the addition of PDBu; and the I500/Ip was 0.22 ± 0.04 with BIM alone and 0.24 ± 0.04 in the presence of BIM and PDBu. Solid bars represent τi and hatched bars indicate I500/Ip. Relative to controls, the asterisks indicate P≤ 0.05.
Physiological activation of PKC slows Kv3.4 channel inactivation in a membrane-delimited manner
To determine whether physiological activation of PKC may also slow fast Kv3.4 channel inactivation, we tested a G-protein coupled receptor (GPCR) agonist a cocktail including 10 μm serotonin, 100 μm histamine and 100 μm bradykinin to activate the corresponding GPCR signalling cascades. Under control conditions (no GPCR activation), Fig. 5A shows a current from a macropatch that clearly contained Kv3.4 channels (representing 83% of the patches), and Fig. 5B depicts another current that only exhibits very slow activation (representing 17% of the patches), indicating the absence of Kv3 channels. If only the extracellular side of the patch was exposed to GPCR agonists (by inclusion of the cocktail in the pipette solution only), there were three distinct fast-activating current phenotypes: a rapidly inactivating IAHV, a slowly inactivating IAHV (>2 SD from the mean τi of controls) and a fast-activating delayed rectifier current (27%, 27% and 45% of the patches, respectively; Fig. 5C). The τi of IAHV in the six patches exhibiting an inactivating component is significantly slower (37.9 ± 16.5 ms) than that of control patches (P = 0.01, Student's t test; Fig. 5F). In addition, compared to all patches recorded in the absence of GPCR agonists, the GPCR agonist cocktail in the patch pipette significantly increased the I500/Ip from 0.47 ± 0.04 (n = 92) to 0.77 ± 0.17 (n = 11; P = 0.03, Student's t test; Fig. 5F). Thus, upon physiological activation of PKC, 72% of the patches display currents that activate quickly and either inactivate slowly or do not inactivate appreciably, indicating modulation of Kv3.4 channel inactivation. To show that this GPCR-mediated modulation results from the activation of PKC, we pre-incubated the neurons with BIM for 5 min and subsequently patch-clamped with the GPCR agonist cocktail in the patch pipette only. Following this treatment, the vast majority of the patches (86%) displayed fast-activating and fast-inactivating currents (Fig. 5D). Furthermore, pretreatment with BIM inhibited the increase in I500/Ip by the GPCR agonists (0.13 ± 0.05, P = 0.01, Student's independent t test; Fig. 5F). A slowly activating current was excluded from this analysis as an outlier (1/7) because its I500/Ip was 2.4 and >2 SD from the mean. In all conditions (control, pipette GPCR agonists and bath BIM), there was no change in the peak amplitude of the total current indicating that there was no loss of channel activity (data not shown). When the agonist cocktail was present in the bath solution and absent in the pipette solution (extracellular side of the patch was not exposed to GPCR agonists), inactivation of Kv3.4 channels remained unaffected (Fig. 5E and G). Since GPCR activation in the patch is necessary and sufficient to modulate fast Kv3.4 channel inactivation, the data suggest a membrane-delimited modulatory mechanism in small-diameter DRG nociceptors.
Figure 5. GPCR agonists slow Kv3.4 channel inactivation in a PKC-dependent and membrane-delimited manner.
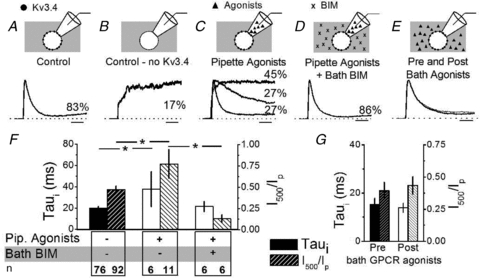
A–E, representative normalized current traces elicited as described in Fig. 4 legend. Scale bars indicate 10 ms. Percentages indicate the fraction of patches corresponding to the represented current phenotypes under the indicated conditions. When no Kv3.4 currents were seen in control patches a slow-activating outward current was seen (B). When GPCR agonists were added to the patch pipette (C), three current phenotypes were seen: a fast-activating delayed rectifier (no inactivation), a slow-inactivating A-type (τi > 2 SD from the control mean), and a fast-inactivating A-type (τi≤ 2 SD from the control mean). Addition of BIM to the bath eliminated the effect of GPCR agonists (D, one recording exhibiting only a slow-activating outward current was not included). In E, currents were recorded before (thick line) and after (thin line) perfusion of GPCR agonists into the bath (pipette solution contained no GPCR agonists). F and G, summary of pipette GPCR agonists compared to controls and BIM in the bath (F) and the addition of GPCR agonists into the bath only (G). All non-inactivating currents were excluded from τi comparisons. For wash-in experiments, τi was 15 ± 3 ms pre- and 14 ± 2 ms post-GPCR agonist application to the bath and the current at 500 ms over the peak current (I500/Ip) was 0.39 ± 0.07 pre- and 0.43 ± 0.07 post-GPCR agonist application (P > 0.05 and n = 5 for both). Solid and hatched bars represent τi and I500/Ip, respectively. The asterisks indicate P≤ 0.05.
