

Abstract
The activity of the tumor suppressor p53 is tightly controlled by its main negative regulator, Mdm2, which inhibits p53's transcriptional activity and targets it for degradation via the proteasome pathway. The closely related Mdm2 homolog, MdmX, is also considered to be a general inhibitor of transactivation by p53, through binding to the p53 activation domain. We show here that, unexpectedly, upon DNA damage and ribosomal stress, MdmX plays a positive role in p53-mediated activation of the Mdm2 gene, but not of numerous other p53 target genes including p21. Downregulation of MdmX results in lower levels of mature and nascent Mdm2 transcripts following cellular stress. This correlates with a longer p53 half-life following DNA damage. In vitro, Mdm2 inhibits the binding of p53 to DNA to a much greater extent than does MdmX, although MdmX does not stimulate p53 interaction with Mdm2 promoter DNA. Strikingly, however, MdmX is required for optimal p53 binding to the Mdm2 promoter in vivo. Thus, we have described a new mechanism by which MdmX can suppress p53, which is through transcriptional activation of p53's principal negative regulator, Mdm2.
INTRODUCTION
The tumor suppressor p53 is a transcription factor that responds to cellular stress conditions such as DNA damage, ribosomal stress, and oncogene activation by regulating the expression of key target genes. This leads to various cellular outcomes, including cell cycle arrest, apoptosis, and senescence (67). As important as the activation of these processes upon damage is for tumor suppression, it is crucial that their induction is inhibited in normal unstressed growth conditions.
p53 is mutated in over 50% of cancers, and other aspects of its network in the remaining group are deregulated frequently via amplification and overexpression of Mdm2 and MdmX (35, 40, 45). p53 is normally inhibited by its principal negative regulator, Mdm2, through multiple mechanisms. Mdm2 prevents recruitment of transcriptional coactivators to target promoters by binding to the p53 activation domain (46). Furthermore, Mdm2 is an E3 ubiquitin ligase, and it promotes p53 polyubiquitination and degradation through the ubiquitin-proteasome pathway (17, 18). MdmX, a structural homolog of Mdm2, also functions as a negative regulator of p53-mediated transcription (60, 61). However, unlike Mdm2, the MdmX RING domain does not possess detectable E3 ligase activity (61). The importance of both Mdm2 and MdmX in negative regulation of p53 was demonstrated in mice, where homozygous deletion of either Mdm gene leads to embryonic lethality unless p53 is codeleted, suggesting nonredundant functions of the two proteins in p53 inhibition during development (23, 37, 41, 49).
Following DNA damage, p53, MdmX, and Mdm2 undergo multiple posttranslational modifications leading to Mdm2-mediated ubiquitination and degradation of MdmX in nuclei (8, 22, 25, 43, 47, 50, 70) as well as stabilization and activation of p53. Additionally, these modifications contribute to p53 target gene selectivity (reviewed in references 26 and 68). How p53 chooses its target genes is an actively studied and still open question. In addition to p53 modifications, target gene promoter architecture and p53 differential protein binding or cofactor recruitment have been reported to affect the transcriptional outcome of p53 activation (42, 57, 67). This suggests that different p53 target genes may require a particular combination of transcriptional activators and a specific modification state to be activated.
One unique target of p53 is the Mdm2 gene itself, thus forming a negative-feedback loop upon p53 activation. Mdm2 is controlled by two promoters: the P1 promoter, which is constitutively active in most cells, though at low levels, and the p53-responsive P2 promoter, located within Mdm2's first intron, adjacent to its transcription start site (2). The Mdm2 P2 promoter contains two p53 binding sites and is activated by p53 in response to various cellular stresses (71).
In this study, we have examined the effects of MdmX on the transcription of p53 target genes. We found that full expression of MdmX is necessary for enabling p53 to activate Mdm2 maximally following stress in multiple cell lines (although some cell lines tested did not display this phenotype). We further investigated the mechanism by which MdmX exerts this effect and showed that MdmX enhances p53 binding to the Mdm2 promoter in cells after stress. The defect in Mdm2 activation following MdmX downregulation results in prolonged p53 stability at times when the cellular p53 response normally decreases. Thus, we have identified a novel mechanism through which MdmX represses p53, by promoting the activation of its chief inhibitor, Mdm2.
MATERIALS AND METHODS
Cell culture.
MCF7, U2OS, and SK-HEP-1 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Drug treatments were as follows: neocarzinostatin (NCS) (300 ng/ml; Kayaku Co., Tokyo, Japan) was added for 4 h or as indicated in the figures, while 5-fluorouracil (5FU) (500 nM; Sigma-Aldrich, St. Louis, MO), actinomycin D (ActD) (4 nM; Calbiochem, San Diego, CA), and doxorubicin (Doxo) (100 nM; Sigma-Aldrich, St. Louis, MO) were administered for 8 h. Cycloheximide (100 μg/ml; Sigma-Aldrich, St. Louis, MO) was given to cells for the times indicated, and Nutlin-3 (10 μM; Sigma-Aldrich, St. Louis, MO) was administered for 16 h.
Transfection.
Small interfering RNA (siRNA) duplexes were obtained from Qiagen (Valencia, CA) and Invitrogen (Carlsbad, CA) and transfected into cells with DharmaFECT 1 reagent (Dharmacon, Thermo Fisher Scientific, Lafayette, CO) for 48 h. siRNA sequences were as follows. The sequences for siRNA directed against luciferase (siLuc) (65) and siRNA directed against MdmX (siMdmX) (8) were published previously. siMdmX 2 refers to Hs_MDM4_4 FlexiTube siRNA (Qiagen, Valencia, CA). For siMdmX 3, the sense strand was AGGAUCACAGUAUGGAUAUUU, while the antisense strand was AUAUCCAUACUGUGAUCCUGU. For siMdmX 4, the sense strand was GGAUAUUCCAAGUCAAGACUU, while the antisense strand was GUCUUGACUUGGAAUAUCCAU.
Quantitative reverse transcription-PCR (qRT-PCR) analysis.
RNA was isolated from cultured cells using the Qiagen RNeasy minikit and reverse transcribed into cDNA with QuantiTect reverse transcription kit (Qiagen, Valencia, CA). PCR was performed with either Prism 7300 or StepOnePlus real-time PCR system using power SYBR green PCR master mix (Applied Biosystems, Foster City, CA). Relative mRNA levels were calculated by the ΔΔCT method (CT stands for threshold cycle), normalized first to the levels of control RPL32 mRNA and then to the levels of the plotted mRNA in untreated siLuc control sample. Primer sequences are available upon request. Graphs are representative of multiple independent experiments, with error bars representing technical PCR replicates.
Immunoblot (Western blot) analysis.
Whole-cell lysates were analyzed by standard immunoblotting procedure as described previously (65). Briefly, the cells were harvested by scraping in phosphate-buffered saline (PBS) and lysed in TEGN buffer (10 mM Tris [pH 8], 1 mM EDTA, 10% glycerol, 0.5% NP-40, 400 mM NaCl) supplemented with protease inhibitors, incubated on ice for 20 min, and cleared by centrifugation (13,000 rpm for 10 min at 4°C). The total protein concentration was measured using the Bio-Rad protein assay (Life Science Research, Hercules, CA). Equal amounts of total protein were separated on 9% polyacrylamide gel and transferred to a nitrocellulose membrane, which was then blocked for 1 h in PBS containing 0.1% Tween 20 (Sigma-Aldrich, St. Louis, MO) and 5% nonfat dry milk. Commercially obtained antibodies used in this study were MdmX A300-287A (Bethyl Laboratories, Montgomery, TX); MdmX (G-10) sc-74467, p21 (C-19) sc-397, and Mdm2 (N-20) sc-813 (Santa Cruz Biotechnology, Santa Cruz, CA). The following mouse monoclonal antibodies were used as hybridoma supernatants: p53 (DO-1 and 1801) and Mdm2 (3G5, 5B10, and 4B11).
IP assay.
