

Abstract
Introduction
Osteogenesis imperfecta (OI) is a genetic disorder characterized by bone fragility and fractures. Patients with OI have clinical features that may range from mild symptoms to severe bone deformities and neonatal lethality. Numerous approaches for the classification of OI have been published. The Sillence classification is the most commonly used. In this study, we aimed at developing a more refined sub-classification by applying a proposed scoring system for the quantitative assessment of clinical severity in different types of OI.
Subjects and methods
This study included 43 patients with OI. Clinical examination and radiological studies were conducted for all patients. Cases were classified according to the Sillence classification into types I–IV. The proposed scoring system included five major criteria of high clinical value: number of fractures per year, motor milestones, long bone deformities, length/height standard deviation score (SDS), and bone mineral density (BMD). Each criterion was assigned a score from 1 to 4, and each patient was marked on a scale from 1 to 20 according to these five criteria.
Results
Applying the proposed clinical scoring system showed that all 11 patients with Sillence type I (100%) had a score between 6 and 10, denoting mild affection. The only patient with Sillence type II had a score of 19, denoting severe affection. In Sillence type III, 7 patients (31.8%) were moderately affected and 15 patients (68.2%) were severely affected. Almost all patients with Sillence type IV (88.9%) were moderately affected.
Conclusions
Applying the proposed scoring system can quantitatively reflect the degree of clinical severity in OI patients and can be used in complement with the Sillence classification and molecular studies.
Keywords: Osteogenesis imperfecta, Sillence classification, Clinical scoring system, Radiological manifestations, Genetics
Introduction
Osteogenesis imperfecta (OI) is one of the most common genetic connective tissue disorders that primarily affect bone, resulting in recurrent fractures. Patients with OI have clinical features that may range from mild symptoms with a scant number of fractures to severe bone deformities and neonatal lethality [1].
Most cases with OI follow a dominant inheritance pattern caused by mutations in either of the two genes encoding type I collagen, COL1A1 and COL1A2. Paternal age effect for increased risk of new mutations has been documented [2]. Studies of the clinically unaffected parents in some families who had more than one child with dominant OI have shown that the cause of the occurrence is parental germ line mosaicism. Clinically, the mosaic carriers are normal or minimally affected. They are most often identified by having more than one child with the fully manifesting heterozygous condition [3].
Recent investigations have generated a new paradigm for OI incorporating many of the prototypical features that distinguish dominant and recessive conditions within a type I collagen framework [4]. The list of genes linked to autosomal recessive (AR)-OI to date includes collagen-modifying enzymes and chaperones CRTAP, FKBP10, LEPRE1, PPIB, and SERPINH1 [5–11]. It also includes SERPINF1 [12] and the transcription factor Sp7/Osterix [13]. Most recently, a mutation causing deficient BMP/mTLD proteolytic activity was identified in an AR-OI family [14]. The discovery of these genes has provided new insights into the bone biology and pathogenetic mechanisms of OI.
Several classification systems were proposed for OI, based on clinical, radiological, and, recently, molecular findings [15–20]. The most commonly used classification proposed by Sillence et al. [17] included four types. This classification has been expanded to include a greater range of subgroups of patients (OI types V–IX; OMIM: 610967, 610968, 610682, 610915, 259440) [21]. van Dijk et al. [22] proposed a revised classification of OI with the exclusion of OI types VII and VIII, since these have been added because of their AR inheritance, while the clinical and radiological features are indistinguishable from OI types II–IV. In the latest nosology and classification of genetic skeletal disorders, Warman et al. [20] stated that the genetic complexity of the molecular bases of OI has been revealed with clear documentation of the extensive phenotypic variation arising from single loci. The authors agreed upon retaining the Sillence classification as the prototypic and universally accepted way to classify the degree of severity.
Given the high heterogeneity in this group of patients and from our experience at the Limb Malformations and Skeletal Dysplasia Clinic (LMSDC) at the National Research Centre (NRC), a referral centre for OI patients, overlap can occur between the different Sillence types, as the degree of clinical severity of the disease is not expressed quantitatively. In this study, we propose a refined sub-classification by applying a scoring system for the quantitative assessment of clinical severity in 43 Egyptian patients with OI.