Modulation of Kv3.4 channel inactivation by PKC regulates AP repolarization in small-diameter nociceptors
The results so far show that the Kv3.4 channel is the dominant high-voltage Kv channel in small-diameter DRG nociceptors, and that pharmacological or physiological activation of PKC substantially slows Kv3.4 channel inactivation. Thus, phosphorylated Kv3.4 channels may influence the rate of AP repolarization more effectively and possibly contribute to the AHP. To test this hypothesis, we applied whole-cell current clamping to characterize the APs of small-diameter nociceptors under basal conditions and upon activating PKC. Somatic APs were triggered by applying a 0.5 ms current injection pulse. Table 2 summarizes the passive and active properties of the neurons. The APs are typically long-duration and display an inflexion in the repolarizing phase that is characteristic of small-diameter nociceptors (Traub & Mendell, 1988; Fig. 6A). Since other TEA-hypersensitive non-Kv3 channels my also shape these APs under basal conditions (Safronov et al. 1996; Scholz et al. 1998; Beekwilder et al. 2003; Chi & Nicol, 2007; Zhang et al. 2010), we exposed the neurons to dendrotoxin-K (DTX-K) and iberiotoxin (IbTX), specific blockers of Kv1.1 and big-conductance Ca2+-activated K+ (BK) channels, respectively. APs from control and toxin-exposed neurons are indistinguishable (Table 2). In the light of these results, we combined the results obtained in the absence and presence of toxins and examined the effect of PDBu on the AP waveform. Based on paired comparisons, the PDBu treatment significantly influences the AP waveform in various ways (Fig. 6A and B, Table 2): it (1) reduces the overshoot to 86 ± 6% of control; (2) increases the amplitude of the AHP to 108 ± 2% of control; (3) decreases the APD50 to 86 ± 4% of control; (4) increases threshold to 111 ± 5% of control; and (5) increases the rate of repolarization to 122 ± 7% of control.
Figure 6. Activation of PKC accelerates AP repolarization in putative nociceptors.
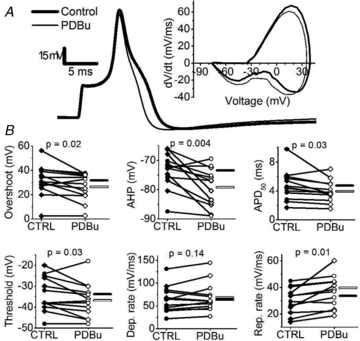
A, representative AP elicited by a 0.5 ms current injection pulse in the absence (thick line) and presence of 300 nm PDBu (thin line). Inset shows the corresponding phase-plane plots. B, summary of paired scatter plots of AP properties before (filled symbols) and after PDBu (open symbols). Lines join individual paired pre and post experiments. Experiments were conducted without DTX-K and IbTX (diamonds) and with 100 nm each of DTX-K and IbTX (circles). Horizontal bars indicate the mean (filled bar = CTRL and open bar = PDBu). Means ± SEM of all parameters are displayed in Table 2.
We also noted that on average the PDBu treatment hyperpolarized the resting membrane potential by 5.4 mV (P = 0.01, Student's paired t test; Table 2). This hyperpolarization could be the result of activating a resting K+ conductance. Thus, we tested the effect of forced hyperpolarization to ask whether the changes induced by PDBu on the AP waveform could be secondary to hyperpolarization of the resting membrane potential. On average, a 17 mV hyperpolarization caused the following significant changes: it (1) increases the rate of AP depolarization to 143 ± 17% of control; (2) increases the rate of AP repolarization to 120 ± 8% of control; and (3) increases the AHP amplitude to 108 ± 3% of control. The increases in the rate of repolarization and the AHP coincide with those induced by PDBu (Table 2); however, a 3-fold larger forced hyperpolarization of the resting membrane potential was necessary to observe them.
Activation of PKC by PDBu most significantly increased the rate of repolarization, decreased the APD50 and increased the amplitude AHP, which conceivably is a consequence of switching the phenotype of Kv3.4 channels from A-type to delayed rectifier-type by PKC-dependent phosphorylation of the channel's NTID. Since GPCR agonists are also capable of inducing this modulation (Fig. 5), physiological activation of PKC should recapitulate the effects of PDBu on the AP waveform. To test this prediction, we examined the properties of the AP waveform before and after exposing the neurons to the GPCR agonist cocktail (see above). Consistently, application of GPCR agonists also increased the rate of repolarization (128 ± 7% of control) and decreased the APD50 (71 ± 5% of control) (Fig. 7A and B, Table 2). In 5/6 neurons, the AHP also appeared increased, but this change does not reach statistical significance (Fig. 7B, Table 2). The effects of GPCR agonists are likely to be PKC dependent because we observed no effects on AP repolarization upon bath application of GPCR agonists in the presence of BIM (n = 4, data not shown).
Figure 7. GPCR agonists accelerate AP repolarization.
