

Abstract
Nicotine is a natural alkaloid produced by tobacco plants, and the mechanisms of its catabolism by microorganisms are diverse. In the present study, we reported the mutation, cloning, and identification of two novel genes involved in nicotine degradation from the newly isolated Pseudomonas sp. strain HZN6. Transposon mutagenesis identified a HZN6 mutant in which the nicotine-degrading pathway was blocked at pseudooxynicotine. A 3,874-bp DNA fragment flanking the transposon insertion site was obtained through self-formed adaptor PCR. Two open reading frames (designated pao and sap) were analyzed, and the deduced amino acid sequences shared 29% identity with 6-hydroxy-l-nicotine oxidase from Arthrobacter nicotinovorans and 49% identity with an aldehyde dehydrogenase from Bartonella henselae. Both pao and sap were cloned and functionally expressed in recombinant Escherichia coli BL21. The pao gene encoded a novel pseudooxynicotine amine oxidase with noncovalently bound flavin adenine dinucleotide (FAD) and exhibited substrate specificity removing the methylamine from pseudooxynicotine with the formation of 3-succinoylsemialdehyde-pyridine and hydrogen dioxide. The sap gene encoded a NADP+-dependent 3-succinoylsemialdehyde-pyridine dehydrogenase that catalyzed the dehydrogenation of 3-succinoylsemialdehyde-pyridine to 3-succinoyl-pyridine. Genetic analyses indicated that the pao gene played an essential role in nicotine or pseudooxynicotine mineralization in strain HZN6, whereas the sap gene did not. This study provides novel insight into the nicotine-degrading mechanism at the genetic level in Pseudomonas spp.
INTRODUCTION
Nicotine is a natural alkaloid produced by tobacco plants that was once used as a pesticide and is essential to the tobacco industry. Tobacco smoking is the leading cause of premature mortality, with more than 6 million tobacco-related deaths per year worldwide (3). Tobacco smoking is also a significant source of primary indoor air pollutants (24). Thirdhand smoke may pose additional indoor health risks (19). The nicotine detoxification of tobacco industry waste and the removal of nicotine from tobacco products by microbes have recently received increasing attention (20, 29).
Various pathways and genes have been reported for nicotine degradation involving an initial attack at either the pyridine or pyrrolidine rings (5, 16, 17). In the Gram-positive strain Arthrobacter nicotinovorans, the degradation pathway (the pyridine pathway) and the related genes and enzymes have been well elucidated (2, 5–7, 10–12). Although aerobic nicotine degradation has been studied in different Gram-negative Pseudomonas strains, the genes encoding enzymes in the pyrrolidine pathway involved in nicotine degradation have yet to be analyzed in detail (16). The only fully elucidated nicotine degradation pathway is the aerobic route from Pseudomonas putida S16 (31). Two functional genes were cloned and analyzed: the nicA gene converts nicotine to 3-succinoyl-pyridine (SP) via pseudooxynicotine (PN), while the hsp gene catalyzes 6-hydroxy-3-succinoyl-pyridine (HSP) to 2,5-dihydroxypyridine (DHP) (26, 27). However, no other functional genes or enzymes were found to be responsible for the degradation of nicotine in Pseudomonas until now.
In our previous work, we isolated a newly nicotine-degrading Pseudomonas sp. strain HZN6 with the pyrrolidine pathway (21). The sirA2 gene of strain HZN6 was disrupted by the Tn5 transposon and identified as essential for SP hydroxylation. However, the sirA2 cluster was different from that of other known Pseudomonas strains, indicating that the HZN6 strain might carry different genetic information. In the present study, we report the cloning, expression, and functional identification of two novel genes encoding pseudooxynicotine amine oxidase (PNAO) and 3-succinoylsemialdehyde-pyridine dehydrogenase (SAPD), which catalyze the second and third enzymatic steps of nicotine degradation in Pseudomonas sp. HZN6. This study should enhance our understanding of the genetic and biochemical diversity of nicotine degradation in Pseudomonas. This is the first study to confirm that a theoretical intermediate metabolite, 3-succinoylsemialdehyde-pyridine (SAP), is involved in nicotine degradation (31).
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains, vectors, and recombinant plasmids used in this study are listed in Table 1. Pseudomonas sp. strain HZN6 was isolated and identified as a nicotine-degrading bacterium and was deposited in the China Center for Type Culture Collection (CCTCC 2010196) (21). The Pseudomonas strains and their derivatives were grown aerobically at 30°C in LB or mineral salts medium (MSM) as previously described (21). The Escherichia coli strains were routinely cultured in LB medium at 37°C. The following antibiotics and concentrations were used: ampicillin (Ap), 100 mg/liter; chloramphenicol, (Cm), 34 mg/liter; kanamycin (Km), 50 mg/liter; and gentamicin (Gm), 50 mg/liter.
