

Abstract
Previous studies have indicated that the adenovirus type 5 E1B 55-kDa protein facilitates viral DNA synthesis in normal human foreskin fibroblasts (HFFs) but not in primary epithelial cells. To investigate this apparent difference further, viral DNA accumulation was examined in primary human fibroblasts and epithelial cells infected by the mutant AdEasyE1Δ2347, which carries the Hr6 frameshift mutation that prevents production of the E1B 55-kDa protein, in an E1-containing derivative of AdEasy. Impaired viral DNA synthesis was observed in normal HFFs but not in normal human bronchial epithelial cells infected by this mutant. However, acceleration of progression through the early phase, which is significantly slower in HFFs than in epithelial cells, eliminated the dependence of efficient viral DNA synthesis in HFFs on the E1B 55-kDa protein. These observations suggest that timely synthesis of the E1B 55-kDa protein protects normal cells against a host defense that inhibits adenoviral genome replication. One such defense is mediated by the Mre11-Rad50-Nbs1 complex. Nevertheless, examination of the localization of Mre11 and viral proteins by immunofluorescence suggested that this complex is inactivated similarly in AdEasyE1Δ2347 mutant-infected and AdEasyE1-infected HFFs.
INTRODUCTION
The E1B gene of species C human adenoviruses, such as adenovirus type 5 (Ad5), encodes unrelated proteins of 19 and 55 kDa that contribute to optimizing the environment for efficient viral replication within infected cells. The 19-kDa protein blocks apoptosis in infected cells (24, 65, 86, 99) and was the first viral homologue of cellular anti-apoptotic proteins to be identified (20, 85, 88). The E1B 55-kDa protein also counteracts cellular responses to infection that would be detrimental to efficient virus reproduction. One of the first properties to be ascribed to the E1B 55-kDa protein was interaction with the cellular tumor suppressor p53 (76). In rodent cells transformed by E1A and E1B gene products, this interaction can sequester p53 in juxtanuclear cytoplasmic structures (13, 42, 107). Binding of the E1B 55-kDa protein to the N-terminal activation domain of p53 has also been reported to inhibit p53-dependent transcription both in in vitro reactions and in transient expression systems (54, 105, 106). Insertions or substitutions in the E1B protein that impaired E1B-dependent transcriptional repression were observed to reduce the ability of the E1B protein to cooperate with E1A gene products in transformation of rodent cells (55, 89, 90, 106). Inhibition of p53-dependent transcription and transforming activity has also been reported to be reduced by a substitution that prevents sumoylation of the E1B 55-kDa protein at Lys104 (28), whereas substitutions that block shuttling of this protein between the nucleus and cytoplasm (25, 47) stimulate both activities (27). These observations indicate that inhibition of the transcriptional function of p53 and, presumably, of induction of apoptosis by this cellular protein are important for the transforming activity of the E1B 55-kDa protein.
In adenovirus-infected cells, the E1B 55-kDa protein is necessary for degradation of p53, as is the E4 Orf6 protein (15, 16, 36, 61, 63, 68, 69, 74, 77, 81). The p53 protein is a substrate of the viral/cellular E3 ubiquitin ligase (the Ad E3 Ub ligase) formed by assembly of the E1B 55-kDa and E4 Orf6 proteins with the cellular proteins cullin 5, elongins B and C, and Rbx1 and is targeted for proteasomal degradation by the action of this enzyme (19, 39, 53, 68). Although the Ad E3 Ub ligase is necessary to prevent accumulation of p53, the results of both genome-wide analyses of cellular gene expression (58) and examination of expression of subsets of p53-responsive genes (41, 63) indicate that, in several different cell types, the p53 protein that accumulates is transcriptionally inactive; nor does it induce apoptosis (16, 63). It therefore appears that in infected cells, one or more additional viral gene products function redundantly with the Ad E3 Ub ligase to ensure that p53 cannot trigger apoptosis or G1 arrest. One such gene product is the E4 Orf3 protein, which has been reported to induce inhibition of p53-dependent transcription in infected small airway epithelial cells (SAECs) (79).
Another function of the E1B 55-kDa protein is induction of selective export of viral late mRNAs from the nucleus to the cytoplasm (66, 100). Such selective export depends on the interaction of the E1B 55-kDa protein with E4 Orf6 (see references 12 and 31) and assembly of the Ad E3 Ub ligase (14, 103), although the relevant substrates have not yet been identified. Other known substrates of this enzyme include integrin α3, degradation of which may facilitate release of viral particles at the end of the infectious cycle (22), DNA ligase IV (7), and the Mre11 and Rad50 proteins (82). The latter two proteins and Nbs1 form the Mre11-Rad50-Nbs1 (MRN) complex, which detects and initiates signaling in response to double-stranded breaks in the genome, ultimately leading to nonhomologous end joining or recombinational repair (reviewed in references 23, 50, 72, 84, 95). The function of the MRN complex is also blocked by the E4 Orf3 protein of species C adenoviruses, which induces recruitment of Mre11 and Rad50 to intranuclear, track-like structures that also contain cellular proteins reorganized from promyelocytic leukemia (PML) bodies (17, 26, 30, 51, 82, 83). The E4 Orf3 protein has also been reported to colocalize with Mre11 in juxtanuclear cytoplasmic structures with the properties of aggresomes (3, 52). When both the formation of the Ad E3 Ub ligase and relocalization of Mre11 by the E4 Orf3 protein are prevented by mutation, viral DNA synthesis is impaired (29, 48, 56), and large concatemers of randomly orientated copies of the viral genome accumulate very late in infection (82, 98). Because concatemers are far too large to be packaged into capsids, their formation presumably reduces production of progeny virus particles. This phenomenon would also impair initiation of viral DNA synthesis by sequestration of the terminal origins of replication at internal positions within concatemers. Nevertheless, several lines of evidence indicate that the inhibition of viral DNA synthesis observed when MRN components are not inactivated in Ad5-infected cells is not the result of formation of concatemers (29, 48, 78). Although the severe defects in viral DNA synthesis observed in infected cells when MRN components cannot be degraded or sequestered are relieved in cells that lack Mre11 or Nbs1 (30, 48, 56), the mechanism by which MRN components inhibit viral DNA synthesis is not yet well understood. When not relocalized and targeted for proteasomal degradation, Mre11 has been observed to associate with viral genomes in viral replication centers (56, 57, 82). It is therefore possible that recruitment of this and other damage response proteins to viral genomes blocks recognition of viral origins of replication or subsequent reactions in viral DNA synthesis.
