

Abstract
Genome sequences of transmitted/founder (T/F) HIV-1 have been inferred by analyzing single genome amplicons of acute infection plasma viral RNA in the context of a mathematical model of random virus evolution; however, few of these T/F sequences have been molecularly cloned and biologically characterized. Here, we describe the derivation and biological analysis of ten infectious molecular clones, each representing a T/F genome responsible for productive HIV-1 clade B clinical infection. Each of the T/F viruses primarily utilized the CCR5 coreceptor for entry and replicated efficiently in primary human CD4+ T lymphocytes. This result supports the conclusion that single genome amplification-derived sequences from acute infection allow for the inference of T/F viral genomes that are consistently replication competent. Studies with monocyte-derived macrophages (MDM) demonstrated various levels of replication among the T/F viruses. Although all T/F viruses replicated in MDM, the overall replication efficiency was significantly lower compared to prototypic “highly macrophage-tropic” virus strains. This phenotype was transferable by expressing the env genes in an isogenic proviral DNA backbone, indicating that T/F virus macrophage tropism mapped to Env. Furthermore, significantly higher concentrations of soluble CD4 were required to inhibit T/F virus infection compared to prototypic macrophage-tropic virus strains. Our findings suggest that the acquisition of clinical HIV-1 subtype B infection occurs by mucosal exposure to virus that is not highly macrophage tropic and that the generation and initial biological characterization of 10 clade B T/F infectious molecular clones provides new opportunities to probe virus-host interactions involved in HIV-1 transmission.
INTRODUCTION
Understanding the process of HIV-1 sexual transmission on a molecular level may reveal vulnerabilities of the virus and identify specific host immune defense mechanisms critical for early interdiction, thus informing the rational development of intervention strategies, including the discovery and clinical testing of sterilizing vaccines. Several earlier studies investigated genetic and biologic properties of viruses or viral sequences isolated within the first weeks or months following clinical infection. Unique biological properties (N-linked glycan restriction, compact variable loops, and neutralization sensitivity) were observed for env genes derived following infections with clade A and C strains of HIV-1 (12, 15, 53) but not for clade B strains (12, 30). However, limitations of these studies included the analysis of virus samples obtained one to 10 months following clinical infection, and PCR methods that did not use single genome amplification (SGA) techniques (12, 15, 30, 53). Thus, in vitro artifacts of sequence recombination and base substitution could not be excluded (54), and the identity of bona fide transmitted/founder (T/F) viruses remained elusive. Recently, Salazar-Gonzalez et al. (54) and Keele et al. (39) identified nucleotide sequences of env genes from viruses responsible for the acquisition of HIV-1 clinical infection, by definition, the T/F virus. Using viral RNA (vRNA) from plasma obtained during the earliest stages of infection, direct sequencing of uncloned single-genome amplicons (SG amplicons), and phylogenetic inference based on a model of random virus evolution (43), Keele et al. demonstrated that in nearly 80% of the cases of HIV-1 sexual transmission, SGA sequences coalesced to just one ancestral sequence, the T/F sequence. Subsequent studies that included cohorts infected with clade A, B, and C viruses reached similar conclusions (1, 36, 54). In cohorts of men who have sex with men (MSM) and injecting drug users (IDU), multivariant transmission is more common (4, 44). HIV-1 deep sequencing (28) and studies in simian models (40, 45) have further validated the methods for unambiguous identification of T/F viruses and a precise assessment of their numbers.
Employing the same experimental strategy, we have more recently reported on the sequence of complete T/F virus genomes identified in subjects with clade B or C HIV-1 infection (55). All inferred genomes were intact and comprised of nine genes with open reading frames (LTR-gag-pol-vif-vpr-tat-rev-vpu-env-nef-LTR). Studies of these T/F virus sequences and progeny arising from them have given new fundamental insights into early virus evolution and the ontogeny of the antiviral immune response (1, 26, 31, 34, 46, 56, 66). We further hypothesized that the derivation of T/F infectious molecular clones, and the biological characterization of progeny virus would be both informative for understanding HIV-1 transmission and provide new opportunities to elucidate virus-host interactions that occur during the earliest stages of HIV-1 transmission. This current study describes the generation of a panel of 10 HIV-1 infectious molecular clones (IMCs) representing T/F viruses inferred from 10 subjects with early/acute clade B infection. We demonstrate that these viruses utilize the CCR5 coreceptor for entry and replicate efficiently in cultures of activated primary T lymphocytes. Furthermore, our results show that T/F viruses replicate in monocyte-derived macrophages (MDM), although with significantly reduced efficiency compared to prototypic “highly” macrophage-tropic HIV-1 strains. The findings of the present study are significant to our understanding of HIV-1 transmission and T/F virus biology.
MATERIALS AND METHODS
Study subjects, clinical specimens, and laboratory staging of acute HIV-1 infection.
Plasma or peripheral blood mononuclear cells (PBMC) were obtained from 10 adult subjects with informed consent and the approval of the institutional review board. Blood was collected in acid-citrate-dextrose, and plasma was separated and stored at −70°C. PBMC were cryopreserved in vapor-phase liquid nitrogen. Plasma samples from each subject were tested for HIV-1 RNA, p24 antigen, and virus-specific antibodies as previously described (39) and then classified according to the system of Fiebig et al. (27). Fiebig stage II plasmas were vRNA and p24 antigen positive but antibody negative, Fiebig stage III plasmas were enzyme-linked immunosorbent assay (ELISA) antibody positive but Western blot negative, and Fiebig stage V plasmas were ELISA and Western blot positive but integrase antibody negative.
Identification of transmitted/founder HIV-1 genomes.
Detailed methods for single-genome PCR amplification of vRNA/cDNA and analysis of single-genome amplicons (SG amplicons) using a mathematical model of early virus replication and evolution to infer the identity of single variant T/F viruses responsible for HIV-1 infection were reported previously (39, 43, 44, 54, 55). The methods involved the generation and analysis of 9 kb, near-genome-length SG amplicons, as well as ∼800-bp SG amplicons encompassing 60 nucleotides (nt) at the R-U5 junction, for which the T/F sequence was obscured by the 9-kb SGA primers (Fig. 1A and see Fig. S2 in the supplemental material). As previously reported, this approach allowed for the inference of the complete T/F genome sequence from several subjects, among them WITO4160, 700010040, 700010058, 700010077, TRJO4551, and SUMA0874 (55), for which we describe here the generation of T/F IMCs. For this study, four additional T/F virus genomes were identified from subjects with early HIV-1 infection, including 700010106, RHPA4259, THRO4156, and REJO4541. For these subjects, analysis of between 19 and 31 complete gp160 env sequences derived by SGA had already determined that HIV-1 acquisition had likely resulted from transmission of one viral genome; this was based on power calculations that indicated a 95% likelihood of detecting sequences represented by as little as 15% in the sampled population for n = 20 (see Fig. S9 in Keele et al. [39]). Based on this information, and applying methodology described earlier (55), we could then analyze as few as four to nine near-complete (9-kb) genome sequences (see Fig. S1 in the supplemental material), none of which contained shared polymorphisms, to infer a likely T/F genome with a high degree of confidence. One caveat is that if all of the sampled sequences shared stochastic mutations resulting from recent common ancestry that differed from the transmitted sequence, these could not be distinguished. However, this occurrence is unlikely based on mathematical modeling (43).
Fig 1.

Highlighter analysis of near-full-length HIV-1 genomes cloned from 9-kb SG amplicon. (A) The SG amplicon is illustrated in relation to the proviral genome (top) and the viral RNA genome (middle). Gray shading indicates the regions corresponding to the two HXB2 primers, each 30 nt in length, used to generate SG amplicons. (B) Highlighter analysis of SG amplicon sequences from the vRNA/cDNA of subject WITO. The T/F sequence (consensus, CON) is indicated as a line at the top of the plot. The sequences of 9 SG amplicons are shown with nucleotide differences indicated by colored tick marks. Dark blue ticks indicate sites with mixed bases in sequencing chromatograms, following the International Union of Pure and Applied Chemistry (IUPAC) nomenclature. (C) Highlighter analyses of the near-full-length genomes cloned from the SG amplicon WITO SGA_E1. The E1 sequence was identical to the inferred T/F genome and was selected for cloning. (D to F) Highlighter analyses of the near-full-length genomes cloned from CH040 (D), CH058 (E), and CH077 (F) SG amplicons. Each Highlighter plot depicts the T/F sequence at the top and the SG amplicon that was selected for cloning. The SG amplicon sequences start at nucleotide position 582 in the 5′ LTR (U5 region) and extend to position 9606 in the 3′LTR (R region), based on the HXB2 numbering system. Thus, the scale bar at the bottom of the highlighter plots (C to F) starts at nucleotide position 1 as the first nucleotide of U3.
DNA sequencing.
Nucleotide sequence analysis, including that of uncloned SG amplicons, used BigDye Terminator v3.1 chemistry (Applied Biosystems, Foster City, CA), and an ABI 3730xl DNA analyzer (Applied Biosystems), as described previously (39, 54, 55). Sequences were edited with the Sequencher program (version 4.7; Gene Codes, Ann Arbor, MI) as previously described.
Generation of T/F IMCs from near-full-length SG amplicons derived from vRNA.
The IMCs for four subjects (WITO4160, 700010040, 700010058, and 700010077) were generated from vRNA. vRNA was extracted from plasma by using a QIAamp viral RNA minikit (Qiagen, Valencia, CA), and cDNA copies of the vRNA were generated by reverse transcription using the SuperScript III protocol (Invitrogen Life Technologies, Carlsbad, CA), both as previously described (55). Samples were heat inactivated at 70°C for 15 min and then digested with RNase H at 37°C for 20 min (Invitrogen Life Technologies, Carlsbad, CA). For each subject/cDNA, multiple SG amplicons spanning from the 5′ U5 through the 3′ R were generated and directly analyzed by nucleotide sequencing (55). For each of the four subjects, the SG amplicon containing the fewest mutations compared to T/F sequences was selected for TA cloning into the pCR-XL-TOPO vector (Invitrogen). For this, the ∼9-kb PCR product (i.e., SG-amplicon) was purified utilizing a UV-Free Gel purification kit (Invitrogen). An adenine overhang was added by adding 0.5 μl of Taq polymerase with 1× buffer (Promega) and 0.2 mM concentrations of each deoxynucleoside triphosphate (dNTP), followed by incubation at 94°C for 2 min and then a single extension at 72°C for 10 min. The prepared DNA molecules were TA ligated into the pCR-XL-TOPO vector by using a TOPO XL PCR cloning kit (Invitrogen) according to the manufacturer's protocol. After transformation into One-Shot Top10 competent cells, bacteria were plated on lysogeny broth (LB) agar plates supplemented with kanamycin and grown overnight at 33°C. For each TA ligation, 9 to 150 colonies were obtained. Single colonies were selected and grown overnight in LB broth at 33°C with 200 rpm shaking, followed by miniprep plasmid DNA isolation (Qiagen). The miniprep DNAs were screened for successful insertion of a 9-kb fragment by checking sizes of undigested and EcoRI-digested samples; EcoRI was present at both flanking sites of the TA cloned viral sequence. Nine to fifteen clones, from each SG amplicon, displaying the correct EcoRI digestion pattern were analyzed by nucleotide sequencing.