Subjects and methods
The study included 43 patients (17 males, 26 females) with classic OI. All were recruited from the LMSDC, NRC, at their first visit. Ethical approval and appropriate informed consent was obtained from all subjects or their parents included in this study.
Each patient was subjected to complete history, including the onset and disease course, as well as the average number of fractures per year, three-generation pedigree construction, and clinical examination with special emphasis on motor milestones and the skeletal system. Radiological evaluation was carried out for all patients. Molecular studies were done for selected patients: offspring of consanguineous parents with a history of affected sibs. Assessment was done prior to the initiation of bisphosphonate therapy or any surgical correction.
According to the age of presentation, severity of the disease, and color of the sclera, patients were classified following the Sillence classification [17] into four types. We proposed a scoring system for the assessment of clinical severity including five major criteria, namely, average number of fractures per year, motor milestones, long bone deformities, length/height standard deviation score (SDS), and z-score of the bone mineral density (BMD). Each criterion was assigned a score from 1 to 4, with 4 being the most severe (Table 1).
Table 1.
Proposed scoring system of osteogenesis imperfecta (OI) patients
| Criterion/points | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Average number of fractures/year | 0–1 | 2–5 | 6–10 | >11 |
| Gross motor development (modified from Daly et al. [23] | Normal | Delayed with catchup | Delayed with no catchup | Abnormal/arrested |
| Degree of long bones deformities −/+ scoliosis | No deformity | Mild (noticed only by X-rays), no scoliosis | Moderate (noticed by both clinical examination and X-rays affecting isolated long bones) −/+ mild scoliosis | Severe (clearly visible by clinical examination and X-rays affecting all long bones), +severe scoliosis |
| Height SDS | <−2.0 | −2.1 to −3.0 | −3.1 to −5.0 | >−5.0 |
| BMD (z-score) | Normal | Osteopenia z-score: −1.5 to −1.9 |
Severe osteopenia z-score: −2.0 to −2.4 |
Osteoporosis z-score: ≤−2.5 |
| Score and degree of severity | 5–10: mild | 11–15: moderate | 16–20: severe |
SDS standard deviation score; BMD bone mineral density measured by dual-energy X-ray absorptiometry (DEXA)
Gross motor development
This was assessed in each patient according to Daly et al. [23], with some modification. Four degrees were considered: (1) normal development; (2) delayed with catchup: the age of achieving milestones was delayed but independent walking was achieved eventually; (3) delayed with no catchup: the time of passing milestones was delayed from infancy and progress had been arrested at varying stages; (4) abnormal/arrested: in these cases, milestones were not passed in the defined order and development was arrested at the sitting, crawling, or standing stages.
Length or standing height
Length was measured from birth up to 24 months of age or, if the patient was unable to stand, using a measuring table with a perpendicular piece and a movable piece. Height was measured by a stadiometer. The patient stood straight so that the heels, buttocks, and shoulders were in contact with the wall. The head was positioned in the Frankfort plane. In cases of leg length discrepancy or contractures, the longest leg was used in all measurements. Length and height measurements in children with bone deformities are difficult to perform. Therefore, all measurements were performed by personnel experienced in dealing with subjects with deforming bone disorders. Scores were given according to the length/height SDS, as shown in Table 1.
Long bone deformities
These were assessed in each patient both clinically and radiologically. Patients were divided into four groups (score from 1 to 4) as follows: (1) no visible long bone deformities both clinically and radiologically; (2) mild long bone deformities seen by X-rays only (Fig. 1a); (3) moderate deformities limited to one or two long bones detected both clinically and radiollogically with or without mild scoliosis (Fig. 1b); (4) severe deformities of all long bones, with severe scoliosis (Fig. 1c).
Fig. 1.
