

Abstract
We describe an unusual xeroderma pigmentosum (XP) patient with a mutation in XP complementation group G, representing only the third reported Japanese XP-G patient. A 40-year-old male (XP3HM), born from consanguineous parents experienced sun sensitivity and pigmentary changes of sun-exposed skin since childhood. He developed a squamous cell carcinoma on his lower lip at the age of 40. He has neither neurological abnormalities nor Cockayne syndrome. The primary fibroblasts of the patient were hypersensitive to killing by UV (D0=0.6 J/m2) and the post-UV unscheduled DNA synthesis was 8 % of normal. Host cell reactivation complementation analysis implicated XP complementation group G. We identified a novel homozygous mutation (c.194T>C) in a conserved portion of the XPG(ERCC5) gene, resulting in a predicted amino acid change; p.L65P. We confirmed that this genetic change reduced DNA repair thus linking this mutation to increased skin cancer.
Keywords: DNA repair, mutation, skin cancer, ultraviolet, xeroderma pigmentosum group G
LETTER TO THE EDITOR
BACKGROUND
Xeroderma pigmentosum (XP) is a rare autosomal recessive disease characterized by severe photosensitivity, abnormal pigmentation and a more than 10,000- fold increase in the frequency of cancers of sun exposed skin and eyes[1, 2, 3]. There are 7 genetically different complementation groups, XP-A through XP-G, with defective nucleotide excision repair (NER) and XP variant with deficient translesion DNA synthesis. In Japan, 55% of XP patients belong to XP-A, a severe form with marked neurological degeneration. In contrast, in the US and Europe, 40% of XP cases are XP-C, in which patients only have cutaneous and ocular symptoms [4, 5]. There have been only 14 XP-G cases reported, including two Japanese XP-G cases [6, 7]. Most of the XP-G patients also have clinical features of Cockayne syndrome (XP/CS complex) or XP neurologic symptoms as well as cutaneous XP lesions [8, 9], while two Japanese patients only had cutaneous lesions [7, 10].
QUESTIONS ADDRESSED
Here we report clinical and laboratory features of a new Japanese XP-G patient with a novel XPG mutation with no features of XP/CS complex or XP neurological disease.
EXPERIMENTAL DESIGN
Post-UV DNA repair and molecular studies using fibroblasts from the skin biopsy specimen of this patient were performed as described [11]. Cultured fibroblasts XP3HM (this case), XP2OS(XP-A), XP20BE (XP-G) and N-3 (3-year-old normal donor) [6, 11], were used.
RESULTS
CASE REPORT
A 40-year-old male (XP3HM), with consanguineous parents, had marked freckling and telangiectasia on his face, upper chest and the dorsal aspects of both hands, and the recent appearance of squamous cell carcinoma in his lower lip (Figure 1). There was sparing of the buttocks and axilla. He had lifelong sun sensitivity but he never had a blistering sunburn. Pigmentary changes appeared at the age of 10. His physical and intellectual development were normal and neither neurological nor ocular abnormalities had developed. He had no difficulty in hearing. His minimal erythema dose was reduced (20 mJ/cm2) (normal ; 50–120mJ/cm2) [12].
Figure 1. Clinical features of the face of the present case; XP3HM.
