

Abstract
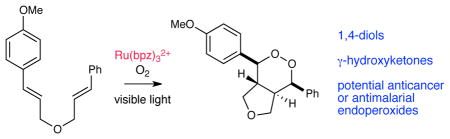
Structurally novel endoperoxides can be sythesized by the photocatalytic cyclotrimerization of bis(styrene) substrates with molecular oxygen. The optimal catalyst for this process is Ru(bpz)32+, which is a markedly more efficient catalyst for these photooxygention reactions than conventional organic photosensitizers. The 1,2-dioxolane products are amenable to synthetic manipulation and can be easily processed to 1,4-diols and γ-hydroxyketones. An initial screen of the biological activity of these compounds reveals promising inhibition of cancer cell growth.
A number of endoperoxides from both natural and synthetic sources have been identified as promising antimalarial, anticancer, and antiviral agents.1 Many studies of the mechanism of action of these remarkable compounds suggest that the endoperoxide moiety is the key pharmacophore; homolytic cleavage of the oxygen–oxygen bond by endogenous reductants produces free radicals that are believed to be responsible for the biological activity of this class of compounds.2 In addition, fragmentation reactions of 1,2-dioxanes provide access to 1,4-diols, γ-ketoalcohols, and similar substitution patterns that are difficult to assemble using standard enolate chemistry.3 As a result of both the biological activity and synthetic utility of this class of compounds, there has been considerable interest in the development of methods for the synthesis of structurally novel 1,2-dioxanes.4
We recently reported that polypyridyl ruthenium(II) photocatalysts can generate radical cations from olefins upon irradiation with visible light, and we showed that these intermediates could undergo intramolecular [2+2] cycloadditions to afford cyclobutane products.5,6,7 In the course of exploring this reaction, we observed that irradiation of bis(styrene) 1 in the presence of a tris(bipyrazyl) ruthenium(II) complex (Ru(bpz)3(PF)2, 2•(PF6)2)8,9 under an atmosphere of oxygen produced the expected [2+2] cycloadduct 3 as well as a by-product that we identified as endoperoxide 4. Intrigued by this result, we initiated an investigation to optimize the reaction conditions for production of 4.
We reasoned that we should be able to partition the reaction towards the [2+2+2] product by increasing the concentration of oxygen. Indeed, by increasing the pressure of oxygen to 4 atm, we were able to improve the ratio of 4 to 3 to 2.8:1 (Table 1, entries 2 and 3). Next, we found that lowering the reaction concentration led to improved yields of the desired endoperoxide to 77% (entry 5). We also investigated the effect of temperature on the reaction and discovered that lowering the reaction temperature to 5 °C completely suppressed formation of the [2+2] cycloadduct without noticeably affecting the rate of endoperoxide formation (entry 6). At this concentration and temperature, we further found that the catalyst loading could be lowered to 0.5 mol% without adverse effect (entry 7). We also attempted the same reaction using the tris(bipyridyl) complex Ru(bpy)32+ instead of Ru(bpz)32+ and observed no reaction, indicating that the identity of the bipyrazyl ligands is critical to the success of the reaction (entry 8).
Table 1.
Optimization and control studies.a
| entry | catalyst (mol%) | O2 | concn (M) | temp (°C) | yield (3/4)b |
|---|---|---|---|---|---|
| 1c | Ru(bpz)3(PF6)2 (5) | 1 atm | 0.1 | 23 | 29%/30% |
| 2c | Ru(bpz)3(PF6)2 (5) | 3 atm | 0.1 | 23 | 22%/35% |
| 3c | Ru(bpz)3(PF6)2 (5) | 4 atm | 0.1 | 23 | 16%/44% |
| 4c | Ru(bpz)3(PF6)2 (5) | 4 atm | 0.05 | 23 | 16%/49% |
| 5 | Ru(bpz)3(PF6)2 (5) | 4 atm | 0.02 | 23 | 11%/77% |
| 6 | Ru(bpz)3(PF6)2 (5) | 4 atm | 0.02 | 5 | <5%/79% |
| 7 | Ru(bpz)3(PF6)2 (0.5) | 4 atm | 0.02 | 5 | <5%/75%d |
| 8 | Ru(bpy)3(PF6)2 (0.5) | 4 atm | 0.02 | 5 | <5%/<5% |
| 9 | tetraphenylporphyrin (5) | 4 atm | 0.02 | 5 | <5%/<5% |
| 10 | 9,10-dicyanoanthracene (5) | 4 atm | 0.02 | 5 | <5%/<5% |
| 11 | triphenylpyrylium•BF4 (5) | 4 atm | 0.02 | 5 | <5%/20% |
Reactions were conducted in a 135 mL glass pressure vessel and irradiated for 30 min with a 200 W incandescent light bulb unless otherwise noted.