The cells were harvested by scraping in PBS, lysed in low-salt immunoprecipitation (IP) buffer (25 mM Tris-HCl [pH 7.5], 137 mM NaCl, 2.7 mM KCl, and 0.5% Nonidet P-40) and cleared by centrifugation (13,000 rpm for 10 min at 4°C). The total protein concentration was measured using the Bio-Rad protein assay (Life Science Research, Hercules, CA). All the following steps were performed at 4°C. One milligram of total protein was taken for each immunoprecipitation sample and precleared by rocking with 20 μl protein G beads (GE Healthcare, Little Chalfont, United Kingdom) for 1 h. Purified monoclonal p53 antibody (1801; 2 μg) was added to the precleared lysate and rocked for 2 h. Protein G beads (35 μl) that were blocked overnight in IP buffer containing bovine serum albumin (BSA) (1 mg/ml; New England BioLabs, Ipswich, MA) were added for an additional 1-h incubation. Following 4 washes with IP buffer, protein sample buffer was added, and the proteins were eluted by incubation at 95°C for 10 min. Immunoblot analysis was performed as described above.
ChIP assay.
The chromatin immunoprecipitation (ChIP) assay was performed as described previously (3, 16). The cells were cross-linked in PBS containing 1% formaldehyde for 15 min and quenched with glycine (final concentration of 125 mM) for 5 min at room temperature (RT). Following 2 washes with ice-cold PBS, the cells were harvested in PBS by scraping. All the following steps were performed at 4°C. Cell lysis was performed using radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris-HCl [pH 8], 5 mM EDTA) supplemented with protease inhibitors. The DNA was then sheared by sonication to fragments of approximately 500 bp, and the lysates were cleared by centrifugation (13,000 rpm for 10 min at 4°C). The total protein concentration was measured using the Bio-Rad protein assay kit (Life Science Research, Hercules, CA), and samples were normalized to ensure equal amounts of protein. The samples were precleared for 1 h with a mixture (15 μl) of protein A beads (Thermo Scientific, Hudson, NH) and protein G beads (GE Healthcare, Little Chalfont, United Kingdom). Immunoprecipitation was performed overnight with mixed protein A/G beads (35 μl) that were blocked overnight at 4°C with RIPA buffer containing 1 mg/ml BSA (New England BioLabs, Ipswich, MA) and 0.3 mg/ml salmon sperm DNA (Invitrogen Corporation, Carlsbad, CA). For p53 ChIP experiments, the beads were incubated for 4 h with anti-p53 DO-1/1801 hybridoma supernatant mixture and washed 3 times in RIPA buffer. For MdmX ChIP experiments (and IgG controls), blocked beads were added to the lysates together with either MdmX A300-287A (Bethyl Laboratories, Montgomery, TX) or mouse IgG (Sigma-Aldrich, St. Louis, MO). Eight washes were performed, with 5 min rocking after each wash. The beads were first washed twice with RIPA buffer, then 4 times with ChIP wash buffer (100 mM Tris-HCl [pH 8.5], 500 mM LiCl, 1% Nonidet P-40, 1% deoxycholic acid), followed by additional two washes with RIPA buffer. Finally, the beads were washed twice briefly with TE (10 mM Tris-HCl [pH 8], 1 mM EDTA). The immunocomplexes were eluted by incubation at 65°C for 10 min in TE supplemented with SDS up to 1%. Cross-linking was reversed by adding NaCl to a final concentration of 200 mM and incubating at 65°C for 5 h. DNA was purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA). ChIP-enriched DNA was quantitated by using either the Prism 7300 or StepOnePlus real-time PCR system with power SYBR green PCR master mix (Applied Biosystems, Foster City, CA), using the absolute quantification method, in which ChIP DNA PCRs were run alongside a standard curve of genomic DNA. Regions amplified included the p53 response elements (REs) in the p21 promoter and the Mdm2 P2 promoters (28). For a negative control, we used a nontranscribed region located within chromosome 5 (27).
EMSA.
The electrophoretic mobility shift assay (EMSA) was performed as previously published (31) with the following double-stranded probes (sense strand presented here): for Mdm2, 5′-AGCTGGTCAAGTTCAGACACGTTCCGAAACTGCAGTAAAAGGAGTTAAGTCCTGACTTGTCTCCAG-3′; for p21, 5′-TCAGGAACATGTCCCAACATGTTGAGCTCTGGCATAGAAGAGGCTGGTGGCTATTTTGTCCTTGGG-3′; for p21 mutant competitor, 5′-TCAGGAAtATaTCCcAAtATaTTgAGCTCTGGCATAGAAGAGGCTGGTGGCTATTTTGTCCTTGGG (mutated residues are in lowercase). p53 (50 ng) was preincubated with mutant p21 oligonucleotide and Mdm2 or MdmX (0 to 400 ng as indicated in the figures) in EMSA buffer (12.5 mM Tris-HCl [pH 6.8], 25 mM KCl, 10% glycerol, 0.05% Triton X-100, 0.5 mg/ml BSA, 1 mM dithiothreitol [DTT]) for 20 min. 32P-labeled Mdm2 or p21 probe (labeled at the end) was added for 10 min, and the protein-DNA complexes were resolved on 4% native polyacrylamide gels, which were then transferred to blotting paper, dried, quantitated by a phosphorimager (GE Healthcare, Little Chalfont, United Kingdom), and analyzed by ImageQuant software (GE Healthcare, Little Chalfont, United Kingdom).
Protein purification.
MdmX and Mdm2 proteins were purified from baculovirus-infected insect cells as described before (75). Insect Sf9 cells were infected with recombinant baculovirus expressing Flag-tagged Mdm2 (Flag-Mdm2) or hemagglutinin-tagged MdmX (HA-MdmX). The cells were lysed by sonication in buffer A (50 mM Tris-HCl [pH 8], 150 mM NaCl, 10% glycerol) supplemented with protease inhibitors and cleared by centrifugation. Flag-Mdm2 was incubated with anti-Flag M2 affinity gel (Sigma-Aldrich, St. Louis, MO), and HA-MdmX was incubated with anti-HA affinity matrix (Roche, Indianapolis, IN). The beads were then washed in high-salt buffer A containing 300 mM salt. Flag-Mdm2 was eluted with 1 mg/ml Flag peptide (Sigma-Aldrich, St. Louis, MO) in buffer A. HA-MdmX was eluted with 1 mg/ml HA peptide (Roche, Indianapolis, IN) at 30°C. Histidine-tagged p53 (His-p53) protein was purified from Escherichia coli as published previously (59). E. coli BL21 cells were transformed with His-p53 expression vector and induced with isopropyl-β-d-thiogalactopyranoside (IPTG) for 2 h at RT. The cells were lysed by one freeze-thaw cycle followed by sonication in lysis buffer (0.3 M NaCl, 50 mM NaH2PO4 [pH 8], 20% glycerol, 1% Nonidet P-40, 10 mM β-mercaptoethanol, 0.5 mM phenylmethylsulfonyl fluoride [PMSF]). The lysates were cleared by centrifugation, and the supernatant was loaded onto a nickel-nitrilotriacetic acid (Ni-NTA) column (Qiagen, Valencia, CA). The column was washed with lysis buffer lacking Nonidet P-40 but supplemented with 40 mM imidazole, and protein was eluted with 0.25 M imidazole followed by dialysis for 30 min in storage buffer (20 mM Tris [pH 8], 0.2 mM EDTA, 20% glycerol, 0.5 mM DTT, and 0.5 mM PMSF).
ABCD assay.
