

Abstract
Escherichia coli has the potential to be a powerful biocatalyst for the conversion of lignocellulosic biomass into useful materials such as biofuels and polymers. One important challenge in using E. coli for the transformation of biomass sugars is diauxie, or sequential utilization of different types of sugars. We demonstrate that, by increasing the intracellular levels of the transcription factor XylR, the preferential consumption of arabinose before xylose can be eliminated. In addition, XylR augmentation must be finely tuned for robust coutilization of these two hemicellulosic sugars. Using a novel technique for scarless gene insertion, an additional copy of xylR was inserted into the araBAD operon. The resulting strain was superior at cometabolizing mixtures of arabinose and xylose and was able to produce at least 36% more ethanol than wild-type strains. This strain is a useful starting point for the development of an E. coli biocatalyst that can simultaneously convert all biomass sugars.
INTRODUCTION
Lignocellulosic biomass is the most abundant renewable organic feedstock on the planet, with production of at least 1.3 billion tons as a feasible, short-term goal (8). Through the action of whole-cell biocatalysts such as Escherichia coli and Saccharomyces cerevisiae, biomass sugars can be converted into liquid fuels capable of replacing petroleum-based fuels such as gasoline (17) and diesel (19). As a feedstock for the chemical industry, these sugars can be transformed into polymers, fine chemicals, and drugs (23). In addition, growth of cellulosic biomass removes CO2 from the atmosphere, potentially slowing anthropogenic climate change (8).
Plants are composed mainly of three types of polymers: cellulose, lignin, and hemicellulose. Cellulose consists of linear chains of glucose arranged in β(1→4) linkages (14). Lignin is composed of phenylpropanoids such as sinapinic acid and coniferyl alcohol (2). Hemicellulose, comprising as much as 45% (12) of the dry weight of some plants, is a mixed polysaccharide consisting of xylose (X), mannose, galactose, and arabinose (A) (8). Of these, xylose and arabinose are the most abundant, representing up to 95% of the total hemicellulose sugars in hardwood and biofuel crops such as switchgrass.
For lignocellulosic feedstocks to displace fossil fuels, it is essential to develop biocatalysts that efficiently metabolize each type of lignocellulosic sugar (8). E. coli is a promising candidate for the production of biofuel, because it is capable of consuming all sugars present in plant material. However, E. coli displays a preference among sugars known as diauxie. In a culture containing xylose and arabinose, arabinose is consumed first, followed by xylose. This complicates the use of natural E. coli strains for the complete bioconversion of hemicellulose sugar mixtures into advanced fuels in fed-batch processes. The presence of arabinose inhibits the consumption of xylose, which builds up in the medium rather than converting to the desired product. Thus, the development of strains that coutilize hemicellulose sugars would be an important milestone in the effort to develop efficient methods for using lignocellulosic material as a feedstock for chemicals and fuels (12, 21).
Most previous work done to develop strains capable of sugar coutilization has focused on glucose and xylose (5, 7). These are the two most abundant sugars in lignocellulosic biomass (16). Wild-type (WT) E. coli strains consume glucose and then xylose (5). Disrupting the ptsG gene, which codes for the transmembrane domain of the glucose transporter, allows coconsumption of these sugars by activating adenylate cyclase. Mutants with a constitutively active cyclic AMP (cAMP) receptor protein (CRP) are also able to simultaneously consume glucose and xylose (11). With the broader goal of cometabolizing all biomass sugars, it would also be necessary to eliminate the diauxie between xylose and arabinose, especially since arabinose represses consumption of the more abundant xylose. It was shown that this phenomenon is caused by the interaction between AraC and xyl-associated promoters (3), but at the time that this was written, no one had yet developed a strain that coutilizes both sugars.
Xylose and arabinose are metabolized in similar fashions. Each sugar is imported into the cell by a high-affinity ABC transporter: AraFGH for arabinose (6) and XylFGH for xylose (18). In addition, E. coli has two other transporters for arabinose and xylose, AraE and XylE, respectively, that are low-affinity, H+-sugar symporters. These proteins have been shown to be less important for sugar metabolism than the ABC transporters (10). The genes in the araBAD regulon encode the isomerases and kinase necessary to convert intracellular arabinose to the pentose-phosphate intermediate xylulose-5-phosphate (X5P). Similarly, the two proteins encoded by the xylAB operon are responsible for metabolizing xylose to X5P. Finally, transcription of the genes necessary for the consumption of each sugar is regulated by a sugar-responsive transcription factor, XylR for xylose and AraC for arabinose. These sugar-responsive transcription factors also play a role in arabinose-xylose diauxie. Desai and Rao (3) demonstrated that E. coli's preference for arabinose over xylose is mediated by AraC (Fig. 1). In addition to those present in the arabinose promoters, such as ParaB, there are AraC binding sites in the promoters of genes for xylose consumption, including xylAB. Presumably, AraC binds these xylose promoters in the presence of arabinose and sterically occludes the binding of XylR, preventing transcription of the transporters and enzymes required for xylose utilization.
Fig 1.

Mechanism for arabinose-xylose diauxie. Arabinose preference is mediated by AraC. In E. coli growing in xylose, the xylose-XylR complex binds to xyl promoters and activates transcription of genes required for xylose consumption. In the presence of xylose and arabinose, arabinose-bound AraC displaces XylR from xyl promoters and represses those genes.
