

Abstract
Epithelial myosin light chain kinase (MLCK)-dependent barrier dysfunction contributes to the pathogenesis of inflammatory bowel diseases (IBD). We reported that epithelial GM-CSF–STAT5 signalling is essential for intestinal homeostatic response to gut injury. However, mechanism, redundancy by STAT5 or cell types involved remained foggy. We here generated intestinal epithelial cell (IEC)-specific STAT5 knockout mice, these mice exhibited a delayed mucosal wound healing and dysfunctional intestinal barrier characterized by elevated levels of NF-κB activation and MLCK, and a reduction of zonula occludens expression in IECs. Deletion of MLCK restored intestinal barrier function in STAT5 knockout mice, and facilitated mucosal wound healing. Consistently, knockdown of stat5 in IEC monolayers led to increased NF-κB DNA binding to MLCK promoter, myosin light chain phosphorylation and tight junction (TJ) permeability, which were potentiated by administration of tumour necrosis factor-α (TNF-α), and prevented by concurrent NF-κB knockdown. Collectively, enterocyte STAT5 signalling protects against TJ barrier dysfunction and promotes intestinal mucosal wound healing via an interaction with NF-κB to suppress MLCK. Targeting IEC STAT5 signalling may be a novel therapeutic approach for treating intestinal barrier dysfunction in IBD.
Keywords: inflammatory bowel disease (IBD), myosin light chain kinase (MLCK), nuclear factor-κB (NF-κB), signals transducers and activators of transcription (STAT) 5, tight junction (TJ)
INTRODUCTION
Inflammatory bowel diseases (IBD), Crohn's disease (CD) and ulcerative colitis (UC), are believed to be caused by complex interactions among genetic susceptibility, environmental triggers and immune-mediated tissue injury leading to chronic relapsing intestinal inflammation (Bouma & Strober, 2003). A balance between IEC proliferation and apoptosis dynamically maintains intestinal epithelial cell (IEC) barrier homeostatic function; however, single cell or highly localized IEC apoptosis alone is insufficient to cause global barrier dysfunction, leading to IBD (Clayburgh et al, 2005; Turner, 2009). In contrast, intestinal tight junction (TJ) barrier dysfunction can accelerate the onset and severity of colitis, and it has been recognized as a major etiologic factor in IBD (Hollander et al, 1986; Su et al, 2009).
Activation of actomyosin contraction, as assessed by phosphorylation of the myosin II regulatory light chain (MLC), has been implicated in the regulation of TJ permeability (Clayburgh et al, 2004). MLC phosphorylation can be regulated by myosin light chain kinase (MLCK) (Wang et al, 2005). Increased MLCK activity has been identified in IECs of IBD patients, and tumour necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) induce IEC barrier dysfunction by up-regulating MLCK expression (Blair et al, 2006; Wang et al, 2005). Long MLCK is the principal form expressed in IECs, and constitutively active MLCK transgenic mice (CA-MLCK Tg) exhibit significant IEC barrier dysfunction (Su et al, 2009). More importantly, long MLCK upregulation is correlated with mucosal inflammation and IBD disease activity (Blair et al, 2006). Conversely, inhibition or depletion of long MLCK protects mice from TNF-α-dependent intestinal epithelial MLC phosphorylation, barrier loss and diarrhea (Clayburgh et al, 2005). Thus, inducible expression of epithelial long MLCK may be a critical regulator of TJ barrier function and may underlie IBD pathogenesis. Identification of the mechanism by which regulation of MLCK activity preserves the TJ barrier may lead to a new therapeutic intervention for IBD.
Recent genome-wide association studies have shown that genetic variation in Janus-associated kinase (JAK) 2 contributes to IBD pathogenesis (Anderson et al, 2011; Barrett et al, 2008; Ferguson et al, 2010). Several cytokines and growth factors utilize JAK2 to activate STAT5, which regulates cell specification, proliferation, differentiation and survival (Hennighausen & Robinson, 2008). Two members of this family, STAT5a and STAT5b (collectively referred to as STAT5), share more than 95% homology (Snow et al, 2003). The absence of STAT5a impairs mammary epithelial adhesion (Miyoshi et al, 2001), suggesting a possible role for STAT5 in regulating cellular junction. In addition, we have previously reported that STAT5 activation is reduced in inflamed colonic IECs in CD and that the administration of growth hormone (GH) and neutralization of TNF-α ameliorates mucosal inflammation in experimental colitis through activation of STAT5 in IECs (Han et al, 2006; Han et al, 2007). Furthermore, stat5b-deficient mice exhibit increased susceptibility to experimental colitis (Han et al, 2009a). Although the mechanism remains unclear, these studies suggest that STAT5 plays a protective role in both intestinal inflammation and barrier homeostatic function.