Because the viral genomes cloned from 9-kb SG amplicons were incomplete and contained nucleotides not matching the inferred T/F sequences (see Results), the missing long terminal repeat (LTR) fragments were repaired and the nucleotide mutations were restored to those of the respective T/F sequence. This required a unique molecular approach for each TA-cloned T/F genome; thus, a detailed description of all of the molecular manipulations required for the generation of each IMC is not provided. Briefly, the missing U3 and R elements of the 5′ LTR were reconstituted by PCR using the TA-cloned genome as a template to amplify 5′ U3- and 3′ R-containing DNA fragments. The 5′-end PCR DNA product extended into the untranslated region, encompassing a naturally existing unique restriction enzyme site. The two amplified DNA fragments (U3-R and U5) contained a region of complementarity at the R-U5 junction, which allowed by a subsequent PCR the generation of the complete 5′ U3-R-U5 LTR. By PCR primer design, a unique restriction enzyme site was introduced 5′ of U3 for ligation into the pCR-XL-TOPO vector that contained the cognate 9-kb genome. To restore the missing U5 region of the 3′ LTR, a U3-R DNA fragment was PCR amplified from the 3′ end of the cognate TA-cloned viral genome, wherein the sense primer was complementary to the sequence upstream of a naturally occurring unique restriction enzyme site, and the antisense primer extended beyond R encompassing T/F sequence-matching nucleotides in the 5′ end of U5. Then, a U5-containing DNA fragment was PCR amplified from the 5′ end of the cognate TA clone using a sense primer complementary to the 5′ end of U5 and an antisense primer that introduced a unique restriction enzyme site 3′ of U5 for cloning into the pCR-XL-TOPO vector. In a subsequent PCR, the two PCR products (U3-R and U5) were used to generate the complete LTR (U3-R-U5), which was then ligated into the 3′ end of the cognate, TA-cloned, viral genome. Correction of Taq polymerase mutations was achieved by either ligating together DNA fragments matching the T/F sequence from several different TA-cloned genomes when unique endonuclease restriction enzyme sites were available, by PCR amplification and cloning of DNA fragments wherein primers could be used to correct specific mutations, and/or by correcting specific nucleotide mutations via the QuikChange method.
The following reagents were used: the PCR polymerases used were either Phusion Hot Start high-fidelity DNA polymerase (New England Biolabs) or the Roche Long Template PCR System (Roche Applied Science). Primers for PCR amplification, including those for mutagenic PCR to correct nucleotides not matching the T/F sequence, were purchased from IDT (Coralville, IA). Some nucleotides mismatched with the respective T/F sequences were corrected by using the QuikChange XL kit (Stratagene). Restriction enzymes were purchased from Promega and New England Biolabs. Purification of DNA fragments after PCR or gel electrophoresis was conducted with either a QIAquick PCR purification kit or a QIAquick gel purification kit (both from Qiagen). DNA ligations were performed using T4 DNA ligase from either Promega (catalog no. M180A) or New England Biolabs (catalog no. M0202S). T4 DNA ligase-ligated plasmids were transformed into either One-Shot Stbl3 chemically competent E. coli or MAX Efficiency Stbl2 competent cells (Invitrogen). All bacterial cultures were grown at 33°C.
Generation of T/F IMCs from proviral DNA.
For six subjects (700010106, THRO4156, RHPA4259, REJO4541, SUMA0874, and TRJO4551), IMCs were generated from integrated proviral DNA. The methodology developed for these six clade B-infected subjects has also been applied, with variations, to one clade B T/F virus in a linked MSM transmission study (44) and two clade C T/F viruses (55). Derivation of IMCs from proviral DNA offered important advantages: amplicons contained complete 5′ and 3′ LTRs, and a shorter PCR amplicon length (overlapping half genomes) allowed the use of a high-fidelity DNA polymerase (Phusion Hot Start high-fidelity DNA polymerase [New England Biolabs]).This significantly reduced the effort required for correcting mutations introduced into SG amplicons by Taq polymerase when cloning from vRNA/cDNA. High-molecular-weight genomic DNA (gDNA) was extracted from either uncultured patient PBMC or, for subject 700010106, from a CD4+/CCR5+ recombinant cell line incubated for 24 h with HIV-1+ patient plasma, using a Qiagen DNeasy blood and tissue kit. The gDNA samples were endpoint diluted in 96-well plates and, as detailed below, subjected to half-nested PCR amplification using primers specific for each subject to amplify the complete proviral DNA genome in two halves, including an overlapping segment containing a unique restriction enzyme site for ligating the two genome halves together.
To generate the pREJO.c, pTHRO.c, and pRHPA.c IMCs, proviral DNA genomes were amplified in two overlapping fragments, wherein the region of sequence overlap contained a unique SbfI restriction site. The PCR primers matched the T/F sequence of each virus, and MluI and ApaI restriction enzyme sites were added on to the 5′ ends of the sense and antisense primers, respectively. Single-round bulk PCR amplification was performed in 1× Phusion Hot Start HiFi buffer, with 0.2 mM concentrations of each dNTP, 0.5 μM concentrations of each primer, a 3% final concentration of dimethyl sulfoxide, and 0.02 U/μl of Phusion Hot Start high-fidelity polymerase in 50-μl reactions. PCR products were purified, and adenine overhangs were added to each purified DNA as described above. The PCR product comprising each half-genome fragment was then independently TA cloned into a pCR-XL-TOPO vector (see above). Miniprep DNA from clones was screened for correct size of the insert and two to three clones of each genome half were sequenced. Clones had either no or few (2 to 4) nucleotide mutations. Those with the fewest changes were used to generate IMCs. For this purpose, any mismatched nucleotides were corrected by mutagenic PCR. Then, the 5′ genome half was excised and cloned into the 3′ genome-containing TOPO XL vector utilizing the MluI and SbfI restriction sites, thereby generating the full-length clone of each T/F provirus. The REJO genome was not stable in the pCR-XL-TOPO vector and thus was ligated into the modified low-copy-number pBR322 plasmid (55). The maxiprep DNA of each final clone was sequenced and confirmed to match the cognate T/F virus genome.
The pSUMA.c IMC was generated according to a similar protocol except the overlapping region of the two PCR-amplified genome-halves contained a unique DraIII site, and they were initially cloned using the Zero Blunt TOPO PCR cloning kit (Invitrogen) before being ligated into the modified pBR322 plasmid.
For pTRJO.c, three DNA fragments were PCR amplified from provirus. Fragment 1 contained the complete 5′ LTR, gag, and part of pol; fragment 2 contained the 3′ ends of pol, vif, vpr, tat-1, rev-1, and vpu and the 5′ end of env; and fragment 3 contained the 3′ ends of env, tat-2, rev-2, and nef and the 3′ LTR. Fragments 1 and 2 overlapped at the unique SbfI site, and fragments 2 and 3 overlapped at the unique AleI site; each fragment was individually TA cloned into the pCR-XL-TOPO vector. Of the many clones of fragment 3 that were analyzed, all of them contained G-to-A hypermutations, which were corrected by mutagenic PCR. Finally, the three genome fragments, each matching the T/F sequence, were ligated together and inserted into the modified pBR322 vector to yield pTRJO.c.
To generate pCH106.c, vRNA+ plasma from subject 700010106 was used to infect a CD4+/CCR5+ recombinant cell line. Approximately 106 cells were seeded per well in a six-well tissue culture plate, and 25 μl of plasma with 400 μl of Hanks balanced salt solution (HBSS) and 40 μg/ml of DEAE-dextran were added to the cells for 4 h. The supernatant was then removed and 1 ml of complete Dulbecco modified Eagle medium (DMEM) with 20 μg/ml of the HIV fusion inhibitor T20 was added. The next day, the gDNA was isolated from the infected cells using a phenol-chloroform gDNA isolation method and then used as a template for PCR. The PCR-amplified 5′ half of the pCH106.c genome contained the 5′ LTR (minus the first 37 nt of U3 since successful amplification required the use of an internal PCR sense primer), gag, pol, and the 5′ end of vif. The 3′ half of the genome contained the 3′ end of vif, vpr, tat-1, rev-1, vpu, env, tat-2, rev-2, and nef and the 3′ LTR. The 5′ and 3′ halves overlapped at a unique BamHI site, and the IMC was cloned into the pCR-XL-TOPO vector as described above.
Accession numbers and submission to NIH ARRRP.
GenBank accession numbers reporting the inferred T/F proviral DNA sequence (5′-U3 through U5-3′) from each subject are shown in Table 1. Also provided in Table 1 are the accession numbers identifying the complete nucleotide sequences of the 10 corresponding IMCs. In six cases the inferred T/F sequence and the IMC sequence are identical. In four cases (pCH106.c, pCH058.c, pCH040.c, and pRHPA.c) the plasmid-cloned nucleotide sequence is not identical to that of the cognate T/F proviral genome. In each case, differences exist only in the LTR, not in coding regions and, importantly, upon infection of target cells with IMC transfection-derived virus, the sequence of the T/F virus is completely restored through the process of reverse transcription (see the details in Results).
Table 1.