The avidin biotin complex DNA (ABCD) assay conditions were modified from published protocol (12) and performed as follows. The cells were harvested by trypsinization, lysed by sonication in C/M buffer (25 mM HEPES [pH 7.5], 100 mM NaCl, 0.5 mM EDTA, 15% glycerol, 0.3% Triton X-100), passed through a 0.45-μm filter, and normalized for equal amounts of total protein. Biotinylated DNA probes (103 bp) that span either the p53 response element sequences in the Mdm2 promoter (wild type [WT]) or one in which the key residues in the p53 REs have been mutated (mutant) were prepared by PCR using biotinylated forward primers (Integrated DNA Technologies, Inc., Coralville, IA) and purified using a QIAquick gel extraction kit (Qiagen, Valencia, CA). Probe sequences can be found in the supplemental material (see Table S1 in the supplemental material). Dynabeads M-280 streptavidin (Invitrogen, Carlsbad, CA) were preincubated with the biotinylated DNA probes in binding and washing buffer (5 mM Tris-HCl [pH 7.5], 500 nM EDTA, 1 M NaCl) according to the manufacturer's instructions. The beads were then added to the cell filtrate and rocked for 1 h at 4°C together with nonbiotinylated mutant DNA competitor. Following four washes with buffer M (25 mM HEPES [pH 7.5], 20% glycerol, 0.4% Triton X-100, 0.5 mM EDTA), protein sample buffer was added and boiled for 5 min. Proteins pulled down by the DNA probes were analyzed by the immunoblot method.
p53 half-life analysis.
Protein levels were quantitated by immunoblot analysis using the Odyssey system (LI-COR Biosciences, Lincoln, NE). The percentage of protein remaining was calculated, and the data were analyzed with GraphPad Prism program (GraphPad Software, La Jolla, CA). Statistical significance was determined by using the Student t test on the data from three independent experiments.
RESULTS
MdmX ablation has a selective effect on p53 target gene induction following multiple forms of cellular stress.
To gain insight into the functional inhibition of p53 by MdmX, we first determined the impact of MdmX ablation by siRNA in MCF7 breast cancer cells. Following introduction of either control siRNA or siRNA directed against MdmX, MCF7 cells were either left untreated or were treated with the radiomimetic drug neocarzinostatin (NCS). First, MdmX was depleted using two concentrations of siRNA (5 nM and 50 nM), and the induction of p53 target genes Mdm2 and p21 was assessed using real-time quantitative reverse transcription-PCR (qRT-PCR) (Fig. 1A). The results with p21 mRNA were as expected: the basal levels were upregulated by MdmX siRNA, while the levels after NCS treatment were largely unaffected, consistent with the role of MdmX as a p53 inhibitor that becomes degraded after DNA damage (Fig. 1A). Unexpectedly, however, the opposite response to MdmX depletion was seen when we examined Mdm2 induction; its mRNA induction after NCS treatment was compromised by MdmX siRNA in a concentration-dependent manner (6-fold induction by NCS in control siRNA samples versus 2-fold induction in samples with 50 nM MdmX siRNA), while basal Mdm2 mRNA levels were largely unaffected (Fig. 1A). Similarly, introduction of MdmX siRNA into either the osteosarcoma cell line U2OS (Fig. 1B) or the hepatocellular carcinoma cell line SK-HEP-1 (Fig. 1C) led to reduction of Mdm2 mRNA levels after NCS treatment compared to control siRNA, while p21 levels were either increased or unaffected, and p53 protein levels were essentially unchanged by the siRNA treatments. Note that in some other tested cell lines (SJSA, WI-38, and HepG2), there was no significant effect of MdmX siRNA on Mdm2 expression (data not shown), and we cannot conclude that this is a cancer cell-specific effect. Importantly, the results were not an off-target effect of the original siRNA that we used, because four different siRNA sequences targeting the MdmX transcript each behaved identically (Fig. 1D). Overexpression of Myc-tagged MdmX in MCF7 and U2OS cells, on the other hand, did not affect the expression of either Mdm2 or p21 regardless of NCS application (see Fig. S1 in the supplemental material).
Fig 1.

MdmX is required for induction of Mdm2 but not p21 after DNA damage. (A) MCF7 cells were transfected with increasing amounts of siRNA to MdmX as indicated below the bars, with a total siRNA concentration of 50 nM in all samples balanced with control siRNA (siRNA to Luc), followed by either no treatment (NT) or 4-hour treatment with NCS (300 ng/ml) prior to lysis of cells and preparation of RNA for analysis as described in Materials and Methods. MdmX, Mdm2, and p21 mRNA levels were measured by qRT-PCR, and the graphs show the levels of the indicated mRNAs relative to values for siLuc controls from untreated cells. (B and C) U2OS (B) or SK-HEP-1 (C) cells were transfected with 50 nM control (Luc) or MdmX siRNA and either not treated (NT) or treated with NCS (300 ng/ml) for 4 h. The bar graphs show Mdm2 and p21 mRNA levels measured as described above for panel A. (Right) Immunoblot analysis of the experiment shown with antibodies against MdmX, Mdm2, p53, p21, and actin as indicated to the left of the blots. (D) MCF7 cells were transfected with either control (Luc) or 4 different MdmX siRNAs (50 nM each) as described above for panel A and either not treated (NT) or treated with NCS (300 ng/ml) for 4 h after which mRNA was prepared and quantified by qRT-PCR amplifying MdmX, Mdm2, and p21 as in panel A.
We next examined whether MdmX is required for Mdm2 induction in response to p53-inducing cell stressors other than NCS. MCF7 cells were transfected with control or MdmX siRNA followed by treatment with either NCS, the thymidylate synthase inhibitor 5-fluorouracil (5FU), the RNA and DNA synthesis inhibitor actinomycin D (ActD), or a topoisomerase II inhibitor doxorubicin (Doxo) (Fig. 2A). In each case, knockdown of MdmX compromised Mdm2 mRNA levels while not affecting p21 levels (Fig. 2A). Even though p53 levels were modestly compromised in the siMdmX-treated cells after some forms of stress, as indicated by the lower levels of p53 protein (Fig. 2B), these results, nevertheless, show that MdmX is required for Mdm2 induction in response to a variety of cellular stress signals, including transient DNA damage (NCS), prolonged DNA damage (Doxo), and ribosomal stress (5FU and ActD).
Fig 2.

MdmX is required for full induction of Mdm2 after multiple cellular stresses. (A) MCF7 cells were transfected with MdmX siRNA for 48 hours followed by either no treatment (NT), treatment for 4 h with NCS (300 ng/ml), or treatment for 8 h with 5-fluorouracil (5FU) (500 nM), actinomycin D (ActD) (4 nM), or doxorubicin (Doxo) (100 nM). p21 and Mdm2 mRNA levels were quantified by qRT-PCR analysis. (B) Immunoblot analysis of the experiment in panel A was performed using antibodies against MdmX, p53, Mdm2, and actin.
MdmX is required for optimal expression of Mdm2 and Wip1 but not other tested p53 target genes.
We then extended our observation to examine the effect of MdmX siRNA on other p53 target genes. MCF7 cells were transfected with control or MdmX siRNA and were either untreated or treated with NCS for 2 or 4 h. mRNA levels of a panel of p53 target genes were evaluated using qRT-PCR (Fig. 3). In untreated cells, following MdmX depletion, several targets (p21, cyclin G, BAX, and PIG3) were induced, while others were not (Mdm2, Wip1, 14-3-3σ, and Noxa). Following NCS treatment, however, most target genes (14-3-3σ, cyclin G1, BAX, PIG3, and Noxa) behaved like the p21 gene, with MdmX not playing a positive role in their expression following DNA damage (Fig. 3A). Interestingly, like Mdm2, the p53-induced phosphatase 1 gene (Wip1) (14) required MdmX for optimal induction after NCS treatment. Immunoblot analysis confirmed that MdmX knockdown was efficient (Fig. 3B). We mention the significance of these findings in Discussion.
Fig 3.

MdmX is required for the induction of Mdm2 and Wip1 but not of other tested p53 target genes. MCF7 cells were transfected with 50 nM control (Luc) or MdmX siRNA and either untreated (0) or treated with NCS (300 ng/ml) for 2 and 4 hours. (A) mRNA levels of p53 target genes were quantitated by qRT-PCR and averaged over three experiments. (B) Immunoblot analysis of a representative experiment with antibodies against MdmX, Mdm2, p53, and actin.
MdmX siRNA reduces the maximum induction of mature and nascent Mdm2 mRNAs.