Table 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Characteristics | Reference or source |
|---|---|---|
| Pseudomonas strains | ||
| Pseudomonas sp. strain HZN6 | Apr; wild type, nicotine degrader; G− | 21 |
| N6mC8 | Apr Kmr; pao mutant of strain HZN6 by Tn5 transposon | This study |
| N6Δpao | Apr Kmr; pao::Kmr mutant of HZN6 | This study |
| N6Δsap | Apr Kmr; sap::Kmr mutant of HZN6 | This study |
| N6ΔpaoC | Apr Kmr Gmr; strain N6Δpao containing pBB-pao | This study |
| Escherichia coli strains | ||
| DH5α | F−recA1 endA1 thi-1 supE44 relA1 deoRΔ(lacZYA-argF)U169 ϕ80lacZΔM15 | TaKaRa |
| BL21(DE3) | F−ompT hsdS(rB− mB−)gal dcm lacY1(DE3) | Novagen |
| SM10λpir | thi thr leu tonA lacY supE recA::RP-4-Tc::Mu (λpir) | Lab stock |
| Plasmids | ||
| pMD18-T | Apr; T-A clone vector | TaKaRa |
| pET29a(+) | Kmr; expression vector | Novagen |
| pSC123 | Cmr Kmr; Mariner transposon | Lab stock |
| pJQ200SK | Gmr; mob+orip15A, lacZα+sacB; suicide vector | 22 |
| pBBR1-MCS5 | Gmr; broad-host-range cloning vector | 14 |
| pJQΔpao | Gmr; BamHI-SpeI fragment containing pao inserted into pJQ200SK where pao was disrupted by the kanamycin resistance gene | This study |
| pJQΔsap | Gmr; BamHI-SpeI fragment containing sap inserted into pJQ200SK where sap was disrupted by the kanamycin resistance gene | This study |
| pBB-pao | Gmr; BamHI-SpeI fragment containing pao inserted into pBBR1-MCS5 | This study |
| pETpao | Kmr; NdeI-XhoI fragment containing pao inserted into pET29a(+) | This study |
| pETsap | Kmr; NdeI-XhoI fragment containing sap inserted into pET29a(+) | This study |
Chemicals and analytical methods.
(S)-Nicotine (>99%) was obtained from Sigma-Aldrich. PN (98%) and SP (98%) were purchased from Toronto Research Chemicals, Inc. (Canada). All other chemicals were of analytical grade and commercially available.
The intermediate metabolites (PN, SAP, and SP) of nicotine were identified by liquid chromatography-mass spectrometry (LC-MS) with a Thermo DECA-60000 XLCQ Deca XP Plus instrument with an UV detector set at 263 nm. The Thermo instrument was equipped with a Grace Alltima C18 column (4.6 by 250 mm; 5 μm). The mobile phase was an acetonitrile-H2O (8 mM HCOOH) mixture (10:90 [vol/vol]; flow rate, 0.6 ml/min), and the column temperature was 30°C. The samples were ionized by electrospray with a positive polarity. The concentrations of nicotine, PN, and SP were determined by high-performance liquid chromatography (HPLC) analysis, comparing the retention times and peak areas with those of standards. HPLC was performed on a JASCO 2000 Plus HPLC (Japan). The mobile phase was a methanol-H2O (1 mM H2SO4) mixture (13:87 [vol/vol]; flow rate, 0.6 ml/min), and the other conditions were the same as for LC-MS.
DNA manipulations.
DNA techniques, such as the preparation of total DNA, plasmid isolation, restriction, ligation, and the transformation of E. coli, were performed according to standard protocols (23).
Selection for nicotine growth-deficient mutants and cloning of the mutant genes.
The generation and selection of nicotine growth-deficient mutants of Pseudomonas sp. HZN6 and the cloning of mutant genes were performed according to methods described previously (21). The mutant gene was amplified by self-formed adaptor PCR (SEFA-PCR) (30) and assembled using the Omega 2.0 software. Analysis of open reading frames (ORFs) and comparisons of amino acid or nucleotide sequences were performed with the ORF finder and BLAST programs on the NCBI website.
Gene cloning, expression, and purification.
The ORFs of the pao and sap genes without their translation stop codon were amplified by PCR (primers paoEF-NdeI/paoER-XhoI and sapEF-NdeI/sapER-XhoI; see Table S1 in the supplemental material) and inserted into the NdeI-XhoI sites of pET29a(+) (Novagen) to produce pETpao and pETsap, respectively. The sequences were verified by DNA sequencing to ensure that no mutations had been introduced during the PCR. E. coli BL21(DE3) strains carrying the resulting plasmids were grown in LB at 37°C to an optical density at 600 nm (OD600) of 0.7 to 0.8 and subsequently induced for 20 to 24 h by the addition of 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 20°C. Harvested cells were washed and disrupted by sonication. Cell debris and insoluble proteins were removed by centrifugation (12,000 × g for 15 min). The cell extracts were loaded onto a His-Bind resin (Novagen). The His6-tagged PNAO and SAPD were eluted with 120 mM imidazole after elution of the nontarget proteins with 40 mM imidazole. The enzyme was dialyzed against potassium phosphate buffer (phosphate-buffered saline [PBS], 50 mM, pH 8.0; 500 ml/time, 6 times in 24 h) using 14-kDa-molecular-size-cutoff dialysis tubing and concentrated with 10-kDa Amicon ultrafiltration tubes. All purification steps were performed at 4°C. The protein concentration was quantified by the Bradford method using bovine serum albumin as the standard (4). The native molecular masses of the purified enzymes were estimated by gel filtration chromatography with catalase (240 kDa), myosin (200 kDa), β-galactosidase (116 kDa), bovine serum albumin (66 kDa), and ovalbumin (44 kDa) as calibration standards. Experiments were performed at a flow rate of 0.2 ml/min using an AKTA purifier fast-performance liquid chromatography system (Amersham Pharmacia Biotech) and a Superdex G-200 column (Amersham Pharmacia Biotech). The buffer used was 50 mM Tris-HCl buffer, pH 8.0.