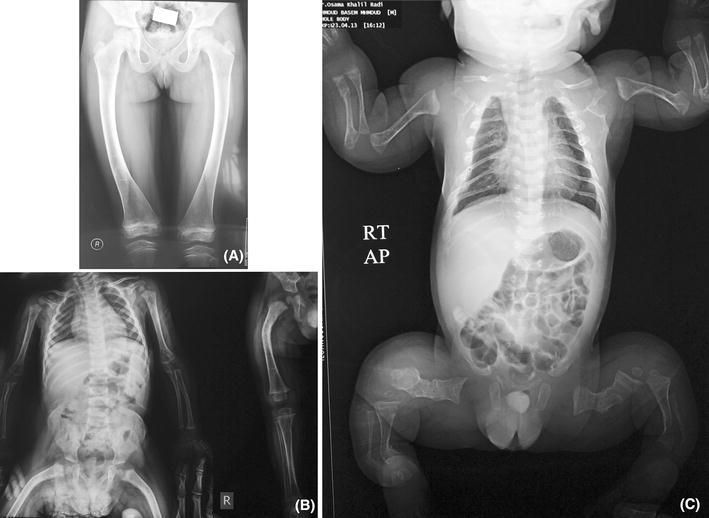
Degrees of long bone deformities in the X-rays of osteogenesis imperfecta (OI) patients. a X-ray of the lower limbs of a patient with OI-I showing bilateral bowing of both femora with a healed fracture of the lower end of the right femur, not visible clinically (score 2). b X-ray showing deformed thin femora with callus formation and mild scoliosis seen both clinically and radiologically, with no deformities of the long bones of the upper limbs in a patient with OI-IV (score 3). c A babygram of a patient with OI-II showing generalized decreased bone density, narrow chest, fracture of ribs with callus formation, platyspondyly, and deformed long bones with crumpled femora and curved humeri, ulnae, radii, tibiae, and fibulae (score 4)
Bone mineral density
The BMD was measured for all cases. Dual-energy X-ray absorptiometry (DEXA) (Norland XR-46), software version 3.9.6, was used to determine the BMD. DEXA was done at two sites; hip bone (left femoral neck) and lumbar spine from L2–L4. The equipment converts the information received by the detector into an image. The results are reported as a total amount of BMD of the hip or lumbar spine, which is the amount of bone per unit of skeleton area. The DEXA results were evaluated with those given in the Norland XR-46 database for the Caucasian population. The mean value of the BMD was expressed in g/cm2. The BMD was expressed as z-score values, which is the difference in the number of standard deviations (SDs) between the mean BMD value of the individual and a group of people of the same age and sex. As bone density varies greatly with age, the densitometry z-score is used in the pediatric population and not the T-score usually used in adults. According to the results of the BMD at the femur and lumbar spine, patients were divided into groups as follows: (1) normal (z-score −1.4 or greater); (2) osteopenia (z-score between −1.5 and −1.9 SD); (3) severe osteopenia (z-score between −2.0 and −2.4 SD); and (4) osteoporosis (z-score −2.5 or less).
The proposed scoring system put all the studied OI patients on a scale in an attempt to cover the wide range of phenotype variations and provide a calculated score for each case. Each patient was put on a scale from 1 to 20. Patients were classified into three categories: scores from 5 to 10 indicate mild condition, scores from 11 to 15 reflect a moderate condition, and scores from 16 to 20 indicate a severe condition.
Statistical analysis
Evaluation of the length and height was done by the use of the z-score method. They were expressed as the SDS using Egyptian references [24]. Values above or below 2.5 SDS were considered to be abnormal. Comparison between different OI types regarding the average number of fractures per year, mean length or height SDS, and mean BMD z-scores was carried out using Student’s t-test and one way analysis of variance between groups (ANOVA). A P-value was considered to be significant if it was ≤0.05. Statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS version 16).
Results
The characteristics of the studied patients are shown in Table 2. Eight patients, with OI type I, were consistent with AD inheritance or represent a de novo dominant mutation; four of them had an affected parent. In OI type II, our single patient represented a de novo mutation with a high paternal age (50 years old). In OI type III, ten patients represented AD inheritance or de novo mutation; six of them had parental affection. In OI type IV, five cases represented a de novo mutation.
Table 2.