We then tested whether MdmX siRNA affects peak induction of Mdm2 transcript levels or, alternately, whether the results were due to change in induction kinetics. To approach this, we conducted a time course experiment where MCF7 cells transfected with control or MdmX siRNA were treated with NCS for 0, 2, 4, or 6 h (Fig. 4). In cells containing control siRNA, both Mdm2 and p21 were maximally induced 4 h after NCS treatment. In cells treated with siRNA directed at MdmX, Mdm2 also peaked 4 h after NCS treatment, but the peak induction was compromised (∼6-fold induction in siLuc samples versus ∼3-fold induction in siMdmX samples). p21 mRNA was induced as expected, and its peak induction at 4 h after NCS treatment was unaffected by MdmX siRNA (Fig. 4A). In agreement with the mRNA data, Mdm2 protein levels were maximally induced 4 h after NCS in both control and MdmX siRNA-treated cells, but the levels in siMdmX-treated cells were lower (Fig. 4B, top blot). To check whether a change in p53 levels may account for the differences in gene expression, p53 protein levels were quantitated (Fig. 4B, bottom graph). While the Mdm2 mRNA levels were reduced by 52% following MdmX siRNA treatment (from 6.53 to 3.16 relative levels), p53 protein levels were reduced by only 18% (from 2.65 to 2.17 relative levels) and were thus unlikely to account for our observations. An examination of nascent Mdm2 transcript levels by performing qRT-PCR with intronic primers both confirmed that activation was at the level of transcription and also showed maximum induction after 2 h NCS treatment that was reduced in cells transfected with MdmX siRNA (8.5-fold induction in siLuc samples versus 5.5-fold in siMdmX samples) (Fig. 4C). These results demonstrate that MdmX siRNA compromises the maximal induction of Mdm2 while not affecting the kinetics of induction following stress.
Fig 4.

MdmX is required for maximum induction of mature and nascent Mdm2 transcripts after NCS treatment but does not affect the kinetics of activation. (A) MCF7 cells were transfected with MdmX siRNA (50 nM) for 48 h followed by treatment with NCS (300 ng/ml) for the times indicated in the figure. Mdm2 and p21 mRNA levels were determined using qRT-PCR as in the legend to Fig. 1. Graphs show qRT-PCR amplifying Mdm2 and p21 mRNA. (B) (Top) Immunoblot analysis of experiment shown in panel A using antibodies against Mdm2, MdmX, p53, p21, and actin. (Bottom) p53 protein levels were quantitated using the Odyssey software, and the levels relative to untreated siLuc were graphed. (C) Experiment was performed as described above for panel A, but nascent Mdm2 transcript was measured by qRT-PCR using primers that anneal within intron 3 at nucleotide +2576.
p53 levels increase rapidly after NCS treatment, peaking at 2 h, and sharply decrease thereafter (Fig. 4B). Mdm2 and p21 mRNA, however, peak at 4 h NCS treatment, coinciding with the time when MdmX siRNA affects Mdm2 mRNA levels (Fig. 4A). Therefore, unless stated otherwise, NCS treatment in this paper was carried out for 4 h. Note that at this time point, p53 levels are decreasing, and hence, the levels may not appear to be induced in some cases because p53 protein has returned to or is approaching basal state.
The half-life of p53 after DNA damage is longer in siMdmX-treated cells.
We next examined the functional consequences of the compromised Mdm2 induction following NCS treatment in siMdmX-treated cells. The p53-Mdm2 feedback loop is essential for turning off the p53 response. We hypothesized that the p53 degradation that normally occurs following p53 activation would be compromised in the presence of MdmX siRNA. To test this, we measured p53 stability in the presence of cycloheximide 4 h after NCS application (Fig. 5), a time point at which Mdm2 is maximally induced, forming a negative-feedback loop, and degrading p53 (Fig. 4). Following NCS treatment, the calculated p53 half-life was significantly longer (P = 0.04) in cells treated with siMdmX (21.7 min) than in control cells treated with siRNA (7.2 min) (Fig. 5A). MdmX was shown to promote the ubiquitination of p53 by Mdm2 (24, 44) by altering its substrate specificity. To exclude the possibility that this activity of MdmX is responsible for the effect of siMdmX on p53 stability after NCS treatment, we conducted a cycloheximide chase experiment in cells that were not treated with NCS. Although we did see a trend in which MdmX siRNA increased p53 half-life in untreated cells (about a twofold increase, from 22.25 min in siLuc cells to 48.08 min in siMdmX compared to a threefold increase after NCS treatment), the fold difference was lower than in NCS-treated cells, and the effect was not statistically significant (Fig. 5B). Thus, the effects of MdmX ablation on Mdm2-mediated ubiquitination of p53 may contribute to the lower p53 half-life we see after NCS treatment in siMdmX-treated cells but are not the primary mechanism. These data indicate that in the absence of MdmX, impaired Mdm2 induction prevents efficient functioning of the p53-Mdm2 feedback loop and thereby slows down the degradation of p53 as the stress response is being shut off.
Fig 5.

Ablation of MdmX extends the half-life of p53 after DNA damage. MCF7 cells were transfected with control siLuc or MdmX siRNA for 48 h and then treated with NCS for 4 h (A) or left untreated (B). The cells were then exposed to cycloheximide (CHX) (100 μg/ml) for the indicated times (in minutes) and flash frozen. Protein levels were analyzed by immunoblotting for MdmX, p53, and actin as indicated (top) and quantitated using Odyssey software (bottom). The bar graph shows the percentage of protein remaining plotted as a function of time based on 3 independent experiments, and the curve was fitted and the half-life was calculated with GraphPad Prism software. Statistical significance was calculated with a Student t test (P < 0.05).
The effect of MdmX on Mdm2 expression does not require the Mdm2-p53 interaction.
Mdm2 inhibits the interaction of p53 with DNA in vitro and in cell culture through its interaction with the C terminus of p53 (53). This raises the possible hypothesis that MdmX functions to activate Mdm2 transcription by preventing Mdm2 from inhibiting p53. Therefore, to study how MdmX affects p53-mediated transcription of Mdm2 after NCS treatment, we examined whether MdmX functions through competing Mdm2 off p53 by testing whether disruption of the p53-Mdm2 complex alleviates the effects of MdmX siRNA. After transfecting MCF7 cells with either control or MdmX siRNA, we pretreated them with Nutlin-3 (66) for 12 h to dissociate p53 from Mdm2. We then treated the cells with NCS and/or Nutlin-3 for 4 h and examined mRNA levels by qRT-PCR (see Fig. S2A in the supplemental material) and protein levels by Western blotting (Fig. S2B). Mdm2 mRNA levels in control siRNA-treated cells were induced 3-fold by NCS and 17-fold by Nutlin-3 and were further increased to 24-fold by Nutlin-3 followed by NCS treatment (Nutlin-NCS) (see Fig. S2A). We observed that Mdm2 mRNA levels were reduced by siMdmX in Nutlin-3–NCS-treated cells (15% reduction). This was not the case in cells treated with Nutlin-3 alone, where Mdm2 mRNA was slightly elevated by siMdmX. With the same trend, siMdmX increased p21 mRNA levels in all treatment conditions (see Fig. S2A). This is expected based on the inhibitory function of MdmX on p53-mediated transcription and the reported synergism between MdmX siRNA and Nutlin-3 treatment on p53 activation (19). Although the effect of MdmX ablation on Mdm2 expression in Nutlin-3–NCS-treated cells was modest, it was statistically significant and consistently seen in multiple experiments, like the failure to see such a reduction with p21.
As further support for Mdm2 not being involved in MdmX's effect on p53-mediated Mdm2 transcription, the p53-Mdm2 interaction was not affected by MdmX siRNA in a coimmunoprecipitation assay (see Fig. S2C in the supplemental material). Equal amounts of Mdm2 were bound by p53 after NCS treatment in both control and MdmX siRNA-treated MCF7 cells. Together, these results provide evidence that the requirement of MdmX for Mdm2 induction is very likely independent of Mdm2's inhibition of the recruitment of p53 to DNA.
MdmX is not required for p53 interaction with DNA spanning the p53 binding site from the Mdm2 P2 promoter in vitro.