Determination of the cofactor of PNAO.
To identify the flavin compound, the purified PNAO was treated with 10% (vol/vol) trichloroacetic acid or boiled for 5 min. Both of these procedures precipitated the protein and released the flavin into solution. After the removal of the precipitate formed by centrifugation, the supernatant was used to identify the flavin compound by HPLC with a Symmetry C18 column (4.6 by 250 mm; 5 μm). The mobile phase was 10 mM ammonium acetate containing 30% methanol (vol/vol). The flow rate was 0.5 ml/min. Flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) were monitored by determining the absorbance at 265 nm. The FAD content of the sample was calculated from a standard curve in the range of 0 to 0.1 mM.
Determination of the enzyme products of PNAO.
To determine the products of PNAO, 10 μg of purified enzyme was mixed with 1 mM PN in 1 ml of PBS (50 mM, pH 8.0), and the reaction mixture was incubated at 35°C. The qualitative and quantitative analyses of H2O2 product were performed with an H2O2 quantitative assay kit (water compatible; Sangon Biotech, Shanghai, China), following the manufacturer's instructions.
To determine whether the H2O2 product was dependent on oxygen, the reaction was run in both aerobic and anaerobic conditions. PBS (20 ml, containing 10 mg/liter PNAO) was purged with high-purity nitrogen (99.99% N2) for 20 min to remove oxygen before adding the PN substrate (1 mM). An additional 20 ml of PBS without the oxygen removed served as a positive control. Samples were removed periodically to measure H2O2 formation.
The methylamine produced in the enzyme assay with PNAO was identified using a thin-layer chromatography (TLC) method similar to that of Chiribau, with silica gel HF254 and n-butanol–acetone–acetic acid–H2O (10:15:3:12 [vol/vol/vol/vol]) as the mobile phase (6). The plates were developed by spraying with a 0.1% (vol/vol) ninhydrin solution in acetone. The concentration of methylamine produced in the enzyme assay was determined by gas chromatography-mass spectrometry (GC-MS) analysis after derivatization with pentafluorobenzaldehyde (PFB) as previously described (18). In brief, the derivatization conditions were as follows: the pH of the enzyme product (1 ml) was adjusted to 12 with 2 N NaOH, and subsequently, 0.5 ml of PFB solution (10 mg/ml, in acetonitrile) was added. The mixtures were allowed to react for 30 min at 24°C and subsequently immersed in ice water for 3 min to stop the reaction. Hexane (1 ml) was added to extract the derivatives. Finally, the extracts were shaken for 1 min with 0.1 N NaOH (1 ml) to convert the PFB to a geminal diol, which could be removed by aqueous phase partitioning. The samples were analyzed by GC-MS (Agilent 7890A-5975C) with an HP-5 MS column (30 m; 0.25-mm inner diameter [i.d.]; 0.25-μm film thickness), and the detection conditions were the same as previously reported (18). Standard methylamine solutions of 0.2, 0.4, 0.6, 0.8, and 1.0 mM were used for quantitative analysis after derivatization with PFB using the same procedure.
Enzyme assays.
PNAO activity was determined by measuring the formation of H2O2. The reaction mixture contained purified PNAO (1 μg) and PBS (50 mM, pH 8.0) in a final volume of 1 ml. The reaction was started by the addition of PN. For kinetic parameter studies, the PN was diluted into at least 10 different concentrations around the dissociation constant (Km) value. The kinetic value was obtained from the Michaelis-Menten equation against various H2O2 concentrations.
A coupled PNAO-SAPD reaction was performed in 1 ml of PBS (50 mM, pH 8.0). The reaction mixture contained purified PNAO (10 μg), NADP+ (50 μM), and an appropriate amount of cell extracts of induced E. coli BL21(DE3) cells carrying pETsap. The reaction was initiated by the addition of PN (50 μM), and the reduction of NADP+ was monitored at 340 nm with a UV-visible (UV-vis) spectrophotometer (JASCO V-550; Japan). A molar extinction coefficient of 6.22 mM−1 cm−1 at 340 nm was used for NADPH. Cell extracts replaced with induced E. coli BL21(DE3) strains carrying pET29a(+) served as negative controls.
Gene disruption and complementation in Pseudomonas sp. HZN6.
The in-frame disruption of the pao and sap genes in Pseudomonas sp. strain HZN6 was performed using suicide plasmid pJQ200SK and a two-step homologous recombination method, as described previously (21). Briefly, to disrupt the pao gene, a 1.749-kb fusion product was constructed by PCR, which consisted of an upstream sequence of the pao gene, kanamycin resistance gene, and downstream sequence using primers paoUF-BamHI/paoUR, KmF/KmR, and paoDF/paoDR-SpeI (see Table S1 in the supplemental material), respectively. Similarly, a 1.731-kb fusion product was constructed by PCR to disrupt the sap gene using primers sapUF-BamHI/sapUR, KmF/KmR, and sapDF/sapDR-SpeI (Table S1). These two fusion products were ligated into pJQ200SK at the SpeI-BamHI site, producing plasmids pJQΔpao and pJQΔsap. The resultant plasmids were introduced into the Pseudomonas sp. HZN6 strain via SM10λpir. Single-crossover clones were selected on LB plates containing Ap, Km, and Gm. Double-crossover mutants were screened on LB plates containing 10% (wt/vol) sucrose and Ap and Km. The disruption of the pao and sap genes in these strains was confirmed by PCR. The pao and sap in-frame-disrupted strains were designated N6Δpao and N6Δsap, respectively.