Subject demographics and laboratory information
| Subject identifiera | T/F virus designation | IMC (plasmid clone)b designation | Accession nos. (T/F; IMC)c | Gender | Riskd | Fiebig stagee | Source of genome for cloning | IMC 3′ LTR structure | Cloning plasmid |
|---|---|---|---|---|---|---|---|---|---|
| WITO4160 | WITO.c | pWITO.c | JN944938; JN944948 | Male | HSX | II | vRNA | U3-R-U5 | pCR-XL-TOPO |
| 700010040 | CH040.c | pCH040.c | JN944905; JN944939 | Male | MSM | II/V | vRNA | U3-R | pCR-XL-TOPO |
| 700010058 | CH058.c | pCH058.c | JN944907; JN944940 | Male | MSM | II/III | vRNA | U3-R | pCR-XL-TOPO |
| 700010077 | CH077.t | pCH077.t | JN944909; JN944941 | Male | MSM | II/V | vRNA | U3-R-U5 | pCR-XL-TOPO |
| 700010106 | CH106.c | pCH106.c | JN944897; JN944942 | Male | MSWM | II | Provirus | U3-R-U5 | pCR-XL-TOPO |
| THRO4156 | THRO.c | pTHRO.c | JN944930; JN944946 | Male | MSM | V | Provirus | U3-R-U5 | pCR-XL-TOPO |
| RHPA4259 | RHPA.c | pRHPA.c | JN944917; JN944944 | Female | HSX | V | Provirus | U3-R-U5 | pCR-XL-TOPO |
| REJO4541 | REJO.c | pREJO.c | JN944911; JN944943 | Male | HSX | V | Provirus | U3-R-U5 | pBR322 |
| SUMA0874 | SUMA.c | pSUMA.c | JN944928; JN944945 | Male | MSM | II | Provirus | U3-R-U5 | pBR322 |
| TRJO4551 | TRJO.c | pTRJO.c | JN944936; JN944947 | Male | MSM | II | Provirus | U3-R-U5 | pBR322 |
All subjects were from the United States and were infected with subtype B HIV-1 by a single-variant transmission.
Plasmid DNAs have been submitted to the NIH ARRRP, catalog no. 11919.
T/F, accession number representing the proviral DNA sequence of each T/F virus; IMC, accession number representing plasmid-cloned DNA, referred to as the infectious molecular clone (IMC).
HSX, heterosexual; MSM, men who have sex with men; MSWM, men who have sex with women and men.
The stage of HIV-1 infection, as defined by Fiebig et al. (27), at which samples were obtained for inference of the T/F sequence and construction of the IMCs.
We also submitted to GenBank the sequences of all near-genome-length 9-kb SG amplicons not previously reported by Salazar-Gonzales (55), i.e., those derived from subjects 700010106 (JN944898 to JN944903), RHPA4259 (JN944918 to JN944926), THRO4156 (JN944929 to JN944934), and REJO4541 (JN944911 to JN944915). Furthermore, the consensus sequences for all 10 R-U5-encompassing ∼800-bp SG amplicons have been submitted to GenBank (with the following accession numbers: CH106_RU5_con, JN944896; CH40_RU5_con, JN944904; CH58_RU5_con, JN944906; CH77_RU5_con, JN944908; REJO_RU5_con, JN944910; RHPA_RU5_con, JN944916; SUMA_RU5_con, JN944927; THRO_RU5_con, JN944929; TRJO_RU5_con, JN944935; and WITO_RU5_con, JN944937).
Each of the IMCs (Table 1) has been submitted to the NIH AIDS Research Reference and Reagent Program, and the complete nucleotide sequence of the proviral DNA-containing plasmids, including annotation, can be found in the Reagent Data Sheet for Catalogue 11919 (https://www.aidsreagent.org/reagentdetail.cfm?t=molecular_clones%id=540).
Viruses.
HIV-1 virus isolates, including BaL (32), SF162 (10), and ADA (33), and molecular clones, including JRCSF (41) and YU-2 (Li et al., unpublished data), were utilized as experimental controls. In certain studies, phenotypes conferred by T/F Envs were analyzed by expressing T/F env genes in an isogenic proviral DNA backbone that is replication competent and stably expresses Renilla luciferase (LucR). This proviral backbone enables expression of exogenous Envs in cis (22) and was used for expressing the env ectodomain-encoding region of the following viruses: BaL, SF162, WITO.c, CH040.c, CH058.c, CH077.t, SUMA.c, CH106.c, REJO.c, THRO.c, and YU-2. The recombinant Env-IMC-LucR viruses were named NL-LucR.T2A-BaL.ecto, NL-LucR.T2A-SF162.ecto, NL-LucR.T2A-WITO.ecto, NL-LucR.T2A-CH040.ecto, NL-LucR.T2A-CH058.ecto, and NL-LucR.T2A-CH077.ecto (all described in reference 22), as well as NL-LucR.T2A-SUMA.ecto, NL-LucR.T2A-CH106.ecto, NL-LucR.T2A-REJO.ecto, NL-LucR.T2A-THRO.ecto, and NL-LucR.T2A-YU-2.ecto. The identity of each construct was confirmed by nucleotide sequence analysis.
Cells.
The TZM-bl cell line was used to analyze virus infectivity, as described previously (62, 63). 293T/17 cells were obtained from the American Type Culture Collection (CRL-11268). Both cell lines were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 U/ml). Primary human PBMC were isolated by Ficoll gradient centrifugation from buffy coats of healthy HIV-1-seronegative donors. Buffy coats were obtained from Research Blood Components (Brighton, MA) or provided by Tom Denny (Duke University) and the CAVD CTC-VIMC (funded by the Bill and Melinda Gates Foundation) in accordance with approved IRB protocols. For certain studies, CD4+ T lymphocytes were positively selected from PBMC with anti-CD4 magnetic beads (Miltenyi Biotec). The cells were then incubated in 10-cm polystyrene tissue culture plates for 2 h at 37°C in HBSS with 10 mM Ca2+ and Mg2+ to remove adherent monocytes. Nonadherent cells were collected, washed in RPMI 1640 supplemented with 15% FBS, and resuspended at 106 cells/ml in RPMI 1640 plus 15% FBS and 3 μg/ml of staphylococcal enterotoxin B (SEB; Sigma-Aldrich) for 48 h at 37°C to activate the lymphocytes. MDM were isolated by using one of two methods. For the first, PBMC were plated at 3 × 106 cells per well in a 24-well plate in HBSS plus 10 mM Ca2+ and Mg2+ plus 10% human AB serum (Sigma-Aldrich) and then incubated at 37°C for 2 h. Nonadherent cells were removed, and DMEM with 10% giant cell tumor (GCT)-conditioned medium (Irvine Scientific) plus 10% human AB serum was added to wells with 5 U/ml of rhMCSF (R&D Systems). After 3 days of incubation, the cells were washed vigorously with phosphate-buffered saline (PBS) three times, and medium containing DMEM with 10% GCT, 10% FBS, and 5 U/ml of rhMCSF was added to the wells. After 3 additional days in culture, the macrophages were utilized for experiments. Alternatively, monocytes were positively selected from PBMC with anti-CD14 magnetic beads (Miltenyi Biotec). The CD14-positive cells were plated onto petri dishes and allowed to adhere for 1 h in serum-free DMEM supplemented with penicillin (100 U/ml) and streptomycin (100 U/ml). After 1 h of incubation, the unattached cells were removed, and the adherent monocytes were incubated overnight in DMEM supplemented with either 10% FBS or 10% autologous serum. After 24 h, the medium was changed, and the cells were allowed to differentiate to macrophages for an additional 5 to 6 days. Cultures were fed with fresh medium every 2 to 3 days. At 1 to 2 days prior to infection, the macrophages were washed with PBS, incubated for 10 min in trypsin at 37°C, gently scraped from the petri dishes, and seeded into appropriately sized wells for experiments.
Generation of virus stocks.
Virus stocks were either generated by proviral DNA transfection of 293T cells using FuGENE 6 according to the manufacturer's protocol (Roche Applied Science) or by propagating the virus in PBMC. Viral supernatants were harvested from 293T cells at 60 h posttransfection and from PBMC at 5 to 7 days postinfection. The supernatants were clarified at 1,800 × g for 10 min, filtered through 0.45-μm-pore-size filters, and frozen at −70°C. The clarified supernatants were analyzed by ELISA (Perkin-Elmer) for HIV-1 p24 antigen concentration. Virus stocks were also titered on the TZM-bl reporter cell line by enumeration of β-galactosidase (β-Gal)-stained cells as described previously (62). Titers are expressed as TZM-bl infectious units (IU) per ml.
Determination of viral coreceptor usage.
Coreceptor utilization was analyzed on TZM-bl cells seeded 1 day prior to infection at 8,300 cells/well in 96-well plates. Medium was removed the next day, and the cells were pretreated for 1 h without or with either AMD3100 (1.2 μM), TAK-779 (10 μM), or a combination of both. Portions (2 × 103 TZM-bl IU) of each virus stock were added in the presence of DEAE-dextran (40 μg/ml) and, after incubation for 48 h at 37°C, the cells were analyzed for firefly luciferase activity (Promega). Relative light units (RLU) were determined using a Tropix luminometer and WinGlow version 1.24 software.
Analysis of viral infectivity and replication in CD4+ T cells and MDM.
Virus replication was analyzed by measuring p24 antigen in cultures of CD4+ T lymphocytes and MDM. Cells were infected for 15 to 18 h with virus normalized to either 50 ng of p24 or 5 × 104 TZM-bl IU per well. The cells were then washed three times and resuspended in fresh medium. Every 3 days, supernatants were collected from the cultures and stored at −70°C until samples were analyzed. The remaining medium in each well was completely removed on each sample day and replaced. Virus released into the supernatant was quantified by HIV p24 ELISA (Perkin-Elmer, catalog no. NEK050); the lower limit of detection for the kit, and therefore for that of undiluted sample, is 10 to 12.5 pg/ml of p24.
Virus infectivity and replication was also assessed in MDM infected with Env-IMC-LucR viruses by analyzing Renilla luciferase (LucR) expression. For analysis of infectivity, MDM seeded at 5 × 104 cells per well in 96-well plates were either infected overnight or for 4 h (donor 4) at an MOI of 1 (5 × 104 TZM-bl IU; donors 1, 2, 3, and 6), 3 (1.5 × 105 TZM-bl IU; donors 4 and 5), or 5 (2.5 × 105 TZM-bl IU; donor 7). The virus was then removed, and fresh medium was added to the cells. The medium was removed from the cells on the day of lysis (5 to 7 days postinfection), and 100 μl of 1× Renilla luciferase assay lysis buffer (Promega catalog no. E2820) was added to each well. To determine virus replication kinetics, MDM were seeded in 96-well plates at 4.5 × 104 cells per well, replicas were infected with 2.25 × 105 TZM-bl IU (MOI of 5) of each virus for 8 h and then washed, and the medium was replaced. On days 1, 2, and 6 postinfection, the cells were lysed and stored at −70°C. Samples were freeze-thawed twice, and then 20 μl of each cell lysate was analyzed for LucR activity using a Victor 3 luminometer (Perkin-Elmer) programmed to inject 100 μl of luciferase assay reagent per well with an exposure time of 10 s/well. All LucR values are corrected for background/blank (typically, 120 to 160 RLU).