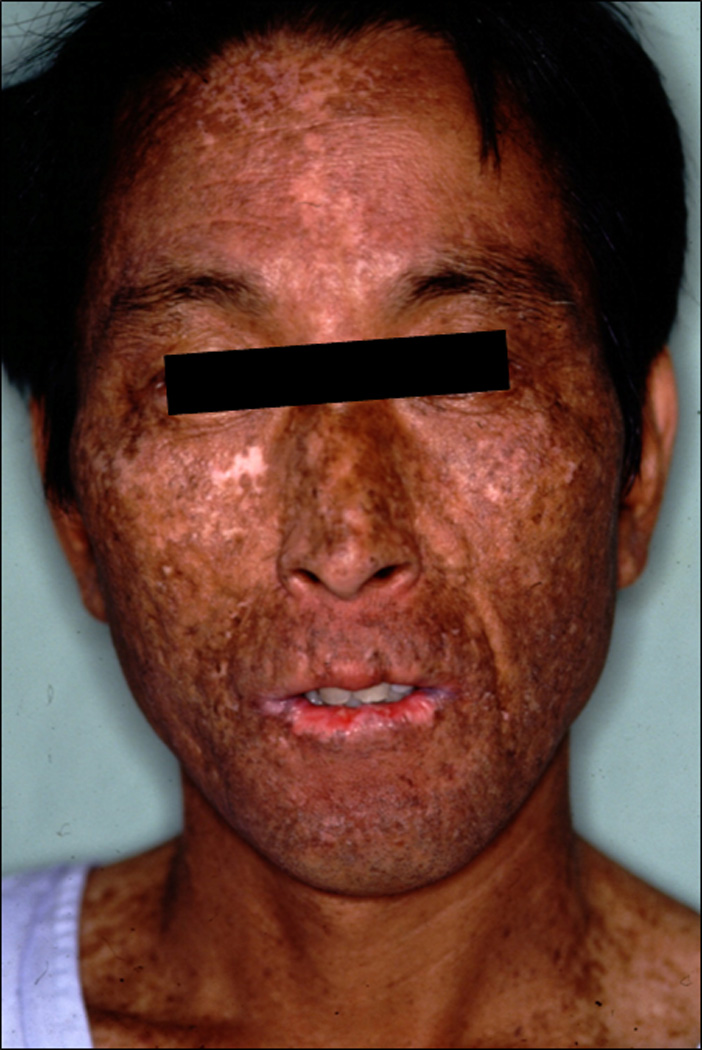
Forty year old patient had marked hyperpigmentation and hypopigmentation of face and sun exposed portion of the shoulders with some sparing upper neck. He also had marked cheilitis and atrophy of lips, however he could open his mouth fully. A squamous cell carcinoma was removed from his lower lip.
CELL STUDIES
The sensitivity to killing by UV of XP3HM cells (D0=0.6 J/m2) [11] was much greater than that of normal N-3 cells (D0=5 J/m2) but not as great as XP-A cells (Figure 2a). The level of UV-induced unscheduled DNA synthesis (UDS) in XP3HM cells was only 8 % of normal [11]. Host cell reactivation assay [11] indicates that XP3HM cells are in XP complementation group G (Figure 2b). Genomic DNA sequencing [13] revealed a novel homozygous T>C change in exon 2 of the XPG gene (c.194T>C, Genbank reference sequence X69978.1), resulting in a predicted amino acid change (p.L65P) (Figure 2c). We constructed an expression vector (pXPGT194C) to determine whether this homozygous missense mutation is the cause of reduced DNA repair. After the transfection of pcDNA3-XPGT194C into XP20BE cells (harboring different XPG mutations) [6], the DNA repair capacity was not restored to the normal level, while the expression of the wild-type XPG cDNA increased the DNA repair capacity of the XP20BE cells (Figure 2d).
Figure 2. Laboratory analysis of XP3HM cells.
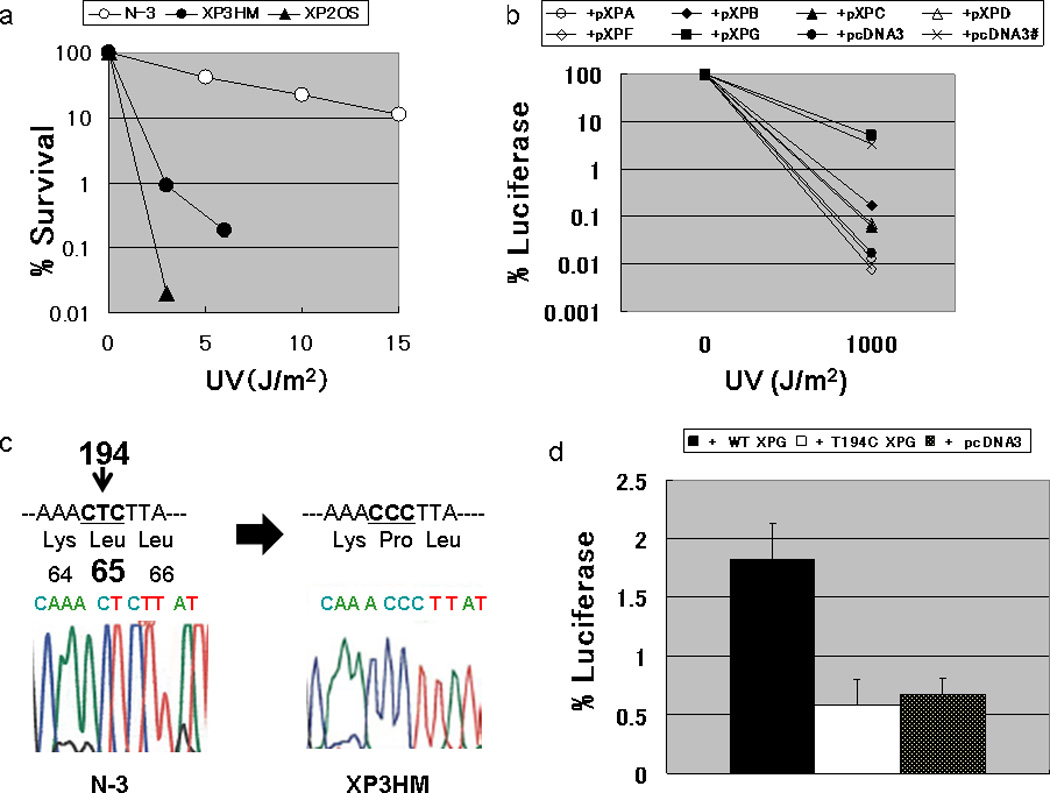
(a) Post-UV survival of the cells: The D0 value (a UV dose that results in 37% cell survival) of XP3HM cells (0.6 J/m2) was much lower than that of the normal N-3 cells (D0 value; 5.0 J/m2). However, these cells were less sensitive than XP2OS (XP-A) (D0 value; 0.3 J/m2). (b) Host cell reactivation assay for the assignment of XP complementation group G in XP3HM cells: The cells were co-transfected with a UV-damaged luciferase gene expression vector along with expression vectors harboring cloned wild-type XP cDNA. Increased luciferase activity was observed when the wild type XPG cDNA expression plasmid was transfected into the cells from the patient, while luciferase activity was still very low after the transfection of expression vectors harboring other XP cDNA (wt XPA, XPB, XPC, XPD, XPF) or the empty vector (pcDNA3). The restored DNA repair capacity after transfecting the wild type XPG cDNA into the XP3HM cells reached the