The pBB-pao plasmid was constructed for the complementation of the N6Δpao strain. The pao gene was amplified using primers paoF-BamHI/paoR-SpeI (see Table S1 in the supplemental material), the PCR product was digested using BamHI and SpeI, and the fragments were ligated into BamHI-XbaI-digested pBBR1MCS-5, yielding the pBB-pao plasmid (see Fig. 1). The pBB-pao plasmid was transferred into the N6Δpao strain by biparental mating to generate the complementation strain N6ΔpaoC.
Fig 1.

Organization of the 3,874-bp DNA fragment of Pseudomonas sp. strain HZN6. The gray pencil shapes indicate the sizes and directions of the pao and sap genes. The short black vertical-pointing arrow above the pao gene indicates the Tn5 insert site. The restriction sites BamHI (B) and SpeI (S) are shown.
Nucleotide sequence accession number.
The nucleotide sequence of the 3,874-bp DNA fragment from Pseudomonas sp. strain HZN6 was deposited in the GenBank database under the accession number JN391188.
RESULTS
Isolation and phenotypic characterization of nicotine growth-deficient mutant N6mC8.
Approximately 8,000 transconjugants of the Tn5-mutageneized genome library were generated. One mutant (designated N6mC8), which was unable to use nicotine as the sole carbon source, was selected for further research. To determine which step in the pathway of nicotine was blocked, mutant N6mC8 was added to MSM containing 1 mM nicotine with a large inoculum size (30% [vol/vol]). After 4 h, nicotine was completely degraded, and one intermediate accumulated and was not further degraded (see Fig. S1 in the supplemental material). The HPLC and LC-MS results showed that the intermediate was PN (0.94 ± 0.02 mM). The ratio of the consumption of nicotine to the accumulation of PN was approximately 1:1. The presence of PN was also reconfirmed with the retention time of the authentic sample.
Cloning, sequencing, and analysis of the interrupted gene of mutant N6mC8.
SEFA-PCR was performed to amplify the 5′ and 3′ flanking sequences of the Tn5 transposon. A 3,874-bp DNA fragment was amplified and assembled (Fig. 1). The Tn5 transposon was inserted at the 1,607-bp site. DNA sequencing identified a 1,494-bp ORF (positions 347 to 1840) flanked by the insert site of the Tn5 transposon. This ORF, designated pao, encodes a 497-residue protein with a predicted molecular mass of 54.101 kDa. The G+C content was 50.67%. Compared with the known enzymes available from the Protein Data Bank (PDB, NCBI database), the predicted protein showed the highest similarity with several flavin-containing amine oxidases, e.g., the 6-hydroxy-l-nicotine oxidase (6HLNO) from Arthrobacter nicotinovorans (29% identity) (11, 12), the monoamine oxidase from Aspergillus niger (28% identity) (1), the human monoamine oxidase A (23% identity) (9), l-amino acid oxidase from Calloselasma rhodostoma (22% identity) (13), and the polyamine oxidase Fms1 from Saccharomyces cerevisiae (24% identity) (15).
In the 3,874-bp sequence, an additional 1,434-bp ORF (positions 1833 to 3266) was discovered (Fig. 1). This ORF, designated sap, was similar to aldehyde dehydrogenase family protein genes encoding a 477-residue protein with a predicted molecular mass of 51.246 kDa. The G+C content was 53.98% and showed 49% and 41% amino acid sequences identities to aldehyde dehydrogenase from Bartonella henselae (PDB 3I44_A) and Mycobacterium tuberculosis (PDB 3B4W_A), respectively. In addition, there was an 8-bp overlap region between the pao and sap genes (Fig. 1).
On the basis of the known reactions catalyzed by amine oxidase and aldehyde dehydrogenase and the nicotine degradation pathway, we hypothesize that the pao gene converts PN to SAP and that the sap gene converts SAP to SP.
Overexpression and purification of PNAO and SAPD.
The recombinant PNAO and SAPD were overexpressed in E. coli BL21(DE3) as C-terminal His6-tagged fusion proteins and purified from the crude extract using Ni-nitrilotriacetic acid affinity chromatography. Single bands at apparent molecular masses of 55 kDa and 52 kDa were detected by SDS-PAGE, corresponding to the molecular masses of His6-tagged PNAO and SAPD, respectively, as deduced from the amino acid sequences (see Fig. S2 in the supplemental material). The purified PNAO was yellow colored, indicating that it was a flavoprotein. After the sample was precipitated with trichloroacetic acid and centrifuged, it formed a white protein pellet and a yellow supernatant, indicating that the flavin cofactor was noncovalently bound to the protein. The purified His6-tagged SAPD was colorless, indicating that there was no bound flavin prosthetic group. As determined by gel filtration chromatography, the PNAO was considered a homodimer and the SAPD was most like a homotetramer.
FAD is a PNAO cofactor.
The spectrum of the His6-tagged PNAO showed the characteristics of typical flavoprotein. The visible and UV absorption spectra of the purified enzyme exhibited absorption maxima centered at 277, 376, and 460 nm (Fig. 2). The visible spectrum of the flavin released from the enzyme after boiling for 5 min was identical to that of FAD. HPLC analysis also revealed that the flavin was FAD; no FMN was detected. The content of FAD was determined to be 0.51 ± 0.01 mol of FAD per mol of subunit (average of 10 determinations). In addition, 1 mM FAD was added to the cultures during the final 2 h of induction, but the ratio of FAD to enzyme did not change.