Characteristics of the studied patients
| Sillence type | Number/percentage (%) | Male:female | Mean age at presentation in years (range) | Parental consanguinity | Sporadic (de novo) cases | Familial cases | Paternal age above 35 years | |
|---|---|---|---|---|---|---|---|---|
| Affected parent | Affected sibs | |||||||
| OI-I | 11 (25.5%) | 2:9 | 6.6 (3.3–12.6) | 3/11 | 4/11 | 4/11 | 3/11 | 4/11 |
| OI-II | 1 (2.3%) | 1 Female | 0.5 | 0/1 | 1/1 | 0/1 | 0/1 | 1/1 |
| OI-III | 22 (51.1%) | 11:11 | 5.5 (1.4–17.8) | 13/22 | 4/22 | 6/22 | 12/22 | 5/22 |
| OI-IV | 9 (20.9%) | 4:5 | 10.0 (3.9–20.0) | 6/9 | 5/9 | 0/9 | 4/9 | 4/9 |
| Total | 43 (100%) | 17:26 | 5.7 (0.05–20.0) | 22/43 | 14/43 | 10/43 | 19/43 | 14/43 |
Molecular studies of 14 consanguineous families with a history of affected sibs including 19 available patients revealed novel mutations in the known AR-OI causative genes in six families with seven available patients (unpublished data) and the reporting of new AR-OI causative genes in two families with three available patients [13, 14]. No mutations in the known AR-OI causative genes were identified in six families with nine available patients, suggesting parental germ line mosaicism or the possibility of the presence of other unknown AR-OI causative genes.
By applying the proposed scoring system to the ten proven patients with AR-OI, a score between 16 and 19 denoting severe affection was calculated for nine patients, classified as OI-III, with PPIB, CRTAP, LEPRE1, SERPINF1, BMP1 novel mutations. The only patient with OSX mutation classified as OI-IV had a score of 14 (moderate severity).
Comparing the patients’ findings according to their Sillence type revealed the following in each criterion:
Average number of fractures/year The onset of fractures in the studied cases varied. More than 50% of patients with type I fractures started in early childhood, fractures in OI-III started at birth in 40% of cases and during infancy in 60% of cases, and 77% of patients with OI-IV fractures had fractures that started from birth to infancy. Lower limb fractures were more common in all types. Although not statistically significant, the mean number of fractures per year was increased in OI types III (8.5) and IV (5.75) compared to type I (3.89).
Gross motor development Nine cases with OI-I had normal motor development (81.8%) and only two cases had history of mild delay with catchup (18.1%). The only patient with OI-II died at the age of 6 months with no head support. In OI-III, only one case (4.5%) had mild delay with catchup, 11 cases (50%) had delayed motor development with no catchup, and ten cases (45.4%) had abnormal/arrested development and were not able to move independently. In OI-IV, five cases (55.5%) had normal motor development, one case (11.1%) had history of mild delay then catchup, and three cases (33.3%) had history of abnormal/arrested development.
Degree of long bone deformities Long bone deformities in the form of malunion, bowing, angulation, loss of bone modeling, and epiphyseal calcifications were predominant findings in all patients with OI-III, the only patient with OI-II, 45.5% of OI-I patients, and 88.8% of OI-IV patients. Scoliosis was more common in the studied patients with types III and IV (55.5% and 45.4%, respectively).
Length/height SDS Almost all (91.3%) cases with OI-III had short stature compared to 30% of OI-I and 75% of OI-IV patients. The mean length/height SDS for types I, III, and IV were −1.8, −5.6, and −4.23, respectively. The results of ANOVA showed a highly significant difference between different types (P value = 0.0001).
Bone mineral density The mean z-score value of the BMD for all cases was −2.83 SDS, indicating osteoporosis. However, the mean values were significantly decreased in types III and IV (−3.37 and −2.73, respectively) compared to type I (−1.55). ANOVA showed a highly significant difference in the DEXA mean z-score values between different types (P value = 0.009).
The degree of severity in Sillence OI types according to the proposed scoring system is shown in Table 3.
Table 3.
Degree of severity in OI types according to the proposed scoring system
| Sillence type | Mild (score: 5–10) | Moderate (score: 11–15) | Severe (score: 16–20) |
|---|---|---|---|
| OI-I | 11 cases (100%) (range: 6–10) | 0 | 0 |
| OI-II | 0 | 0 | 1 case (score: 19) |
| OI-III | 0 | 7 cases (31.8%) (range: 11–15) | 15 cases (68.2%)a (range: 16–19) |
| OI-IV | 1 case (11.1%) (score: 10) | 8 cases (88.9%)b (range: 11–15) | 0 |
aIncluding nine patients with autosomal recessive osteogenesis imperfecta (AR-OI)
bIncluding one patient with AR-OI (OSX mutation)
Discussion
Osteogenesis imperfecta (OMIM: 166200, 166210, 259420, 166220) [21] is a bone-related genetic disorder characterized by low bone mass and bone fragility that is clinically and genetically heterogeneous. In an attempt to provide a more refined sub-classification, we selected five major criteria of high clinical value and proposed a score for each. This scoring system was applied to the studied patients, classified according to Sillence types, to test its significance and applicability for clinical purposes. The selected criteria included the following.
