

Abstract
G-protein coupled receptors (GPCRs) are found to be attractive drug targets for the treatment of various neuronal diseases. Allosteric modulators have their role in enhancing or suppressing the effect of glutamate on mGluRs. Structure of mGluR1 was generated with the help of Modeller software by considering human B2-adrenergic GPCR protein as template. Structure of various already known drug molecules were used for similarity search in the ZINC database and a large number of similar molecules were obtained, than filtering of these molecules were done by applying drug features. Molecules were screened by Molegro Virtual Docking program and numbers of novel molecules were generated by using LigBuilder software. Finally 16 novel drug candidates were selected, which were showing better results than the seed molecule and previously known modulators. These results will help in designing and synthesis of better drugs against diseases like Epilepsy and Parkinson's.
Keywords: GPCRs, allosteric ligands, homology, modulators, drug features
Background
GPCRs are the large protein families that are found only in eukaryotes. They are the membrane spanning proteins that traverse the membrane several times [1]. There are a variety of ligands that can activate these receptors like hormones, pheromones, neurotransmitters and many others. GPCRs are now a day's used as potent targets in the process of many modern drug developments. GPCRs play important role in causing many diseases like neuronal dysfunction including seizures, Parkinson's disease, night blindness and epilepsy [2]. Antagonists of mGluRs may exert anticonvulsant effect in contrast to soman-induced seizures in rats challenged with soman [3]. Glutamate plays a very crucial role of excitatory neurotransmitter in brain. mGluRs are a member of GPCR super family. Metabotropic receptors are a type of GPCRs as their activity involves in a series of intracellular events which involves the activation of G-protein and often signal transduction pathway. Glutamate which is an amino acid acts as a neurotransmitter [4]. Swanson C. J. et al indicated that mGluR are interesting new targets to treat human's anxiety and stress disorders [5]. Among known eight classes of mGluRs the most extensively studied receptor is the metabotropic glutamate receptor mGluR1 because of their pharmacological, physiological, and anatomical as well as biochemical characteristics [1]. There are two states of GPCR proteins i.e. active state and inactive states, and these states maintains equilibrium with each other and can switch over to each other on sensitization of any ligand or external signals [1]. Active structure of mGluR1 was modeled by using human B2– adrenergic G protein-coupled receptor as template [6].
Materials and Methods
Obtaining template sequence from PDB:
mGluR proteins previously used the structure of Rhodopsin protein as a template to model the structure of other GPCRs as stated by Melherbe et al [4]. In this work human B2-adrenergic G protein-coupled receptor protein was selected as template strand because it has similar active sites and activation mechanism of mGluR protein [6]. The structure of human B2- adrenergic G protein-coupled receptor was taken from PDB database having the PDB ID 2RH1 in the activated state and the structure was generated for mGlur1 its sequence was obtained from NCBI,GenBank [7, 8].
Modelling and Evaluation:
Homology model of mGlur1 protein was generated by using Modeller 9v7 version and finally structures were evaluated by using Procheck program [9, 10].
Similarity searching on ZINC database:
ZINC database was used to obtain similar molecules on the basis of the structures of previously known ligands, similar molecules screened based on drug likeliness parameters and also based on Lipinski's rule of 5 [11].
Pharmacophore generation:
The pharmacophore structure was generated by extracting the information about the common interacting atoms among 15 already known ligand molecules with the help of Pharmagist online server [12, 13].
Docking and interaction study:
The Molegro docking program was used for analyzing the binding between receptor and ligand molecules to study the existing interaction between the receptor and the ligand molecule Ligplot was used [14, 15].
Designing Novel drug like molecules:
The de novo method of ligand designing process was done using the Ligbuilder software (Figure 2) in which Pocket, Grow and Process modules have been used to develop novel drug candidates [16, 17].
Figure 2.
Showing the basis of preparing the seed
Results
The Ramachandran plot in case of active mGluR1 protein was showing that 90.5% residues were falling in the core region, 8.8% of the total residues in the allowed region and 0.7% in the generously allowed region. By analyzing Naveena et al., 2008 work allosteric modulators were used and developed pharmacophore (Figure 3), based on this pharmacophore Zinc database has been screened (Figure 1) and finally 200 molecules obtained [1].
Figure 3.