Yields determined by 1H NMR spectroscopy using an internal standard, unless noted.
Irradiated for 2 h.
Isolated yield.
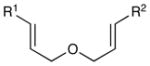
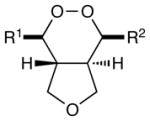
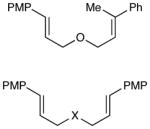
The scope of the photocatalytic endoperoxide synthesis using 2•(PF6)2 is summarized in Table 2. An examination of substituent effects (entries 1–6) revealed that the presence of an electron-donating substituent at the ortho or para position of one of the styrenes is required for successful reaction. As in the case of the [2+2] cycloadditions we previously reported, we suspect that one-electron oxidation of the styrene is the initial step of this process; the failure of less electron-rich substrates such as unsubstituted or m-methoxy-substituted styrenes to initiate cycloaddition is consistent with this hypothesis (entries 2 and 4). Significant variation of the substitution pattern, however, is possible; electron-withdrawing halogen substituents are tolerated at the meta position, and other electron-donating moieties such as silyloxy and carbamate can be used to activate the styrene (entries 7– 9). The reacting partner cannot be an aliphatic olefin (entry 10), but olefins tolerated in this role include styrenes bearing both electron-donating and – withdrawing substituents as well as enynes (entries 11– 14). Substitution on the α position of the olefin is also tolerated (entry 15), although β substitution results in lower yield and poorer diastereoselectivity. Finally, while we were unable use these conditions to conduct efficient photooxidation of untethered styrenes, a variety of tethering groups can be used in this process (entries 16–18).
Table 2.
Scope of the endoperoxide synthesis.a
| entry | substrate | product | time | yieldb | drb |
|---|---|---|---|---|---|
|
|
||||
| 1 | R1 = 4-MeOC6H4 (PMP) | R2 = Ph | 30 min | 92% | 6:1 |
| 2 | R1 = 3-MeOC6H4 | R2 = Ph | 24 h | 0% | -- |
| 3c | R1 = 2-MeOC6H4 | R2 = Ph | 24 h | 45% | 10:1 |
| 4 | R1 = Ph | R2 = Ph | 24 h | 0% | -- |
| 5 | R 1 = 2,4-(MeO)2C6H3 | R2 = Ph | 2 h | 58% | 4:1 |
| 6 | R 1 = 3,4-(MeO)2C6H3 | R2 = Ph | 1 h | 91% | 7:1 |
| 7c | R 1 = 3-Br-4-MeOC6H3 | R2 = Ph | 12 h | 65% | 8:1 |
| 8 | 4-TIPSO-C6H4 | R2 = Ph | 2 h | 75% | 10:1 |
| 9 | 4-BocNH-C6H4 | R2 = Ph | 30 min | 96% | 6:1 |
| 10 | R1 = PMP | R2 = Me | 24 h | <5% | -- |
| 11 | R1 = PMP | R2 = PMP | 30 min | 75% | >10:1 |
| 12 | R1 = PMP | R2 = 2-CF3 C6H4 | 2 h | 82% | 4:1 |
| 13 | R1 = PMP | R2 = 4-ClC6H4 | 2 h | 86% | 5:1 |
| 14 | R1 = PMP | R2 =-C=C-Ph | 2 h | 82% | 2:1 |
| 15c |
|

|
2 h | 81% | >10:1 |
| 16 | X = NTs | 2 h | 73% | >10:1 | |
| 17 | X = C(CO2Et)2 | 2 h | 92% | >10:1 | |
| 18 | X = CH 2 | 2 h | 80% | >10:1 | |
Unless otherwise noted, reactions were conducted using 0.5 mol % Ru(bpz)3(PF6)2 in a glass pressure vessel and irradiated with a 200 W incandescent light bulb.