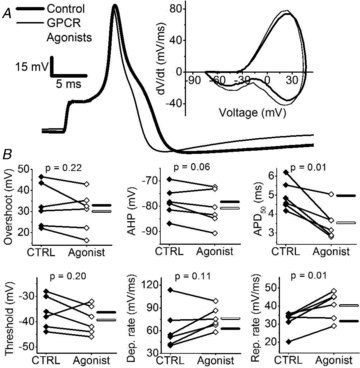
A, representative effect of GPCR agonists applied to the bath as done in Fig. 6. B, summary of paired scatter plots of AP properties before (filled symbols) and after GPCR agonists (open symbols). Lines join individual paired pre and post experiments. Horizontal bars indicate mean (filled bar = CTRL and open bar = GPCR agonists). Means ± SEM of all parameters are displayed in Table 2.
If the effects of PKC activation by PDBu on the AP waveform mainly result from modulating inactivation of Kv3.4 channels, low doses of TEA should reverse these effects. Accordingly, under sustained activation of PKC, the application of 100 μm TEA decreased the rate of repolarization by 38 ± 10% (P = 0.02, Student's paired t test) and returned the APD50 to near control levels (Table 2). However, low doses of TEA may also block other K+ channels (Gutman et al. 2005). Thus, to show more conclusively that Kv3.4 channels are indeed key players in the regulation of the AP by PKC activation, we transfected the neurons with Kv3.4-specific siRNAs to specifically knock down IAHV (Fig. 3). Under basal conditions, the Kv3.4 siRNA treatment decreased the rate of repolarization to 78 ± 10% of control APs recorded from neurons 24 h after plating and the APD50 increased to 126 ± 16% of controls (Fig. 8A and B, Table 3). However, only changes in APD50 could be conclusively associated with Kv3.4 knock down (Table 3). Moreover, the treatment with Kv3.4 siRNA eliminated the effects of GPCR agonists on the rate of repolarization, APD50 and AHP (Fig. 9A and B, Table 3); and furthermore, the responses of control siRNA transfected neurons to GPCR agonists were indistinguishable from those of control neurons (Table 3 and Fig. S3). Therefore, the data strongly suggest that the modulation of Kv3.4 channel N-type inactivation by PKC is a major regulatory mechanism of the AP waveform in nociceptors.
Figure 8. Kv3.4 siRNA slows repolarization of nociceptor AP.
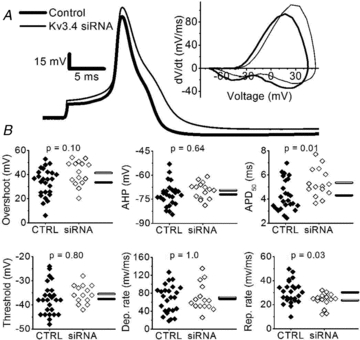
A, representative overlaid comparison of a control AP recorded >24 h after plating (thick trace) against an AP from a Kv3.4 siRNA-transfected neuron (thin trace). The corresponding phase-plane plots are shown in the inset. B, summary scatter plots of AP properties in controls (filled symbols, CTRL) and Kv3.4 siRNA-transfected neurons (open symbols, siRNA). Controls were cultured for >24 h to account for possible changes induced by culture conditions. Horizontal bars indicate the mean (filled bar = CTRL and open bar = Kv3.4 siRNA). P values are from post hoc Bonferonni-corrected t tests after an ANOVA of >24 h control neurons, control siRNA transfected neurons and Kv3.4 siRNA transfected neurons. Means ± SEM of all parameters (including control siRNA-transfected neurons) are displayed in Table 3.
Table 3.
Passive and active properties of APs from siRNA transfected neurons and the effect PKC activation
| CTRL siRNA + GPCR Agonists | Kv3.4 siRNA + GPCR Agonists | ||||||
|---|---|---|---|---|---|---|---|
| >24 h Controla | CTRL siRNAa | Kv3.4 siRNAa | Pre | Post | Pre | Post | |
| RMP (mV) | −58.4 ± 1.4 | −61.9 ± 1.7 | −57.3 ± 2.1 | −63.4 ± 2.4 | −67.8 ± 4.4 | −59.2 ± 3.1 | −60.5 ± 4.4 |
| Input resistance (GΩ) | 1.0 ± 0.1 | 0.8 ± 0.4 | 0.7 ± 0.1 | 0.7 ± 0.3 | 0.5 ± 0.1 | 0.6 ± 0.2 | 0.8 ± 0.2 |
| Capacitance (pF) | 13.2 ± 1.1 | 17.1 ± 1.9 | 18.4 ± 1.9*† | 15.2 ± 1.7 | — | 20.7 ± 3.1 | — |