Therefore, one possible strategy to eliminate preferential consumption of arabinose is to modify E. coli so that AraC no longer binds to xyl promoters. There are two ways to accomplish this goal: one could mutate AraC so that it no longer recognizes the xyl promoter sequences or mutate the AraC or XylR binding site in each xyl promoter so that it retains the ability to bind XylR but not AraC. As AraC-DNA interaction is complex, involving DNA looping and binding-site switching (6), we focused on the second strategy. In initial experiments, we were able to generate mutant xyl promoters that were not repressed in the presence of arabinose but were still activated by xylose. However, these modifications changed the strength of each promoter and ultimately led to poorer xylose utilization.
In this context, we investigated the role of the proteins required for xylose utilization. The central issue in this study was to determine which of these repressed genes is responsible for the arabinose-xylose diauxie. To address that issue, xylAB, xylFGH, and xylR were expressed in separate strains of E. coli growing in minimal medium supplemented with arabinose and xylose as the sole carbon sources. Here we demonstrate that production of XylR at the appropriate level relieves the arabinose-xylose diauxie and allows E. coli to utilize both sugars simultaneously. Finally, we show that a strain coutilizing arabinose and xylose is more effective at converting hemicellulose sugar mixtures into ethanol.
MATERIALS AND METHODS
Plasmids and strains.
Escherichia coli DH10b [F− endA1 recA1 galE15 galK16 nupG rpsL ΔlacX74 ϕ80dlacZΔM15 araD139 Δ(ara,leu)7697 mcrA Δ(mrr-hsdRMS-mcrBC) λ−] was used for plasmid construction and propagation (Table 1). All strains used for sugar consumption assays and green fluorescent protein (GFP) transcription assays were isogenic derivatives of E. coli MG1655 λrph-1 (WT). Cells were maintained in Luria broth (LB). For plasmid selection, LB was supplemented with 100 μg/ml carbenicillin or 50 μg/ml kanamycin where appropriate. MOPS (morpholinepropanesulfonic acid) minimal medium (MMM) contained 1× MOPS and 1× K2HPO4 solutions from Teknova (Hollister, CA) supplemented with 0.4% (wt/vol) sugar and carbenicillin (100 μg/ml) or kanamycin (50 μg/ml) where appropriate. All plasmid construction was performed using Gibson isothermal DNA assembly (4) unless noted otherwise. All PCRs were performed using Phusion DNA polymerase from New England Biolabs (Ipswich, MA).
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| MG1655 | λrph-1 (WT) | Coli Genetic Stock Center |
| ΔaraD | MG1655 araD::kanr | This study |
| araD-xylR | MG1655 araD-xylR | This study |
| Mut12 | MG1655 araD-xylR-(SD Opt) | This study |
| Plasmids | ||
| pUA66 | SC101, Kanr | 22 |
| pBbS8A-RFP | SC101, Ampr, ParaBAD-mRFP | 13 |
| pBbS8A-xylR | SC101, Ampr, ParaBAD-xylR | This study |
| pBbS8A-xylFGH | SC101, Ampr, ParaBAD-xylFGH | This study |
| pUA66-GFP | SC101, Kanr, PxylF-const-gfpmut2 | This study |
| pUA66-xylR | SC101, Kanr, PxylF-const-xylR | This study |
| pUA66-xylFGH | SC101, Kanr, PxylF-const-xylFGH | This study |
| pUA66-xylAB | SC101, Kanr, PxylF-const-xylAB | This study |
| pKD46 | repA101ts, Ampr, ParaBAD-lambda Red | 1 |
| pBb-araD-xylR | SC101, Ampr, araD-xylR | This study |
| pBb-araD-xylR-Opt | SC101, Ampr, araD-SD-OPT-xylR | This study |
| pUA66-ParaB | SC101, Kanr, ParaB-gfpmut2 | 22 |
| pUA66-PxylA | SC101, Kanr, PxylA-gfpmut2 | This study |
| pKS13 | BBR, Tetr, Plac-UV5-pdc-ahdB | 19 |
SD Opt, Shine-Delgado optimized; Kanr, kanamycin resistance; Ampr, ampicillin resistance; Tetr, tetracycline resistance, BBR, broad-host-range origin.
The plasmid for arabinose-driven gene expression was constructed from the BglBrick vector pBbS8A-RFP[18], which has an araBAD promoter (PBAD), a pSC101 origin of replication, and a β-lactamase marker. Red fluorescent protein (RFP) was replaced with xylR or xylFGH, amplified from the genome of E. coli MG1655 by the use of colony PCR with cells grown overnight in LB medium. The vector for constitutive transcription was created from pUA66, which has a pSC101 origin and a kanamycin resistance marker (22). The native xylAB promoter between the BamHI and XhoI restriction sites was replaced with a constitutive derivative of the xylFGH promoter with mutated −35 and −10 regions as follows: GATAAAAATCTGTAATTGACAAGCCCTGTTCAGTTGCTAAAT (the −35 and −10 regions are underlined).