With few exceptions, the studies summarized in previous paragraphs were performed using HeLa or other established lines of human cells as hosts. Such cells, most of which were derived from human tumors, are, by definition, immortal and proliferate rapidly and under conditions (e.g., contact inhibition) in which normal cells do not. Furthermore, they are genetically abnormal, for example, carrying mutations that contribute to bypass of the circuits that regulate cell cycle progression and checkpoint responses, and likely to differ in genotype from one another. These properties raise the possibility that some functions of adenoviral proteins necessary for efficient replication in normal host cells may be dispensable in transformed cells. Consistent with this view, the 243R E1A protein is required for efficient viral DNA synthesis in normal human lung fibroblasts but not in HeLa cells (80). We therefore initiated investigations of the roles played by the E1B 55-kDa protein during Ad5 replication in normal human cells. One unexpected observation was that, in the absence of this protein, viral DNA synthesis was impaired in proliferating human fibroblasts (32), although it is not in HeLa and other lines of transformed human cells (5, 34, 38, 66, 100). Furthermore, McCormick and colleagues had previously reported that no differences in viral DNA synthesis were observed in quiescent small airway epithelial cells infected by wild-type virus or the E1B 55-kDa null mutant ONYX-015 (also known as dl1520) (63). The studies reported here were initiated in an attempt to resolve this apparent discrepancy.
MATERIALS AND METHODS
Cells and viruses.
293 cells and HFFs were grown as monolayer cultures in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% and 10% fetal calf serum, respectively. Primary normal human bronchial/tracheal epithelial cells (NHBECs) were obtained from BioWhittaker, Inc. and cultured using bronchial epithelial cell growth medium (BEGM), and growth conditions according to the manufacturer's recommendations. Cells were considered to be in the proliferative phase when ≤90% confluent, whereas cells in the quiescent state were obtained by prolonged incubation (≥4 days) after contact inhibition was observed. The construction of a phenotypically wild-type derivative of AdEasy (40) containing the E1A and E1B genes (AdEasy E1) was described previously (44). To introduce a green fluorescent protein (GFP) reporter gene into this background, the segment of pShuttleE1 (44) from the BamHI site downstream of the left arm (40) to the NotI site at bp 364 (40) was replaced with the corresponding fragment of pAdTrack-CMV (40). The resulting plasmid, pShuttle E1-G, contains the expression cassette comprising the human cytomegalovirus (HCMV) immediate early (IE) promoter/enhancer, the enhanced green fluorescent protein (EGFP) coding sequence, and a poly(A) addition site from pADTrack-CMV immediately upstream of and in inverse orientation to the E1A transcription unit. Recovery of this modified E1A region into the AdEasy-1 genome to create AdEasyE1-G by homologous recombination in Escherichia coli, introduction of the Hr6 frameshift mutation (deletion of bp 2347 in Ad5 DNA [100]) into this background, and isolation of viruses were as described for AdEasyE1 and the AdEasyE1Δ2347 mutant (44). Phenotypically wild-type (AdEasyE1 and AdEasyE1-G) and the E1B 55-kDa null mutants (AdEasyE1Δ2347 and AdEasyE1Δ2347-G) were propagated in monolayers of 293 cells. Viruses were titrated by plaque assay on these same cells as described previously (101).
Analysis of accumulation of viral DNA.
Proliferating or quiescent cells in 6-well dishes were infected in parallel with wild-type virus and the corresponding E1B 55-kDa null mutant (e.g., AdEasyE1 and the AdEasyE1Δ2347 mutant) and harvested after increasing periods of infection. DNA was purified from nuclei isolated as described previously (32) or by using the DNeasy tissue kit (Qiagen) according to the manufacturer's protocol. Quantitative real-time PCR was carried out using the ABI PRISM 7900HT sequence detection system and SYBR green detection of an amplicon within the major late (ML) transcription units, 90 bp long (nucleotides 7128 to 7218). The primer set was as follows: ML Fwd, 5′-ACT CTT CGC GGT TCC AGT ACT C-3′, and ML Rev, 5′-CAG GCC GTC ACC CAG TTC TAC-3′. Reaction mixtures contained 2 to 4 μl sample DNA (diluted as necessary), 300 nM each primer, and Power SYBR green master mix (Applied Biosystems). To provide an internal control, concentrations of cellular DNA were determined in parallel, using primers for an amplicon within the promoter of the human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter. The forward primer corresponded to positions 6513800 to 6513820 of human chromosome 12 reference assembly, GenBank accession number NC_000012.10 (5′-TACTAGCGGTTTTACGGGCG-3′), and the reverse primer was complementary to positions 6513942 to 6513965 (5′-TCGAACAGGAGGAGCAGAGAGCA-3′). PCR cycles were programmed as follows: two initial steps at 50°C for 2 min and 95°C for 10 min and then 40 cycles of 95°C for 15 s and 60°C for 60 s. Relative DNA concentrations were determined by the standard curve method. All measurements were performed in triplicate.
Immunoblotting.
HFFs or NHBECs at approximately 75 to 80% confluence were infected with wild-type or E1B 55-kDa null mutant viruses. Cells were harvested at the times after infection indicated, washed with phosphate-buffered saline (PBS), and extracted with 25 mM Tris HCl (pH 8.0) containing 50 mM NaCl, 0.5% (wt/vol) sodium deoxycholate, 0.5% (vol/vol) Nonidet P-40 (NP-40), and 1 mM phenylmethylsulfonyl fluoride for 30 min at 4°C. Extracts were incubated with 125 units Benzonase nuclease (Sigma) for 30 min at 37°C, and cell debris were removed by centrifugation at 10,000 × g at 4°C for 5 min. The extracts were analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and immunoblotting as described previously (33). The E2 DNA-binding protein (DBP) was detected with the monoclonal antibodies (MAb) B6 (70) and cellular β-actin, as an internal control, with a horseradish peroxidase (HRP)-labeled anti-β-actin MAb (Abcam).
Immunofluorescence.
HFFs grown on coverslips to approximately 90% confluence were mock infected, or infected with wild-type or E1B 55-kDa null mutant viruses for various periods, and the cells were processed for immunofluorescence as described previously (33). The viral E2 DBP was visualized using the B6 antibody (70) and donkey anti-mouse IgG labeled with Alexa Fluor 488 (Jackson Immuno Research Laboratories Inc.) and the E4 Orf3 protein by using the rat monoclonal antibody 6A11 (62) and Alexa Flour 568-conjugated goat anti-mouse IgG (Invitrogen). The cellular Mre11 protein was examined using a rabbit polyclonal antibody (GeneTex) and Cy5-donkey anti-rabbit IgG (Jackson Immuno Research Laboratories Inc.) secondary antibody. The coverslips were mounted on glass slides in Aqua Polymount (Polysciences Inc.), and samples were examined by confocal microscopy as described previously (33). Images were organized using Adobe Photoshop 7.0.
RESULTS
The dependence of viral DNA synthesis in normal human fibroblasts and epithelial cells on the E1B 55-kDa protein.
In initial studies of viral replication in normal human cells, we observed that accumulation of viral DNA was impaired in HFFs infected by the E1B 55-kDa null mutant Hr6 (32). In subsequent experiments, Hr6-infected normal cell nuclei have been found to contain 20- to 30-fold higher concentration of viral DNA at 2 h postinfection (p.i.) than nuclei from cells infected in parallel with an equal multiplicity of Ad5 (S. Kato, J. S. Chahal, and S. J. Flint, unpublished data). This property, and the subsequent extensive degradation of Hr6 genomes, precluded meaningful interpretation and comparison of temporal changes in the concentrations of viral DNA in Ad5- and Hr6-infected cells. To investigate further the contribution of the E1B 55-kDa protein to viral DNA synthesis in normal human cells, we therefore exploited a mutant (AdEasyE1Δ2347) that carries the Hr6 frameshift mutation in the background of a phenotypically wild-type derivative of AdEasy (40), which includes the E1A and E1B genes (AdEasyE1) (44). As reported elsewhere (44), no E1B 55-kDa protein can be detected in HeLa cells or HFFs infected by the AdEasyE1Δ2347 mutant, as expected, and this mutant reproduced the defects in viral late gene expression observed in cells infected by other E1B 55-kDa null mutants (6, 35, 38, 66, 100).