| Diameter (μm) | 17.7 ± 0.3 | 17.9 ± 0.4 | 18.1 ± 0.4 | 18.8 ± 0.9 | — | 17.7 ± 0.6 | — |
| Rheobase (pA) | 40 ± 17 | 70 ± 14 | 48 ± 11 | 87 ± 25 | 116 ± 32 | 64 ± 17 | 68 ± 22 |
| Threshold (mV) | −37.6 ± 1.3 | −37.1 ± 1.3 | −35.5 ± 1.0 | −36.4 ± 1.6 | −39.1 ± 2.3 | −35.1 ± 1.7 | −38.2 ± 1.5 |
| Overshoot (mV) | 33.4 ± 2.3 | 34.0 ± 2.5 | 41.3 ± 2.6 | 32.4 ± 2.9 | 36.0 ± 3.3 | 46.3 ± 2.6 | 39.2 ± 3.5** |
| AHP (mV) | −72.0 ± 1.4 | −71.2 ± 1.2 | −69.5 ± 1.3 | −71.4 ± 2.3 | −76.4 ± 2.3* | −69.8 ± 2.0 | −71.1 ± 3.7 |
| APD50 (ms) | 4.3 ± 0.3 | 4.3 ± 0.1 | 5.4 ± 0.3*‡ | 4.5 ± 0.2 | 4.0 ± 0.2* | 5.3 ± 0.4 | 4.9 ± 0.3 |
| APD90 (ms) | 1.21 ± 0.06 | 1.26 ± 0.04 | 1.38 ± 0.1 | 1.28 ± 0.05 | 1.12 ± 0.05 | 1.36 ± 0.08 | 1.28 ± 0.08 |
| Max depolarization rate (mV ms−1) | 67.3 ± 6.2 | 69.7 ± 5.8 | 70.3 ± 7.8 | 60.0 ± 4.9 | 73.4 ± 3.8 | 70.1 ± 7.8 | 78.8 ± 9.5 |
| Max repolarization rate (mV ms−1) | 30.2 ± 1.9 | 25.4 ± 1.2 | 23.5 ± 1.6*† | 24.2 ± 1.9 | 28.8 ± 1.5* | 25.0 ± 1.6 | 24.0 ± 2.3 |
| n | 26 | 19 | 15 | 9 | 8 | ||
AP properties were measured as described in Methods and Student's paired t test used for comparisons of paired pre and post data.
>24 h control neurons, control siRNA transfected neurons and Kv3.4 siRNA transfected neurons were compared using ANOVA with post hoc Bonferroni-corrected t tests. There was no difference between >24 controls and control siRNA.
P≤ 0.05
P≤ 0.01
difference exists between Kv3.4 siRNA transfected neurons and >24 h control neurons
difference exists between Kv3.4 siRNA transfected neurons and both >24 h control and control siRNA transfected neurons
RMP = resting membrane potential.
Figure 9. Kv3.4 siRNA abolishes the effects of GPCR agonists.
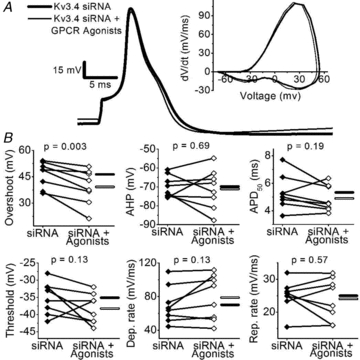
A, representative APs before (thick trace) and after the application of GPCR agonists (thin trace) in a Kv3.4 siRNA-transfected neuron. The corresponding phase-plane plots are shown in the inset. B, summary of paired scatter plots of AP properties from Kv3.4 siRNA-transfected neurons before (filled symbols) and after GPCR agonists (open symbols). Lines join individual paired pre and post experiments. Horizontal bars indicate mean (filled bar = siRNA and open bar = siRNA + GPCR agonists). Means ± SEM of all parameters are displayed in Table 3.
Discussion
We applied a combination of biophysical, molecular and pharmacological approaches to establish that Kv3.4 channels underlie the IAHV in small-diameter DRG nociceptors. Further confirming this conclusion, we demonstrate that upon pharmacological or physiological activation of PKC, native Kv3.4 channels undergo a characteristic dramatic slowing of fast N-type inactivation. Then, to probe the electrophysiological impact of this modulation, we determined that PKC activation selectively narrows the AP, accelerates AP repolarization, and hyperpolarizes the AHP. These changes show that, as a result of slowing fast Kv3.4 channel inactivation, the influence of a high voltage-activated K+ conductance on the AP waveform is enhanced. Finally, specific siRNAs silence Kv3.4 channel expression and, consequently, eliminate the effects of PKC activation on the AP waveform. From this study, the high voltage-activated Kv3.4 channel and its modulation of N-type inactivation by PKC emerge as major factors shaping the repolarization of the AP in nociceptors.