The gfpmut2 reporter gene on pUA66-GFP was replaced with xylR, xylAB, or xylFGH. pUA66-PxylA was constructed by amplifying the genomic region between xylF and xylA, which contains the xylA promoter (PxylA), and ligating this DNA fragment into the BamHI and XhoI sites. pBb-araD-xylR was constructed by amplifying araD and placing it 5′ of xylR in pBbS8A-xylR. pBb-araD-xylR-Opt was made with quick-change mutagenesis to alter the ribosome binding site for xylR to the Shine-Dalgarno (SD) consensus sequence, AGGAGG.
To integrate xylR into the araBAD operon on the chromosome, the native araD was first deleted. It was replaced with a kanamycin resistance cassette via lambda Red recombination to generate E. coli MG1655 araD::kanr(ΔaraD) (1). araD mutants were confirmed by genomic PCR and loss of the ability to grown on arabinose. These cells were maintained at 30°C with ampicillin selection to prevent loss of pKD46. Next, the ΔaraD cells transformed with pKD46 were grown in LB medium with 0.4% arabinose at 30°C until an optical density at 600 nm (OD600) of 0.6 was reached and were then rinsed three times with ice-cold 15% glycerol to generate electrocompetent cells expressing lambda Red recombinase.
A DNA fragment containing araD and xylR was generated by PCR using pBbS8A-araD-xylR and pBbS8A-araD-xylR-Opt as templates. The PCR primers introduced on each side of the construct 40 bp of DNA homologous to the genome sequence flanking the kanamycin resistance cassette. The amount of template was titrated using 10-fold serial dilutions to determine the smallest amount necessary to produce a PCR product visible on a gel after 35 cycles. The PCR product generated from the most dilute template was treated with DpnI for 3 h at 37°C and then transformed into ΔaraD electrocompetent cells. After an overnight recovery at 23°C, the cells were plated onto 2% Bacto agar MMM plates with 0.4% arabinose. Cells in which the kanamycin cassette had been replaced with the araD-xylR construct regained the ability to grow on arabinose. Mutants were screened to verify that they were now susceptible to kanamycin. The sequence of the araD-xylR construct was verified by genomic PCR and DNA sequencing.
Sugar consumption assays.
Growth was monitored as the optical density at 600 nm (OD600). E. coli MG1655 and its derivative strains were grown overnight for at least 20 h in MMM supplemented with 0.4% glucose and any relevant antibiotics. The next day, the stationary-phase cultures were diluted 1:20 to an OD600 of 0.1 in 50-ml cultures of fresh MMM supplemented with 0.4% total sugar containing mixtures of xylose and arabinose and relevant antibiotics. These cultures were incubated at 37°C with agitation at 200 rpm. At regular time intervals, samples were removed for sugar analysis and stored at −20°C. Sampling continued for 14 h or until growth ceased. Carbohydrate (xylose-arabinose) separation was conducted on an Aminex HPX-87H column (Bio-Rad, Hercules, CA) (300-mm length, 7.8-mm internal diameter, 9-μm particle size, and 8% cross-linkage) using an Agilent 1200 Series high-performance liquid chromatography (HPLC) system equipped with a binary solvent delivery system and a refractive index (RI) detector (Agilent Technologies Inc., Santa Clara, CA). The temperatures of the column compartment and sample tray were set to 50°C and 4°C, respectively. Analytes were separated using isocratic elution with a mobile phase of 4 mM sulfuric acid in HPLC-grade water. A flow rate of 0.6 ml/min was used throughout.
Protein quantification.
E. coli cells grown overnight in MMM with 0.4% glucose were diluted 1:20 into fresh MMM supplemented with 0.2% arabinose, 0.2% xylose, and any appropriate antibiotics. Cells were grown approximately 7 h, until the mid-log phase at OD = 0.7, and then harvested by centrifugation at 6,000 × g for 10 min and stored at −80°C. Total protein was extracted by chloroform-methanol precipitation (20), dried, and reconstituted in 100 mM ammonium bicarbonate with 10% methanol. Protein concentrations were determined using DC protein reagent (Bio-Rad, Hercules, CA). A 100-μg volume of protein was reduced with 5 mM tris(2-carboxyethyl)phosphine (TCEP) for 30 min at room temperature. The reduced cysteines were blocked by iodoacetic acid (200 mM [dissolved in 100 mM NaOH]) followed by digestion with trypsin (1:50 [trypsin:protein] at 37°C overnight). The relative concentrations of the proteins involved in xylose and arabinose utilization in each sample were determined using targeted proteomics and normalized to spiked bovine serum albumin (BSA) peptides as an external standard as described by Redding-Johanson et al. (15). Briefly, samples were analyzed on a 4000 Q-Trap mass spectrometer (Applied Biosystems, Foster City, CA) running in single-reaction monitoring (SRM) mode coupled to an Eksigent TEMPO nanoLC-2D system. Samples were loaded onto a PepMap 100 u-guard column (Dionex-LC Packings) and separated on a PepMap 100 C18 analytical column (75-μm inner diameter, 150-mm length, 100-Å pore size, 3-μm-diameter beads). The column was equilibrated with 95% buffer A (2% acetonitrile, 0.1% formic acid) and 5% buffer B (98% acetonitrile, 0.1% formic acid) for 2 min followed by a 15-min ramp up to 70% buffer B, during which the peptides were separated. The remaining peptides were washed off the column by ramping up to 80% buffer B in 3 min, where the reaction was held for 10 min, after which it was quickly ramped down to 5% buffer B, where it was held for 13 min to allow the column to reequilibrate for the next sample.