Mdm2 has been shown to inhibit the interaction of p53 with DNA by multiple assays (11, 53, 74). We entertained the possibility that, in contrast to the inhibitory effect of Mdm2, MdmX might stimulate p53 binding to DNA sequences within the Mdm2 promoter. Two separate approaches were used to determine how Mdm2 and MdmX affect p53 binding to DNA in vitro. In the first approach, we performed an electrophoretic mobility shift assay with purified components in which increasing amounts of either Mdm2 or MdmX protein were added to reaction mixtures containing p53 protein and a 66-nucleotide end-labeled probe spanning the p53 binding sites in either the p21 or Mdm2 promoter (Fig. 6A). For each Mdm protein concentration, the p53-DNA complex band intensity was normalized to the value of binding of p53 alone (Fig. 6A). We confirmed that Mdm2 inhibits the p53-DNA interaction. Interestingly, this effect was more severe with the Mdm2 probe (60% reduction) than with the p21 probe (40% reduction). Nevertheless, the addition of MdmX also reduced the p53-DNA interaction, albeit to a much lesser extent and only at the higher concentrations (20% reduction for both probes). This result is in agreement with the recently published observation that, as opposed to a copurified Flag-Mdm2–p53 complex that fails to bind exogenous DNA, a Flag-MdmX–p53 complex retains most binding activity (11) but does not support the hypothesis that MdmX is required for full p53 binding to naked Mdm2 DNA.
Fig 6.

MdmX is not required for p53 binding to DNA in vitro. (A) Electrophoretic mobility shift assay (EMSA) was performed with purified components. Reaction mixtures containing p53 (50 ng), increasing amounts of Mdm2 or MdmX (0 or 100 to 400 ng as indicated in the graphs), and 32P-labeled 66-nucleotide double-stranded DNA probes (labeled at the end) spanning the p53 REs from either the p21 (p21 probe) or Mdm2 (Mdm2 probe) promoter sequences were incubated as described in Materials and Methods and then separated by native PAGE. (Top) Phosphorimager images of the p53-DNA complexes shifted up with the following samples: no p53 (control) (lane 1), p53 alone (lane 2), p53 together with increasing amounts (100 to 400 ng) of Mdm2 (lanes 3 to 6), p53 alone (lane 7), p53 together with increasing amounts (100 to 400 ng) of MdmX (lanes 8 to 11). Note that Mdm2 and MdmX panels are from the same gel. (Bottom) Quantification of the DNA-protein complexes in which the intensity of each complex shifted up was quantitated, and relative binding was plotted as band intensity normalized to intensity of the no-Mdm protein band. (B) Avidin-biotin complex DNA (ABCD) assay. MCF7 cells were transfected with control siRNA or MdmX siRNA for 48 h, followed by NCS (300 ng/ml) treatment for 90 min. The cells were harvested and lysed in a low-salt buffer by sonication. The lysates were added to streptavidin Dynabeads prebound to biotinylated DNA spanning either the wild-type or mutated p53 binding sites in the Mdm2 promoter and incubated for 1 h. Input material and DNA-bound proteins were resolved on polyacrylamide gels and identified by immunoblot analysis with antibodies against p53, Mdm2, MdmX, and actin as indicated.
The second approach examined the effect of reducing MdmX levels with siRNA on the interaction of p53 with biotinylated DNA probes added to whole-cell extracts in an avidin biotin complex DNA (ABCD) assay (12). This strategy was taken because we speculated that other factors present in the cell extract might influence the ability of p53 to interact with DNA and the effects of Mdm2 and MdmX thereon. MCF7 cells were treated with control or MdmX siRNA followed by mock or NCS treatment. The cells were then lysed in low-salt buffer and incubated with Dynabeads prebound to DNA containing either wild-type (WT) or mutated p53 binding sites within the Mdm2 promoter DNA probe as indicated in Fig. 6B. Proteins bound to DNA on beads were visualized by immunoblotting (Fig. 6B). The conditions of the assay were set such that p53 bound to the DNA probes in a strictly sequence-specific manner. Nevertheless, siRNA against MdmX did not alter the amount of p53 bound to the WT DNA probe. For a control, we showed that under our assay conditions (2 μg total protein per reaction mixture), the binding capacity of the DNA-bound beads was not reached (see Fig. S3 in the supplemental material). It is also noteworthy that in this assay, virtually no detectable Mdm2 or MdmX cobound with p53 to the WT DNA probe.
Taken together, two different assays showed that neither addition of MdmX nor reduction of MdmX levels significantly affected p53 interaction with naked DNA. Yet, we showed that MdmX positively affects p53-mediated transactivation of the Mdm2 gene. Since these experiments were performed with DNA fragments in vitro, we went on to determine whether MdmX might still regulate p53 interactions in vivo with intact Mdm2 and p21 promoters present in their endogenous locations.
Recruitment of p53 to the Mdm2 promoter is inhibited by MdmX siRNA.
We used a chromatin immunoprecipitation (ChIP) assay to assess whether MdmX is required for recruitment of p53 to the Mdm2 promoter. p53 recruitment to the Mdm2 and p21 promoters peaks after 90 min of NCS treatment (data not shown). MCF7 cells were therefore transfected with control or MdmX siRNA, followed by 90 min of NCS treatment prior to processing for the ChIP assay. Quantitative real-time PCR was used to determine the amount of DNA bound, amplifying the p53 binding site regions/response elements (REs) from the Mdm2 P2 promoter, the p21 5′ RE, and a negative-control region downstream of the p21 gene (nucleotide position +11443 [data not shown]) (Fig. 7). Note that in this experiment as well as the experiments described in the legend to Fig. S2C in the supplemental material and in the legend to Fig. 6B, NCS treatment was performed for 90 min, a time point prior to Mdm2 expression, and therefore, the impact of MdmX knockdown on Mdm2 protein levels was not apparent. As expected, p53 binding to both promoters was stimulated by treatment with NCS. Remarkably, MdmX siRNA strongly inhibited p53 recruitment to the Mdm2 but not to the p21 promoter. This suggests an explanation for the reduced Mdm2 expression seen upon MdmX knockdown: impaired p53 recruitment to the Mdm2 promoter in the absence of MdmX results in lower Mdm2 mRNA and protein levels while not affecting p21 gene expression. This is supported by the observation that Mdm2 mRNA expression correlates with p53 interaction with the Mdm2 promoter (62). Since we observed the effect of MdmX on p53 interaction with DNA in cells and not in vitro, we conclude that one or more aspects of the cellular milieu (such as chromatin) must be important for the requirement of MdmX for maximal expression of Mdm2.
Fig 7.
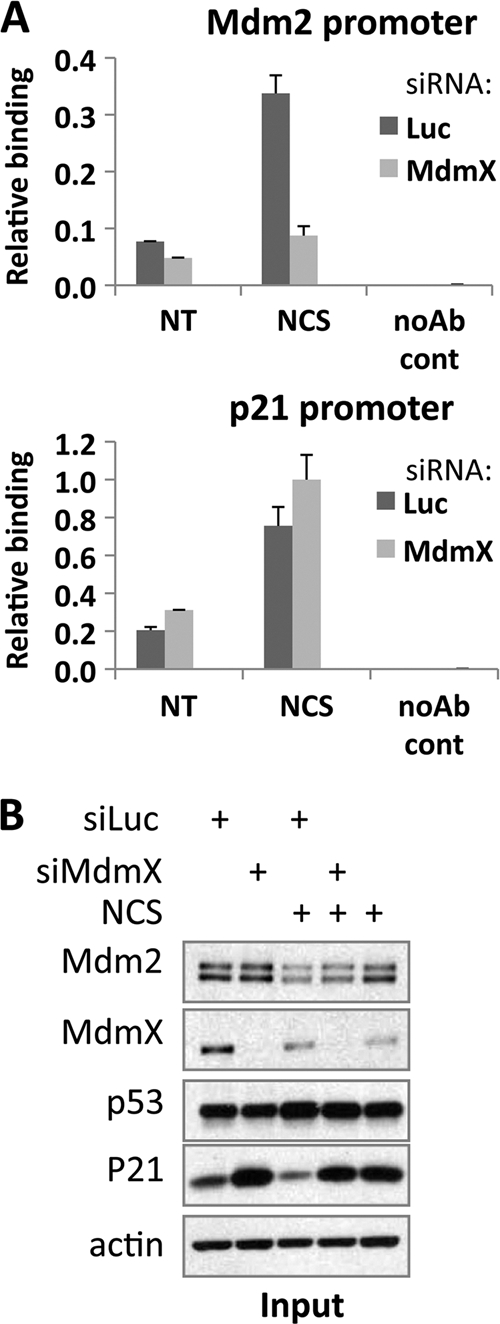
Recruitment of p53 to the Mdm2 promoter in vivo requires MdmX. A chromatin immunoprecipitation (ChIP) assay was performed. MCF7 cells were transfected with control (Luc) siRNA or MdmX siRNA for 48 h. The cells were then treated with NCS for 90 min and subjected to the ChIP protocol as described in Materials and Methods. noAb cont, no-antibody control. (A) ChIP-enriched DNA was quantitated by real-time PCR with primers amplifying Mdm2 P2 promoter (top panel) or p21 5′ RE (bottom). (B) Immunoblot of input material in a typical experiment using antibodies against Mdm2, MdmX, p53, p21, and actin as indicated.