The molecular basis of high voltage-activated A-type K+ currents in DRG neurons
Several studies have characterized the A-type K+ channels in rodent DRG neurons (Gold et al. 1996b; Rasband et al. 2001; Winkelman et al. 2005; Chien et al. 2007; Phuket & Covarrubias, 2009). Consistently, these studies report a low voltage-activated A-type K+ current, which most likely is mediated by Kv channels composed of either Kv4.x subunits (Winkelman et al. 2005; Phuket & Covarrubias, 2009) or Kv1.4 subunits (Rasband et al. 2001). In contrast, much less was known about IAHV in DRG neurons. Gold et al. (1996b) used whole-cell patch-clamping and a subtraction strategy to isolate a high voltage-activated transient outward current that is hypersensitive to TEA and has gating properties similar to those of heterologously expressed Kv3.4 currents. The molecular identity and function of this IAHV, however, remained uncertain. Here, we show that under minimally invasive conditions (cell-attached patch-clamping) in small-diameter nociceptors, it is possible to directly isolate a robust IAHV bearing hallmark properties of Kv3.4 channels: (1) depolarized voltage dependence of activation and inactivation; (2) fast deactivation; (3) fast inactivation; (4) slow recovery from inactivation; and (5) high sensitivity to TEA. Single-cell qPCR and siRNA knock down experiments provide further strong support for the presence of Kv3.4 channels as the most significant Kv3.x isoform in these neurons. Additionally, Chien et al. (2007) demonstrated robust anti-Kv3.4 immuno-staining in small-diameter IB4- and IB4+ nociceptors, which is in excellent agreement with our electrophysiological results (Fig. S1). Thus, most likely Kv3.4 homotetramers underlie IAHV in small-diameter nociceptors from rats, but the possibility of some heterotetramers including Kv3.3 subunits cannot be ruled out entirely because Kv3.3 transcripts are also found in a majority of investigated neurons (Fig. 2C). Based on the biophysical, molecular and pharmacological evidence, independent contributions of other A-type K+ channels to the IAHV in nociceptors are not likely. Kv4.x, Kv1.4 and Kv12.1 channels are not hypersensitive to TEA (Trudeau et al. 1999; Gutman et al. 2005). Kv1.1 and BK channels associated with β-subunits that induce fast inactivation are also TEA-sensitive. However, their biophysical properties do not agree with those of the nociceptor IAHV, and currents from these channels are down-regulated upon activation of PKC (Boland & Jackson, 1999; Zhou et al. 2010).
Membrane-delimited modulation of Kv3.4 channel inactivation in DRG nociceptors
The molecular mechanism underlying the elimination of fast Kv3.4 channel inactivation by PKC-dependent phosphorylation of the channel's NTID is understood at the atomic level. Essentially, our previous work combined biophysical and high-resolution structural analyses to show that the non-additive effects of phosphorylating four N-terminal serines (S8, S9, S15 and S21) cause a major conformational change in the NTID, which renders it mostly unstructured and unable to occlude the channel's internal mouth (Beck et al. 1998; Antz et al. 1999). For many years, however, the signalling mechanism and physiological impact of this dramatic modulation remained unknown. The results reported here provide multiple lines of evidence to establish that neuronal Kv3.4 channels expressed in nociceptors undergo dramatic slowing of inactivation upon pharmacological and physiological activation of PKC. In particular, the modulation induced by PDBu occurs in non-membrane-delimited manner, whereas that induced by GPCR agonists occurs in a membrane-delimited manner (Brown, 1993). That is, PDBu present in the external bath but absent in the recording patch electrode enters the neuron and helps translocate PKC globally in the cell to phosphorylate Kv3.4 channels. In contrast, GPCR agonists applied to the external bath may only activate PKC in a membrane-associated signalling complex that is not protected by the recording patch electrode, and thus modulation of Kv3.4 channels in the cell-attached patch is not observed. The dramatic modulation of inactivation only occurs when the GPCR agonists are present inside the patch electrode, suggesting that GPCRs, the necessary second messenger molecules, PKC and Kv3.4 channels are in the cell-attached patch forming a membrane-associated signalling complex. We hypothesize that the proposed signalling complex is similar to those identified for other voltage-gated ion channels in excitable tissues (Levitan, 2006; Dai et al. 2009).
The modulation observed here confirms that Kv channel N-type inactivation is highly malleable. Several other A-type K+ channels (Kv1.4, Kv1.1+β1-subunit and Kv3.3) undergo reversible modulation of fast inactivation through several distinct physiological mechanisms including phosphorylation, oxidation/reduction and phosphatidylinositol 4,5-bisphosphate (PIP2) interactions (Ruppersberg et al. 1991; Oliver et al. 2004; Desai et al. 2008; Pan et al. 2011). To the best of our knowledge, this study shows for the first time experimentally how modulation of Kv channel N-type inactivation may impact neuronal excitability.
Kv3.4 channels govern the repolarization of the nociceptor AP
The important contribution of Kv3.4 channels to the repolarization of the AP in small-diameter DRG neurons is strongly supported by converging results showing: (1) prolonged AP duration in neurons transfected with Kv3.4 siRNA; (2) accelerated rate of repolarization and reduced width upon pharmacological and physiological activations of PKC; (3) reversibility of the PKC effects upon application of submillimolar doses of TEA; and (4) elimination of the PKC effects in neurons transfected with Kv3.4 siRNA. These results indicate that, upon phosphorylation of the Kv3.4 NTID, the transformation of Kv3.4 channels from rapidly inactivating A-type to delayed rectifier-type enhances their influence on the repolarization of the AP. Also, consistent with increasing this influence of Kv3.4 channels upon activation of PKC, the AHP is slightly hyperpolarized.