Fig 2.

Visible and UV absorption (Abs) spectra of purified PNAO (dotted line), FAD released from the PNAO (continuous black line), and authentic FAD (dashed line). The inset shows enlarged spectra.
PNAO catalyzes the deamination of PN to SAP.
The purified PNAO was added to PBS to catalyze the biotransformation of PN. After 2 h, the reaction mixture was applied to TLC, and methylamine was identified (Fig. 3). The product methylamine was further confirmed by GC-MS after derivatization with PFB. The characteristic fragment ion peaks of the derivative were at m/z = 209, 208, 161, and 117 (see Fig. S3 in the supplemental material), which were identical to those in a previous report (18). The final concentrations of methylamine and H2O2 were determined to be 0.89 ± 0.03 mM and 0.93 ± 0.02 mM, respectively. Additionally, little or no H2O2 was detected under anaerobic conditions, indicating that the reaction was oxygen dependent (Fig. S4). HPLC results showed that PN was degraded completely, and one intermediate was accumulated (Fig. S5a). The LC-MS results indicated that it was SAP (molecular ion at m/z 164.2[M+H]+, fragment ions at m/z 136.2[M-CHO]+) (Fig. S5b). This intermediate was unstable and was easily transformed into other compounds after 6 h in the reaction mixture. The ratio of the PN consumption to the production of methylamine and H2O2 was approximately 1:1:1.
Fig 3.
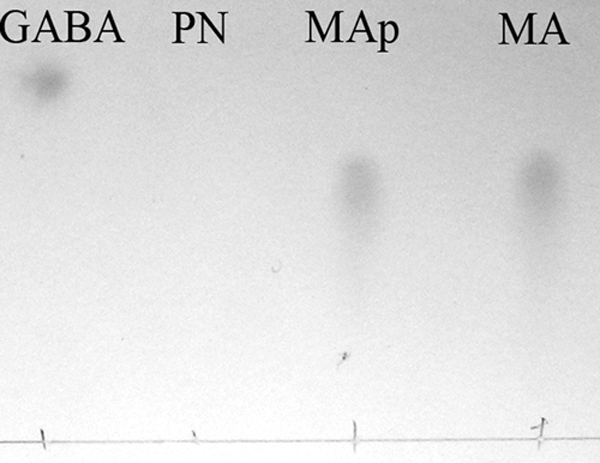
TLC analysis of the product of the enzyme reaction and standards. GABA, γ-aminobutyrate; PN, pseudooxynicotine, which does not react with ninhydrin; MAp, methylamine formed in the PNAO reaction with PN as a substrate; MA, methylamine.
The optimal pH of PNAO was observed to be approximately 8.0, and the optimal temperature was 35°C. The apparent Km and kcat values for PN were determined to be 0.247 ± 0.019 mM and 151 ± 14 s−1, respectively. The enzymatic activity was strongly inhibited by Ag+, Co2+, Cu2+, and Hg2+. In contrast, Ca2+, Mg2+, Mn2+, Ni2+, and Zn2+ had no significant effects on the enzyme. PNAO had no activities with the following com-pounds tested as substrates: nicotine, γ-N-methylaminobutyrate, γ-aminobutyrate, spermidine, spermine, sarcosine, methylamine, dimethylamine, dimethylglycine, and 3-(methylamino)propyl-amine, indicating that the PNAO activity was specific for PN.
SAPD catalyzes the dehydrogenation of SAP to SP.
Cell extracts of E. coli BL21(pETsap) were found to contain 3-succinoylsemialdehyde-pyridine dehydrogenase activities. The consumption of PN and NADP+ (250 nm) and the accumulation of NADPH (340 nm) during the coupled reaction are shown in Fig. 4. HPLC analysis also showed that an approximately equimolar amount of SP (48 ± 3 μM) was produced from PN. Neither SP nor NADPH was detected in the negative controls, when the expression vector contained no insert. No activity was detected when NADP+ was replaced with NAD+, indicating that SAPD was NADP+ dependent. During the coupled enzymatic reaction, little or no SAP was detected, indicating that SAP was converted into SP immediately after it was produced by PNAO (see Fig. S6 in the supplemental material). According to these results, we could conclude that SAPD catalyzes the dehydrogenation of SAP to SP. In addition, the cell extracts of E. coli BL21(pETsap) also utilized formaldehyde and acetaldehyde as substrates.
Fig 4.

Spectrophotometric changes during formation of NADPH from PN by PNAO and cell extracts of E. coli BL21(pETsap). The black arrows indicate the directions of spectral changes.
Genetic and functional analyses of the pao and sap genes in strain HZN6.