MdmX preferentially associates with the Mdm2 promoter.
One possible hypothesis explaining how MdmX affects the recruitment of p53 to DNA is that MdmX is bound to the Mdm2 promoter where it may function in recruiting p53 or is otherwise facilitating Mdm2 transcription. To assess this possibility, MCF7 cells were transfected with control or MdmX siRNA followed by ChIP analysis of MdmX or p53 association with the Mdm2 and p21 promoters (Fig. 8). Quantitative PCR of ChIP-enriched DNA showed that indeed significantly more MdmX was bound to the Mdm2 promoter than to the p21 promoter (Fig. 8A). On the other hand, a p53 ChIP performed in parallel showed more p53 at the p21 promoter than at Mdm2 (Fig. 8B). The lower ChIP signal in siMdmX-treated cells compared to siLuc control confirmed the specificity of the MdmX antibody used in the MdmX ChIP (Fig. 8C). This result, while providing only correlative support at this point, provides a framework for future investigation of the mechanism by which MdmX selectively facilitates Mdm2 transcription in cells undergoing genotoxic stress.
Fig 8.

More MdmX is bound to the Mdm2 promoter than to the p21 promoter. MCF7 cells were transfected with Luc control siRNA (A, B, and C) or MdmX siRNA (C) for 48 h. ChIP was performed with antibodies against MdmX (A and C) or p53 (B) or total mouse IgG (A and B) as indicated. ChIP-enriched DNA was quantitated by real-time PCR with primers amplifying the p53 REs from the Mdm2 or p21 promoters or a negative-control region (control). (C) (Top) The cells containing siLuc or siMdmX were subjected to MdmX ChIP analysis as described above for panel A. (Bottom) Immunoblot analysis of input material using antibodies against MdmX, p53, and actin.
DISCUSSION
To gain more insight into the relationship between p53, Mdm2, and MdmX, it will be necessary to fully describe the complex interactions between these three proteins and how alterations in the level or activity of each affects p53 function. The relative levels of Mdm2, MdmX, and p53 are critical for their function. For example, Mdm2 or MdmX heterozygous mice are viable, but the double heterozygotes die in utero and are rescued by deletion of a single p53 allele (64). Furthermore, alterations in the amount of Mdm2 protein expressed in mice result in a shift in p53 response, with high Mdm2 levels permitting tumorigenesis and normal levels supporting tissue homeostasis, while further reductions in Mdm2 levels lead to tumor suppression, tissue-specific apoptosis, and eventually embryonic lethality due to unleashed p53 activity (36, 54). Hence, we set forth to examine how altering the levels of MdmX in cells affects the p53 response. Our experiments revealed an additional mechanism of p53 regulation by MdmX, in which MdmX is required for the activation of Mdm2 and Wip1 expression after a variety of cellular stresses (Fig. 9).
Fig 9.
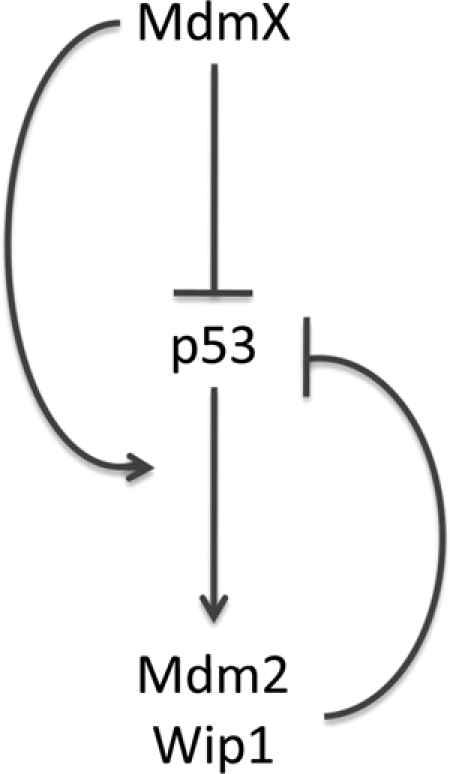
A model for the inhibitory effects MdmX exerts on p53. MdmX inhibits p53 both directly, through binding to its trans-activation domain and preventing it from recruiting transcriptional coactivators and indirectly, through promoting the activation of the p53 inhibitors Mdm2 and Wip1.
It is interesting that of several p53 targets assayed, only Mdm2 and Wip1 require full expression of MdmX to be induced by p53 in stressed cells. In contrast to the great majority of p53 target genes that mediate cellular outcomes that are consistent with p53 tumor suppression (cell cycle arrest, death, etc.), these two proteins actually serve to restrain and repress p53 functionally. Mdm2 is a well-described negative regulator of p53 activity and protein levels, while Wip1, a serine/threonine phosphatase, counteracts p53 by dephosphorylating its activators such as ATM (ataxia-telangiectasia mutated) and Chk2 among others (33). Further, like Mdm2 and MdmX, Wip1 is overexpressed in many tumor types (29). Thus, MdmX contributes to the establishment of two p53 negative-feedback loops: Mdm2-mediated inhibition and degradation of p53, and Wip1-mediated downregulation of the cellular stress response (32).
p53 recruitment to the Mdm2 promoter after treating cells with NCS is reduced in the absence of MdmX. To gain mechanistic insight, multiple hypotheses can be considered. First, on the basis of our observation that Mdm2 inhibits p53 interaction with DNA, an idea supported by previous work (11, 53, 74), we hypothesized that MdmX acts in the nucleoplasm to release p53 from Mdm2. If that were the case, we would expect to find more p53 bound to Mdm2 in cells treated with MdmX siRNA. However, coimmunoprecipitation experiments showed no difference in the amount of p53 bound to Mdm2, suggesting this is not likely to be the mechanism of action. Alternately, MdmX may function on chromatin, aiding the recruitment of p53 to the Mdm2 promoter. Indeed, ChIP experiments showed more MdmX bound at the Mdm2 than at the p21 promoter.
How chromatin-bound MdmX would exert a positive effect on p53 binding to the Mdm2 promoter, however, is unclear. One possibility could be that a specific chromatin structure or modification state renders it accessible to MdmX, which would then promote p53 binding to its RE within the region through protein-protein interaction. In that regard, some proteins that interact with the p53 N terminus (as does MdmX) such as transcription factor IID (TFIID) (10) or N-terminus-specific p53 antibodies (7) can stabilize p53 interactions with DNA. It is also conceivable that a subpopulation of MdmX is recruited to DNA together with p53. In this context, it is appealing to hypothesize that MdmX influences p53's selectivity toward interaction with the Mdm2 promoter elements, adding MdmX to the collection of p53-binding partners that affect promoter selectivity, such as the ASPP (apoptosis-stimulating proteins of p53) family proteins (4, 58).