HFFs or NHBECs were infected in parallel with 30 PFU/cell or 5 PFU/cell, respectively, AdEasyE1 or the AdEasyE1Δ2347 mutant, and the concentrations of viral DNA entering nuclei by 2 h after infection measured as described in Materials and Methods. Similar concentrations of intranuclear DNA were observed in wild-type-infected and mutant-infected HFFs or NHBECs (Table 1), indicating that a mutation other than the E1B 55-kDa coding sequence frameshift mutation (deletion of bp 2347) is responsible for the poor infectivity of Hr6 virus particles. The closely similar infectivities of AdEasyE1 and the AdEasyE1Δ2347 mutant were therefore exploited to assess unambiguously the impact of failure to produce the E1B 55-kDa protein on viral DNA synthesis in normal human fibroblasts and epithelial cells.
Table 1.
Comparison of viral DNA concentrations entering AdEasyE1-infected and AdEasyΔ2347-infected cells
| Cell type | Entering DNAa |
||
|---|---|---|---|
| AdEasyE1Δ2347 | AdEasyE1 | Mutant/wild type | |
| HFF | |||
| Expt 1 | 0.236 | 0.197 | 1.20 |
| Expt 2 | 0.099 | 0.07 | 1.41 |
| NHBEC | |||
| Expt 1 | 0.345 | 0.318 | 1.08 |
| Expt 2 | 0.175 | 0.264 | 1.51 |
Viral DNA concentrations (arbitrary units) 2 h after infection of proliferating cells with 30 PFU/cell (HFFs) or 5 PFU/cell (NHBECs) relative to those of GAPDH DNA.
Proliferating HFFs were infected with the wild-type and mutant viruses, and the concentrations of intranuclear DNA were measured after increasing periods of infection by quantitative PCR, as described in Materials and Methods. The concentration of viral DNA was observed to decrease somewhat between 2 and 18 h after infection but by a similar factor in AdEasyE1-infected and AdEasyE1Δ2347 mutant-infected cells (Fig. 1A). In both cases, viral DNA concentrations increased thereafter, in agreement with results of previous analysis of the kinetics of the viral infectious cycle in HFFs (32). However, viral DNA synthesis was less efficient in AdEasyE1Δ2347 mutant-infected cells, which contained a 10-fold lower concentration of viral DNA than did wild-type-infected cells at 36 h after infection (Fig. 1A). As illustrated in Fig. 1B, the difference in the accumulation of viral DNA in AdEasyE1Δ2347 mutant-infected compared to wild-type-infected cells was lower later in infection (44 h p.i.) than at around the time of the onset of viral DNA synthesis (22 to 24 h p.i.), suggesting that this process is delayed in HFFs in the absence of the E1B 55-kDa protein. A similar impairment in viral DNA synthesis was observed when quiescent HFFs were infected by the E1B 55-kDa null mutant (Fig. 1C).
Fig 1.

Viral DNA synthesis in AdEasyE1-infected and AdEasyE1Δ2347 mutant-infected HFFs. (A and B) Proliferating HFFs at ∼70% confluence were infected with 50 PFU/cell AdEasyE1 or the AdEasyE1Δ2347 mutant. At the times indicated, viral DNA concentrations were determined by quantitative PCR. These values were corrected for concentrations of GAPDH DNA measured in parallel and are expressed in arbitrary units (A) or relative to the value measured 2 h after infection (B). The values shown represent the mean of two independent experiments, with the average deviations indicated by the error bars. (C) As in panel B, except that quiescent HFFs were infected. In all panels: WT, AdEasyE1; Δ2347, the AdEasyE1Δ2347 mutant.
In an alternative approach to assess viral DNA synthesis, the formation of viral replication centers containing the E2 DNA binding protein (DBP) was compared in HFFs infected by AdEasyE1 or the AdEasyE1Δ2347 mutant. In adenovirus-infected cell nuclei, the DBP forms two morphologically distinct structures, small dot-like foci, and larger, globular ring-like structures (87, 96). The small foci appear early in infection, and their formation is independent of viral DNA synthesis. In contrast, the ring-like structures, which are associated with newly synthesized viral DNA (60, 67, 96), do not appear when viral DNA synthesis is blocked by drugs or mutations (87, 96). We have reported previously that synthesis of the DBP is not impaired in AdEasyE1Δ2347 mutant-infected HFFs (44). Replication centers were therefore examined by immunofluorescence as described in Materials and Methods. Striking differences were observed: the majority of AdEasyE1-infected cells contained enlarged rings of DBP, which are formed upon initiation of viral DNA synthesis (87, 96), whereas DBP was observed either in small foci or as diffuse nuclear staining in most AdEasyE1Δ2347 mutant-infected cells (Fig. 2A). Quantification of the different patterns of intranuclear localization of DBP indicated that the enlarged ring-like structures characteristic of replicating viral DNA developed in over 90% of AdEasyE1-infected cells but in less than 30% of those infected by the mutant (Fig. 2B). These observations indicate that the E1B 55-kDa protein is required for efficient genome replication in Ad5-infected HFFs.
Fig 2.

Formation of viral replication centers in AdEasyE1- and AdEasyE1Δ2347 mutant-infected HFFs. (A) The E2 DBP was examined by immunofluorescence (as described in Materials and Methods) 25 h after infection of HFFs with 50 PFU/cell AdEasy E1 (WT), the AdEasyE1Δ2347 mutant (Δ2347), or mock-infected cells (M). Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) (blue). (B) The appearance of DBP only as diffuse nuclear staining (diffuse), in small dot-like foci with or without diffuse DBP (small foci), or in enlarged ring-like structures (large rings) was counted in ≥100 cells infected by AdEasyE1 or the AdEasyE1Δ2347 mutant. The percentage of the total number of infected cells containing each form of DBP are shown.
We next compared the temporal changes in viral DNA concentration in normal human bronchial/tracheal cells (NHBECs) infected by AdEasyE1 or the AdEasyE1Δ2347 mutant. As subgroup C adenoviruses, such as Ad5, are associated with upper respiratory tract infections (reviewed in reference 102), these cells seemed likely to provide a closer facsimile of natural host cells than either HFFs or SAECs used in other studies (63), which were derived from the lower respiratory tract. Proliferating NHBECs, which are significantly more infectible than HFFs (32), were infected with 5 PFU/cell, and viral DNA concentrations were measured at various times thereafter. In these cells, significantly higher concentrations of viral DNA were detected than in HFFs (compare Fig. 3A and 1A), a property also observed in SAECs (data not shown). In contrast to the results obtained in HFFs, the kinetics and efficiency of viral DNA synthesis were essentially indistinguishable in AdEasyE1-infected and AdEasyE1Δ2347 mutant-infected cells (Fig. 3A), nor was any significant difference detected when quiescent NHBECs were infected (Fig. 3B).