Average number of fractures per year
Recurrent fractures were the most common and persistent finding in the studied patients. Chavassieux et al. [25] stated that, for most patients, the presence of multiple fractures early in life, with or without blue sclera, is usually sufficient to establish the diagnosis.
Rauch et al. [26] found no significant relationship between the presence of limb deformities and fractures at birth with either the type of affected collagen 1 alpha chain or the substituting amino acid in AD OI patients. However, recurrent fractures result in bone deformities and can lead to significant physical handicap. The mean number of fractures per year in the studied patients was increased in OI-III and OI-IV compared to OI-I. Correlations between the number of fractures per year and height measurements indicated that height was decreased with the increased number of fractures.
Gross motor development
Delayed motor milestones were one of the most troublesome complaints for our patients that disturbed their lifestyles. Usually, OI patients have normal mentality but their inability to walk limits their school performance and social activities. The severity of affection in the studied patients correlated with the degree of delay in physical milestones. The early achievement of motor milestones contributes to the ability of independent walking. According to our study, most patients with OI-III and some of the OI-IV patients were unable to walk independently.
A few studies have examined the relationship between the achievement of developmental function and eventual mobility status. In infants with OI types III or IV, developmental milestones are delayed and the order of achievement of milestones differs from that normally expected [27]. Engelbert et al. [28] stated that the type of OI was strongly associated with current walking ability. Patients of type III and IV had a lower chance of ultimately walking compared with those of type I. The authors reported that Dutch children under the age of seven and half years with types III and IV OI scored more than two standard deviations below the median in the mobility domain.
Degree of long bones deformities −/+ scoliosis
From our results, it was noted that even patients with OI type I can present with bone deformities, usually due to repeated fractures, bad management, and malunion. The degree of deformities is usually correlated to the severity of the disease. About half of the patients with OI-I, all patients with OI-III, the only patient with OI-II, and 88.8% of patients with OI-IV were deformed in different degrees related to the severity of each case. Kyphosis and/or scoliosis were detected in almost half of the patients with OI types III and IV and only one patient with OI type I. Bauze et al. [16] divided their 42 patients with OI into mild, moderate, and severe groups according to the deformity of long bones. None of the patients in the mild group had scoliosis.
Length/height
Growth deficiency is a key feature of severe OI and a frequent feature of mild to moderate forms of the disease [29]. Lund et al. [30] stated that the mean standing height was reduced in all groups of OI patients, short stature was frequent in type I, and severe in types III and IV. We found a parallel relation between the severity and degree of short stature. Nearly 90% of OI-III patients had short stature compared to 75% of OI-IV and 30% of OI-I cases. Rowe et al. [31] reported a spectrum of disease severity within a five-generation family; those most severely affected exhibited more severe short stature and scoliosis relative to those who were less severely affected. In a genotype–phenotype study of 192 OI patients, Rauch et al. [32] noted that patients with haploinsufficiency mutations were, on average, taller than patients with helical mutations in the alpha 1 or alpha 2 chains.
Bone mineral density
The BMD was significantly decreased in the studied patients with OI-III and OI-IV compared to OI-I. Correlations between the number of fractures per year and bone density indicated that the number of fractures increased with decreased BMD. Osteopenia was more severe in patients with OI-I, while osteoporosis was more severe in types II, III, and IV. Rauch et al. [32] found that the mean lumbar spine BMD z-scores were higher in the haploinsufficiency group compared to both helical mutation groups. They also found no significant association between the lumbar spine BMD z-scores and the affected alpha chain, the substituting amino acid, or the position of the mutation on the triple helical mutations.