Promoter activation assays.
pUA66-ParaB and pUA66-PxylA coupled GFP expression with the activity of promoters associated with arabinose and xylose utilization, respectively. E. coli strains transformed with promoter reporter plasmids were grown overnight, for at least 20 h, in MMM supplemented with 0.4% glucose and kanamycin. The following morning, cells were diluted 1:100 into fresh MMM supplemented with 0.4% total sugar and kanamycin. Freshly inoculated medium (500 μl) was placed into each well of a 24-well, clear plastic plate (BD Biosciences, San Jose, CA). Plates were incubated in a Tecan F200 plate reader maintained at 37°C with linear agitation at 90 rpm. OD600 was measured at 10-min intervals with a 595-nm filter. GFP fluorescence was measured using an excitation wavelength of 485 nm with an emission wavelength of 535 nm and was normalized to OD600. Values from two biological replicates were averaged together, with replicate values typically differing by less than 10%.
Production of ethanol from hemicellulose sugar blends.
E. coli MG1655 and Mut12 were transformed with plasmid pKS13, which carries the genes encoding pyruvate decarboxylase (pdc) and alcohol dehydrogenase (adhB), both from Zymomonas mobilis, under the control of a lacUV5 promoter, endowing them with the ability to produce ethanol. Both strains were grown overnight in EZ rich medium (Teknova) supplemented with 0.4% glucose, 15 μg/ml tetracycline, and 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) (19). The next day, the stationary-phase cultures were diluted 1:20 to an OD600 of 0.1 in EZ rich medium supplemented with 4% total sugar containing mixtures of xylose and arabinose and 15 μg/ml tetracycline. Replicate experiments for ethanol measurements were performed using 5-ml cultures in glass culture tubes. For the sugar consumption measurements, 35-ml cultures in 250-ml flasks were used. These cultures were incubated at 37°C with agitation at 200 rpm. At regular time intervals, samples were removed for sugar analysis and stored at −20°C. Sampling continued for 72 h. Sugar concentrations were analyzed as described above, and ethanol concentrations were analyzed in separate HPLC runs using the same protocol.
RESULTS
Identification of genes reversing diauxie.
It was previously demonstrated that arabinose is used preferentially by E. coli, because arabinose actively represses expression of the genes for xylose metabolism (3). To determine if the expression of any of the xylose metabolism components could restore xylose utilization despite the presence of arabinose, xylR and xylFGH were expressed under the control of PBAD from low-copy vectors pBbS8A-xylR and pBbS8A-xylFGH, respectively. In addition, xylR, xylAB, and xylFGH were expressed from a weak constitutive promoter on pUA66. Cells expressing xylFGH had enhanced simultaneous consumption of both sugars but only if expressed at a high level from PBAD (see Fig. S1 in the supplemental material). Overexpression of xylAB did not improve coutilization. Interestingly, high-level expression of xylR led to robust xylose utilization, but arabinose consumption was arrested in these cells. This correlated with a prematurely arrested log phase (Fig. 2). We hypothesize that this behavior occurs because high levels of XylR repress expression of proteins necessary for arabinose utilization. Accordingly, augmentation with lower levels of XylR, when expressed from pUA66-xylR, led to nearly identical rates of consumption of both sugars (Fig. 2).
Fig 2.
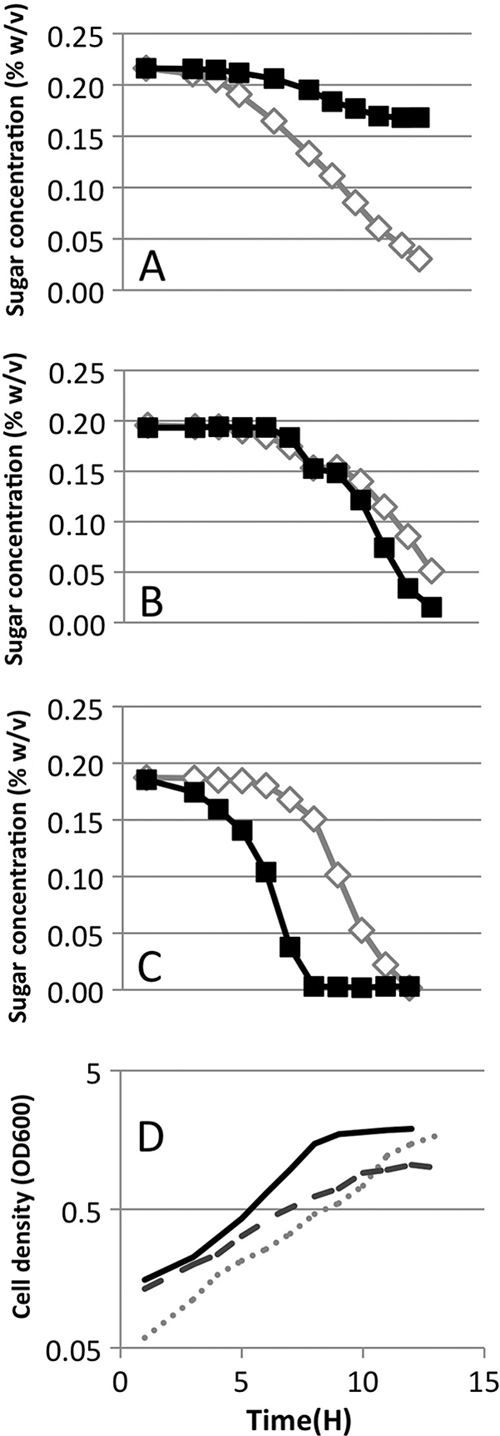
Sugar consumption by E. coli with elevated levels of XylR. (A to C) Sugar consumption by E. coli MG1655 transformed with plasmids pBbS8A-xylR (A), pUA66-xylR (B), and pUA66-GFP (C) and grown on 0.2% arabinose (filled squares) and 0.2% xylose (hollow diamonds). (D) Growth of E. coli MG1655 transformed with pBbS8A-xylR (dashed line), pUA66-xylR (dotted line), and pUA66-GFP (solid line) is shown.