Analysis of HIV-1 cDNA synthesis.
MDM were seeded at 2.5 × 105 cells per well in 24-well plates and infected with 1.25 × 106 TZM-bl IU (MOI of 5) of PBMC-derived virus stocks, except where otherwise specified. At either 24 or 72 h postinfection, the cells were washed three times, and the total DNA was extracted with a QIAamp DNA blood minikit. Early cDNA products of reverse transcription were analyzed by using semiquantitative methods as previously described (67, 68). The primer pair used for PCR consists of a sense primer (M667) corresponding to a sequence in the R region and an antisense primer (AA55) corresponding to a segment of the U5 LTR (68). As a control for quantitation, the GAPDH housekeeping gene was PCR amplified from the same samples. All samples were DpnI digested prior to amplification to ensure there was no plasmid DNA contamination in the cultures. The quantity of each PCR amplicon was measured in agarose gels using a Fujifilm LAS-4000 minigel documentation system.
RESULTS
Patient demographics.
Single-variant T/F IMCs were generated from either acute plasma vRNA or PBMC obtained from 10 adult subjects during the acute phase of HIV-1 infection (Table 1). One female and two male subjects reported heterosexual (HSX) exposure as their only HIV-1 risk factor, six subjects were men who had sex with men (MSM), and one male subject reported HSX, as well as MSM, as risk factors. Each study subject was infected with a clade B strain of HIV-1.
Identification of T/F genome sequences.
Knowledge of complete proviral T/F genome sequences is a prerequisite for the generation of T/F IMCs. We previously described in detail how the identity of single-variant T/F viruses was inferred for 6 of the 10 subjects analyzed in the present study, including WITO4160, 700010040, 700010058, 700010077, TRJO4551, and SUMA0874 (55) (referred to as WITO, CH040, CH058, CH077, TRJO, and SUMA, respectively, throughout the Results). For this study, T/F virus genomes were identified from four additional subjects, 700010106, RHPA4259, THRO4156, and REJO4541 (referred to as CH106, RHPA, THRO, and REJO, respectively) by a similar strategy (see Fig. S1 in the supplemental material). A caveat of the 9-kb SGA PCR strategy is that a portion of the T/F sequence is obscured by the use of HIV-1/HXB2-specific amplification primers corresponding to the first 30 nucleotides of the 5′ U5 and the last 30 nucleotides of the 3′ R, respectively (Fig. 1A). Therefore, the T/F virus sequence of these regions was determined separately by generating SG amplicons comprising a 5′ R-U5-containing DNA segment of the genome from vRNA/cDNA and then deriving the consensus sequence (RU5_con; see Fig. S2 in the supplemental material). In concordance with the mathematical model (39, 43, 54), the consensus sequence was identified as the actual T/F sequence in each case. An alignment of the ∼800-bp SG amplicon consensus sequences beyond the nucleotides missing from the 9-kb SG amplicon analysis with cognate T/F sequences demonstrated 100% identity. This is illustrated for each of the 10 subjects in Fig. S2 in the supplemental material for the first 200 nt of the amplicons, including sequence comprising the R-U5 junction.
Nucleotide sequence analysis of clones derived from 9-kb near-genome-length SG amplicons of plasma vRNA.
WITO was the first subject from which a T/F IMC was generated by cloning near-full-length genomes from SG amplicon. Previous Highlighter analysis of the nucleotide sequence obtained by direct sequencing of SG amplicons derived from WITO identified nine amplicons that had all open reading frames intact (55). Each of the nine differed from the others by at least one nucleotide and, with the exception of E1, the genomes contained between 1 and 7 nt that did not match that of the T/F sequence (Fig. 1B). Therefore, the E1 SG amplicon was selected for cloning to create a T/F IMC. DNA molecules comprising the E1 SG amplicon were cloned by ligation into the pCR-XL-TOPO vector, and complete nucleotide sequence analysis of nine clones demonstrated that none were identical to either the WITO T/F sequence or each other (Fig. 1C). The number of mutations per clone ranged from 2 to 14, with an average of 8.
Using an identical approach, molecular clones were derived from SG amplicons of subjects CH040, CH058, and CH077. A 9-kb SG amplicon with only one T/F-mismatched nucleotide was identified for each subject and thus was selected for cloning (Fig. 1D to F). Among 10 clones derived from the selected SG amplicon of each subject, sequence analysis revealed multiple mutations introduced by Taq polymerase error compared to the T/F nucleotide sequence: the numbers of mutations per clone ranged from 9 to 17, 5 to 20, and 7 to 17, with averages of 10.9, 11.7, and 11.5, respectively (Fig. 1D to F). Direct (uncloned) sequencing of SG-amplicons does not detect these errors unless introduced by Taq polymerase during the first one to two cycles of PCR, in which case they manifest as mixed bases on the chromatogram (54). Our results reiterate that SG amplicons comprise mixed populations of HIV-1 sequences. At the clonal level, the variants are detected with a greater frequency and complicate the derivation of nucleotide exact T/F virus IMCs.
Generation of full-length T/F IMCs from plasma vRNA/cDNA.
The SG amplicons generated from vRNA/cDNA, and thus clones derived from them, are incomplete genomes, lacking the 5′ U3-R and the 3′ U5 elements comprised in proviral DNA (Fig. 1A). To generate infectious, replication-competent molecular clones that produce virus identical to T/F viruses, we had to reconstitute the LTRs as well as correct the Taq polymerase mutations found scattered throughout the cloned genomes. The latter was achieved by a combination of molecular approaches, including the ligation of subgenomic fragments matching the T/F sequence from several different TA-cloned genomes, mutagenic PCR amplification and recloning of corrected fragments, and the QuikChange method. The approach to reconstitute the 5′ and 3′ LTRs, is detailed in Materials and Methods. Briefly, a DNA fragment encompassing the complete T/F 5′ LTR and the adjacent untranslated region sequence was first generated by PCR and ligated into the pCR-XL-TOPO vector containing the cognate 9-kb genome. Second, for WITO and CH077, we also restored the U3-R-U5 structure of the 3′ LTR by PCR methods.
Nucleotide sequence analysis confirmed that the completed molecular clones derived from WITO and CH077, designated pWITO.c and pCH077.t, respectively, were identical to their cognate T/F genome sequences; these plasmid clones comprise all genetic elements of a complete provirus. In the case of CH058 and CH040, the 3′ LTR of the molecular clones (designated pCH058.c and pCH040.c, respectively) does not contain U5; however, it is restored in the first cycle of virus replication since the 3′ U5 is synthesized by the reverse transcriptase using the 5′ LTR U5 cDNA as a template (59). The pCH040.c clone also contains a single nucleotide mutation (T) at the third nucleotide position prior to the end of the 3′ R that was unintentionally introduced by PCR during the cloning process. The transmitted sequence for CH040 has an A at this position. However, since the 5′ R cDNA is used as a template by the reverse transcriptase to synthesize the 3′ R during reverse transcription, the wild-type nucleotide (A) is restored in the first cycle of virus replication. Therefore, the integrated proviruses and the proteomes of CH040.c and CH058.c are identical to those of the respective T/F viruses.
Derivation of full-length T/F IMCs from proviral DNA.
The examples of WITO, CH077, CH040, and CH058 indicate the labor-intensive nature of generating IMCs from plasma vRNA/cDNA. Reasoning there would be minimal sequence diversity of proviral DNA during early/acute infection, we explored an alternative approach involving PCR amplification of proviral DNA from uncultured PBMC obtained from subjects for which T/F sequences had already been deduced. This strategy offered two significant advantages. First, reconstitution of the LTRs would not be necessary and, second, a high-fidelity DNA polymerase (Phusion) could be used, obviating a significant effort required for correcting mutations introduced in SG amplicons by Taq polymerase. Briefly, gDNA was extracted from PBMC obtained from five subjects: TRJO4551, REJO4541, RHPA4259, SUMA0874, and THRO4156 (referred to here as TRJO, REJO, RHPA, SUMA, and THRO). PBMC were not available for subject 700010106 (referred to here as CH106), and therefore vRNA+ plasma from acute infection was used to infect a CD4+/CCR5+ recombinant cell line, from which gDNA was isolated.
The proviral DNA genomes were amplified in two halves, amplicons were TA-ligated into the pCR-XL-TOPO vector, and clones of each fragment were sequenced and compared to the T/F sequence. With the exception of TRJO, the number of nucleotide differences throughout the genome was relatively small, between 2 and 4. These nucleotide mismatches were corrected by PCR using mutagenic primers. In the case of TRJO, all of the clones of the 3′ half contained multiple G-to-A substitutions, indicative of the APOBEC hypermutation. The clone with the fewest mutations, which were localized between nt 8225 and 8382, was restored to the T/F nucleotide sequence using a long mutagenic primer and PCR. Full-length IMCs were generated by ligation of the cognate 5′ and 3′ pieces, facilitated by the internal unique restriction site. The proviral clones of TRJO, REJO, and SUMA were not stable in the pCR-XL-TOPO vector and were therefore ligated into a modified low-copy-number pBR322 plasmid (55). In the case of CH106, the 5′ PCR amplicon, and thus the IMC, lacked the first 37 nt of the 5′ LTR since successful amplification required the use of an internal PCR sense primer. The cloned and sequence-confirmed T/F IMCs were designated pCH106.c, pTHRO.c, pRHPA.c, pREJO.c, pSUMA.c, and pTRJO.c (Table 1).
Infectivity of T/F IMC virus stocks.