In conclusion, all patients with OI-I were mildly affected and the only patient with OI-II had severe affection. In OI-III, slightly more than two-thirds of the patients were severely affected, while less than one-third were moderately affected. Almost all patients with OI-IV were moderately affected. According to the proposed scoring system, almost all patients with proved AR inheritance had a severe clinical affection. This study clarified that applying the proposed scoring system can quantitatively reflect the degree of clinical severity and provide a finer sub-classification to the commonly used Sillence types. It might also help in the follow-up after the initiation of therapy to measure the patient’s response to treatment. Although molecular studies are important for genetic counseling and the understanding of the disease pathogenesis, they can add to the complexity of disease classification. We recommend the application of the proposed clinical scoring system in complement with the Sillence classification and molecular studies.
References
- 1.Martin E, Shapiro JR. Osteogenesis imperfecta: epidemiology and pathophysiology. Curr Osteoporos Rep. 2007;5:91–97. doi: 10.1007/s11914-007-0023-z. [DOI] [PubMed] [Google Scholar]
- 2.Blumsohn A, McAllion SJ, Paterson CR. Excess paternal age in apparently sporadic osteogenesis imperfecta. Am J Med Genet. 2001;100:280–286. doi: 10.1002/ajmg.1269. [DOI] [PubMed] [Google Scholar]
- 3.Cabral WA, Marini JC. High proportion of mutant osteoblasts is compatible with normal skeletal function in mosaic carriers of osteogenesis imperfecta. Am J Hum Genet. 2004;74:752–760. doi: 10.1086/383252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marini JC, Cabral WA, Barnes AM. Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. 2010;339:59–70. doi: 10.1007/s00441-009-0872-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR, Leikin S, Makareeva E, Kuznetsova N, Uveges TE, Ashok A, Flor AW, Mulvihill JJ, Wilson PL, Sundaram UT, Lee B, Marini JC. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med. 2006;355:2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, Castagnola P, Rauch F, Glorieux FH, Vranka J, Bächinger HP, Pace JM, Schwarze U, Byers PH, Weis M, Fernandes RJ, Eyre DR, Yao Z, Boyce BF, Lee B. CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 7.Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, Bulas DI, Kozma C, Smith PA, Eyre DR, Marini JC. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drögemüller C, Becker D, Brunner A, Haase B, Kircher P, Seeliger F, Fehr M, Baumann U, Lindblad-Toh K, Leeb T. A missense mutation in the SERPINH1 gene in Dachshunds with osteogenesis imperfecta. PLoS Genet. 2009;5:e1000579. doi: 10.1371/journal.pgen.1000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PGJ, Piersma SR, Fratantoni SA, Jimenez CR, Huizer M, Morsman AC, Cobben JM, van Roij MHH, Elting MW, Verbeke JI, Wijnaendts LC, Shaw NJ, Högler W, McKeown C, Sistermans EA, Dalton A, Meijers-Heijboer H, Pals G. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, Alikasifoglu M, Tuncbilek E, Orhan D, Bakar FT, Zabel B, Superti-Furga A, Bruckner-Tuderman L, Curry CJ, Pyott S, Byers PH, Eyre DR, Baldridge D, Lee B, Merrill AE, Davis EC, Cohn DH, Akarsu N, Krakow D. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, Pepin MG, Weis MA, Eyre DR, Byers PH. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker J, Semler O, Gilissen C, Li Y, Bolz HJ, Giunta C, Bergmann C, Rohrbach M, Koerber F, Zimmermann K, de Vries P, Wirth B, Schoenau E, Wollnik B, Veltman JA, Hoischen A, Netzer C. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011;88:362–371. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lapunzina P, Aglan M, Temtamy S, Caparrós-Martín JA, Valencia M, Letón R, Martínez-Glez V, Elhossini R, Amr K, Vilaboa N, Ruiz-Perez VL. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010;87:110–114. doi: 10.1016/j.ajhg.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martínez-Glez V, Valencia M, Caparrós-Martín JA, Aglan M, Temtamy S, Tenorio J, Pulido V, Lindert U, Rohrbach M, Eyre D, Giunta C, Lapunzina P, Ruiz-Perez VL. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat. 2012;33:343–350. doi: 10.1002/humu.21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falvo KA, Root L, Bullough PG. Osteogenesis imperfecta: clinical evaluation and management. J Bone Joint Surg Am. 1974;56:783–793. [PubMed] [Google Scholar]