Scarless knock-in of araD-xylR.
Ultimately, we wanted to create a strain capable of coutilizing hemicellulosic sugars without the aid of genes on a plasmid, necessitating the integration of any genes required to eliminate the ara-xyl diauxie into the chromosome. Due to the decrease in copy number associated with moving genes from a plasmid to chromosome, one can expect a significant decrease in protein production. Because decreased expression of xylose metabolism or transport genes might reduce the xylose utilization rate, chromosomal expression of xylR from the PBAD promoter seemed to be the best method for balancing coutilization of xylose and arabinose.
To develop a strain that cometabolized arabinose and xylose, an additional copy of xylR was placed on the chromosome. As AraC is responsible for repression of xyl gene expression, transcriptional repression was alleviated by placing xylR 3′ of araD in the araBAD regulon (Fig. 3). In these strains, arabinose would drive XylR expression, allowing dynamic regulation of XylR levels. With arabinose and xylose present in the medium, XylR would be produced at a high level and bind xylose; the XylR-xylose complex would then compete with arabinose-bound AraC for XylR binding sites in xyl promoters. When xylose is alone or at a much higher level than arabinose in the medium, less XylR would be needed to compete with the lower levels of arabinose-bound AraC; sufficient XylR could be produced from the native xylR. In addition, this avoids the deleterious effects of constitutively expressing xylR at high levels.
Fig 3.

Integration of xylR into the araBAD operon allows dynamic regulation of XylR levels. When arabinose and xylose are present in Mut12 cells, XylR is produced at a high level to compete with arabinose-bound AraC for XylR binding sites in xyl promoters. When xylose is present alone or at a much higher level than arabinose in the medium, XylR produced from the native locus is sufficient to compete with the lower levels of arabinose-bound AraC.
araD was disrupted using the efficient protocol of Datsenko and Wanner (1). The ΔaraD mutants generated during this step were no longer able to metabolize arabinose. In the second step, growth on arabinose was restored by integrating the araD-xylR construct into the chromosome, creating araBAD-xylR (Fig. 4). Since the sequence flanking araD-xylR was identical to that of the WT cells, the knock-in procedure was scarless. Two steps were employed to minimize background from the template plasmid. The minimum template necessary for a successful PCR was used, and the PCRs were treated with DpnI for 3 h to digest the remaining template. This strategy was successful for five separate constructs, and for each construct the integration frequency for 25 colonies was at least 40%.
Fig 4.

Scarless addition of genes to araBAD regulon by the use of a two-step process. araD is disrupted and then restored to its native locus with an additional gene using homologous recombination. Both steps are mediated by lambda Red recombinase.
Optimization of XylR levels for hemicellulose coutilization.
It is clear that there is a range of XylR augmentation that enables cometabolism of hemicellulosic sugars (Fig. 2). Too much XylR in the cell disrupted arabinose consumption. Too little XylR led to sequential utilization of arabinose and xylose. It is evident that, in the initial araBAD-xylR construct (E. coli araD-xylR), xylR expression was too low and xylose consumption lagged behind arabinose utilization, even though it was closer to the timing of arabinose consumption than that seen with E. coli MG1655 (Fig. 5A and B). In E. coli Mut12, an additional copy of xylR was integrated 3′ of araD and translated using a ribosome binding site stronger than that in E. coli araD-xylR. In this strain, arabinose and xylose were consumed at nearly identical rates during most of the growth of these cultures (Fig. 5C), although arabinose consumption was arrested prematurely at a quarter of the starting arabinose concentration in the medium.
Fig 5.

Optimal XylR augmentation leads to coutilization. (A to C) Arabinose (filled symbols) and xylose (open symbols) consumption by E. coli MG1655 (A), E. coli araD-xylR (B), and E. coli Mut12 (C). (D) Relative concentrations of XylR in various strains. (E) Relative concentrations of other xylose- and arabinose-associated proteins. XylA concentration, green; XylB concentration, blue; XylG concentration, yellow; AraA concentration, red; AraB concentration, black.
Quantification of proteins involved in pentose metabolism.