To investigate the functions of the pao and sap genes in PN degradation in vivo, derivatives of strain HZN6 with in-frame deletions were individually constructed and functionally analyzed. Strain N6Δpao was unable to utilize nicotine or PN as the sole carbon source but could convert nicotine into PN, which was the same phenotypic characterization as for mutant N6mC8 (see Fig. S1 in the supplemental material). The complemented strain N6ΔpaoC recovered the ability to degrade PN at the wild-type (WT) level (Fig. 5). Although strain N6Δsap was still able to grow on PN, the cell growth (a growth rate of 0.010 ± 0.002 h−1 compared with 0.017 ± 0.002 h−1 for the WT strain) and the rate of PN removal (0.107 ± 0.013 mM h−1 for PN degradation compared with 0.141 ± 0.016 mM h−1 for the WT strain) were initially slightly reduced. However, after the N6Δsap strain was cultured with PN for several generations, the PN degradation characteristics reached the wild-type level (Fig. 5).
Fig 5.

Time course of PN degradation and cell growth in cultures of strain HZN6 and its derivatives. PN concentrations (left y axis) (black symbols) in strain HZN6 (■), strain N6Δsap (▲), N6Δsap (●) after culturing with PN for several generations (dotted line), and N6ΔpaoC (▼) (dash dot line) are shown. Cell growth (right y axis [lg, log]) (open symbols) of HZN6 (□), N6Δsap (▵), N6Δsap (○) after culturing with PN for several generations (dotted line), and N6ΔpaoC (▿) (dash dot line) is shown.
DISCUSSION
The degradation mechanism of nicotine in Pseudomonas has been studied for more than 50 years (21, 28). However, there have been few studies at the genetic level reported so far. In the present study, we cloned two novel genes involved in the initial steps of nicotine degradation by strain HZN6, which differ from any other reported mechanisms (Fig. 6D). The pao gene was cloned and expressed in E. coli, and the purified enzyme was shown to be a novel pseudooxynicotine amine oxidase, which catalyzes the conversion of PN to SAP. The sap gene was expressed, and the cell extracts showed NADP+-dependent 3-succinoylsemialdehyde-pyridine dehydrogenase activity, which was responsible for the transformation of SAP to SP.
Fig 6.

Proposed pathways for nicotine degradation by microorganisms. (A) Demethylation pathway used by Aspergillus oryzae 112822. (B) Pyridine pathway used by Arthrobacter nicotinovorans plasmid pAO1. (C) Pyrrolidine pathway used by Pseudomonas putida S16. (D) Initial steps of nicotine degradation by Pseudomonas sp. strain HZN6. The enzyme steps catalyzed by PNAO and SAPD are indicated in the box. G+ bacterium, Gram-positive bacterium; G−, Gram-negative bacterium.
On the basis of our results, we concluded that the stoichiometry of the pseudooxynicotine amine oxidase reaction is as follows: PN was oxidatively deaminated using the noncovalent cofactor FAD in a reaction consuming O2 and H2O and producing SAP, methylamine, and H2O2 (Fig. 6D). According to the enzymatic reaction, PNAO was shown to be a typical amine oxidase. By searching in the Enzyme Database (http://www.brenda-enzymes.org/), we classified PNAO as EC 1.5.3.-, which is a group of enzymes that act on the CH-NH group of donors and use the O2 group as the accepter.
The stoichiometry of FAD to the PNAO subunit in all preparations obtained thus far is only approximately 0.5. Findings of 0.5 mol of FAD/mol of protein monomer have been reported for glycerol-3-phosphate dehydrogenase from E. coli (25) and protoporphyrinogen oxidase from Myxococcus xanthus (8). This FAD content is not a whole number, probably because the cells are unable to synthesize sufficient FAD or because there is a partial loss of FAD in the purification process. However, similar to the protoporphyrinogen oxidase from Myxococcus xanthus, the FAD content of the purified enzyme did not discernibly change after the addition of 1 mM FAD to the culture during the last 2 h of induction. Furthermore, the activity of the purified PNAO also did not significantly increase after the addition of a 5-fold excess of exogenous FAD. Understanding why the content of FAD was 0.5 mol/monomer requires further study.
The gene disruption and complementation results indicated that the pao gene was essential for the cell growth of strain HZN6. However, when the sap gene was knocked out, the derivative strain N6Δsap could still use PN as the sole carbon source. The most reasonable explanation was that SAP was easily oxidized to SP by other aldehyde dehydrogenases in the mutant strain. These results might also explain why no SAP was detected during the degradation of nicotine by strain HZN6 or its derivatives.
Three different nicotine degradation pathways by microorganisms have been reported thus far: the demethylation pathway (Fig. 6A), the pyridine pathway (Fig. 6B), and the pyrrolidine pathway (Fig. 6C). The demethylation pathway used by the fungal strain Aspergillus oryzae 112822 starts with the elimination of the methyl group from the pyrrolidine ring to produce nornicotine (17). Subsequently, N-methylnicotinamide and 2,3-dihydroxypyridine are produced after pyrrolidine ring cleavage and side chain removal. No genes or enzymes have been reported to be involved in this pathway. In addition, no compounds were shared between the pathways degraded by the A. oryzae 112822 strain and strain HZN6.