- 16.Bauze RJ, Smith R, Francis MJ. A new look at osteogenesis imperfecta. a clinical, radiological and biochemical study of forty-two patients. J Bone Joint Surg Am. 1975;57:2–12. [PubMed] [Google Scholar]
- 17.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanscom DA, Winter RB, Lutter L, Lonstein JE, Bloom BA, Bradford DS. Osteogenesis imperfecta. Radiographic classification, natural history, and treatment of spinal deformities. J Bone Joint Surg Am. 1992;74:598–616. [PubMed] [Google Scholar]
- 19.Plotkin H. Syndromes with congenital brittle bones. BMC Pediatr. 2004;4:16. doi: 10.1186/1471-2431-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, Mortier G, Mundlos S, Nishimura G, Rimoin DL, Robertson S, Savarirayan R, Sillence D, Spranger J, Unger S, Zabel B, Superti-Furga A. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Online Mendelian Inheritance in Man (OMIM). McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD), and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD). Home page at: http://www.ncbi.nlm.nih.gov/omim
- 22.van Dijk FS, Pals G, Van Rijn RR, Nikkels PG, Cobben JM. Classification of osteogenesis imperfecta revisited. Eur J Med Genet. 2010;53:1–5. doi: 10.1016/j.ejmg.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Daly K, Wisbeach A, Sanpera I, Jr, Fixsen JA. The prognosis for walking in osteogenesis imperfecta. J Bone Joint Surg Br. 1996;78:477–480. [PubMed] [Google Scholar]
- 24.Ghalli I, Salah N, Hussien F, Erfan M, El-Ruby M, Mazen I, Aglan MS, Hosny L, Zaki M, Ismail S, Elgammal M, Abd El-Dayem S et al. In: Sartorio A, Buckler JMH, Marazzi N (eds) Proceedings of the 1st National Congress for Egyptian Growth Curves, Cairo University, Cairo, Egypt, 11 December 2003. Published in Cresceve Nelmondo (2008). Ferring Company. Egyptian Growth Curves 2002 for Infants, Children and Adolescents
- 25.Chavassieux P, Seeman E, Delmas PD. Insights into material and structural basis of bone fragility from diseases associated with fractures: how determinants of the biomechanical properties of bone are compromised by disease. Endocr Rev. 2007;28:151–164. doi: 10.1210/er.2006-0029. [DOI] [PubMed] [Google Scholar]
- 26.Rauch F, Lalic L, Roughley P, Glorieux FH. Genotype–phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I. Eur J Hum Genet. 2010;18:642–647. doi: 10.1038/ejhg.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruck-Gibis J, Plotkin H, Hanley J, Wood-Dauphinee S. Reliability of the gross motor function measure for children with osteogenesis imperfecta. Pediatr Phys Ther. 2001;13:10–17. doi: 10.1097/00001577-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 28.Engelbert RH, Uiterwaal CS, Gulmans VA, Pruijs H, Helders PJ. Osteogenesis imperfecta in childhood: prognosis for walking. J Pediatr. 2000;137:397–402. doi: 10.1067/mpd.2000.107892. [DOI] [PubMed] [Google Scholar]
- 29.National Institutes of Health (NIH) Clinical Center (CC) (2007) Growth Hormone Therapy in Osteogenesis Imperfecta. National Institutes of Health Clinical Center, 9000 Rockville Pike, Bethesda, MD, prpl@mail.cc.nih.gov
- 30.Lund AM, Müller J, Skovby F. Anthropometry of patients with osteogenesis imperfecta. Arch Dis Child. 1999;80:524–528. doi: 10.1136/adc.80.6.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowe DW, Shapiro JR, Poirier M, Schlesinger S. Diminished type I collagen synthesis and reduced alpha 1(I) collagen messenger RNA in cultured fibroblasts from patients with dominantly inherited (type I) osteogenesis imperfecta. J Clin Invest. 1985;76:604–611. doi: 10.1172/JCI112012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rauch F, Lalic L, Roughley P, Glorieux FH. Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J Bone Miner Res. 2010;25:1367–1374. doi: 10.1359/jbmr.091109. [DOI] [PubMed] [Google Scholar]