Fig 3.

Viral DNA synthesis in AdEasyE1-infected and AdEasyE1Δ2347 mutant-infected NHBECs. Proliferating NHBECs at ∼60% confluence (A) or quiescent NHBECs (B) were infected with 5 PFU/cell AdEasy E1 or the AdEasyE1Δ2347 mutant, and viral DNA concentrations were measured at the times indicated (see Materials and Methods). The values shown represent the mean of two independent experiments, and the error bars indicate average deviations. In all panels: WT, AdEasyE1; Δ2347, the AdEasyE1Δ2347 mutant.
Acceleration of early phase progression in HFFs restores efficient genome replication in the absence of the E1B 55-kDa protein.
Comparison of genome replication of AdEasyE1 in HFFs and NHBEs demonstrated not only more efficient viral DNA synthesis in the latter cell type but also differences in the kinetics of this process. In wild-type-infected NHBECs, viral DNA synthesis was well under way by 16 h p.i., when the relative viral DNA concentration had increased some 200-fold (Fig. 3A). In contrast, no genome replication was detected at this time after AdEasyE1 infection of HFFs, and by 24 h p.i., the quantity of intranuclear viral DNA had increased by a factor of only 4 (Fig. 1A). The delayed onset of viral DNA synthesis in HFFs is consistent with our previous observations that in these cells viral immediate early (IE) E1A proteins cannot be detected until 14 to 16 h after infection (32). Indeed, the E2 DBP does not accumulate to a significant concentration until 24 h after infection (Fig. 4A). In contrast, this viral replication protein was readily detected by 12 h after infection of NHBECs with AdEasy E1 (Fig. 4B), consistent with the earlier onset of viral DNA synthesis in these cells (Fig. 3A). Because of this difference in the rate of progression through the early phase, it was not clear whether the E1B 55-kDa protein was required to promote viral DNA synthesis specifically in fibroblasts or whether some inhibitory mechanism, blocked by this viral protein, became activated during the extended period before initiation of genome replication in HFFs. To distinguish between these possibilities, we exploited derivatives of AdEasyE1 and the AdEasyE1Δ2347 mutant that carry the coding sequence for EGFP under the control of the HCMV IE promoter/enhancer upstream of and in inverse orientation to the E1A transcription unit. As illustrated in Fig. 4C, synthesis of the E2 DBP was evident by 9 h after infection of HFFs by the wild-type derivative, AdEasyE1-G, and had accumulated to a much higher concentration by 12 h p.i. These data indicate that insertion of the expression cassette led to considerably accelerated progression through the early phase of infection in HFFs, presumably as a result of activation of E1A transcription by the HCMV IE enhancer. We therefore compared viral DNA synthesis in proliferating HFFs infected with AdEasyE1-G or the AdEasyE1Δ2347-G mutant, by the methods described previously. In contrast to the results shown in Fig. 1, no differences were observed in the kinetics or efficiency of viral DNA synthesis in mutant-infected compared to wild-type-infected cells, and in both cases, an increase in intranuclear viral DNA concentration, albeit modest, was detected by 16 h p.i. (Fig. 5). Although accelerated synthesis of viral early proteins eliminated the dependence of viral DNA synthesis in HFFs on the E1B protein, the relative quantity of viral DNA made by 36 h p.i. remained lower than observed in NHBECs (compare Fig. 3A and 5), suggesting that cell type-specific differences govern the degree of amplification of the viral genome.
Fig 4.
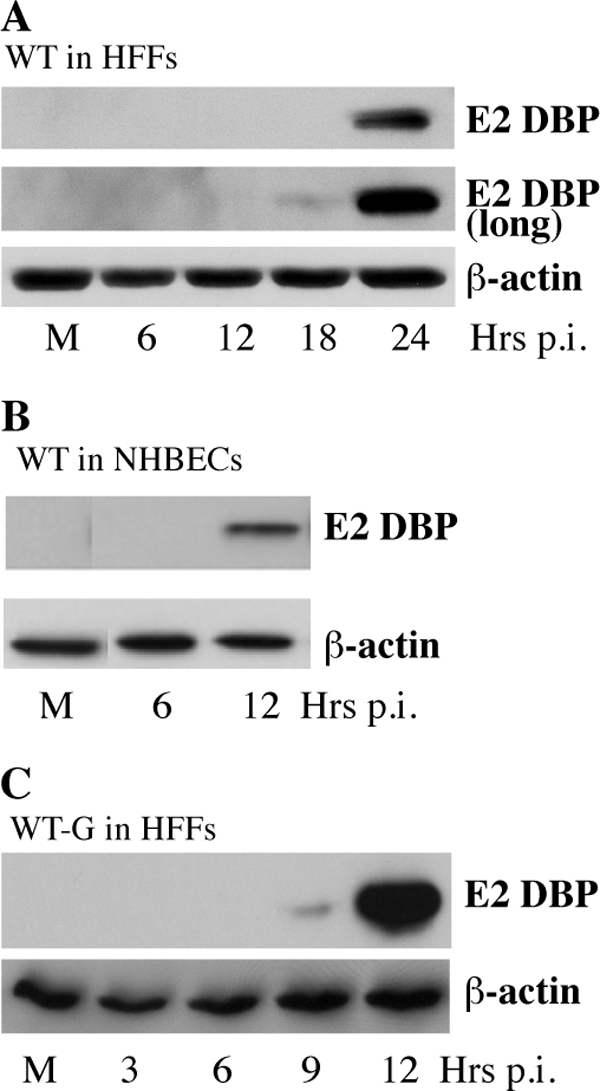
Synthesis of early proteins in normal human cells infected by AdEasyE1 or AdEasy E1-G. (A) HFFs at ∼70% confluence were infected with 50 PFU/cell AdEasyE1 for the periods indicated or mock infected (M), and the concentrations of DBP and β-actin were examined by immunoblotting. (B) As in panel A, except that NHBECs at ∼70% confluence were infected with 5 PFU/cell Ad5, or mock-infected (M). (C). As in panel A, except that HFFs at ∼70% confluence were infected with 50 PFU/cell AdEasyE1-G, which contains the HCMV IE promoter/enhancer immediately upstream of the E1A transcription unit. Note the different time scales in the different panels.
Fig 5.