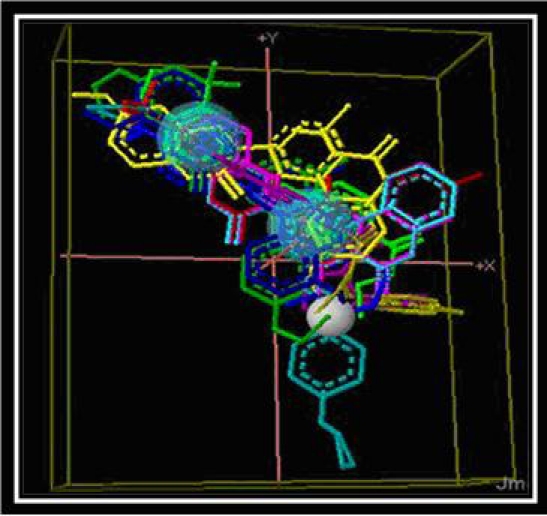
Pharmacophore molecule generated
Figure 1.
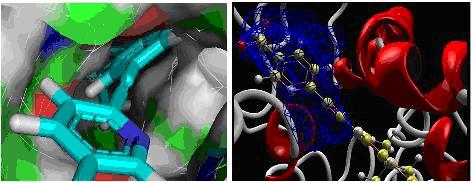
Showing the docked position of ligand (IDZINC24890) in the cavity in two different forms.
On the basis of docking energies 12 molecules were selected. In the other method de novo ligand designing done and results into 200 novel molecules, from this finally 16 potent molecules were screened based on binding energies and other parameters Table 3 (see supplementary material). After docking the molecules obtained by de novo and similarity search method both are showing better results than the previously known drug molecules Table 2a & Table 2b (see supplementary material).Total energy of 10 best ligands obtained by both de novo method and similarity search method were compared (Figure 4), it shows ligands generated by de novo designing method have greater energy values and they may be a good potential drug candidates.
Figure 4.
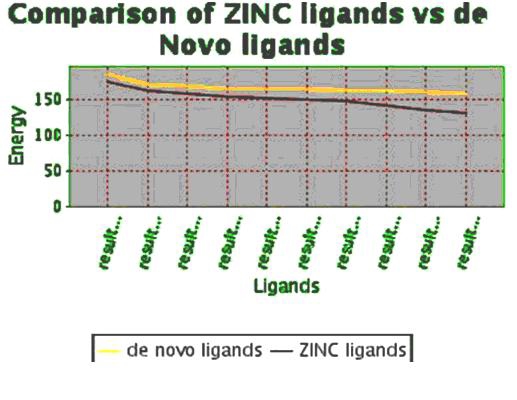
Comparison of the energy values between ligands generated by de novo method and ligands obtained by similarity search method by pharmacophore based on ZINC database.
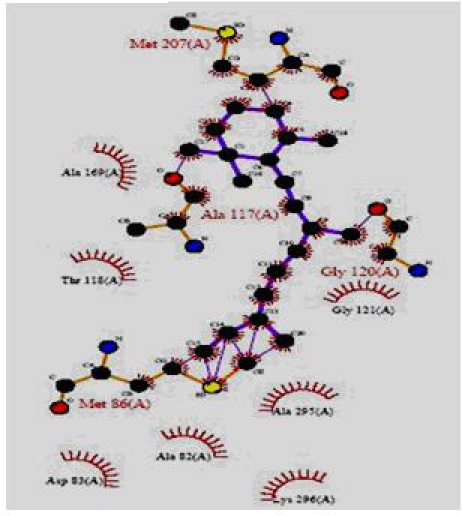
The total energy value of the pivot molecule was found to be - 132.576; it has been compared by the ligands generated by the de novo method. The graph shows that (Figure 5) the energy comparison of the drug like molecules generated by LigBuilder and the energy of pivot molecule (Figure 6) i.e. -132.576, it is observed the de novo method based developed ligands are better than the pivot molecule. The residues involved in the close interaction (Glycine 120, Alanine 117, Methionine 207 and Methionine 86 while the residues that are distantly interacted with the ligands are Asp 83, Lys 296, Ala 83, Thr 118 and Ala 169) were obtained by Ligplot analysis and a pivot molecule (ZINC ID: [00] ZINC24890) was selected based on pharmacophoric characters. A graph has been generated by comparing the energy values of 10 best previously known ligands and the ligands generated by de novo method.
Figure 5.

Comparison of the energy values between ligands generated by de novo method and the pivot molecule (ZINC ID: [00] ZINC24890).