The pyridine pathway used by Arthrobacter nicotinovorans plasmid pAO1 is shown in Fig. 6B. Nicotine was hydroxylated at position 6 of the pyridine ring, and the pyrrolidine ring was later cleaved. Comparing the two different degradation processes, the corresponding steps catalyzed by PNAO and SAPD in strain HZN6 were the deamination of γ-N-methylaminobutyrate by an amine oxidase (AO) and the dehydrogenation of succinic semialdehyde by succinic semialdehyde dehydrogenase (SsaDH) in A. nicotinovorans pAO1, respectively (6). Similar to the AO of pAO1, the FAD cofactor of PNAO was noncovalently bound to the apoprotein, and the PNAO activity was also substrate specific. However, the C-terminal fingerprint sequences SGGCY and AGGAY were not found in PNAO. Database searches revealed that the amino acid sequence of PNAO showed 29% identity to the 6HLNO from Arthrobacter nicotinovorans pAO1. The 6HLNO also uses a noncovalent FAD as the cofactor. According to the amino acid sequence alignment results, the most similar regions between PNAO and 6HLNO were the three FAD binding domains, which are the residues 1 to 81, 200 to 285, and 361 to 425 of 6HLNO (12) and the corresponding residues 59 to 135, 258 to 344, and 425 to 489 of PNAO, respectively. Although they shared 29% sequence identity with one another, the enzymatic reactions catalyzed by 6HLNO and PNAO were distinct. The pyrrolidine ring of 6-hydroxy-l-nicotine was oxidatively cleaved by 6HLNO, yielding 6-hydroxypseudooxynicotine. The secondary-amine-containing compound was the product of 6HLNO while serving as a substrate of PNAO. In addition, similar to SsaDH, the SAPD activity was NADP+ dependent and not substrate specific (6).
Because the PNAO showed no activity to nicotine, according to the nicotine-degrading mechanism of Arthrobacter nicotinovorans pAO1 (5, 6), we can infer that the initial steps of nicotine degradation are catalyzed by the products of three individual genes in Pseudomonas sp. strain HZN6 (Fig. 6D). The first enzymatic step was catalyzed by an amine oxidase, similar to the step catalyzed by 6HLNO (11, 12), converting nicotine to N-methylmyosmine and spontaneously yielding PN. The second and third enzymatic steps were catalyzed by PNAO and SAPD, respectively.
The pyrrolidine pathway was well determined in Pseudomonas putida S16 (Fig. 6C). Additionally, we determined that strain HZN6 also followed the pyrrolidine pathway. However, in contrast to strain HZN6, nicotine was converted directly into SP by nicotine oxidoreductase (NicA) in Pseudomonas putida S16 strain (26). One protein catalyzed three enzymatic steps. The amino acid sequence of NicA showed 40% similarity to NADH dehydrogenase subunit I and cytochrome c oxidase subunit I from eukaryotes, indicating that NicA contained oxidase and dehydrogenase activities. The oxidase activity of NicA might be equal to the first unknown amine oxidase and PNAO activities of strain HZN6, catalyzing nicotine to PN and SAP. Moreover, the dehydrogenase activity of NicA was the counterpart of SAPD activity, converting SAP into SP. Therefore, we may conclude that there is a novel metabolic mechanism of nicotine at the genetic level in Pseudomonas sp. strain HZN6, which is different from any other reported mechanisms. Furthermore, cloning the gene responsible for the first step, i.e., catalyzing nicotine to PN, is currently in progress.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to three anonymous reviewers for helpful comments on the manuscript. We also acknowledge Roderich Brandsch (Institute of Biochemistry and Molecular Biology, University of Freiburg) and Weiwu Wang (Department of Microbiology, Nanjing Agricultural University) for useful suggestions on the enzyme study and Quandong Yuan and Jihong Jiang (Research Center of Environmental Science, Zhejiang University of Technology) for excellent technical assistance.
This work was supported by the National Natural Science Foundation of China (grants 20837002 and 21007058) and the National Basic Research Program of China (grant 2009CB421603).
Footnotes
Published ahead of print 20 January 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Atkin KE, et al. 2008. The structure of monoamine oxidase from Aspergillus niger provides a molecular context for improvements in activity obtained by directed evolution. J. Mol. Biol. 384: 1218–1231 [DOI] [PubMed] [Google Scholar]
- 2.Baitsch D, Sandu C, Brandsch R, Igloi GL. 2001. Gene cluster on pAO1 of Arthrobacter nicotinovorans involved in degradation of the plant alkaloid nicotine: cloning, purification, and characterization of 2,6-dihydroxypyridine 3-hydroxylase. J. Bacteriol. 183: 5262–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benowitz NL. 2010. Nicotine addiction. N. Engl. J. Med. 362: 2295–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254 [DOI] [PubMed] [Google Scholar]
- 5.Brandsch R. 2006. Microbiology and biochemistry of nicotine degradation. Appl. Microbiol. Biotechnol. 69: 493–498 [DOI] [PubMed] [Google Scholar]
- 6.Chiribau CB, et al. 2006. Final steps in the catabolism of nicotine. Deamination versus demethylation of gamma-N-methylaminobutyrate. FEBS J. 273: 1528–1536 [DOI] [PubMed] [Google Scholar]