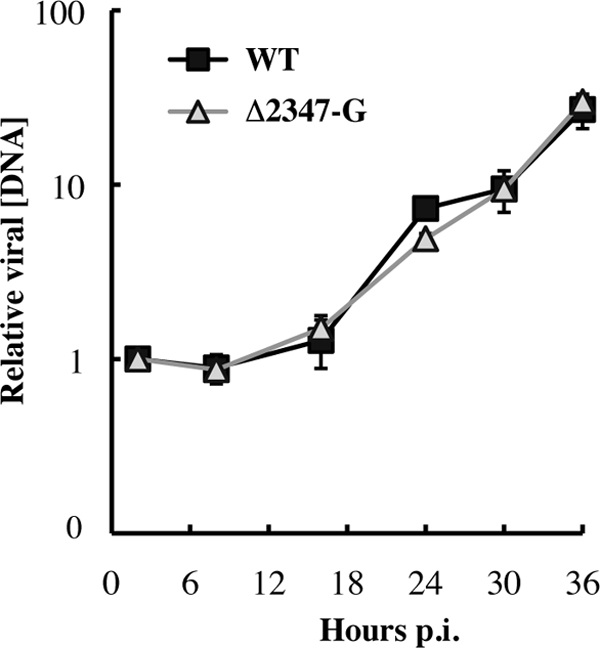
Viral DNA synthesis in HFFs infected by AdEasyE1-G. HFFs at 70% confluence were infected with 50 PFU/cell AdEasyE1-G (WT-G) or the AdEasyE1Δ2347-G mutant (Δ2347-G), and the concentrations of intranuclear viral DNA relative to the input value (2 h p.i.) were determined by quantitative PCR as described in Fig. 1. Values shown represent the average of two independent experiments, and error bars indicate average deviations.
Localization of Mre11 in infected HFFs.
The E1B 55-kDa protein neither participates directly in viral DNA synthesis (10) nor facilitates production of viral replication proteins during the early phase of infection in transformed or normal human cells (4, 35, 44, 49, 73). The results described above therefore suggest that, when synthesized in timely fashion, this early protein protects against inhibition of viral DNA synthesis by a cellular defense mechanism, such as the double-stranded DNA break repair response. As discussed in the introduction, the activity of the Ad E3 Ub ligase targets proteins of the cellular MRN complex for proteasomal degradation, and viral DNA synthesis is inhibited in established lines of human cells when both assembly of this infected cell-specific enzyme and synthesis of the viral E4 Orf3 protein are prevented by mutation. Consistent with these previous studies, the steady-state concentration Mre11 was observed to decrease more slowly in HFFs infected by the AdEasyE1Δ2347 mutant than in AdEasyE1-infected cells (44). We therefore wish to compare the relocalization of Mre11 by E4 Orf3 in normal human cells in the presence and absence of the E1B 55-kDa protein. Prolifera-ting HFFs were infected in parallel with AdEasyE1 or the AdEasyE1Δ2347 mutant for 24 h, and viral replication centers (E2 DBP), the E4 Orf3 protein, and Mre11 were visualized by immunofluorescence using mouse, rat, and rabbit primary antibodies, respectively, as described in Materials and Methods.
In uninfected HFFs, Mre11 was concentrated in nuclei, where it was excluded from nucleoli (Fig. 6a and b). Upon infection, the nuclear Mre11 signal was reduced (compare Fig. 6g and l with b), as expected (see the introduction). In AdEasyE1-infected cells, the cellular protein was observed in discrete fleck-like structures not present in uninfected cells (Fig. 6g, arrows). In these structures, Mre11 was localized with the E4 Orf3 protein and was not associated with viral replication centers (Fig. 6g, h, i, and j), as initially observed in Ad5-infected established human cell lines (30, 82). These same changes in the properties of Mre11, reduced intranuclear concentration, reorganization to structures that contained the E4 Orf3 protein, and lack of association with viral replication centers, were observed in HFFs infected by the AdEasyE1Δ2347 mutant (Fig. 6l to o). Furthermore, examination of ∼100 cells infected by the mutant or its wild-type parent indicated that the number of infected cells in which Mre11 was localized with the E4 Orf3 proteins was not reduced in the absence of the E1B 55-kDa protein but rather increased somewhat from 51.2% to 73.6%. This difference is probably a consequence of the delayed degradation of Mre11 when the Ad E3 Ub ligase is not present in infected cells.
Fig 6.

Localization of Mre11, E2 DBP, and the E4 Orf3 proteins in infected HFFs. HFFs at ∼70% confluence were infected with 50 PFU/cell AdEasyE1 (WT), the AdEasyE1Δ2347 mutant (Δ2347), or mock-infected cells (M) for 24 h. They were then processed for immunofluorescence, and Mre11, E2 DBP, and E4 Orf3 were visualized as described in Materials and Methods. The E4 Orf3 protein signal is false-colored in blue. Nuclei stained with DAPI are shown false-colored in cyan. The merged images do not include the nuclear stain.
DISCUSSION
The apparent discrepancies in the dependence of viral DNA synthesis in normal human cells on the E1B 55-kDa protein reported previously (32, 63) have been investigated further by exploiting a mutant (AdEasyE1Δ2347) that carries the Hr6 E1B frameshift mutation in the AdEasyE1 genome (44): this mutant reproduces such phenotypes of Hr6 (and other E1B 55-kDa null mutants) as impaired expression of viral late genes (44) but does not exhibit the low infectivity of Hr6 (Table 1), which will be described elsewhere (Kato, Chahal, and Flint, unpublished). Comparison of the accumulation of viral DNA in cells infected by this mutant and its wild-type parent indicated that viral genome replication is impaired in proliferating and quiescent normal human fibroblasts in the absence of the E1B 55-kDa protein (Fig. 1), consistent with our previous observation that viral DNA synthesis was reduced in HFFs infected not only by Hr6 but also by a mutant (H224) that carries a 4-amino-acid insertion mutation in the E1B 55-kDa protein coding sequence (32). However, no such defect was detected in NHBECs (Fig. 3). It has been reported previously that viral DNA synthesis is not defective in small airway epithelial cells infected by the E1B 55-kDa null mutant ONYX-015 (dl1520) (63). Nevertheless, the difference in the dependence of viral DNA synthesis on the E1B protein observed between normal HFFs and epithelial cells does not appear to represent yet another example of the well-documented variation in the efficiency of replication of E1B 55-kDa null mutants with host cell type (11, 35, 38, 75, 91): when introduction of the HMCV IE promoter/enhancer into the viral genome accelerated expression of early genes and progression through the early phase in infected HFFs (Fig. 4), no defect in viral DNA synthesis was observed in the absence of the E1B 55-kDa protein (Fig. 5). We therefore propose that synthesis of this protein within a prescribed period after initiation of the infectious cycle is necessary to allow maximally efficient viral DNA synthesis, most probably by countering a cellular defense mechanism.
When the Ad E3 Ub ligase cannot assemble in transformed human cells, relocalization of the cellular Mre11, Rad50, and Nbs1 protein by the species C adenovirus E4 Orf3 protein (83) is necessary to block inhibition of viral DNA synthesis by the MRN complex (30). If the E4 Orf3 protein is also absent, Mre11 and other MRN proteins become localized with E2 DBP-containing viral replication centers (56, 57, 82), where Mre11 binds to viral DNA (57). Although degradation of Mre11 is delayed in HFFs infected by the AdEasyE1Δ2347 mutant compared to its phenotypically wild-type parent (44), this cellular protein was observed to be sequestered with E4 Orf3 in both wild-type-infected and mutant-infected cells (Fig. 6). As such relocalization of Mre11 is sufficient to prevent inhibition of viral DNA by the MRN complex (30), it is highly unlikely that this complex is responsible for the impaired genome replication observed in AdEasyE1Δ2347 mutant-infected HFFs.