Figure 6.
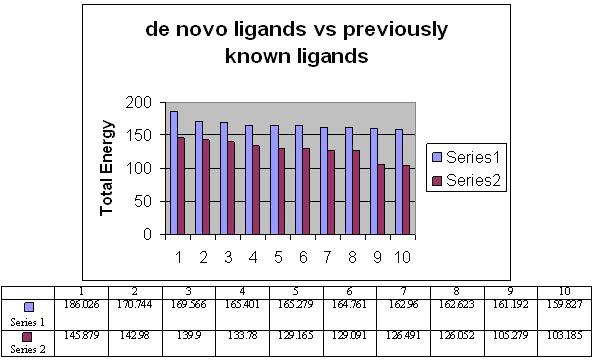
Comparison of the energy values between ligands generated by de novo method and the pivot molecule. Series one indicates energy of molecule generated by de novo and series two indicates energy of molecule generated by pivote molecule. The blue color represents the de novo ligands while red one indicates already known ligands.
Discussion
Our goal was to generate suitable drug candidate which can be used for the treatment of neural dysfunction diseases. We got insight from previous work of Melherbe et al [4] who used structure of Rhodopsin protein as a template to model the structure of other GPCRs. Human B2-adrenergic GPCR template because it has similar active sites and activation mechanism of mGluR protein [6]. Human B2-adrenergic G protein-coupled receptor structure was taken from PDB in the activated state and the structure was generated for mGlur1 [7, 8]. Homology model of mGlur1 protein was generated and finally structures were evaluated [09, 10]. ZINC database was used to obtain similar molecules on the basis of the structures of previously known ligands, similar molecules screened based on drug likeliness parameters and also based on Lipinski's rule of 5 [11]. Pharmacophore structure was generated by extracting the information about the common interacting atoms among 15 already known ligand molecules [12, 13]. Analyzed the binding between receptor and ligand molecule [14].The de novo method of ligand designing process was also performed to develop novel drug candidates [16]. It was found de novo based drugs showed better binding than pivot molecule.
Conclusion
However the ligands generated through our method are showing better binding to mGluR glutamate receptor protein as compared to previous work. Hence they may be better modulators for the activity of mGluR1 protein. They may help to treat various neural dysfunctions like Epilepsy and Parkinson's disease, but these results may be proved by experimental evaluation.
Supplementary material
Acknowledgments
The authors are thankful to Dr. M. D. Tiwari, Director, IIIT, and Allahabad for providing a supportive environment for research and infrastructural support.
Footnotes
Citation:Omer & Prasad, Bioinformation 8(4): 170-174 (2012)
References
- 1.N Yanamala, et al. BMC Bioinformatics. 2008;1471:2105. [Google Scholar]
- 2.Filmore, David American Chemical Society. 2004;24:28. [Google Scholar]
- 3.T Myhrer, et al. Eur J Pharmacol. 2010;636:82. doi: 10.1016/j.ejphar.2010.02.047. [DOI] [PubMed] [Google Scholar]
- 4.P Malherbe, et al. Mol Pharmacol. 2003;823:32. [Google Scholar]
- 5.CJ Swanson, et al. Nat Rev Drug Discov. 2005;131:44. doi: 10.1038/nrd1630. [DOI] [PubMed] [Google Scholar]
- 6.C Vadim, et al. Science. 1258:65. [Google Scholar]
- 7. http://www.rcsb.org/pdb/home/home.do.
- 8. http://www.ncbi.nlm.nih.gov/
- 9.A Sali, et al. Proteins. 1995;23:318. [Google Scholar]
- 10.DE Clark, et al. Drug Discovery World. 2004;37:41. [Google Scholar]
- 11. http://bioinfo3d.cs.tau.ac.il/PharmaGist/
- 12. http://www.biochem.ucl.ac.uk/bsm/ligplot/ligplot.html.
- 13. www.ebi.ac.uk/thornton-srv/software/PROCHECK/
- 14. http://bioinfo3d.cs.tau.ac.il/PharmaGist/
- 15.Gehlhaar, et al. Proceedings of the Fourth International Conference on Evolutionary Programming. 1995;615:62. [Google Scholar]
- 16. http://www.biochem.ucl.ac.uk/bsm/ligpl ot/ligplot.html.
- 17.R Wang, et al. Journal of Molecular Modeling. 2000;498:516. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.