Yields and diastereomer ratios represent the averaged results of two reproducible experiments.
Conducted using 2 mol % Ru(bpz)3(PF6)2.
The photochemical synthesis of 1,2-endoperoxides by [2+2+2] aerobic cycloaddition of olefins is most commonly achieved by irradiation of 1,1-diarylalkenes in the presence of oxygen and 9,10-dicyanoanthracene (DCA) as a photosensitizer.10 The mechanism is believed to involve photoinduced one-electron oxidation of the alkene to the corresponding radical cation (e.g., 1 to 1•+, Scheme 2). This reactive intermediate undergoes [2+2+2] cyclooxygenation with triplet oxygen to afford an endoperoxide radical cation (5•+) that gives the 1,2-dioxolane product (5) upon reduction by either the reduced sensitizer or another equivalent of substrate.10g The scope of the [2+2+2] cycloaddition, however, is quite limited when DCA is used as a photosensitizer, and only extremely electron rich 1,1-disubstituted styrenes (e.g., 1,1-bis(p-methoxyphenyl) ethylene) have been shown to give good yields of 1,2-dioxolane. This has been attributed to the ability of DCA to sensitize the formation of superoxide radical.11 The formation of this reactive oxygen species is problematic because (1) superoxide reacts with alkene radical cations to produce oxidative cleavage products,10b,12 which are the main products when less electron-rich olefins are utilized, and (2) competitive quenching of the photosensitizer by oxygen reduces the efficiency of the desired cyclooxygenation by decreasing the rate of formation of the key alkene radical cation.
Scheme 2.

Proposed mechanism for endoperoxide formation.
Compared to standard organic photosensitizers, Ru(bpz)32+ is a superior photocatalyst for the formation of endoperoxides and significantly extends the scope of this reaction. Table 1, entries 9–11, show that the photooxygenation of 1 using tetraphenylporphyrin or DCA is not successful, and that the yield of photooxidation is dramatically reduced using triphenylpyrylium tetrafluoroborate (TPT) even when the catalyst loading is increased to 5 mol%. We attribute these observations to several factors. (1) The homoleptic bipyrazyl complex Ru(bpz)32+ is significantly more electron-deficient than Ru(bpy)32+. Thus, its photoexcited state, Ru*(bpz)32+ (+1.4 V vs SCE) can oxidize 1 (+1.1 V) readily to generate the key radical cation intermediate, while Ru*(bpy)32+ (+0.8 V) cannot. (2) The observation that tetraphenylporphyrin fails to generate any of the desired endoperoxide suggests that singlet oxygen is not involved in the formation of endoperoxide 5. (3) Like photoexcited TPT*, Ru*(bpz)32+ does not generate superoxide that might lead to oxidative cleavage of the olefin.11 (4) Ruthenium polypyridyl complexes possess a longer excited state lifetime than common organic PET sensitizers. For example, the excited state lifetime of *TPT is only 3 ns,13 while the lifetime of Ru*(bpz)32+ has been reported to be 740 ns in MeCN.14
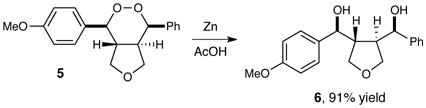
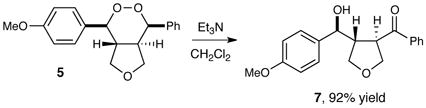
In addition to their biological activity, endoperoxide structures are valuable as versatile synthetic intermediates. When endoperoxide 5 is treated with zinc metal in acetic acid, the O–O bond undergoes reductive cleavage in high yields to afford 1,4-diol 6, which retains all four contiguous stereocenters set in the photooxygenation reaction (eq 1). When 5 is treated with triethylamine, the endoperoxide undergoes a highly regioselective Kornblum–DeLaMare rearrangement15 to afford γ-hydroxyketone 7 (eq 2). We attribute the high selectivity to better stereoelectronic overlap between the equatorial C–H bond and the O–O σ* antibonding orbital (Figure 1). Thus, the products of this new photocatalytic process enable the construction of stereochemically well-defined 1,4-diols as well as γ-hydroxyketones that are difficult to assemble using alternate methods.