level observed in normal cells (x) after transfecting the empty vector (pcDNA3)., (c) Nucleotide sequence analysis of the XPG gene in XP3HM cells: We identified a homozygous T to C change in exon 2 of the XPG cDNA (c.194T>C) with predicted amino acid change (p.L65P). (d) Host cell reactivation assay for analysis of the DNA repair function of the c.T194C mutation in exon 2 of the XPG gene. We constructed a pXPGT194C plasmid using a pXPG plasmid and QuikChange Site-Directed Mutagenesis Kit (Stratagene, CA, USA). After the transfection of pXPGT194C into the XP20BE cells, the luciferase activity (or the DNA repair capacity; DRC) was not significantly different from the control empty vector plasmid (pcDNA3), while the wild type XPG cDNA increased the DRC of the patient’s cells.
CONCLUSIONS
XP-G was first reported in 1979 [14]. Of the 15 reported XP-G patients including the present case, 7 had XP/CS complex, 2 had XP neurological symptoms and 6 had only cutaneous XP symptoms [6, 15]. Skin cancers were reported in 3 of these 6, all of whom were middle-aged Japanese patients, and in one Western XP/CS complex case, XPCS4RO [6]. All 3 Japanese XP-G patients (XP3HM, XP52HM and XP31KO) had similar clinical phenotypes of mild cutaneous features compared to those seen in XP-A or XP-C patients [7, 10]. They had their first skin cancers (squamous cell carcinoma of the lip, melanoma on the shoulder and basal cell carcinoma on the face), at the ages of 40, 54 and 32, respectively. This is older than many XP patients [1] but younger than the average age of skin cancer incidences in the Japanese general population [16, 17, 18. 19]. In addition, these 3 XP-G patients had no neurological symptoms or any evidence of CS. In contrast, 7 of the 12 non-Japanese patients had clinical features of XP/CS complex and two had severe or a late-onset XP neurological phenotype [6].
The levels of post-UV UDS in the other Japanese XP-G patients (XP52HM and XP31KO) were 50% and 25 % of normal, which was substantially higher than that observed in severe cases of XP-G or in the present case (8%). The mild XP symptoms in these 3 Japanese XP-G patients therefore may not be explained by the residual NER capacity, estimated by the level of UDS. The diversity of the phenotypes of XP-G may be due to different ethnic groups or to the different types and sites of mutations in the XPG gene.