A virus stock of each IMC was generated by DNA transfection of 293T cells. The production of HIV-1 p24 antigen was similar among the different T/F, and control viruses, including YU-2, BaL, JRCSF, and SF162 (Fig. 2A). This result indicated that neither the lack of the first 37 nucleotides of the 5′ LTR (CH106.c) nor the absence of the 3′ U5 LTR element (CH040.c and CH058.c) had a marked negative effect on the generation of virions in transfected 293T cells. We confirmed infectivity of transfection-derived virus stocks on TZMbl cells (data not shown) before inoculating PBMC cultures in order to analyze the relative infectivity of T/F viruses produced in primary cells. Among the 10 T/F viruses, p24 antigen concentrations at 5 days postinfection ranged from 80 to 350 ng/ml. PBMC-derived viruses were also titered on TZM-bl cells, and the infectivity ranged from about 104 to 105 IU/ng of p24 (Fig. 2B). The median infectivity value of the T/F IMCs (5.7 × 104 IU/ng of p24) was similar to that of BaL, SF162, JRCSF, and YU-2 (3.2 × 104 IU/ng of p24). These results indicate that the T/F viruses productively infect PBMC and that progeny virus is similar in infectivity on TZM-bl cells to that of the control viruses.
Fig 2.

Virus production and relative infectivity of T/F viruses. PBMC were infected for 4 h at an MOI of 1 (TZM-bl IU) with transfection-derived IMC virus stocks. The cells were washed, and after 5 days the culture supernatants were collected, clarified by centrifugation, and cryostored. The samples were analyzed for p24 antigen concentration (A) and infectivity on TZM-bl cells relative to 1 ng of p24 (IU/ng-p24) (B).
Analysis of coreceptor usage.
HIV-1 generally utilizes either CXCR4 or CCR5 as a coreceptor; however, CCR5-tropic viruses are almost exclusively transmitted. We tested the coreceptor usage of the T/F viruses on TZM-bl cells infected in the presence of the coreceptor antagonist TAK779 (CCR5), AMD3100 (CXCR4), or a combination of both. The CXCR4-tropic virus NL4-3 and the CCR5-tropic viruses YU-2 and ADA were included as controls. Each of the T/F viruses demonstrated high levels of infection in the presence of AMD3100 and sensitivity to TAK779, indicating they primarily utilize the CCR5 coreceptor for infection of TZM-bl cells (Fig. 3). The ability to utilize CCR5 was in accordance with the findings of Wilen et al. (64), who analyzed pseudovirions for coreceptor usage in NP2-CD4+/CCR5+ cells. Interestingly, pseudovirions containing the CH077 T/F Env efficiently infected NP2-CD4+/CXCR4+ cells, suggesting utilization of the CXCR4 coreceptor (data not shown). In addition, a subset of IMC-derived T/F viruses was tested in PBMC from a donor carrying the delta32-CCR5 mutation, and CH077.t, but not CH040.c, CH058.c, or THRO.c, was able to replicate (data not shown).
Fig 3.

Analysis of T/F virus coreceptor usage. TZM-bl cells preincubated for 1 h with 1.2 μM AMD3100, 10 μM TAK-779, or both were infected with 2 × 103 TZM-bl IU (MOI = 0.24) of each virus. After a 48-h incubation period, the cells were analyzed for firefly luciferase expression. The data are plotted as the percent infection relative to infected cells not preincubated with TAK-779 or AMD3100.
Analysis of macrophage tropism.
For historical reasons, CCR5 tropism is still often equated with macrophage tropism. However, R5-tropic viruses exhibit considerable variation in their capacity to infect primary macrophages (50–52). Recently, Duncan and Sattentau (18) approached the concept of tropism is both categorical (replication or not), and continuous (the spectrum of replication capacity) terms and introduced the terms “macrophage tropic” (mac-tropic) and “macrophage replication capacity” (MRC) to distinguish more clearly the distinct qualitative and quantitative aspects, respectively, of the tropism phenotype. Generally, R5 Envs from brain tissue demonstrate a high MRC, whereas those derived from lymph nodes, blood, and semen are generally either not mac-tropic or exhibit a low MRC (inefficient infection) (18, 52). Because of the significance for understanding mucosal HIV-1 transmission, we examined the panel of 10 clade B T/F viruses for infection and replication in cultures of primary CD4+ T lymphocytes and autologous MDM. In T lymphocyte cultures, viral T/F virus p24 concentrations increased over a 10-day period, with kinetics similar to those of the YU-2, BaL, and ADA control viruses (Fig. 4A). In MDM, however, replication of the T/F virus group did not resemble that of the control viruses (Fig. 4C). At day 4, p24 produced from control viruses reached 18 to 74 ng/ml, while that of the T/F viruses ranged from 0.1 to 1 ng/ml, with the exception of CH058.c, which remained below the assay limit of detection in this particular donor PBMC. The amount of p24 produced in MDM cultures infected with control viruses continued to rise on days 7 and 10. While MDM infected with the T/F viruses also continued to release p24, the levels were up to 2 orders of magnitude lower than for the control viruses with high MRC. As illustrated in Fig. 4B and D, on day 10 the p24 concentrations produced by T/F and control viruses in T lymphocytes were similar (median values of 1,824 and 1,783 ng/ml, respectively), whereas in MDM the median values (4.4 and 211 ng/ml, respectively) were significantly different.
Fig 4.

T/F virus replication in CD4+ T lymphocytes and MDM. CD4+ T lymphocytes (A) and autologous MDM (C) were incubated overnight with 5 × 104 TZM-bl IU of the control viruses YU-2, ADA, and BaL (labeled as M-tropic) and a panel of T/F viruses. Cell cultures were washed three times on day 1 and resuspended in 1 ml of fresh medium. On days 4, 7, and 10, the culture medium was completely removed for analysis of p24 antigen, and fresh medium was added back. Scatter plots show p24 antigen concentrations on day 10 for CD4+ T lymphocyte (B) and MDM (D) cultures. The median value for each group is indicated by a horizontal bar. Statistical significance of difference between medians was determined by calculating an exact two-tailed P value (Wilcoxon rank-sum test) and is indicated by “**”. NS, not significant.
Virus replication was examined using several different blood donors, and in a representative experiment (Fig. 5), a subset of the T/F viruses was analyzed alongside the JRCSF strain of HIV-1. JRCSF, isolated from the cerebrospinal fluid of an individual with chronic HIV-1 disease, utilizes the CCR5 coreceptor and has been classified as low- or non-mac-tropic (41). Similar to our findings above, this experiment demonstrated that T/F viruses replicate efficiently in CD4+ T lymphocytes but not in MDM in comparison with ADA, YU-2, and BaL. The replication profile of the T/F viruses in both CD4+ T lymphocytes and MDM resembled that of JRCSF (Fig. 5). When the analysis of T/F virus replication in CD4+ T lymphocytes and MDM from the same donor was repeated several times, using a total of four different blood donors, similar results were obtained each time (Fig. 4 and 5 and data not shown). Notably, while the rank order of T/F virus replication varied among different donor MDM, REJO.c and THRO.c replicated to higher titers in most MDM compared to the other T/F viruses. These results indicate that, as a group, T/F viruses do not display a high capacity to replicate in MDM. Nevertheless, our results also suggest that T/F viruses are not a homogeneous group with respect to their macrophage replication capacity.
Fig 5.

T/F virus replication in CD4+ T lymphocytes and MDM. CD4+ T lymphocytes (A) and autologous MDM (C) were incubated overnight with 50 ng of p24 of the control viruses with high MRC, YU-2, ADA, and BaL (labeled as M-tropic), the low/non-mac-tropic control virus JRCSF, and a panel of T/F viruses. Cell cultures were washed three times on day 1 and resuspended in 2 ml of fresh medium. On days 4, 7, and 10, the culture medium was completely removed for analysis of p24 antigen, and fresh medium was added back. Scatter plots show the p24 antigen concentrations on day 10 for CD4+ T lymphocyte (B) and MDM (D) cultures. The median value for each group is indicated by a horizontal bar. Statistical significance of difference between medians of the T/F and M-tropic group was determined by calculating an exact two-tailed P value (Wilcoxon rank-sum test) and is indicated by “*”. NS, not significant.
Sensitivity of T/F virus infection to soluble CD4 (sCD4).
The molecular determinants of HIV-1 Env that define macrophage tropism are not well understood. It has been reported that viruses with a high MRC are more sensitive to inhibition by reagents that interfere with gp120-CD4 interactions (e.g., sCD4) (50). Thus, we analyzed the sensitivity of the T/F viruses to serially diluted sCD4 in comparison to YU-2, SF162, JRCSF, and BaL by using TZM-bl cells as targets of infection. For each virus tested, we observed a dosage-dependent inhibitory effect on infectivity. The median 50% inhibitory concentration (IC50) of sCD4 was significantly lower in the mac-tropic control group than in the T/F group (3.15 μg/ml versus 65.29 μg/ml; P = 0.01) (Fig. 6). The IC50s for most of the T/F viruses were similar to that of the JRCSF control. Notably, there were outliers among the T/F viruses. In particular, REJO.c exhibited a relatively high degree of sensitivity to sCD4, a phenotype consistent with its greater MRC compared to the other T/F viruses. Similarly, THRO.c was overall more infectious in macrophages and more sensitive to sCD4 relative to the other T/F viruses. These results indicate that, as a group, T/F viruses are less sensitive to sCD4 than viruses with a high MRC. However, our results suggest that among the T/F viruses some variants exhibit an intermediate level of replication capacity in macrophages.
Fig 6.