Using targeted proteomics, it was possible to determine the relative intracellular concentrations of XylR, XylA, XylB, XylF, XylG, AraB, and AraA (Fig. 5D and E; see Table S2 in the supplemental material for a table of protein transitions). Of these proteins, the concentrations of XylR differed the most across five different strains exhibiting different sugar consumption physiologies growing in MMM with 0.2% arabinose and xylose. For reference, the relative concentrations of two “housekeeping” proteins, Acs and GroS, varied by less than 25% across all samples (see Fig. S3 in the supplemental material). E. coli MG1655 transformed with plasmid pUA66-xylR and E. coli Mut12 metabolized arabinose and xylose at similar rates (Fig. 2B and Fig. 5D and E). These strains had 27 and 16 times the XylR of E. coli MG1655 in the mid-exponential-growth phase, respectively. Furthermore, this appears to be the optimal range of XylR supplementation for simultaneous metabolism of both sugars. E. coli araD-xylR, which consumes arabinose before xylose, has only 70% more XylR than the parental strain. E. coli MG1655 transformed with pBbS8A-xylR, with 309 times more XylR at midlog than the parent, has severely impaired arabinose consumption. Thus, cells expressing 10 to 30 times more xylR than E. coli MG1655 are primed for the cometabolism of xylose and arabinose.
The proteins required for xylose utilization showed the same trend observed for XylR. E. coli MG1655 had the lowest cellular levels of XylA, XylB, XylF, and XylG, while E. coli MG1655 transformed with pBbS8A-xylR had the highest levels of these proteins. These data also demonstrate a striking natural coordination in levels of these proteins. All four proteins showed nearly the same change in relative concentration within each sample. As an explanation for the observation that XylR antagonizes the consumption of arabinose, the levels of AraB and AraA showed an inverse trend. The highest levels of these arabinose-associated proteins occurred in WT cells, and the lowest levels were observed in cells harboring pBbS8A-xylR.
Consumption of hemicellulose sugar mixes.
E. coli Mut12 exhibited arabinose consumption arrest in mixtures with at least 50% arabinose (Fig. 6A). However, such mixtures are typically not observed for hemicellulose hydrolysates, because xylose is much more abundant in this polymer. To determine how this strain would perform with a sugar mixture representative of hemicellulose, it was grown in medium containing a 3:7 ratio of arabinose to xylose (A:X) (16). In these mixtures, E. coli Mut12 was capable of complete arabinose catabolism (Fig. 6B). These results demonstrate that E. coli Mut12 is a better candidate for biocatalysis of hemicellulose sugars than wild-type cells. For the 1:1 A:X mixture, E. coli Mut12 consumed the same amount of each sugar, while the parental cells consumed about 4 times more arabinose. For the 3:7 A:X mixture, E. coli Mut12 exhausted the arabinose and xylose at much more comparable times than E. coli MG1655.
Fig 6.

Hemicellulose sugar utilization by E. coli MG1655 and Mut12. (A and B) E. coli MG1655 (black symbols and traces) and Mut12 (gray symbols and traces) were grown at 37°C in MMM cultures supplemented with 0.2% arabinose (squares) and 0.2% xylose (diamonds) (1:1) (A) and 0.12% arabinose (squares) and 0.28% xylose (diamonds) (3:7) (B). Filled squares represent the arabinose concentration in the medium, and open diamonds the represent xylose concentration in the medium. (C and D) The cometabolism ratio was calculated as the percentage of xylose (xyl) consumed divided by the percentage of arabinose (ara) consumed for the 1:1 sugars (C) and the 3:7 sugars (D). Gray diamonds, E. coli Mut 12; black squares, E. coli MG1655. (E and F) E. coli MG1655 and Mut12 transformed with promoter reporter plasmids were grown in 24-well plates supplemented with kanamycin and 3:7 arabinose:xylose. Cells were incubated at 37°C in a shaking plate reader, and OD600 measurements (E) and GFP fluorescence measurements (F) were taken at 10-min intervals. Data represent the average results from two biological replicate experiments. Red, E. coli MG1655 transformed with pUA66-ParaB; black, E. coli MG1655 transformed with pUA66-PxylA; blue, E. coli Mut12 transformed with pUA66-ParaB; yellow, E. coli Mut12 transformed with pUA66-PxylA; green, E. coli MG1655 transformed with pUA66 (no promoter).
To quantify the degree to which each strain preferentially utilized one sugar during exponential growth, the cometabolism ratio is plotted in Fig. 6C and D as the ratio of the percentage of xylose consumed to the percentage of arabinose consumed. Values closer to 1 indicate better coutilization. In the 1:1 A:X cultures, E. coli Mut12 had almost perfect cometabolism, with a value close to 1 for the mid-exponential-growth-phase time points between 6 and 8 h, approximately four times greater than that of the wild-type cells, with their clear preference for arabinose. For the 3:7 A:X hemicellulose mixture, E. coli Mut12 had cometabolism ratios double those of wild-type cells across this same time period, indicating that Mut12 was consuming xylose nearly twice as fast as the wild-type cells.