The human XPG (ERCC5) gene, contains 15 exons [13, 20, 21]. The XPG protein functions as a nuclease in the NER, making incisions 3' to the lesion releasing a 25~27 nucleotide DNA fragment containing the photoproduct [3]. Mutations resulting in markedly truncated, inactive XPG proteins are found in XP/CS complex patients, while individuals with XPG without neurological disease have been found to have missense mutations that retain some functional activity [6].
The homozygous c.T194C change in the XPG gene in the present case is predicted to result in an amino acid change; p.L65P. This missense mutation is located in a PIN domain that is highly conserved in eukaryotes (supplemental figure), and can interact with both the XPB and XPD proteins [22]. We confirmed that this mutation was related to decreased NER in the XP3HM cells using post-UV HCR analysis employing mutant XPG cDNA expression vector (Figure 2d). This could explain the sun sensitivity and skin cancers in patient XP3HM. It is possible that p.L65P mutation preserves another function of XPG protein such as transcriptional activity [22, 23] thus preventing neurological degeneration. Genetic analysis has not yet been performed in XP31KO and XP52HM cells to determine their causative mutations. Additional studies will be needed to clarify the molecular basis of the mild clinical features of XPG cutaneous disease in the Japanese XP-G cases.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for the technical assistance of Ms. Makoto Tomida and Ms.Sachiko Nakamura. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
All of the genetic and cellular analyses using the patient’s cells were performed with institutional approval (notification No.25 approved by an Osaka Medical College- Review Board in August 18, 2006) written informed consent from the patient. The study was conducted according to the principles of the Declaration of Helsinki.
Shinichi Moriwaki performed the research under the guidance of Masahiro Takigawa. Naoya Igarashi, Yayoi Nagai, Hiroo Amano and Osamu Ishikawa performed skin surgery, skin biopsy and have been seeing the patient. Sikandar G. Khan and Kenneth H. Kraemer design the research study and analyzed the data. Shinichi Moriwaki, Sikandar G. Khan and Kenneth H. Kraemer wrote the paper.
Footnotes
CONFLICT OF INTEREST
The authors state that they have no conflict of interest.
REFERENCES
- 1.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 2.Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki S, Emmert S, Pike KM, Raziuddin A, Plona TM, Digiovanna JJ, Tucker MA, Kraemer KH. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48:168–176. doi: 10.1136/jmg.2010.083022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kraemer KH, Patrons NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007;145:1388–1396. doi: 10.1016/j.neuroscience.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moriwaki S, Kraemer KH. Xeroderma pigmentosum—bridging a gap between clinic and laboratory. Photodermatol Photoimmunol Photomed. 2001;17:47–54. doi: 10.1034/j.1600-0781.2001.017002047.x. [DOI] [PubMed] [Google Scholar]
- 5.Cleaver JE. Common pathways for ultraviolet skin carcinogenesis in the repair and replication defective groups of xeroderma pigmentosum. J Dermatol Sci. 2000;23:1–11. doi: 10.1016/s0923-1811(99)00088-2. [DOI] [PubMed] [Google Scholar]
- 6.Emmert S, Slor H, Busch DB, Batko S, Albert RB, Coleman D, Khan SG, Abu-Libdeh B, DiGiovanna JJ, Cunningham BB, Lee MM, Crollick J, Inui H, Ueda T, Hedayati M, Grossman L, Shahlavi T, Cleaver JE, Kraemer KH. Relationship of neurologic degeneration to genotype in three xeroderma pigmentosum group G patients. J Invest Dermatol. 2002;118:972–982. doi: 10.1046/j.1523-1747.2002.01782.x. [DOI] [PubMed] [Google Scholar]
- 7.Yoneda K, Moriue J, Matsuoka Y, Moriwaki S, Moriue T, Nakai K, Yokoi I, Nibu N, Demitsu T, Kubota Y. A case of xeroderma pigmentosum complementation G in association with malignant melanoma. Eur J Dermatol. 2007;17:540–541. doi: 10.1684/ejd.2007.0275. [DOI] [PubMed] [Google Scholar]