In agreement with the dominating presence of Kv3.4 channels underlying a robust IAHV and Kv2.x or Kv7.x channels possibly underlying a slow-activating sustained outward current in a majority of cell-attached patches, DTX-K and IbTX did not produce significant effects on the AP waveform. Kv1.1 channels, the main targets of DTX-K, are more typically found in larger DRG neurons not included in this study (Rasband et al. 2001) and when present in small-diameter neurons, Kv1.1 channels do not affect AP duration (Chi & Nicol, 2007). BK channels, the specific targets of IbTX, may not be observed in cell-attached patches under basal conditions and may not have a significant effect on AP waveform as seen in hippocampal mossy fibre boutons (Alle et al. 2011). Also, the expression levels of BK channels in the subpopulation of young neurons investigated here might be variable and very low (Scholz et al. 1998). Could other voltage-gated ion channels contribute to the effects of PKC on the AP waveform? Previous reports show that PKC enhances currents mediated by TTX-resistant Na+ channels and N- and L-type Ca2+ channels in DRG neurons (Gold et al. 1998; Sculptoreanu & de Groat, 2003). If these modulations were the main effect of PKC in small-diameter nociceptors, they would prolong the AP, which is contrary to our findings but has been seen when TEA is present (Werz & Macdonald, 1987). A slight reduction in the AP overshoot in response to GPCR activation in Kv3.4 siRNA-transfected neurons (Fig. 9B) could have resulted from modulating Na+ channels, which is unmasked upon knocking down Kv3.4 channels.
A possible role for Kv3.4 channel N-terminal phosphorylation in nociceptors
We hypothesize that the modulation of Kv3.4 channel N-type inactivation on AP repolarization may dynamically regulate the physiological responses of DRG nociceptors in relevant scenarios. Ca2+-dependent vesicular release of neurotransmitters and neuromodulators could be particularly sensitive to this modulation. For instance, somatic Kv3.4 channels may regulate release of neuromodulators from the somas of DRG neurons (Huang & Neher, 1996; Zheng et al. 2009). In addition, strong Kv3.4 immunoreactivity has been reported in nociceptor nerve terminals of dorsal horn laminas I–III (Brooke et al. 2004; Chien et al. 2007) where the channel may regulate neurotransmitter release. In these locations, the modulation of Kv3.4 channel inactivation by PKC may impact Ca2+ entry and vesicular release by regulating AP duration (McCobb & Beam, 1991). Under normal conditions of acute pain signalling, the slowing of Kv3.4 channel inactivation may underlie a homeostatic response (Zhao et al. 2011) with anti-nociceptive implications. If Kv3.4 channels become phosphorylated, the AP is shortened, Ca2+ entry is reduced and, consequently, Ca2+-dependent neurotransmission and downstream nociceptive signalling are inhibited. During the transition from acute to chronic neuropathic pain, the down-regulation of Kv3.4 channel expression (Chien et al. 2007) may compromise the ability to regulate Ca2+-dependent vesicular release. Testing this working hypothesis will require investigating the contributions of the Kv3.4 channel to neurotransmitter/neuromodulator release, and elucidating the precise physiological processes that activate the nociceptor GPCRs. The latter may involve local factors (in the DRG and dorsal horn) and descending anti-nociceptive pathways from the CNS.
Acknowledgments
This work was supported by a Dubb's MD/PhD Scholar Fellowship and an Eakin's Legacy Fund Fellowship, both from Thomas Jefferson University (D.M.R.). We thank Dr Irwin Levitan for critical comments and members of the Covarrubias lab for their feedback. This work is part of the requirements toward a doctoral degree in Neuroscience from Thomas Jefferson University for D.M.R.
Glossary
- AHP
afterhyperpolarization
- AP
action potential
- APD50
action potential duration at 50% of the spike height
- APD90
action potential duration at 90% of the spike height
- BIM
bisindolylmaleimide II
- BK
Ca2+-activated K+ channel
- DRG
dorsal root ganglia
- DTX-K
dendrotoxin-K
- I75
current amplitude at 75 ms
- I500
current amplitude at 500 ms
- IAHV
high-voltage A-type K+ current
- Ip
peak current amplitude
- IbTX
iberiotoxin
- IB4
isolectin B4
- GPCR
G-protein coupled receptor
- PDBu
phorbol 12,13-dibutyrate
- PKC
protein kinase C
- PMA
phorbol-12-myristate-13-acetate
- τi
time constant of inactivation
- TTX
tetrodotoxin
Author contributions
D.M.R., M.E.O. and M.C. conceived and designed the experiments. D.M.R and C.H. collected the data. D.M.R. and M.C. analysed the data and drafted the article. Experiments were performed in the laboratories of M.E.O. and M.C. at Thomas Jefferson University. All authors approved the final version of this manuscript.