Figure 1.

Origin of regioselectivity in Kornblum–DeLaMare rearrangement of 5.
 |
(1) |
 |
(2) |
Finally, the endoperoxides synthesized in this study have not previously been accessible using other synthetic methods, and we speculated that they might have biological activity consistent with that of other endoperoxides. As an initial exploration of their potential activity, a selection of the compounds reported in Table 2 were assayed for cytotoxicity in human prostate cancer cell lines (Du145). Indeed, these novel endoperoxides exhibited a range of IC50 values, varying from >100 μM for the least potent members (Table 2, entries 1, 5, and 11), to 4.6 μM for the most potent (entry 14).16 We believe, therefore, that this method provides an attractive approach to the production of endoperoxide structures whose biological activity profiles have yet to be fully explored.
In summary, Ru(bpz)32+ is an excellent photocatalyst for the synthesis of endoperoxides by [2+2+2] aerobic photooxygenation of α,ω-dienes and is considerably more effective than organic photosensitizers that have commonly been utilized for photooxygenation. This reaction can rapidly generate structurally complex endoperoxides that cannot be synthesized by other means from relatively simple bis(styrene) starting materials. We expect this method will be useful in the discovery of potential anti-malarial and anticancer compounds, and we are now initiating a program aimed at studying this possibility in greater detail.
Supplementary Material
Scheme 1.

Acknowledgments
Financial support from the NIH (GM095666), Beckman Foundation, Research Corporation, and Sloan Foundation is gratefully acknowledged. The NMR facilities at UW-Madison are funded by the NSF (CHE-9208463, CHE-9629688) and NIH (RR08389-01).
Footnotes
Supporting Information Available. Experimental procedures and spectral data for all new compounds (PDF format) are provided. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews of the biological activity of endoperoxides, see: Casteel DA. Nat Prod Rep. 1992;9:289–312. doi: 10.1039/np9920900289.Casteel DA. Nat Prod Rep. 1999;16:55–73.McCullough KJ, Nojima M. Curr Org Chem. 2001;5:601–636.Tang Y, Dong Y, Vennerstrom JL. Med Res Rev. 2004;24:425–448. doi: 10.1002/med.10066.Posner GH, D’Angelo J, O’Neill PM, Mercer A. Expert Opin Ther Pat. 2006;16:1665–1672.Dembitsky VM. Eur J Med Chem. 2008;43:223–251. doi: 10.1016/j.ejmech.2007.04.019.Hencken CP, Kalinda AS, Solomon A, D’Angelo JG. Annu Rep Med Chem. 2009;44:359–378.
- 2.For recent reviews of the mechanism of action of biologically active endoperoxides, see: Wu YK. Acc Chem Res. 2002;35:255–259. doi: 10.1021/ar000080b.Robert A, Dechy-Cabaret O, Cazelles J, Meunier B. Acc Chem Res. 2002;35:167–174. doi: 10.1021/ar990164o.O’Neill PM, Posner GH. J Med Chem. 2004;47:2945–2964. doi: 10.1021/jm030571c.Posner GH, O’Neill PM. Acc Chem Res. 2004;37:397–404. doi: 10.1021/ar020227u.Krishna S, Uhlemann AC, Haynes RK. Drug Resist Update. 2004;7:233–244. doi: 10.1016/j.drup.2004.07.001.
- 3.For a review of synthetic applications of endoperoxide chemistry, see: Balci M. Chem Rev. 1981;81:91–108.Clennan EL. Tetrahedron. 1991;47:1343–1382.
- 4.Selected examples: Porter NA, Funk MO, Gilmore D, Isaac R, Nixon J. J Am Chem Soc. 1976;98:6000–6005. doi: 10.1021/ja00435a037.Porter NA, Gilmore DW. J Am Chem Soc. 1977;99:3503–3504. doi: 10.1021/ja00452a053.Porter NA, Roe AN, McPhail AT. J Am Chem Soc. 1980;102:7574–7576.Xu XX, Dong HQ. J Org Chem. 1995;60:3039–3044.O’Neill PM, Searle NL, Raynes KJ, Maggs JL, Ward SA, Storr RC, Park BK, Posner GH. Tetrahedron Lett. 1998;39:6065–6068.Ghorai P, Dussault PH, Hu C. Org Lett. 2008;10:2401–2404. doi: 10.1021/ol800657m.Harris JR, Waetzig SR, Woerpel KA. Org Lett. 2009;11:3290–3293. doi: 10.1021/ol901046z.Gemma S, Martí F, Gabellieri E, Campiani G, Novellino E, Butini S. Tetrahedron Lett. 2009;50:5719–5722.