To clarify the mechanism responsible for the differences in sugar metabolism between E. coli Mut12 and E. coli MG1655, both strains were transformed with plasmids that were used to report on the transcription from ParaBAD and PxylA in cells grown in MMM with 3:7 A:X. Those two strains have similar growth but distinct promoter activation characteristics (Fig. 6F). PxylA exhibited biphasic activation: an early, weak transcription and a later, strong transcription. This switch from weak to strong activation coincided with a marked decrease in transcription from ParaBAD, indicating exhaustion of arabinose from the medium. For E. coli Mut12, both PxylA and ParaB showed strong, consistent activation, which is likely the mechanism for robust coutilization.
Production of ethanol from hemicellulose sugar blends.
As a final proof of the utility of using a strain capable of cometabolizing hemicellulose sugars, E. coli MG1655 and Mut12 were transformed with a plasmid (pKS13) that encodes an ethanol production pathway and were analyzed for their ability to produce ethanol from 4% hemicellulose sugar mixtures. The ethanol concentration produced in 72 h from wild-type cells was 0.43% for the 3:7 arabinose:xylose sugar blend and 0.50% for the 1.5:8.5 arabinose:xylose sugar blend. E. coli Mut12 produced 0.59% ethanol from the 3:7 mixture, 37% more than MG1655, and 0.81% ethanol from the 1.5:8.5 mixture, 60% more than MG1655 (Fig. 7A). As shown by these data, E. coli Mut12 is significantly better than MG1655 at converting hemicellulose mixtures into ethanol. It would be worthwhile to determine if this trend holds for production of other chemicals, such as fatty acids (19).
Fig 7.

Ethanol production from 4% hemicellulose sugars by the use of E. coli MG1655 and Mut12 harboring pKS13. Black bars, symbols, and lines, MG1655; gray bars, symbols, and lines, Mut12. (A) Ethanol concentration after 72 h from 3:7 and 1.5:8.5 arabinose:xylose. (B) Sugar consumption for strains grown on 3:7 arabinose:xylose. Squares, arabinose concentration; diamonds, xylose concentration.
In an attempt to understand the origin of this increased ethanol production, sugar consumption was measured in cultures actively producing alcohol from the 3:7 arabinose:xylose mixture (Fig. 7B). These data show that E. coli Mut12 is competent to cometabolize arabinose and xylose at concentrations 10 times higher than previously used. For MG1655, the arabinose was exhausted from the medium after 57 h, but even after 72 h there was 0.45% xylose remaining in the medium. E. coli Mut12 was much more effective at metabolizing xylose in the presence of arabinose; after 72 h there was less than 0.1% of either sugar. The higher titers of ethanol produced by E. coli Mut12 may be the result of better hemicellulose sugar conversion than that produced by the parent strain.
DISCUSSION
The central conclusion from this study is that the E. coli arabinose-xylose diauxie can be controlled by altering the intracellular XylR concentration. This should not be surprising; XylR controls transcription of the genes responsible for xylose metabolism and import and positively regulates its own expression. Intriguingly, this mechanism for diauxie differs from that of other systems, such as the lactose-glucose diauxie (9). In these systems, “inducer exclusion” controls the order of sugar consumption. In this study, we showed that overexpression of the XylFGH xylose transporter was less effective in promoting xylose utilization than overexpression of XylR. It would be interesting to see if this repression of transcriptional activator or “activator repression” mechanism is observed with other diauxies.
Armed with this insight, the next step toward realizing an E. coli strain that cometabolizes the hemicellulose sugars was to increase the concentration of XylR in the cell. By placing it in the ara locus, xylR expression was dynamically regulated by the same chemical cue that represses xyl operons. With arabinose and xylose present in the medium, XylR would be produced at a high level and bind to xylose; the xylose-bound XylR would compete with arabinose-bound AraC for XylR binding sites in xyl promoters. With xylose present alone or at a much higher level than arabinose in the medium, less XylR would be needed to compete with the lower levels of arabinose-bound AraC. An adaptation of the Datsanko-Wanner procedure was employed to perform this genomic knock-in without altering any of the surrounding sequences. This is highly advantageous for further genomic manipulation, because it does not produce any recombination-prone sequences such as Frt sites. In addition, this process can be used iteratively, with repeated cycles of araD disruption followed by the reintroduction of araD with some additional gene or DNA sequence.
This integration procedure allowed us to develop two strains of E. coli that differed only in the ribosome binding site for the araBAD-xylR copy of xylR. The mutant with the weaker Shine-Dalgarno sequence did not efficiently consume both sugars at the same time, while E. coli Mut12, with the stronger ribosome binding site, cometabolized arabinose and xylose, though arabinose consumption stalled in mixtures with high concentrations of this sugar. This experiment highlights the need for precise tuning of the cellular levels of these transcription factors for optimal sugar utilization. Indeed, XylR augmentation must be small enough to avoid interfering with arabinose utilization but large enough to avoid activator repression.
Although E. coli Mut12 does not function ideally in media containing high percentages of arabinose, it consumed xylose twice as fast as wild-type cells in sugar mixtures, reflective of what one would obtain from the hemicellulose fraction of most biomasses. These solutions typically contain 30% arabinose or less. Even at these low concentrations, the wild-type cells still exhibited a preference for arabinose, as judged by the time required for each sugar to be exhausted and the ratio of the sugars consumed at midlog. E. coli MG1655 grown in a sugar mixture with 30% arabinose experienced repression of xyl promoters, while E. coli Mut12 did not. The coutilizing phenotype was still observed in sugar mixtures that were 10 times more concentrated, and this contributes to better biocatalyst performance for ethanol production.