Supplementary material
Figure S1
Figure S2
Figure S3
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Alle H, Kubota H, Geiger JRP. Sparse but highly efficient Kv3 outpace BKCa channels in action potential repolarization at hippocampal mossy fiber boutons. J Neurosci. 2011;31:8001–8012. doi: 10.1523/JNEUROSCI.0972-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Decosterd I, Samad TA, Plumpton C, Tate S, Mannion RJ, Costigan M, Woolf CJ. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- Antz C, Bauer T, Kalbacher H, Frank R, Covarrubias M, Kalbitzer HR, Ruppersberg JP, Baukrowitz T, Fakler B. Control of K+ channel gating by protein phosphorylation: structural switches of the inactivation gate. Nat Struct Biol. 1999;6:146–150. doi: 10.1038/5833. [DOI] [PubMed] [Google Scholar]
- Antz C, Kalbitzer HR, Geyer M, Fakler B, Schott MK, Guy HR, Frank R, Ruppersberg JP. NMR structure of inactivation gates from mammalian voltage-dependent potassium channels. Nature. 1997;385:272–275. doi: 10.1038/385272a0. [DOI] [PubMed] [Google Scholar]
- Baranauskas G, Tkatch T, Nagata K, Yeh JZ, Surmeier DJ. Kv3.4 subunits enhance the repolarizing efficiency of Kv3.1 channels in fast-spiking neurons. Nat Neurosci. 2003;6:258–266. doi: 10.1038/nn1019. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Julius D, Scherrer G. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck EJ, Sorensen RG, Slater SJ, Covarrubias M. Interactions between multiple phosphorylation sites in the inactivation particle of a K+ channel. Insights into the molecular mechanism of protein kinase C action. J Gen Physiol. 1998;112:71–84. doi: 10.1085/jgp.112.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekwilder JP, O'Leary ME, van Den Broek LP, van Kempen GTH, Ypey DL, van Den Berg RJ. Kv1.1 channels of dorsal root ganglion neurons are inhibited by n-butyl-p-aminobenzoate, a promising anesthetic for the treatment of chronic pain. J Pharmacol Exp Ther. 2003;304:531–538. doi: 10.1124/jpet.102.042135. [DOI] [PubMed] [Google Scholar]
- Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22:10277–10290. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland LM, Jackson KA. Protein kinase C inhibits Kv1.1 potassium channel function. Am J Physiol Cell Physiol. 1999;277:C100–110. doi: 10.1152/ajpcell.1999.277.1.C100. [DOI] [PubMed] [Google Scholar]
- Brooke RE, Atkinson L, Batten TFC, Deuchars SA, Deuchars J. Association of potassium channel Kv3.4 subunits with pre- and post-synaptic structures in brainstem and spinal cord. Neuroscience. 2004;126:1001–1010. doi: 10.1016/j.neuroscience.2004.03.051. [DOI] [PubMed] [Google Scholar]
- Brown AM. Membrane-delimited cell signaling complexes: direct ion channel regulation by G proteins. J Membr Biol. 1993;131:93–104. doi: 10.1007/BF02791318. [DOI] [PubMed] [Google Scholar]
- Cardenas CG, Del Mar LP, Scroggs RS. Variation in serotonergic inhibition of calcium channel currents in four types of rat sensory neurons differentiated by membrane properties. J Neurophysiol. 1995;74:1870–1879. doi: 10.1152/jn.1995.74.5.1870. [DOI] [PubMed] [Google Scholar]
- Chi XX, Nicol GD. Manipulation of the potassium channel Kv1.1 and its effect on neuronal excitability in rat sensory neurons. J Neurophysiol. 2007;98:2683–2692. doi: 10.1152/jn.00437.2007. [DOI] [PubMed] [Google Scholar]
- Chien L-Y, Cheng J-K, Chu D, Cheng C-F, Tsaur M-L. Reduced expression of A-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J Neurosci. 2007;27:9855–9865. doi: 10.1523/JNEUROSCI.0604-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M, Wei A, Salkoff L, Vyas TB. Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron. 1994;13:1403–1412. doi: 10.1016/0896-6273(94)90425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Hall DD, Hell JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 2009;89:411–452. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai R, Kronengold J, Mei J, Forman SA, Kaczmarek LK. Protein kinase C modulates inactivation of Kv3.3 channels. J Biol Chem. 2008;283:22283–22294. doi: 10.1074/jbc.M801663200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci U S A. 1998;95:8963–8968. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Dastmalchi S, Levine JD. Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience. 1996a;71:265–275. doi: 10.1016/0306-4522(95)00433-5. [DOI] [PubMed] [Google Scholar]
- Gold MS, Levine JD, Correa AM. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neurosci. 1998;18:10345–10355. doi: 10.1523/JNEUROSCI.18-24-10345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Shuster MJ, Levine JD. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J Neurophysiol. 1996b;75:2629–2646. doi: 10.1152/jn.1996.75.6.2629. [DOI] [PubMed] [Google Scholar]
- Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16:1248–1257. doi: 10.1038/nm.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Lazdunski M, Grissmer S, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stühmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- Harper AA, Lawson SN. Electrical properties of rat dorsal root ganglion neurones with different peripheral nerve conduction velocities. J Physiol. 1985;359:47–63. doi: 10.1113/jphysiol.1985.sp015574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C, O'Leary ME. Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol Cell Neurosci. 2011;46:159–166. doi: 10.1016/j.mcn.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LY, Neher E. Ca2+-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron. 1996;17:135–145. doi: 10.1016/s0896-6273(00)80287-1. [DOI] [PubMed] [Google Scholar]