New roles of Mdm proteins in regulation of transcription are continuously being uncovered. In addition to regulating transcription through its effects on p53, Mdm2 can recruit the corepressor proteins SUV39H1 and EHMT1 to p53-bound promoters, establishing a repressive chromatin state (9). Mdm2 can also bind to p53-responsive promoters and promote monoubiquitination of histone H2B (38, 39). This modification was recently described to be associated with actively transcribed genes, perhaps playing a role in elongation, thus raising the possibility of Mdm2 functioning as a positive and negative transcriptional regulator (38, 39). It is therefore not surprising that the Mdm2 gene is regulated in multiple ways, not only by p53.
The Mdm2 P2 promoter can be regulated by multiple factors. These factors include the estrogen receptor (ER) in ERα-positive cell lines (52), the thyroid hormone receptor in pituitary cell lines (55), the Ras-induced mitogen-activated protein kinase (MAPK) pathway (56) and transforming growth factor β (TGF-β) (1) signaling, all in a p53-independent manner. Furthermore, in MCF7 cells, p53 binding to the Mdm2 promoter does not require the chromatin remodeling protein Brg1, while the p21 promoter does (73), suggesting a difference in the chromatin state between those two promoters, with the chromatin at the Mdm2 promoter being relatively open. This agrees with a report showing that in the murine embryonic fibroblast cell line 10-1, the Mdm2 locus is constitutively nucleosome free (72). We now add MdmX to this list of proteins regulating the expression of the Mdm2 gene. What is unique about the Mdm2 promoter that renders it dependent on such a vast array of regulators is an important question in our quest to understand the regulation of stress responses and tumor suppression.
In different mouse models of MdmX knockout (15), MdmX RING domain deletion (48), or MdmX RING domain mutation (20), p53 is spontaneously activated. In our system, in unstressed cells, following MdmX knockdown, there was selective activation of p53 target genes. Specifically, p21, cyclin G1, Bax, and Pig3 were induced, while Mdm2, Wip1, 14-3-3, and Noxa were not. Interestingly, consistent with our observations, Garcia et al. reported activation of subsets of p53 targets in a tissue-specific manner (15). The differences in p53 stabilization and activation between our experiments in cell lines and the mouse models may result from the contrast between a complete knockout in the mouse versus partial knockdown by siRNA. Alternatively, there may be tissue-specific effects or perhaps even differences between mice and humans. Nonetheless, these studies together suggest that MdmX affects p53 target gene selectivity in a complex manner. In this paper, we showed that MdmX is required for Mdm2 activation after specific cellular stresses in a selection of cell lines. It would be of great interest to see whether the same can be observed in conditional MdmX knockout mice.
MdmX is imported into the nucleus following DNA damage (30). In the nucleus, it interacts with the RING domain of Mdm2 (63), undergoes ubiquitination by Mdm2, and degradation by the 26S proteasome (13). This sequence of events leaves the unanswered question: why would the cell import MdmX into the nucleus following DNA damage? Is it solely in order to degrade it? Our work suggests a potential answer to these questions by revealing a function that MdmX exerts in the nucleus following DNA damage—activation of the Mdm2 gene. MdmX seems to exert its function within 90 min of NCS application, when its effects on p53-DNA binding are observed. Due to the complex regulation of MdmX following damage by modifications, subcellular localization, and degradation, there seems to be a window in which MdmX is modified correctly and at the right level to exert its function before being fully degraded.
Restoration of the p53 pathway activity in tumors is being actively sought through multiple approaches (5). For example, the small molecule PRIMA-1 (p53 reactivation and induction of massive apoptosis) restores mutant p53 to wild-type conformation (6), and both Nutlin-3 (66) and RITA (reactivation of p53 and induction of tumor cell apoptosis) (21) release p53 from Mdm2. Additionally, a peptide dual inhibitor (PDI) was developed to inhibit p53 interaction with both Mdm2 and MdmX (34, 51), and more recently, a specific inhibitor of MdmX expression has been identified (69). Here we have reported an additional layer of complexity in the intricate relationship between Mdm2, MdmX, and p53. MdmX is required for p53 to interact optimally with the Mdm2 promoter after some forms of stress and to activate transcription of this important p53 suppressor. With an increased understanding of the p53-Mdm2-MdmX trio, we will be able to better predict the effects of such pharmaceutical interventions.
Supplementary Material
ACKNOWLEDGMENTS
We thank Megan Keniry and Ramon Parsons for generous help with the ABCD assay, Joaquin Espinosa for providing primer sequences, Oleg Laptenko for help and advice with the purification of proteins for the DNA binding assays, Moshe Oren for helpful discussions and suggestions, Ben Dubin-Thaler and Nili Ostrov for critical reading of the manuscript, and Ella Freulich for providing expert technical assistance.
This work was supported by NIH grant CA58316 to C.P. and GM097174 to J.L.M.
Footnotes
Published ahead of print 30 January 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Araki S, et al. 2010. TGF-beta1-induced expression of human Mdm2 correlates with late-stage metastatic breast cancer. J. Clin. Invest. 120:290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barak Y, Gottlieb E, Juven-Gershon T, Oren M. 1994. Regulation of mdm2 expression by p53: alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 8:1739–1749 [DOI] [PubMed] [Google Scholar]
- 3. Beckerman R, et al. 2009. A role for Chk1 in blocking transcriptional elongation of p21 RNA during the S-phase checkpoint. Genes Dev. 23:1364–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beckerman R, Prives C. 2010. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2:a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. 2009. Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer 9:862–873 [DOI] [PubMed] [Google Scholar]
- 6. Bykov VJ, et al. 2002. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 8:282–288 [DOI] [PubMed] [Google Scholar]
- 7. Cain C, Miller S, Ahn J, Prives C. 2000. The N terminus of p53 regulates its dissociation from DNA. J. Biol. Chem. 275:39944–39953 [DOI] [PubMed] [Google Scholar]
- 8. Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. 2005. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 24:3411–3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen L, et al. 2010. MDM2 recruitment of lysine methyltransferases regulates p53 transcriptional output. EMBO J. 29:2538–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen X, Farmer G, Zhu H, Prywes R, Prives C. 1993. Cooperative DNA binding of p53 with TFIID (TBP): a possible mechanism for transcriptional activation. Genes Dev. 7:1837–1849 [DOI] [PubMed] [Google Scholar]
- 11. Cross B, et al. 2011. Inhibition of p53 DNA binding function by the MDM2 protein acidic domain. J. Biol. Chem. 286:16018–16029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Daitoku H, Yamagata K, Matsuzaki H, Hatta M, Fukamizu A. 2003. Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR. Diabetes 52:642–649 [DOI] [PubMed] [Google Scholar]
- 13. de Graaf P, et al. 2003. Hdmx protein stability is regulated by the ubiquitin ligase activity of Mdm2. J. Biol. Chem. 278:38315–38324 [DOI] [PubMed] [Google Scholar]
- 14. Fiscella M, et al. 1997. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 94:6048–6053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garcia D, et al. 2011. Validation of MdmX as a therapeutic target for reactivating p53 in tumors. Genes Dev. 25:1746–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gomes NP, et al. 2006. Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev. 20:601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haupt Y, Maya R, Kazaz A, Oren M. 1997. Mdm2 promotes the rapid degradation of p53. Nature 387:296–299 [DOI] [PubMed] [Google Scholar]
- 18. Honda R, Tanaka H, Yasuda H. 1997. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 420:25–27 [DOI] [PubMed] [Google Scholar]
- 19. Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. 2006. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J. Biol. Chem. 281:33030–33035 [DOI] [PubMed] [Google Scholar]
- 20. Huang L, et al. 2011. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. U. S. A. 108:12001–12006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Issaeva N, et al. 2004. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 10:1321–1328 [DOI] [PubMed] [Google Scholar]