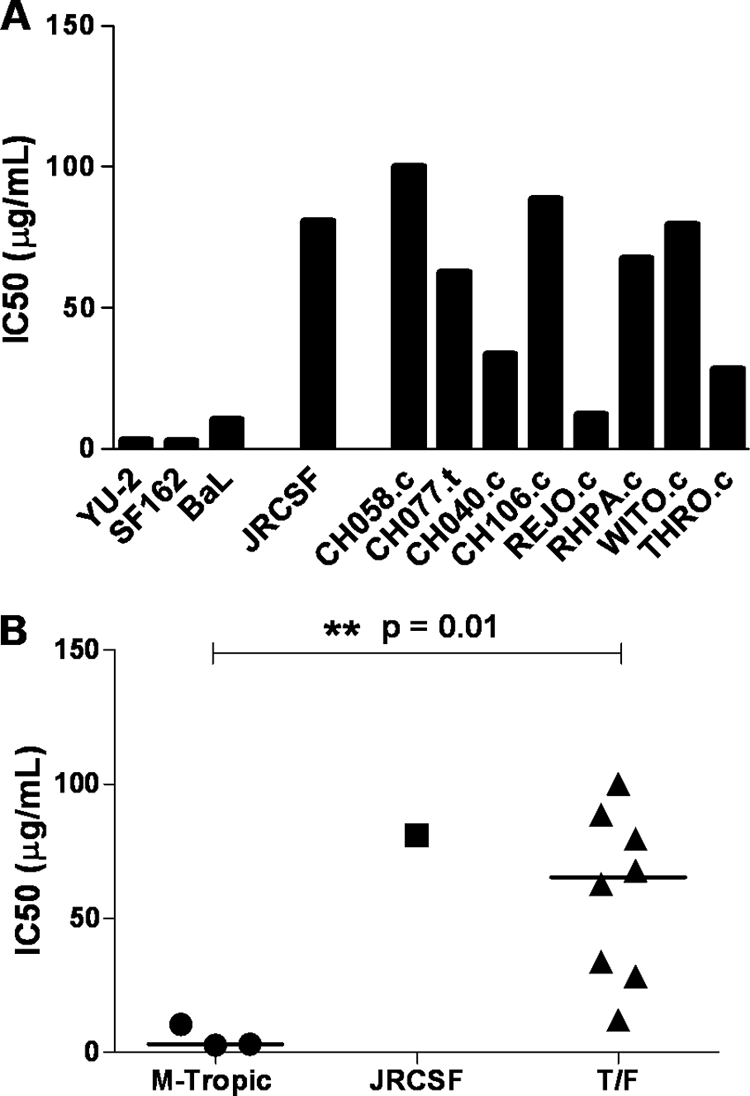
Sensitivity of T/F viruses to sCD4. T/F and control viruses were preincubated with serial dilutions of sCD4 for 1 h prior to infection. The virus/sCD4 mixtures were then placed on TZM-bl cells (MOI of 0.25) in the presence of DEAE-dextran (40 μg/ml) and incubated for 48 h. The cells were then lysed and analyzed for firefly luciferase expression. (A) Bar graph representing the IC50s for the different viruses analyzed. (B) Scatter plot illustrating the differences in IC50s between the T/F viruses and the control viruses with high MRC (labeled as M-tropic). The median IC50 for each group is indicated by a horizontal bar; the associated exact two-tailed P value (Wilcoxon rank-sum test) is shown; significance is indicated by “*”.
Determinants of T/F virus macrophage tropism map to Env.
We examined whether determinants expressed by the env gene were responsible for the poor replication of T/F viruses in macrophages by analyzing T/F env sequences expressed in an isogenic proviral genome. We utilized the previously reported Env-IMC-LucR approach (22), which enables analysis of heterologous env's expressed in cis in a replication-competent, Renilla luciferase (LucR)-expressing backbone. Since the sites for Env receptor and coreceptor interaction are present within the Env ectodomain, this strategy allowed an assessment specific to mechanisms of virus entry and avoided confounding differences among viral proteomes outside of Env that may be affected differently by host/macrophage restriction factors that may act at several different steps of the life cycle (5). MDM were infected with normalized amounts of virus and as early as 24 h postinfection, de novo LucR expression was detected for each of the control Envs (Fig. 7A). The viruses encoding T/F Envs yielded much lower levels of LucR expression; however, for NL-LucR.T2A-REJO.ecto and NL-LucR.T2A-THRO.ecto, the reduction was less pronounced. Detection of LucR at this 24-h time point is indicative of first-round virus infection (22), suggesting that viruses encoding T/F versus control Envs differ at an early step in the virus life cycle. Differences were magnified at day 6, likely by additional rounds of virus replication. LucR expression from viruses encoding T/F Envs from CH040.c, CH058.c, CH106.c, and SUMA.c was at least 100-fold lower compared to the controls, and T/F Envs from REJO.c and THRO.c yielded ∼10-fold lower LucR expression (median for T/F group, 7 × 103 LucR RLU; median for control group, 1.04 × 106 LucR RLU) (Fig. 7A and B).
Fig 7.

Low replication of T/F viruses in macrophage maps to the Env ectodomain. (A) MDM were infected (day 0) at an MOI of 5 (TZM-bl IU) with transfection-derived Env-IMC-LucR viruses expressing either T/F env's from CH040.c, CH058.c, CH106.c, SUMA.c, REJO.c, and THRO.c or control env's from BaL and SF162. The cells were washed, and harvested on days 1, 2, and 6 for analysis of LucR expression. (B) The scatter plot compares virus replication (RLU values) for both virus groups on day 6 (median RLU values are indicated by horizontal bars; the associated exact two-tailed P value [Wilcoxon rank-sum test] is shown). (C) Using a subset of Env-IMC-LucR viruses that expressed env's from WITO.c (♦), CH077.t (■), CH040.c (▼), CH058.c (▲), BaL (□), and SF162 (○), respectively, MDM from seven different donors were infected and analyzed for LucR expression 5 to 7 days postinfection. Scatter plots compare LucR expression in the T/F and control Env-IMC-LucR virus groups for each donor MDM (indicated by numbers 1 through 7).
Using subsets of viruses, the analysis was repeated with MDM derived from seven different donors. In each experiment, less LucR expression was detected from the T/F Env-IMC-LucR viruses compared to controls (Fig. 7C). For each donor, the median day 6 LucR values show markedly lower productive MDM infection with T/F Env-IMC-LucR viruses, and across the seven donors the difference between the T/F versus control viruses was statistically significant, as determined by the exact two-tailed Wilcoxon matched-pairs signed-rank test (P = 0.0156). Notably, although considerable variation was observed in virus replication (i.e., level of LucR expression) among the different donor MDM, a phenotype previously reported (7, 8, 23, 29), in each case there remained a difference of 1 to 3 orders of magnitude between T/F and control viruses. This analysis revealed differences in terms of virus infectivity and replication that are similar to those observed studying full-length viruses. It also recapitulates the intermediate phenotype exhibited by REJO.c and THRO.c. These results suggest that the macrophage tropism phenotype and the MRC of the T/F viruses is due to virus-specific determinants present in the ectodomain of Env glycoproteins from these viruses.
Analysis of T/F virus cDNA synthesis in MDM.
To further analyze whether differences between T/F and control viruses manifest early in the life cycle, MDM were infected with Env-IMC-LucR viruses and analyzed for the synthesis of viral cDNA. Compared to virus expressing the env ectodomain of BaL, less viral cDNA was detected in MDM infected with LucR-reporter virus encoding T/F Env from CH040.c and CH077.t (Fig. 8A). LucR expression levels quantified in a parallel experiment were consistent with the cDNA results (Fig. 8B). To validate these findings, we infected MDM with PBMC-derived full-length T/F viruses. At 24 h after infection, near the peak of cDNA synthesis and prior to secondary infection (57), greater amounts of early viral cDNA (R-U5) accumulated in MDM infected with BaL, SF162, and YU-2 than in cells infected with T/F viruses (Fig. 9). Together, our results indicate that the T/F virus phenotype of low replication capacity in macrophages maps to Env and involves a step in the replication cycle prior to or at the initiation of reverse transcription, most likely virus entry.
Fig 8.
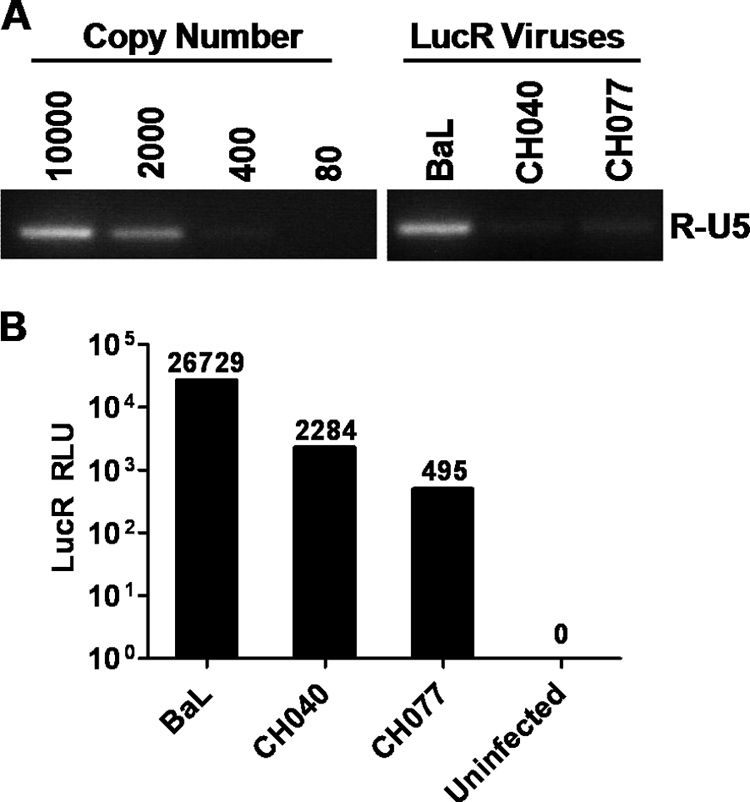
Analysis of viral transcripts in Env-IMC-LucR virus-infected MDM. MDM were infected for 4 h at an MOI of 5 (TZM-bl IU) with transfection-derived Env-IMC-LucR viruses expressing the control env BaL or the T/F env's from CH040.c and CH077.c. The cells were washed after a 4-h incubation and then cultured for 3 days. (A) At 3 days postinoculation, the MDM were washed and analyzed for early (R-U5) viral cDNA products. A 5-fold serial dilution containing known amounts of viral DNA was analyzed in parallel. Prior to amplification, samples were DpnI digested to eliminate potential carryover plasmid DNA. (B) MDM were analyzed in parallel for LucR expression.
Fig 9.

Analysis of T/F virus cDNA synthesis in MDM. MDM were infected for 8 h at an MOI of 5 (TZM-bl IU) with PBMC-derived full-length control and T/F viruses. Due to an overall lower titer of RHPA virus stocks, MDM were infected with an MOI of 2 with RHPA.c. After 8 h, the MDM were washed five times, and fresh medium was added back to each well. (A) At 24 h postinfection, the MDM were washed three times and analyzed for early (R-U5) viral cDNA products. Prior to amplification, samples were DpnI digested to eliminate any potential carryover plasmid DNA. (B) Viral cDNA products were quantified and normalized to the cDNA products of the GAPDH housekeeping gene. The ratio of viral transcripts to transcripts of GAPDH is shown.