The E1B 55-kDa protein-containing Ad E3 Ub ligase targets a number of other cellular proteins for proteasomal degradation (see the introduction) and has been reported more recently to function as a Sumo1 E3 ligase (59, 64). It is therefore possible that timely synthesis of the enzymes that contain this E1B protein in normal human cells is required to induce removal, or inhibition by modification, of an as yet unidentified cellular protein that inhibits genome replication directly or indirectly. However, in HFFs the E1B 55-kDa protein also represses expression of a subset of cellular genes highly enriched for those associated with innate antiviral defenses and immune responses (58). The former class includes a substantial number of interferon (IFN)-inducible genes, as well as several encoding components of signaling pathways that are activated in response to infection, such as Myd88 and Irf7. One mechanism by which adenovirus infection is recognized to initiate innate immune responses is by the pathogen recognition receptor Tlr 9 (2, 8, 9, 18, 104, 108), which signals via the adaptor Myd88, Irf7, and other transcriptional activators to induce production of type I IFN and other proinflammatory cytokines (see references 21, 92, and 97). Repression of expression of these genes by the E1B 55-kDa protein may therefore contribute to antagonism of antiviral defenses induced by type I IFN in Ad5-infected cells, as do the small viral RNA VA-RNAI (45, 46), the E1A proteins (1, 37, 43, 71), and the E4 Orf3 protein (93, 94). Such a function of the E1B protein could account for impaired genome replication when the early phase of infection is prolonged in HFFs infected by E1B 55-kDa null mutants, as the longer period prior to induction of viral DNA synthesis might permit production of quantities of type 1 IFN sufficient to induce an effective antiviral state by autocrine and paracrine mechanisms.
ACKNOWLEDGMENTS
We thank Thomas Dobner for the generous gift of rat anti-E4 Orf3 monoclonal antibody, 6A11, Wenying Huang for expert technical assistance, Mohammed Selman for analysis of viral early protein synthesis in NHBECs, and Ellen Brindle-Clark for assistance with preparation of the manuscript.
This work was supported by Public Health Service grant RO1AI058172 from the National Institute of Allergy and Infectious Disease. Jasdave Chahal was partially supported by a postgraduate scholarship from the National Science and Engineering Research Council of Canada.
Footnotes
Published ahead of print 25 January 2012
REFERENCES
- 1. Ackrill AM, et al. 1991. Inhibition of the cellular response to interferons by products of the adenovirus type 5 E1A oncogene. Nucleic Acids Res. 19:4387–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Appledorn DM, et al. 2008. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J. Immunol. 181:2134–2144 [DOI] [PubMed] [Google Scholar]
- 3. Araujo FD, Stracker TH, Carson CT, Lee DV, Weitzman MD. 2005. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 79:11382–11391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Babiss LE, Ginsberg HS. 1984. Adenovirus type 5 early region 1b gene product is required for efficient shutoff of host protein synthesis. J. Virol. 50:202–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Babiss LE, Ginsberg HS, Fisher PB. 1983. Cold-sensitive expression of transformation by a host-range mutant of type 5 adenovirus. Proc. Natl. Acad. Sci. U. S. A. 80:1352–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Babiss LE, Young CSH, Fisher PP, Ginsberg HS. 1983. Expression of adenovirus E1A and E1B gene products and the Escherichia coli XPRT gene in KB cells. J. Virol. 46:454–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baker A, Rohleder KJ, Hanakahi LA, Ketner G. 2007. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 81:7034–7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barlan AU, Griffin TM, McGuire KA, Wiethoff CM. 2011. Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 85:146–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Basner-Tschakarjan E, et al. 2006. Adenovirus efficiently transduces plasmacytoid dendritic cells resulting in TLR9-dependent maturation and IFN-alpha production. J. Gene Med. 8:1300–1306 [DOI] [PubMed] [Google Scholar]
- 10. Berk AJ. 2007. Adenoviridae: the viruses and their replication, p 2355–2394 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 11. Bischoff JR, et al. 1996. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274:373–376 [DOI] [PubMed] [Google Scholar]
- 12. Blackford AN, Grand RJ. 2009. Adenovirus E1B 55-kilodalton protein: multiple roles in viral infection and cell transformation. J. Virol. 83:4000–4012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blair Zajdel ME, Blair GE. 1988. The intracellular distribution of the transformation-associated protein p53 in adenovirus-transformed rodent cells. Oncogene 2:579–584 [PubMed] [Google Scholar]
- 14. Blanchette P, et al. 2008. Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires E4orf6 ubiquitin ligase activity. J. Virol. 82:2642–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boyer JL, Ketner G. 2000. Genetic analysis of a potential zinc-binding domain of the adenovirus E4 34k protein. J. Biol. Chem. 275:14969–14978 [DOI] [PubMed] [Google Scholar]
- 16. Cardoso FM, Kato SE, Huang W, Flint SJ, Gonzalez RA. 2008. An early function of the adenoviral E1B 55 kDa protein is required for the nuclear relocalization of the cellular p53 protein in adenovirus-infected normal human cells. Virology 378:339–346 [DOI] [PubMed] [Google Scholar]
- 17. Carvalho T, et al. 1995. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J. Cell Biol. 131:45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cerullo V, et al. 2007. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol. Ther. 15:378–385 [DOI] [PubMed] [Google Scholar]
- 19. Cheng CY, Blanchette P, Branton PE. 2007. The adenovirus E4orf6 E3 ubiquitin ligase complex assembles in a novel fashion. Virology 364:36–44 [DOI] [PubMed] [Google Scholar]
- 20. Chiou SK, Tseng CC, Rao L, White E. 1994. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J. Virol. 68:6553–6566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Colonna M. 2007. TLR pathways and IFN-regulatory factors: to each its own. Eur. J. Immunol. 37:306–309 [DOI] [PubMed] [Google Scholar]
- 22. Dallaire F, Blanchette P, Groitl P, Dobner T, Branton PE. 2009. Identification of integrin alpha3 as a new substrate of the adenovirus E4orf6/E1B 55-kilodalton E3 ubiquitin ligase complex. J. Virol. 83:5329–5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. D'Amours D, Jackson SP. 2002. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 3:317–327 [DOI] [PubMed] [Google Scholar]
- 24. Debbas M, White E. 1993. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 7:546–554 [DOI] [PubMed] [Google Scholar]
- 25. Dosch T, et al. 2001. The adenovirus type 5 E1B-55k oncoprotein actively shuttles in virus-infected cells, whereas transport of E4Orf6 is mediated by a CRM1-independent mechanism. J. Virol. 75:5677–5683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doucas V, et al. 1996. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 10:196–207 [DOI] [PubMed] [Google Scholar]
- 27. Endter C, Hartl B, Spruss T, Hauber J, Dobner T. 2005. Blockage of CRM1-dependent nuclear export of the adenovirus type 5 early region 1B 55-kDa protein augments oncogenic transformation of primary rat cells. Oncogene 24:55–64 [DOI] [PubMed] [Google Scholar]