- 22. Jin Y, et al. 2006. 14-3-3gamma binds to MDMX that is phosphorylated by UV-activated Chk1, resulting in p53 activation. EMBO J. 25:1207–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jones SN, Roe AE, Donehower LA, Bradley A. 1995. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378:206–208 [DOI] [PubMed] [Google Scholar]
- 24. Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM. 2007. RING domain-mediated interaction is a requirement for MDM2's E3 ligase activity. Cancer Res. 67:6026–6030 [DOI] [PubMed] [Google Scholar]
- 25. Kawai H, et al. 2003. DNA damage-induced MDMX degradation is mediated by MDM2. J. Biol. Chem. 278:45946–45953 [DOI] [PubMed] [Google Scholar]
- 26. Kruse JP, Gu W. 2009. Modes of p53 regulation. Cell 137:609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Labhart P, et al. 2005. Identification of target genes in breast cancer cells directly regulated by the SRC-3/AIB1 coactivator. Proc. Natl. Acad. Sci. U. S. A. 102:1339–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laptenko O, Beckerman R, Freulich E, Prives C. 2011. p53 binding to nucleosomes within the p21 promoter in vivo leads to nucleosome loss and transcriptional activation. Proc. Natl. Acad. Sci. U. S. A. 108:10385–10390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Le Guezennec X, Bulavin DV. 2010. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem. Sci. 35:109–114 [DOI] [PubMed] [Google Scholar]
- 30. Li C, Chen L, Chen J. 2002. DNA damage induces MDMX nuclear translocation by p53-dependent and -independent mechanisms. Mol. Cell. Biol. 22:7562–7571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lokshin M, Li Y, Gaiddon C, Prives C. 2007. p53 and p73 display common and distinct requirements for sequence specific binding to DNA. Nucleic Acids Res. 35:340–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu X, et al. 2007. The Wip1 phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell 12:342–354 [DOI] [PubMed] [Google Scholar]
- 33. Lu X, Nguyen TA, Zhang X, Donehower LA. 2008. The Wip1 phosphatase and Mdm2: cracking the “Wip” on p53 stability. Cell Cycle 7:164–168 [DOI] [PubMed] [Google Scholar]
- 34. Madden MM, et al. 2011. Synthesis of cell-permeable stapled peptide dual inhibitors of the p53-Mdm2/Mdmx interactions via photoinduced cycloaddition. Bioorg. Med. Chem. Lett. 21:1472–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Manfredi JJ. 2010. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 24:1580–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mendrysa SM, et al. 2006. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 20:16–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Migliorini D, et al. 2002. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol. Cell. Biol. 22:5527–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Minsky N, Oren M. 2004. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol. Cell 16:631–639 [DOI] [PubMed] [Google Scholar]
- 39. Minsky N, et al. 2008. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat. Cell Biol. 10:483–488 [DOI] [PubMed] [Google Scholar]
- 40. Momand J, Jung D, Wilczynski S, Niland J. 1998. The MDM2 gene amplification database. Nucleic Acids Res. 26:3453–3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Montes de Oca Luna R, Wagner DS, Lozano G. 1995. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378:203–206 [DOI] [PubMed] [Google Scholar]
- 42. Murray-Zmijewski F, Slee EA, Lu X. 2008. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 9:702–712 [DOI] [PubMed] [Google Scholar]
- 43. Okamoto K, et al. 2005. DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol. Cell. Biol. 25:9608–9620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Okamoto K, Taya Y, Nakagama H. 2009. Mdmx enhances p53 ubiquitination by altering the substrate preference of the Mdm2 ubiquitin ligase. FEBS Lett. 583:2710–2714 [DOI] [PubMed] [Google Scholar]
- 45. Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. 1992. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358:80–83 [DOI] [PubMed] [Google Scholar]
- 46. Oliner JD, et al. 1993. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362:857–860 [DOI] [PubMed] [Google Scholar]
- 47. Pan Y, Chen J. 2003. MDM2 promotes ubiquitination and degradation of MDMX. Mol. Cell. Biol. 23:5113–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pant V, Xiong S, Iwakuma T, Quintas-Cardama A, Lozano G. 2011. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc. Natl. Acad. Sci. U. S. A. 108:11995–20000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Parant J, et al. 2001. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 29:92–95 [DOI] [PubMed] [Google Scholar]
- 50. Pereg Y, et al. 2005. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc. Natl. Acad. Sci. U. S. A. 102:5056–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Phan J, et al. 2010. Structure-based design of high affinity peptides inhibiting the interaction of p53 with MDM2 and MDMX. J. Biol. Chem. 285:2174–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Phelps M, Darley M, Primrose JN, Blaydes JP. 2003. p53-independent activation of the hdm2-P2 promoter through multiple transcription factor response elements results in elevated hdm2 expression in estrogen receptor alpha-positive breast cancer cells. Cancer Res. 63:2616–2623 [PubMed] [Google Scholar]
- 53. Poyurovsky MV, et al. 2010. The C terminus of p53 binds the N-terminal domain of MDM2. Nat. Struct. Mol. Biol. 17:982–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Poyurovsky MV, Prives C. 2006. Unleashing the power of p53: lessons from mice and men. Genes Dev. 20:125–131 [DOI] [PubMed] [Google Scholar]
- 55. Qi JS, Yuan Y, Desai-Yajnik V, Samuels HH. 1999. Regulation of the mdm2 oncogene by thyroid hormone receptor. Mol. Cell. Biol. 19:864–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ries S, et al. 2000. Opposing effects of Ras on p53: transcriptional activation of mdm2 and induction of p19ARF. Cell 103:321–330 [DOI] [PubMed] [Google Scholar]
- 57. Riley T, Sontag E, Chen P, Levine A. 2008. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9:402–412 [DOI] [PubMed] [Google Scholar]
- 58. Samuels-Lev Y, et al. 2001. ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell 8:781–794 [DOI] [PubMed] [Google Scholar]
- 59. Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. 2000. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 14:289–300 [PMC free article] [PubMed] [Google Scholar]
- 60. Shvarts A, et al. 1996. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 15:5349–5357 [PMC free article] [PubMed] [Google Scholar]
- 61. Stad R, et al. 2000. Hdmx stabilizes Mdm2 and p53. J. Biol. Chem. 275:28039–28044 [DOI] [PubMed] [Google Scholar]
- 62. Szak ST, Mays D, Pietenpol JA. 2001. Kinetics of p53 binding to promoter sites in vivo. Mol. Cell. Biol. 21:3375–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tanimura S, et al. 1999. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 447:5–9 [DOI] [PubMed] [Google Scholar]
- 64. Terzian T, et al. 2007. Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol. Cell. Biol. 27:5479–5485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Urist M, Tanaka T, Poyurovsky MV, Prives C. 2004. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 18:3041–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vassilev LT, et al. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844–848 [DOI] [PubMed] [Google Scholar]
- 67. Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137:413–431 [DOI] [PubMed] [Google Scholar]
- 68. Wade M, Wang YV, Wahl GM. 2010. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 20:299–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang H, Ma X, Ren S, Buolamwini JK, Yan C. 2011. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol. Cancer Ther. 10:69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang YV, et al. 2007. Quantitative analyses reveal the importance of regulated Hdmx degradation for p53 activation. Proc. Natl. Acad. Sci. U. S. A. 104:12365–12370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu X, Bayle JH, Olson D, Levine AJ. 1993. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7:1126–1132 [DOI] [PubMed] [Google Scholar]
- 72. Xiao G, White D, Bargonetti J. 1998. p53 binds to a constitutively nucleosome free region of the mdm2 gene. Oncogene 16:1171–1181 [DOI] [PubMed] [Google Scholar]
- 73. Xu Y, Zhang J, Chen X. 2007. The activity of p53 is differentially regulated by Brm- and Brg1-containing SWI/SNF chromatin remodeling complexes. J. Biol. Chem. 282:37429–37435 [DOI] [PubMed] [Google Scholar]
- 74. Zauberman A, Barak Y, Ragimov N, Levy N, Oren M. 1993. Sequence-specific DNA binding by p53: identification of target sites and lack of binding to p53-MDM2 complexes. EMBO J. 12:2799–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhu Y, et al. 2009. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol. Cell 35:316–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.