DISCUSSION
The nucleotide sequence of HIV-1 genomes responsible for sexual HIV-1 transmission can now be identified (36, 39, 54, 55). Based on the hypothesis that the derivation of full-length, replication-competent molecular clones of T/F viruses will provide new opportunities to elucidate HIV-1 mucosal transmission and inform prevention strategies, we describe here the molecular derivation of 10 clade B IMCs, each expressing the proteome of a single-variant T/F virus. Four of the ten IMCs were derived by cloning 9-kb near-genome-length SG amplicons from plasma vRNA. Each of the four T/F viral genomes had been identified previously by direct sequencing of the SG amplicons (55). Sequence analysis of the molecular clones revealed that individual SG amplicons contained numerous nucleotide substitutions introduced by Superscript III reverse transcriptase and/or Taq polymerase. These mutations are not detected by direct sequencing of uncloned SG amplicons unless polymerase errors occur during cDNA synthesis or the first or second cycle of PCR. Therefore, to create the T/F IMCs from SG amplicons it was necessary to repair numerous mutations and missing LTR sequence. Because of the labor-intensive nature of this effort, an alternative, less labor-intensive strategy was developed and applied to generate six additional T/F IMCs. Here, HIV-1 genomes were cloned from PCR-amplified proviral DNA (gDNA) of either uncultured PBMC obtained from acutely HIV-1-infected subjects or from a CD4+/CCR5+ recombinant cell line that was infected with acute plasma virus. An advantage of cloning from proviral DNA is that a high-fidelity DNA polymerase could be used for PCR amplification, thereby significantly reducing the frequency of polymerase errors. Moreover, the amplicons comprise complete LTR sequences. After constructing each IMC, the entire nucleotide sequence was analyzed to confirm identity with that inferred for the T/F virus by analyzing SG amplicon sequences. In certain instances, we elected not to correct nucleotide mismatches for pCH040.c, pCH058.c, pCH106.c, and pRHPA.c, since they exist in noncoding regions of the viral DNA (either the 5′ U3-R or the 3′ U5 LTR elements) and are replaced with T/F sequence in the very first round of virus infection through the process of reverse transcription (59). Possible negative effects of these non-T/F nucleotides on transcription following DNA transfection into 293T cells and the generation of progeny virus were considered and ruled out empirically. A third approach for generating T/F IMCs involves the chemical synthesis of proviral genome fragments based on nucleotide sequence inferred from SG amplicons of plasma vRNA/cDNA, followed by cloning to generate a complete copy of the proviral genome (44, 55). This approach, while the least labor-intensive, can be cost prohibitive but allows generation of IMCs in situations where patient samples are not available.
Having created the panel of 10 T/F IMCs, the first question we addressed was whether the viruses were infectious and replication competent. As would be expected of T/F viruses causing clinical infection, our analysis demonstrated that all 10 clade B IMCs produced replication-competent virus. This result further validates and supports the mathematical model that was used to infer T/F virus genomes from plasma vRNA-derived sequences (39, 43, 55). Initial biological support had come from two of these studies (44, 55) when the genomes of one clade B virus and three clade C viruses were cloned and found to encode replication-competent virus.
In cultures of activated PBMC the T/F viruses replicated efficiently, exhibiting kinetics similar to control viruses which included “highly” mac-tropic strains, YU-2, BaL, and ADA (i.e., strains with a high MRC [18]). The T/F viruses also replicated in MDM; however, their replication was significantly less efficient compared to YU-2, BaL, and ADA. This phenotype was shown to be a function of the Env glycoprotein and was manifested at or before reverse transcription, likely at virus entry. Notably, the magnitude of replication in MDM varied appreciably among the different T/F viruses. This was especially evident for REJO.c and THRO.c, which were more infectious and replicated more efficiently in most donor MDM. Consistent with this phenotype, REJO.c and THRO.c were more sensitive to inhibition by sCD4 than were the other T/F viruses. In agreement with earlier studies, our results also indicated that MDM from different donors exhibited considerable variation in their susceptibility to productive HIV-1 infection (7, 65). While our panel of 10 clade B T/F viruses exhibited a lower MRC than prototypic mac-tropic control viruses, our analysis clearly demonstrates that as a group they are replication competent in cultures of MDM. These findings provide a clearer picture of the replication properties of clade B T/F viruses in MDM than was previously reported in one study limited to a single clade B T/F virus (55) and one limited to three clade C T/F viruses derived from 2 patients (44).
Determinants of macrophage tropism have been identified in the HIV-1 env gene (17, 19–21, 50, 52). Two well-described env variants often found in mac-tropic viruses displaying high MRC encode amino acid residues N283 and D386. N283 resides in the gp120-CD4-binding site and presumably increases the affinity of gp120 for CD4 (20). D386 lies in V4 and disrupts a potential N-linked glycosylation site that has been postulated to affect macrophage tropism by increasing exposure of the gp120-CD4 binding site (19, 21). Among the 10 clade B T/F viruses, RHPA.c and WITO.c encode N238 and CH058.c and CH077.c express D386. Nonetheless, these viruses exhibited inefficient cDNA synthesis, infection, and replication in MDM. It is noteworthy that determinants of HIV-1 macrophage tropism may also involve host factors that affect replication at different levels of the virus life cycle subsequent to entry (3, 5, 29, 42). However, since the phenotypes of both the T/F IMCs and control viruses (BaL, YU2, SF162, and JRCSF)—as defined by levels of cDNA synthesis, viral gene expression, and replication—were transferable by expression of their respective Env ectodomains in the isogenic Env-IMC-LucR backbone, it seems unlikely that the “low” macrophage tropism phenotype of T/F viruses is due to interactions between other (non-env) viral determinants and macrophage host restriction factors. Therefore, our analyses of env in the Env-IMC-LucR isogenic backbone would argue that a low capacity to replicate in MDM is a function of the T/F env.
Initially, HIV-1 viruses were categorized by their ability to infect either macrophages or replicate efficiently in T cell lines (25, 61). The identification of the CCR5 and CXCR4 coreceptors provided a molecular explanation for this observation (2, 6, 9, 11, 14, 16, 24), eventually leading to a classification of viral tropism as R5-tropic versus X4-tropic, based on the chemokine receptor used for cell entry. This classification, however, does not define the ability of virus to infect a given cell type expressing that receptor. We found that all T/F IMCs utilize CCR5 as a coreceptor, and CH077.t can also use CXCR4 on certain target cell types (64; data not shown). Marked differences exist between R5 viruses in their tropism for primary macrophages (13, 18, 41, 58). HIV-1 R5 variants that exhibit a high MRC can gain entry into cells with low surface expression of CD4, whereas R5 viruses with a low MRC seem to require higher levels for entry (47, 49, 52). Furthermore, a strong correlation exists between high MRC and sensitivity to reagents that inhibit gp120-CD4 interactions (50). Our results are in keeping with this observation in that the T/F viruses infected and replicated in MDM less efficiently than high MRC control viruses and required significantly higher concentrations of sCD4 to inhibit their infection of susceptible cells. Our findings suggest that viral variants with a high replication capacity in MDM are neither selected by nor required for mucosal HIV-1 transmission. Since such variants would arguably have broader cell tropism, a plausible explanation for their absence among the 10 T/F viruses described here is that high MRC variants are not predominant in tissues involved in sexual transmission. Consistent with this interpretation, viruses with a high MRC phenotype tend to be compartmentalized to brain (35, 47, 52, 60), while viruses in blood, lymph nodes, and semen generally do not efficiently infect MDM. Studies demonstrating that “resting” memory CD4+ T cells are likely the principal early targets of HIV-1 and SIV infection in humans and primates, respectively (38, 48, 69, 70), would further argue that macrophages may not be initial targets in HIV-1 transmission. Nevertheless, the sensitivity of specialized tissue cells present at mucosal sites of HIV-1 transmission to T/F viruses remains to be determined. Ongoing studies in our laboratories and others aim to elucidate T/F virus tropism in mucosal cell populations, including tissue macrophages.
Our analysis using endo- and ectocervix explants models suggest that T/F viruses are less infectious or replicative compared to BaL, SF162, and YU-2, which are commonly used strains for in vitro model systems (unpublished). Taken together, our results bear significance for the design of in vitro model systems meant to analyze HIV-1 transmission, since prototypic “highly macrophage-tropic” viruses may not best represent virus-host interactions operative during natural infection. Finally, discovery and validation of effective measures to prevent HIV-1 sexual transmission will require understanding fundamental principles of mucosal immunobiology and virology at the sites of HIV-1 entry (37). Thus, the generation and initial biological characterization of the 10 clade B T/F IMC provide important underpinnings for AIDS/HIV prevention research, in particular that aimed at elucidating virus-host interactions during the earliest stages of HIV-1 clinical infection.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the NIH Center for HIV/AIDS Vaccine Immunology (CHAVI; UO1-AI067854); the Bill and Melinda Gates Foundation's Collaboration for AIDS Vaccine Discovery/Comprehensive Antibody Vaccine Immune Monitoring Consortium (grant 38619); a Bill and Melinda Gates Foundation Grand Challenges grant (grant 37874); the facilities of the Virology, Biostatistics, and Genetic Sequencing cores of the UAB Center for AIDS Research (P30-AI-27767); and the Genetically Defined Microbe and Expression Core of the UAB Mucosal HIV and Immunobiology Center (R24 DK-64400). Research was also supported by a Merit Review Award (J.C.K.) and a Research Career Scientist Award (J.C.K.) from the Department of Veterans Affairs Medical Center, Research Services. T.G.E. is supported by NIH training grant AI007439.
We thank the CHAVI team, especially Kelly Soderberg, and all study participants. We are also grateful to Ashutosh Tamhane and Andy Westfall for help with statistical analysis.