- 28. Endter C, Kzhyshkowska J, Stauber R, Dobner T. 2001. SUMO-1 modification required for transformation by adenovirus type 5 early region 1B 55-kDa oncoprotein. Proc. Natl. Acad. Sci. U. S. A. 98:11312–11317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Evans JD, Hearing P. 2003. Distinct roles of the Adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 77:5295–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evans JD, Hearing P. 2005. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 79:6207–6215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Flint SJ, Gonzalez RA. 2003. Regulation of mRNA production by the adenoviral E1B 55kDa and E4 Orf6 proteins. Curr. Top. Microbiol. Immunol. 272:287–330 [DOI] [PubMed] [Google Scholar]
- 32. Gonzalez R, Huang W, Finnen R, Bragg C, Flint SJ. 2006. Adenovirus E1B 55-kilodalton protein is required for both regulation of mRNA export and efficient entry into the late phase of infection in normal human fibroblasts. J. Virol. 80:964–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gonzalez RA, Flint SJ. 2002. Effects of mutations in the adenoviral E1B 55 kDa protein coding sequence on viral late mRNA metabolism. J. Virol. 76:4507–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goodrum FD, Ornelles DA. 1997. The early region 1B 55-kilodalton oncoprotein of adenovirus relieves growth restrictions imposed on viral replication by the cell cycle. J. Virol. 71:548–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goodrum FD, Ornelles DA. 1999. Roles for the E4 orf6, orf3, and E1B 55-kilodalton proteins in cell cycle-independent adenovirus replication. J. Virol. 73:7474–7488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grand RJ, Grant ML, Gallimore PH. 1994. Enhanced expression of p53 in human cells infected with mutant adenoviruses. Virology 203:229–240 [DOI] [PubMed] [Google Scholar]
- 37. Gutch MJ, Reich NC. 1991. Repression of the interferon signal transduction pathway by the adenovirus E1A oncogene. Proc. Natl. Acad. Sci. U. S. A. 88:7913–7917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harada JN, Berk AJ. 1999. p53-Independent and -dependent requirements for E1B-55K in adenovirus type 5 replication. J. Virol. 73:5333–5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harada JN, Shevchenko A, Pallas DC, Berk AJ. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 76:9194–9206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He TC, et al. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. U. S. A. 95:2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hobom U, Dobbelstein M. 2004. E1B-55-kilodalton protein is not required to block p53-induced transcription during adenovirus infection. J. Virol. 78:7685–7697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hutton FG, Turnell AS, Gallimore PH, Grand RJ. 2000. Consequences of disruption of the interaction between p53 and the larger adenovirus early region 1B protein in adenovirus E1 transformed human cells. Oncogene 19:452–462 [DOI] [PubMed] [Google Scholar]
- 43. Kalvakolanu DV, Bandyopadhyay SK, Harter ML, Sen GC. 1991. Inhibition of interferon-inducible gene expression by adenovirus E1A proteins: block in transcriptional complex formation. Proc. Natl. Acad. Sci. U. S. A. 88:7459–7463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kato SE, Huang W, Flint SJ. 2011. Role of the RNA recognition motif of the E1B 55kDa protein in the adenovirus type 5 infectious cycle. Virology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kitajewski J, et al. 1986. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45:195–200 [DOI] [PubMed] [Google Scholar]
- 46. Kitajewski J, Schneider RJ, Safer B, Shenk T. 1986. An adenovirus mutant unable to express VAI RNA displays different growth responses and sensitivity to interferon in various host cell lines. Mol. Cell. Biol. 6:4493–4498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krätzer F, et al. 2000. The adenovirus type 5 E1B-55K oncoprotein is a highly active shuttle protein and shuttling is independent of E4orf6, p53 and Mdm2. Oncogene 19:850–857 [DOI] [PubMed] [Google Scholar]
- 48. Lakdawala SS, et al. 2008. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J. Virol. 82:8362–8372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lassam NJ, Bayley ST, Graham FL. 1979. Tumor antigens of human Ad5 in transformed cells and in cells infected with transformation defective host range mutants. Cell 18:781–791 [DOI] [PubMed] [Google Scholar]
- 50. Lavin MF. 2007. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 26:7749–7758 [DOI] [PubMed] [Google Scholar]
- 51. Leppard KN, Everett RD. 1999. The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J. Gen. Virol. 80:997–1008 [DOI] [PubMed] [Google Scholar]
- 52. Liu Y, Shevchenko A, Berk AJ. 2005. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 79:14004–14016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Luo K, et al. 2007. Adenovirus E4orf6 assembles with Cullin5-ElonginB-ElonginC E3 ubiquitin ligase through an HIV/SIV Vif-like BC-box to regulate p53. FASEB J. 21:1742–1750 [DOI] [PubMed] [Google Scholar]
- 54. Martin ME, Berk AJ. 1998. Adenovirus E1B 55K represses p53 activation in vitro. J. Virol. 72:3146–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Martin ME, Berk AJ. 1999. Corepressor required for adenovirus E1B 55,000-molecular-weight protein repression of basal transcription. Mol. Cell. Biol. 19:3403–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mathew SS, Bridge E. 2007. The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology 365:346–355 [DOI] [PubMed] [Google Scholar]
- 57. Mathew SS, Bridge E. 2008. Nbs1-dependent binding of Mre11 to adenovirus E4 mutant viral DNA is important for inhibiting DNA replication. Virology 374:11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Miller DL, Rickards B, Mashiba M, Huang W, Flint SJ. 2009. The adenoviral E1B 55-kilodalton protein controls expression of immune response genes but not p53-dependent transcription. J. Virol. 83:3591–3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Muller S, Dobner T. 2008. The adenovirus E1B-55K oncoprotein induces SUMO modification of p53. Cell Cycle 7:754–758 [DOI] [PubMed] [Google Scholar]
- 60. Murti KG, Davis DS, Kitchingman GR. 1990. Localization of adenovirus-encoded DNA replication proteins in the nucleus by immunogold electron microscopy. J. Gen. Virol. 71:2847–2857 [DOI] [PubMed] [Google Scholar]
- 61. Nevels M, Rubenwolf S, Spruss T, Wolf H, Dobner T. 2000. Two distinct activities contribute to the oncogenic potential of the adenovirus type 5 E4orf6 protein. J. Virol. 74:5168–5181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nevels M, et al. 1999. Transforming potential of the adenovirus type 5 E4orf3 protein. J. Virol. 73:1591–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. O'Shea CC, et al. 2004. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 6:611–623 [DOI] [PubMed] [Google Scholar]
- 64. Pennella MA, Liu Y, Woo JL, Kim CA, Berk AJ. 2010. Adenovirus E1B 55-kilodalton protein is a p53-SUMO1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. J. Virol. 84:12210–12225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pilder S, Logan J, Shenk TE. 1984. Deletion of the gene encoding the adenovirus 5 early region 1b 21,000-molecular weight polypeptide leads to degradation of viral and host cell DNA. J. Virol. 52:664–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pilder S, Moore M, Logan J, Shenk T. 1986. The adenovirus E1B-55kd transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol. Cell. Biol. 6:470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Puvion-Dutilleul F, Pedron J, Cajean-Feroldi C. 1984. Identification of intranuclear structures containing the 72K DNA-binding protein of human adenovirus type 5. Eur. J. Cell Biol. 34:313–322 [PubMed] [Google Scholar]