- 5.Ischay MA, Lu Z, Yoon TP. J Am Chem Soc. 2010;132:8572–8574. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For recent reviews of transition metal photoredox catalysis, see: Zeitler K. Angew Chem, Int Ed. 2009;48:9785–9789. doi: 10.1002/anie.200904056.Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527–532. doi: 10.1038/nchem.687.Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102–113. doi: 10.1039/b913880n.Teply F. Collect Czech Chem Commun. 2011;76:859–917.
- 7.For leading references on transition metal photocatalysis from other groups, see: Nicewicz DA, MacMillan DWC. Science. 2008;322:77–80. doi: 10.1126/science.1161976.Pham PV, Nagib DA, MacMillan DWC. Angew Chem Int Ed. 2011;50:6119–6122. doi: 10.1002/anie.201101861.Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582.Furst L, Narayanam JMR, Stephenson CRJ. Angew Chem Int Ed. 2011;50:9655–9659. doi: 10.1002/anie.201103145.Andrews RS, Becker JJ, Gagné MR. Angew Chem Int Ed. 2010;49:7274–7276. doi: 10.1002/anie.201004311.Chen Y, Kamlet AS, Steinman JB, Liu DR. Nat Chem. 2011;3:146–153. doi: 10.1038/nchem.932.Larraufie MH, Pellet R, Fensterbank L, Goddard JP, Lacôte E, Malacria M, Ollivier C. Angew Chem Int Ed. 2011;50:4463–4466. doi: 10.1002/anie.201007571.
- 8.Crutchley RJ, Lever ABP. J Am Chem Soc. 1980;102:7128–7129. [Google Scholar]
- 9.We recently reported radical cation Diels–Alder cycloadditions catalyzed by Ru(bpz)32+. See: Lin S, Ischay MA, Fry CG, Yoon TP. J Am Chem Soc. 2011;133:19350–19353. doi: 10.1021/ja2093579.
- 10.(a) Gollnick K, Schnatterer A. Tetrahedron Lett. 1984;25:185–188. [Google Scholar]; (b) Gollnick K, Schnatterer A. Tetrahedron Lett. 1984;25:2735–2738. [Google Scholar]; (c) Mattes SL, Farid S. J Am Chem Soc. 1986;108:7356–7361. [Google Scholar]; (d) Tamai T, Mizuno K, Hashida I, Otsuji Y. Tetrahedron Lett. 1993;34:2641–2644. [Google Scholar]; (e) Gollnick K, Schnatterer A, Utschick G. J Org Chem. 1993;58:6049–6056. [Google Scholar]; (f) Tamai T, Mizuno K, Hashida I, Otsuji Y, Ishida A, Takamuku S. Chem Lett. 1994:149–152. [Google Scholar]; (g) Mizuno K, Tamai T, Hashida I, Otsuji Y, Kuriyama Y, Tokumaru K. J Org Chem. 1994;59:7329–7334. [Google Scholar]
- 11.In accord with this hypothesis, Mattay has reported that triphenylpyrylium tetrafluoroborate (TPT), which cannot form superoxide, is a more effective photosensitizer for photooxidation of 1,1-diphenylethylene than DCA, although the reported yield was still modest. See: Mattay J, Vondenhof M, Denig R. Chem Ber. 1989;122:951–958.
- 12.Eriksen J, Foote CS. J Am Chem Soc. 1980;102:6083–6088. [Google Scholar]
- 13.Miranda MA, García H. Chem Rev. 1994;94:1063–1089. [Google Scholar]
- 14.Haga M-A, Dodsworth ES, Eryavec G, Seymour P, Lever ABP. Inorg Chem. 1985;24:1901–1906. [Google Scholar]
- 15.Kornblum N, DeLaMare JE. J Am Chem Soc. 1951;73:880–881. [Google Scholar]
- 16.See the Supporting Information for details about these screening studies.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.