- Levitan IB. Signaling protein complexes associated with neuronal ion channels. Nat Neurosci. 2006;9:305–310. doi: 10.1038/nn1647. [DOI] [PubMed] [Google Scholar]
- Martina M, Metz AE, Bean BP. Voltage-dependent potassium currents during fast spikes of rat cerebellar Purkinje neurons: inhibition by BDS-I toxin. J Neurophysiol. 2007;97:563–571. doi: 10.1152/jn.00269.2006. [DOI] [PubMed] [Google Scholar]
- McCobb DP, Beam KG. Action potential waveform voltage-clamp commands reveal striking differences in calcium entry via low and high voltage-activated calcium channels. Neuron. 1991;7:119–127. doi: 10.1016/0896-6273(91)90080-j. [DOI] [PubMed] [Google Scholar]
- Oliver D, Lien C-C, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–270. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- Pan Y, Weng J, Levin EJ, Zhou M. Oxidation of NADPH on Kvβ1 inhibits ball-and-chain type inactivation by restraining the chain. Proc Natl Acad Sci U S A. 2011;108:1–6. doi: 10.1073/pnas.1100316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuket TRN, Covarrubias M. Kv4 channels underlie the subthreshold-operating A-type K-current in nociceptive dorsal root ganglion neurons. Front Mol Neurosci. 2009;2:3. doi: 10.3389/neuro.02.003.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A. 2001;98:13373–13378. doi: 10.1073/pnas.231376298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Wunder F, Stocker M, Lichtinghagen R, Mastiaux F, Beckh S, Kues W, Pedarzani P, Schröter KH, Ruppersberg JP, Veh R, Pongs O. Characterization of a Shaw-related potassium channel family in rat brain. EMBO J. 1992;11:2473–2486. doi: 10.1002/j.1460-2075.1992.tb05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazanski V, Becker A, Chen J, Sochivko D, Lie A, Wiestler OD, Elger CE, Beck H. Functional and molecular analysis of transient voltage-dependent K+ currents in rat hippocampal granule cells. J Physiol. 2001;537:391–406. doi: 10.1111/j.1469-7793.2001.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian IKA channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- Safronov BV, Bischoff U, Vogel W. Single voltage-gated K+ channels and their functions in small dorsal root ganglion neurones of rat. J Physiol. 1996;493:393–408. doi: 10.1113/jphysiol.1996.sp021391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz A, Gruss M, Vogel W. Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J Physiol. 1998;513:55–69. doi: 10.1111/j.1469-7793.1998.055by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sculptoreanu A, de Groat WC. Protein kinase C is involved in neurokinin receptor modulation of N- and L-type Ca2+ channels in DRG neurons of the adult rat. J Neurophysiol. 2003;90:21–31. doi: 10.1152/jn.00108.2003. [DOI] [PubMed] [Google Scholar]
- Traub RJ, Mendell LM. The spinal projection of individual identified A-δ- and C-fibers. J Neurophysiol. 1988;59:41–55. doi: 10.1152/jn.1988.59.1.41. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Titus SA, Branchaw JL, Ganetzky B, Robertson GA. Functional analysis of a mouse brain Elk-type K+ channel. J Neurosci. 1999;19:2906–2918. doi: 10.1523/JNEUROSCI.19-08-02906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco I, Beck EJ, Covarrubias M. Receptor-coupled regulation of K+ channel N-type inactivation. Neurobiology (Bp) 1998;6:23–32. [PubMed] [Google Scholar]
- Villière V, McLachlan EM. Electrophysiological properties of neurons in intact rat dorsal root ganglia classified by conduction velocity and action potential duration. J Neurophysiol. 1996;76:1924–1941. doi: 10.1152/jn.1996.76.3.1924. [DOI] [PubMed] [Google Scholar]
- Werz MA, Macdonald RL. Phorbol esters: voltage-dependent effects on calcium-dependent action potentials of mouse central and peripheral neurons in cell culture. J Neurosci. 1987;7:1639–1647. doi: 10.1523/JNEUROSCI.07-06-01639.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelman DLB, Beck CL, Ypey DL, O'Leary ME. Inhibition of the A-type K+ channels of dorsal root ganglion neurons by the long-duration anesthetic butamben. J Pharmacol Exp Ther. 2005;314:1177–1186. doi: 10.1124/jpet.105.087759. [DOI] [PubMed] [Google Scholar]
- Zhang X-L, Mok L-P, Katz EJ, Gold MS. BKCa currents are enriched in a subpopulation of adult rat cutaneous nociceptive dorsal root ganglion neurons. Eur J Neurosci. 2010;31:450–462. doi: 10.1111/j.1460-9568.2009.07060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Dreosti E, Lagnado L. Homeostatic synaptic plasticity through changes in presynaptic calcium influx. J Neurosci. 2011;31:7492–7496. doi: 10.1523/JNEUROSCI.6636-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Fan J, Xiong W, Zhang C, Wang X-B, Liu T, Liu H-J, Sun L, Wang Y-S, Zheng L-H, Wang B-R, Zhang CX, Zhou Z. Action potential modulates Ca2+-dependent and Ca2+-independent secretion in a sensory neuron. Biophys J. 2009;96:2449–2456. doi: 10.1016/j.bpj.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X-B, Wulfsen I, Utku E, Sausbier U, Sausbier M, Wieland T, Ruth P, Korth M. Dual role of protein kinase C on BK channel regulation. Proc Natl Acad Sci U S A. 2010;107:8005–8010. doi: 10.1073/pnas.0912029107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.