- 68. Querido E, et al. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 15:3104–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Querido E, et al. 1997. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J. Virol. 71:3788–3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Reich NC, Sarnow P, Duprey E, Levine AJ. 1983. Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128:480–484 [DOI] [PubMed] [Google Scholar]
- 71. Reichel R, Kovesdi I, Nevins JR. 1988. Activation of a pre-existing cellular factor as a basis for adenovirus E1A-mediated transcription control. Proc. Natl. Acad. Sci. U. S. A. 85:387–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Riches LC, Lynch AM, Gooderham NJ. 2008. Early events in the mammalian response to DNA double-strand breaks. Mutagenesis 23:331–339 [DOI] [PubMed] [Google Scholar]
- 73. Ross SR, Levine AJ, Galos RS, Williams J, Shenk T. 1980. Early viral proteins in HeLa cells infected with adenovirus type 5 host range mutants. Virology 103:475–492 [DOI] [PubMed] [Google Scholar]
- 74. Roth J, et al. 1998. Inactivation of p53 but not p73 by adenovirus type 5 E1B 55-kilodalton and E4 34-kilodalton oncoproteins. J. Virol. 72:8510–8516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, Hzur Hausen. 1998. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J. Virol. 72:9470–9478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sarnow P, Ho YS, Williams J, Levine AJ. 1982. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 28:387–394 [DOI] [PubMed] [Google Scholar]
- 77. Shen Y, Kitzes G, Nye JA, Fattaey A, Hermiston T. 2001. Analyses of single-amino-acid substitution mutants of adenovirus type 5 E1B-55K protein. J. Virol. 75:4297–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shepard RN, Ornelles DA. 2004. Diverse roles for E4orf3 at late times of infection revealed in an E1B 55-kilodalton protein mutant background. J. Virol. 78:9924–9935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Soria C, Estermann FE, Espantman KC, O'Shea CC. 2010. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 466:1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Spindler KR, Eng CY, Berk AJ. 1985. An adenovirus early region 1A protein is required for maximal viral DNA replication in growth-arrested human cells. J. Virol. 53:742–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Steegenga WT, Riteco N, Jochemsen AG, Fallaux FJ, Bos JL. 1998. The large E1B protein together with the E4orf6 protein target p53 for active degradation in adenovirus infected cells. Oncogene 16:349–357 [DOI] [PubMed] [Google Scholar]
- 82. Stracker TH, Carson CT, Weitzman MD. 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418:348–352 [DOI] [PubMed] [Google Scholar]
- 83. Stracker TH, et al. 2005. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J. Virol. 79:6664–6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Stracker TH, Petrini JH. 2011. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12:90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Subramanian T, Boyd JM, Chinnadurai G. 1995. Functional substitution identifies a cell survival promoting domain common to adenovirus E1B 19 kDa and Bcl-2 proteins. Oncogene 11:2403–2409 [PubMed] [Google Scholar]
- 86. Subramanian T, Kuppuswamy M, Mak S, Chinnadurai G. 1984. Adenovirus cyt+ locus, which controls cell transformation and tumorigenicity, is an allele of lp+ locus, which codes for a 19-kilodalton tumor antigen. J. Virol. 52:336–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sugawara K, Gilead Z, Wold WSM, Green M. 1977. Immunofluorescence study of the adenovirus type 2 single-stranded DNA binding protein in infected and transformed cells. J. Virol. 22:527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tarodi B, Subramanian T, Chinnadurai G. 1993. Functional similarity between adenovirus e1b 19k gene and bcl2 oncogene-mutant complementation and suppression of cell-death induced by DNA-damaging agents. Int. J. Oncol. 3:467–472 [DOI] [PubMed] [Google Scholar]
- 89. Teodoro JG, Branton PE. 1997. Regulation of p53-dependent apoptosis, transcriptional repression, and cell transformation by phosphorylation of the 55-kilodalton E1B protein of human adenovirus type 5. J. Virol. 71:3620–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Teodoro JG, et al. 1994. Phosphorylation at the carboxy terminus of the 55-kilodalton adenovirus type 5 E1B protein regulates transforming activity. J. Virol. 68:776–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Turnell AS, Grand RJ, Gallimore PH. 1999. The replicative capacities of large E1B-null group A and group C adenoviruses are independent of host cell p53 status. J. Virol. 73:2074–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Uematsu S, Akira S. 2007. Toll-like receptors and Type I interferons. J. Biol. Chem. 282:15319–15323 [DOI] [PubMed] [Google Scholar]
- 93. Ullman AJ, Hearing P. 2008. Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J. Virol. 82:7325–7335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ullman AJ, Reich NC, Hearing P. 2007. Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J. Virol. 81:4744–4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. van den Bosch M, Bree RT, Lowndes NF. 2003. The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep. 4:844–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Voelkerding K, Klessig DF. 1986. Identification of two nuclear subclasses of the adenovirus type 5-encoded DNA-binding protein. J. Virol. 60:353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Watters TM, Kenny EF, O'Neill LA. 2007. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol. Cell Biol. 85:411–419 [DOI] [PubMed] [Google Scholar]
- 98. Weiden MD, Ginsberg HS. 1994. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. U. S. A. 91:153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. White E, Grodzicker T, Stillman BW. 1984. Mutations in the gene encoding the adenovirus early region 1B 19,000-molecular-weight tumor antigen cause the degradation of chromosomal DNA. J. Virol. 82:410–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Williams J, et al. 1986. The adenovirus E1B 495R protein plays a role in regulating the transport and stability of the viral late messages. Cancer Cells 4:275–284 [Google Scholar]
- 101. Williams JF. 1970. Enhancement of adenovirus plaque formation on HeLa cells by magnesium chloride. J. Gen. Virol. 9:251–253 [DOI] [PubMed] [Google Scholar]
- 102. Wold WSM, Horwitz MS. 2007. Adenoviruses, p 2395–2436 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 103. Woo JL, Berk AJ. 2007. Adenovirus ubiquitin-protein ligase stimulates viral late mRNA nuclear export. J. Virol. 81:575–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yamaguchi T, et al. 2007. Role of MyD88 and TLR9 in the innate immune response elicited by serotype 5 adenoviral vectors. Hum. Gene Ther. 18:753–762 [DOI] [PubMed] [Google Scholar]
- 105. Yew PR, Berk AJ. 1992. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 357:82–85 [DOI] [PubMed] [Google Scholar]
- 106. Yew PR, Liu X, Berk AJ. 1994. Adenovirus E1B oncoprotein tethers a transcriptional repression domain to p53. Genes Dev. 8:190–202 [DOI] [PubMed] [Google Scholar]
- 107. Zantema A, et al. 1985. Localization of the E1B proteins of adenovirus 5 in transformed cells, as revealed by interaction with monoclonal antibodies. Virology 142:44–58 [DOI] [PubMed] [Google Scholar]
- 108. Zhu J, Huang X, Yang Y. 2007. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J. Virol. 81:3170–3180 [DOI] [PMC free article] [PubMed] [Google Scholar]