Footnotes
Published ahead of print 21 December 2011
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Abrahams MR, et al. 2009. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-Poisson distribution of transmitted variants. J. Virol. 83:3556–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alkhatib G, et al. 1996. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955–1958 [DOI] [PubMed] [Google Scholar]
- 3. Ayinde D, Maudet C, Transy C, Margottin-Goguet F. 2010. Limelight on two HIV/SIV accessory proteins in macrophage infection: is Vpx overshadowing Vpr? Retrovirology 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bar KJ, et al. 2010. Wide variation in the multiplicity of HIV-1 infection among injection drug users. J. Virol. 84:6241–6247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bergamaschi A, Pancino G. 2010. Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology 7:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bjorndal A, et al. 1997. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 71:7478–7487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bol SM, et al. 2009. Donor variation in in vitro HIV-1 susceptibility of monocyte-derived macrophages. Virology 390:205–211 [DOI] [PubMed] [Google Scholar]
- 8. Chang J, et al. 1996. Twin studies demonstrate a host cell genetic effect on productive human immunodeficiency virus infection of human monocytes and macrophages in vitro. J. Virol. 70:7792–7803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng-Mayer C, Liu R, Landau NR, Stamatatos L. 1997. Macrophage tropism of human immunodeficiency virus type 1 and utilization of the CC-CKR5 coreceptor. J. Virol. 71:1657–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng-Mayer C, Quiroga M, Tung JW, Dina D, Levy JA. 1990. Viral determinants of human immunodeficiency virus type 1 T-cell or macrophage tropism, cytopathogenicity, and CD4 antigen modulation. J. Virol. 64:4390–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choe H, et al. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135–1148 [DOI] [PubMed] [Google Scholar]
- 12. Chohan B, et al. 2005. Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1-V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J. Virol. 79:6528–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dejucq N, Simmons G, Clapham PR. 1999. Expanded tropism of primary human immunodeficiency virus type 1 R5 strains to CD4+ T-cell lines determined by the capacity to exploit low concentrations of CCR5. J. Virol. 73:7842–7847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deng H, et al. 1996. Identification of a major coreceptor for primary isolates of HIV-1. Nature 381:661–666 [DOI] [PubMed] [Google Scholar]
- 15. Derdeyn CA, et al. 2004. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303:2019–2022 [DOI] [PubMed] [Google Scholar]
- 16. Dragic T, et al. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381:667–673 [DOI] [PubMed] [Google Scholar]
- 17. Duenas-Decamp MJ, Peters PJ, Burton D, Clapham PR. 2009. Determinants flanking the CD4 binding loop modulate macrophage tropism of human immunodeficiency virus type 1 R5 envelopes. J. Virol. 83:2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duncan CJ, Sattentau QJ. 2011. Viral determinants of HIV-1 macrophage tropism. Viruses 3:2255–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dunfee RL, Thomas ER, Gabuzda D. 2009. Enhanced macrophage tropism of HIV in brain and lymphoid tissues is associated with sensitivity to the broadly neutralizing CD4 binding site antibody b12. Retrovirology 6:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dunfee RL, et al. 2006. The HIV Env variant N283 enhances macrophage tropism and is associated with brain infection and dementia. Proc. Natl. Acad. Sci. U. S. A. 103:15160–15165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dunfee RL, et al. 2007. Loss of the N-linked glycosylation site at position 386 in the HIV envelope V4 region enhances macrophage tropism and is associated with dementia. Virology 367:222–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edmonds TG, et al. 2010. Replication-competent molecular clones of HIV-1 expressing Renilla luciferase facilitate the analysis of antibody inhibition in PBMC. Virology 408:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eisert V, et al. 2001. Analysis of cellular factors influencing the replication of human immunodeficiency virus type I in human macrophages derived from blood of different healthy donors. Virology 286:31–44 [DOI] [PubMed] [Google Scholar]
- 24. Feng Y, Broder CC, Kennedy PE, Berger EA. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872–877 [DOI] [PubMed] [Google Scholar]
- 25. Fenyo EM, Albert J, Asjo B. 1989. Replicative capacity, cytopathic effect and cell tropism of HIV. AIDS 3(Suppl. 1):S5–S12 [DOI] [PubMed] [Google Scholar]
- 26. Ferrari G, et al. Relationship between functional profile of HIV-1 specific CD8 T cells and epitope variability with the selection of escape mutants in acute HIV-1 infection. PLoS Pathog. 7:e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fiebig EW, et al. 2003. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS 17:1871–1879 [DOI] [PubMed] [Google Scholar]
- 28. Fischer W, et al. 2010. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 5:e12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fouchier RA, Brouwer M, Kootstra NA, Huisman HG, Schuitemaker H. 1994. HIV-1 macrophage tropism is determined at multiple levels of the viral replication cycle. J. Clin. Invest. 94:1806–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frost SD, et al. 2005. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc. Natl. Acad. Sci. U. S. A. 102:18514–18519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ganusov VV, et al. Fitness costs and diversity of the cytotoxic T lymphocyte (CTL) response determine the rate of CTL escape during acute and chronic phases of HIV infection. J. Virol. 85:10518–10528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gartner S, et al. 1986. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 233:215–219 [DOI] [PubMed] [Google Scholar]
- 33. Gendelman HE, et al. 1988. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 167:1428–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goonetilleke N, et al. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 206:1253–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gorry PR, et al. 2002. Increased CCR5 affinity and reduced CCR5/CD4 dependence of a neurovirulent primary human immunodeficiency virus type 1 isolate. J. Virol. 76:6277–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haaland RE, et al. 2009. Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS Pathog. 5:e1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haase AT. 2011. Early events in sexual transmission of HIV and SIV and opportunities for interventions. Annu. Rev. Med. 62:127–139 [DOI] [PubMed] [Google Scholar]
- 38. Hladik F, et al. 2007. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity 26:257–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Keele BF, et al. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keele BF, et al. 2009. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J. Exp. Med. 206:1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koyanagi Y, et al. 1987. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 236:819–822 [DOI] [PubMed] [Google Scholar]
- 42. Laguette N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee HY, et al. 2009. Modeling sequence evolution in acute HIV-1 infection. J. Theor. Biol. 261:341–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H, et al. 2010. High multiplicity infection by HIV-1 in men who have sex with men. PLoS Pathog. 6:e1000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44a. Li Y, et al. 1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J. Virol. 65:3973–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu J, et al. 2010. Low-dose mucosal simian immunodeficiency virus infection restricts early replication kinetics and transmitted virus variants in rhesus monkeys. J. Virol. 84:10406–10412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu P, et al. Dynamic antibody specificities and virion concentrations in circulating immune complexes in acute to chronic HIV-1 infection. J. Virol. 85:11196–11207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martin-Garcia J, Cao W, Varela-Rohena A, Plassmeyer ML, Gonzalez-Scarano F. 2006. HIV-1 tropism for the central nervous system: brain-derived envelope glycoproteins with lower CD4 dependence and reduced sensitivity to a fusion inhibitor. Virology 346:169–179 [DOI] [PubMed] [Google Scholar]
- 48. Mattapallil JJ, et al. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434:1093–1097 [DOI] [PubMed] [Google Scholar]
- 49. Peters PJ, et al. 2004. Biological analysis of human immunodeficiency virus type 1 R5 envelopes amplified from brain and lymph node tissues of AIDS patients with neuropathology reveals two distinct tropism phenotypes and identifies envelopes in the brain that confer an enhanced tropism and fusogenicity for macrophages. J. Virol. 78:6915–6926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peters PJ, et al. 2008. Variation in HIV-1 R5 macrophage-tropism correlates with sensitivity to reagents that block envelope: CD4 interactions but not with sensitivity to other entry inhibitors. Retrovirology 5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peters PJ, Duenas-Decamp MJ, Sullivan WM, Clapham PR. 2007. Variation of macrophage tropism among HIV-1 R5 envelopes in brain and other tissues. J. Neuroimmune Pharmacol. 2:32–41 [DOI] [PubMed] [Google Scholar]
- 52. Peters PJ, et al. 2006. Non-macrophage-tropic human immunodeficiency virus type 1 R5 envelopes predominate in blood, lymph nodes, and semen: implications for transmission and pathogenesis. J. Virol. 80:6324–6332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sagar M, Wu X, Lee S, Overbaugh J. 2006. Human immunodeficiency virus type 1 V1-V2 envelope loop sequences expand and add glycosylation sites over the course of infection, and these modifications affect antibody neutralization sensitivity. J. Virol. 80:9586–9598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Salazar-Gonzalez JF, et al. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 82:3952–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Salazar-Gonzalez JF, et al. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206:1273–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Salazar-Gonzalez JF, et al. Origin and evolution of HIV-1 in breast milk determined by single-genome amplification and sequencing. J. Virol. 85:2751–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schmidtmayerova H, Alfano M, Nuovo G, Bukrinsky M. 1998. Human immunodeficiency virus type 1 T-lymphotropic strains enter macrophages via a CD4- and CXCR4-mediated pathway: replication is restricted at a postentry level. J. Virol. 72:4633–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Simmons G, et al. 1996. Primary, syncytium-inducing human immunodeficiency virus type 1 isolates are dualtropic and most can use either Lestr or CCR5 as coreceptors for virus entry. J. Virol. 70:8355–8360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Telesnitsky A, Goff SP. 1997. Reverse transcription and the generation of retroviral DNA, p 121–160 In Coffin JM, et al. (ed), Retroviruses. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 60. Thomas ER, et al. 2007. Macrophage entry mediated by HIV Envs from brain and lymphoid tissues is determined by the capacity to use low CD4 levels and overall efficiency of fusion. Virology 360:105–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Valentin A, Albert J, Fenyo EM, Asjo B. 1990. HIV-1 infection of normal human macrophage cultures: implication for silent infection. Virology 177:790–794 [DOI] [PubMed] [Google Scholar]
- 62. Wei X, et al. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wei X, et al. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307–312 [DOI] [PubMed] [Google Scholar]
- 64. Wilen CB, et al. 2011. Phenotypic and immunologic comparison of clade B transmitted/founder and chronic HIV-1 envelope glycoproteins. J. Virol. 85:8514–8527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Williams LM, Cloyd MW. 1991. Polymorphic human gene(s) determines differential susceptibility of CD4 lymphocytes to infection by certain HIV-1 isolates. Virology 184:723–728 [DOI] [PubMed] [Google Scholar]
- 66. Wood N, et al. 2009. HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog. 5:e1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu X, et al. 1999. Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J. Virol. 73:2126–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zack JA, et al. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213–222 [DOI] [PubMed] [Google Scholar]
- 69. Zhang Z, et al. 1999. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science 286:1353–1357 [DOI] [PubMed] [Google Scholar]
- 70. Zhang ZQ, et al. 2004. Roles of substrate availability and infection of resting and activated CD4+ T cells in transmission and acute simian immunodeficiency virus infection. Proc. Natl. Acad. Sci. U. S. A. 101:5640–5645 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.