- 8.Moriwaki S, Stefanini M, Lehmann AR, Hoeijmakers JH, Robbins JH, Rapin I, Botta E, Tanganelli B, Vermeulen W, Broughton BC, Kraemer KH. DNA repair and ultraviolet mutagenesis in cells from a new patient with xeroderma pigmentosum group G and Cockayne syndrome resemble xeroderma pigmentosum cells. J Invest Dermatol. 1996;107:647–653. doi: 10.1111/1523-1747.ep12584287. [DOI] [PubMed] [Google Scholar]
- 9.Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55:1442–1449. doi: 10.1212/wnl.55.10.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ichihashi M, Fujiwara Y, Uehara Y, Matsumoto A. A mild form of xeroderma pigmentosum assigned to complememtation group G and its repair heterogeneity. J Invest Dermatol. 1985;85:284–287. doi: 10.1111/1523-1747.ep12276776. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi Y, Endo Y, Sugiyama Y, Inoue S, Iijima M, Tomita Y, Kuru S, Takigawa M, Moriwaki S. XPA gene mutations resulting in subtle truncation of protein in xeroderma pigmentosum group A patients with mild skin symptoms. J Invest Dermatol. 2010;130:2481–2488. doi: 10.1038/jid.2010.137. [DOI] [PubMed] [Google Scholar]
- 12.Ito T, Tokura Y, Moriwaki S, Yasuda K, Ohnishi A, Furukawa F, Yamaizumi K, Takigawa M. Rothmund-Thomson syndrome with herpes encephalitis. Eur J Dermatol. 1999;9:354–356. [PubMed] [Google Scholar]
- 13.Emmert S, Schneider TD, Khan SG, Kraemer KH. The human XPG gene: gene architecture, alternative splicing and single nucleotide polymorphisms. Nucleic Acid Res. 2001;297:1143–1152. doi: 10.1093/nar/29.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kreijzer W, Jasper NG, Abraham PJ, Taylor AM, Arlett CF, Zelle B, Takebe H, Kimmont PD, Bootsma A. A seventh complementation group in excision-deficient xeroderma pigmentosum. Mutat Res. 1979;62:183–190. doi: 10.1016/0027-5107(79)90231-8. [DOI] [PubMed] [Google Scholar]
- 15.Schaerer OD. XPG : its products and biological roles. Adv Exp Med Biol. 2008;637:83–92. doi: 10.1007/978-0-387-09599-8_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anzai S, Anan T, Kai Y, Goto M, Arakawa S, Shimizu F, Hatano Y, Sato H, Shibuya H, Katagiri K, Fujiwara S. Skin cancer screening on a fishing island and in an inland agricultural area of Japan. J Dermatol. 2005;32:875–882. doi: 10.1111/j.1346-8138.2005.tb00864.x. [DOI] [PubMed] [Google Scholar]
- 17.Chuang TY, Reizner GT, Elpern DJ, Stone JL, Farmer ER. Nonmelanoma skin cancer in Japanese ethnic Hawaiians in Kauai, Hawaii: an incidence report. J Am Acad Dermatol. 1995;33:422–426. doi: 10.1016/0190-9622(95)91387-4. [DOI] [PubMed] [Google Scholar]
- 18.Ishihara K, Saida T, Otsuka F, Yamazaki N. Statistical profiles of malignant melanoma and other skin cancers in Japan: 2007 update. Int J Clin Oncol. 2008;13:33–41. doi: 10.1007/s10147-007-0751-1. [DOI] [PubMed] [Google Scholar]
- 19.Matsuda T, Marugame T, Kamo K, Katanoda K, Ajiki W, Sobue T. Japan Cancer Surveillance Research Group. Cancer incidence and incidence rates in Japan in 2005: based on data from 12 population-based cancer registries in the Monitoring of Cancer Incidence in Japan (MCIJ) project. Jpn J Clin Oncol. 2011;41:139–147. doi: 10.1093/jjco/hyq169. [DOI] [PubMed] [Google Scholar]
- 20.Mudgett JS, Maclnnes MA. Isolation and functional human excision repair gene ERCC5 by intercosmid recombination. Genomics. 1990;8 doi: 10.1016/0888-7543(90)90248-s. 623-jer33. [DOI] [PubMed] [Google Scholar]
- 21.O’donovan A, Wood RD. Identical defects in DNA repair in xeroderma pigmentosum group G and rodent ERCC group G. Nature. 1993;363:185–188. doi: 10.1038/363185a0. [DOI] [PubMed] [Google Scholar]
- 22.Arab HH, Wani G, Ray A, Shah ZI, Zhu Q, Wani AA. Dissociation of CAK from core TFIIH reveals a functional link between XP-G/CS and the TFIIH disassembly state. PLoS.One. 2010;5:e11007. doi: 10.1371/journal.pone.0011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C, Coin F, Egly JM, Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol.Cell. 2007;26:231–243. doi: 10.1016/j.molcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.