- 7.Chiribau CB, Sandu C, Fraaije M, Schiltz E, Brandsch R. 2004. A novel gamma-N-methylaminobutyrate demethylating oxidase involved in catabolism of the tobacco alkaloid nicotine by Arthrobacter nicotinovorans pAO1. Eur. J. Biochem. 271: 4677–4684 [DOI] [PubMed] [Google Scholar]
- 8.Dailey HA, Dailey TA. 1996. Protoporphyrinogen oxidase of Myxococcus xanthus. Expression, purification, and characterization of the cloned enzyme. J. Biol. Chem. 271: 8714–8718 [DOI] [PubMed] [Google Scholar]
- 9.Hubalek F, et al. 2005. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J. Biol. Chem. 280: 15761–15766 [DOI] [PubMed] [Google Scholar]
- 10.Igloi GL, Brandsch R. 2003. Sequence of the 165-kilobase catabolic plasmid pAO1 from Arthrobacter nicotinovorans and identification of a pAO1-dependent nicotine uptake system. J. Bacteriol. 185: 1976–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kachalova G, Decker K, Holt A, Bartunik HD. 2011. Crystallographic snapshots of the complete reaction cycle of nicotine degradation by an amine oxidase of the monoamine oxidase (MAO) family. Proc. Natl. Acad. Sci. U. S. A. 108: 4800–4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kachalova GS, et al. 2010. Crystal structure analysis of free and substrate-bound 6-hydroxy-l-nicotine oxidase from Arthrobacter nicotinovorans. J. Mol. Biol. 396: 785–799 [DOI] [PubMed] [Google Scholar]
- 13.Kopacz MM, Rovida S, van Duijn E, Fraaije MW, Mattevi A. 2011. Structure-based redesign of cofactor binding in putrescine oxidase. Biochemistry 50: 4209–4217 [DOI] [PubMed] [Google Scholar]
- 14.Kovach ME, et al. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166: 175–176 [DOI] [PubMed] [Google Scholar]
- 15.Landry J, Sternglanz R. 2003. Yeast Fms1 is a FAD-utilizing polyamine oxidase. Biochem. Biophys. Res. Commun. 303: 771–776 [DOI] [PubMed] [Google Scholar]
- 16.Li HJ, Li XM, Duan YQ, Zhang KQ, Yang JK. 2010. Biotransformation of nicotine by microorganism: the case of Pseudomonas spp. Appl. Microbiol. Biotechnol. 86: 11–17 [DOI] [PubMed] [Google Scholar]
- 17.Meng XJ, Lu LL, Gu GF, Xiao M. 2010. A novel pathway for nicotine degradation by Aspergillus oryzae 112822 isolated from tobacco leaves. Res. Microbiol. 161: 626–633 [DOI] [PubMed] [Google Scholar]
- 18.Ngim KK, Ebeler SE, Lew ME, Crosby DG, Wong JW. 2000. Optimized procedures for analyzing primary alkylamines in wines by pentafluorobenzaldehyde derivatization and GC-MS. J. Agric. Food Chem. 48: 3311–3316 [DOI] [PubMed] [Google Scholar]
- 19.Petrick LM, Svidovsky A, Dubowski Y. 2011. Thirdhand smoke: heterogeneous oxidation of nicotine and secondary aerosol formation in the indoor environment. Environ. Sci. Technol. 45: 328–333 [DOI] [PubMed] [Google Scholar]
- 20.Piotrowska-Cyplik A, Olejnik A, Cyplik P, Dach J, Czarnecki Z. 2009. The kinetics of nicotine degradation, enzyme activities and genotoxic potential in the characterization of tobacco waste composting. Bioresource Technol. 100: 5037–5044 [DOI] [PubMed] [Google Scholar]
- 21.Qiu J, et al. 2011. A sirA-like gene, sirA2, is essential for 3-succinoyl-pyridine metabolism in the newly isolated nicotine-degrading Pseudomonas sp. HZN6 strain. Appl. Microbiol. Biotechnol. 92: 1023–1032 [DOI] [PubMed] [Google Scholar]
- 22.Quandt J, Hynes MF. 1993. Versatile suicide vectors which allow direct selection for gene replacement in gram-negative bacteria. Gene 127: 15–21 [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J, Fritsch E, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 24.Schick S, Glantz S. 2005. Scientific analysis of second-hand smoke by the tobacco industry, 1929–1972. Nicotine Tob. Res. 7: 591–612 [DOI] [PubMed] [Google Scholar]
- 25.Schryvers A, Lohmeier E, Weiner JH. 1978. Chemical and functional properties of the native and reconstituted forms of the membrane-bound, aerobic glycerol-3-phosphate dehydrogenase of Escherichia coli. J. Biol. Chem. 253: 783–788 [PubMed] [Google Scholar]
- 26.Tang HZ, et al. 2009. Novel nicotine oxidoreductase-encoding gene involved in nicotine degradation by Pseudomonas putida strain S16. Appl. Environ. Microbiol. 75: 772–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang HZ, et al. 2008. A novel gene, encoding 6-hydroxy-3-succinoylpyridine hydroxylase, involved in nicotine degradation by Pseudomonas putida strain S16. Appl. Environ. Microbiol. 74: 1567–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wada E, Yamasaki K. 1953. Mechanism of microbial degradation of nicotine. Science 117: 152–153 [DOI] [PubMed] [Google Scholar]
- 29.Wang M, Yang G, Min H, Lv Z, Jia X. 2009. Bioaugmentation with the nicotine-degrading bacterium Pseudomonas sp. HF-1 in a sequencing batch reactor treating tobacco wastewater: degradation study and analysis of its mechanisms. Water Res. 43: 4187–4196 [DOI] [PubMed] [Google Scholar]
- 30.Wang SM, He J, Cui ZL, Li SP. 2007. Self-formed adaptor PCR: a simple and efficient method for chromosome walking. Appl. Environ. Microbiol. 73: 5048–5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang SN, Liu Z, Tang HZ, Meng J, Xu P. 2007. Characterization of environmentally friendly nicotine degradation by Pseudomonas putida biotype A strain S16. Microbiology 153: 1556–1565 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.