E. coli Mut12 is capable of robust hemicellulose sugar cometabolism. This strain has the potential to serve as a platform for bioconversion of hemicellulose into useful chemicals and fuels. Such a strain would be extremely valuable for the development of processes that fractionate lignocellulosic biomass into cellulose and hemicellulose fractions. Furthermore, the mutations made to E. coli Mut12 are fully compatible with mutations that eliminate preferential consumption of hexoses before pentoses, such as the disruption of ptsG, the gene that encodes the transporter for glucose (7). An E. coli strain with both sets of mutations should be capable of simultaneously metabolizing the four most important biomass sugars (arabinose, xylose, mannose, and glucose), which is crucial for the development of lignocellulosic biomass as a chemical feedstock to replace petroleum.
Supplementary Material
ACKNOWLEDGMENTS
We thank our colleagues at JBEI, in particular, James Carothers, and S. del Cardayre and B. da Costa at LS9 for many helpful suggestions and assistance with the project.
This work was supported by the University of California Discovery Grant program and LS9 and by the Joint BioEnergy Institute (www.jbei.org) supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy.
Footnotes
Published ahead of print 27 January 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davin LB, Lewis NG. 2005. Lignin primary structures and dirigent sites. Curr. Opin. Biotechnol. 16:407–415 [DOI] [PubMed] [Google Scholar]
- 3. Desai TA, Rao CV. 2010. Regulation of arabinose and xylose metabolism in Escherichia coli. Appl. Environ. Microbiol. 76:1524–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gibson DG, et al. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6:343–345 [DOI] [PubMed] [Google Scholar]
- 5. Gosset G. 2005. Improvement of Escherichia coli production strains by modification of the phosphoenolpyruvate:sugar phosphotransferase system. Microb. Cell Fact. 4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hendrickson W, Flaherty C, Molz L. 1992. Sequence elements in the Escherichia coli araFGH promoter. J. Bacteriol. 174:6862–6871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hernández-Montalvo T, Valle F, Bolivar F, Gosset G. 2001. Characterization of sugar mixtures utilization by an Escherichia coli mutant devoid of the phosphotransferase system. Appl. Microbiol. Biotechnol. 57:186–191 [DOI] [PubMed] [Google Scholar]
- 8. Himmel ME, et al. 2007. Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315:804–807 [DOI] [PubMed] [Google Scholar]
- 9. Inada T, Kimata K, Aiba H. 1996. Mechanism responsible for glucose-lactose diauxie in Escherichia coli: challenge to the cAMP model. Genes Cells 1:293–301 [DOI] [PubMed] [Google Scholar]
- 10. Khankal R, Chin JW, Cirino PC. 2008. Role of xylose transporters in xylitol production from engineered Escherichia coli. J. Biotechnol. 134:246–252 [DOI] [PubMed] [Google Scholar]
- 11. Khankal R, Chin JW, Ghosh D, Cirino PC. 2009. Transcriptional effects of CRP* expression in Escherichia coli. J. Biol. Eng. 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim JH, Block DE, Mills DA. 2010. Simultaneous consumption of pentose and hexose sugars: an optimal microbial phenotype for efficient fermentation of lignocellulosic biomass. Appl. Microbiol. Biotechnol. 88:1077–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee TS, et al. 2011. BglBrick vectors and datasheets: a synthetic biology platform for gene expression. J. Biol. Eng. 5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nishiyama Y, Langan P, Chanzy H. 2002. Crystal structure and hydrogen-bonding system in cellulose Ibeta from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 124:9074–9082 [DOI] [PubMed] [Google Scholar]
- 15. Redding-Johanson AM, et al. 2011. Targeted proteomics for metabolic pathway optimization: application to terpene production. Metab. Eng. 13:194–203 [DOI] [PubMed] [Google Scholar]
- 16. Schädel C, Blöchl A, Richter A, Hoch G. 2010. Quantification and monosaccharide composition of hemicelluloses from different plant functional types. Plant Physiol. Biochem. 48:1–8 [DOI] [PubMed] [Google Scholar]
- 17. Shen CR, et al. 2011. Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl. Environ. Microbiol. 77:2905–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song S, Park C. 1997. Organization and regulation of the D-xylose operons in Escherichia coli K-12: XylR acts as a transcriptional activator. J. Bacteriol. 179:7025–7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Steen EJ, et al. 2010. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463:559–562 [DOI] [PubMed] [Google Scholar]
- 20. Wessel D, Flugge UI. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138:141–143 [DOI] [PubMed] [Google Scholar]
- 21. Yomano LP, York SW, Shanmugam KT, Ingram LO. 2009. Deletion of methylglyoxal synthase gene (mgsA) increased sugar co-metabolism in ethanol-producing Escherichia coli. Biotechnol. Lett. 31:1389–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zaslaver A, et al. 2006. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods 3:623–628 [DOI] [PubMed] [Google Scholar]
- 23. Zhang H, Obias V, Gonyer K, Dennis D. 1994. Production of polyhydroxyalkanoates in sucrose-utilizing recombinant Escherichia coli and Klebsiella strains. Appl. Environ. Microbiol. 60:1198–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.