NF-κB is an important regulator of intestinal homeostatic growth and inflammation (Neurath et al, 1996); however, its effects on various intestinal cells are divergent. Deletion of IKK-γ/NEMO in IECs causes severe spontaneous colitis in mice at a young age (Nenci et al, 2007). Conversely, NF-κB activation in immune cell types promotes chronic intestinal inflammation in both CD and UC (Schreiber et al, 1998). In IECs, TNF-α, IFN-γ and interleukin-1β (IL-1β) induce a progressive increase in MLCK protein expression directly through NF-κB activation, leading to the increases in TJ permeability (Al-Sadi et al, 2009). Hence, tight regulation of NF-κB activity is critical in the intestinal mucosa in order to maintain barrier function during injury.
We have reported that STAT5 maintains colonic barrier integrity by modulating NF-κB activation (Han et al, 2009a); however, the subsequent molecular events and the disease drivers are poorly defined. In our current studies, employing IEC-specific STAT5 knockout mice (STAT5IEC KO) and CA-MLCK Tg and long MLCK knockout (MLCKKO) mice (Clayburgh et al, 2005; Su et al, 2009), we demonstrate for the first time that enterocyte STAT5 signalling stabilizes ZO assembly and protects against IEC barrier dysfunction via an interaction with NF-κB to suppress MLCK; a STAT5⊣ NF-κB → MLCK negative feedback mechanism in IECs regulates IEC TJ barrier function and innate immune responses to intestinal injury.
RESULTS
Depletion of STAT5 in the IECs predisposes mice to intestinal injury
By crossing STAT5 flox mice (Cui et al, 2004) with villin-cre transgenic mice (Supporting information Fig S1A), both stat5a and stat5b genes were specifically depleted from IECs by CRE-mediated recombination (Supporting information Fig S1B). The STAT5IEC KO mice had a normal birth rate, weight gain and intestinal histology similar to littermate STAT5 flox mice (STAT5IEC WT) (Fig 1A). We treated STAT5IEC KO mice with 3% DSS either for 7 days for acute experiments or for 5 days followed by 5 days of water for recovery studies. The STAT5IEC KO mice were more susceptible to acute DSS-induced colitis and also had much poorer colonic wound healing during the water recovery phase than STAT5IEC WT mice (Fig 1A). This worse healing was characterized by a significantly higher histological score (Fig 1B and C), a slower rate of weight gain (Fig 1D), a shorter colon length (Fig 1E), and more severe rectal bleeding, soft stools and worse diarrhea than STAT5IEC WT mice. To confirm our findings, we extended the DSS treatment and water recovery either for 5-day DSS followed by 10-day water recovery or 7-day DSS followed by 14-day water recovery. We consistently found that the STAT5IEC KO mice, followed by either 10-day water recovery or 14-day water recovery, also exhibited much poorer colonic wound healing than STAT5IEC WT mice (Fig 1A and F). The STAT5IEC KO mice were characterized by higher histopathological scores (Fig 1F) and slower rate of weight gain than STAT5IEC WT mice (Supporting information Fig S2A). We have reported that STAT5 activation is reduced in the inflamed IECs in CD (Han et al, 2006). Here, we extend previous findings and we found in particular that STAT5 activation was dramatically upregulated in the adjacent healthy crypts in CD (Fig 1G). In accordance with our previous report (Han et al, 2010), we found the impairment of tyrosine phosphorylated STAT5 in necrotic and inflamed colonic crypts in the DSS-treated controls. Interestingly, STAT5 activation is elevated in the proliferating crypt cells (Supporting information Fig S2B). These findings suggest a protective or repair mechanism. To determine whether these phenotypic responses were limited in colon, we examined whether STAT5IEC KO mice also responded to non-steroidal anti-inflammatory drug (NSAID) administration, which induces a specific ileal injury in immune-deficient mice (Han et al, 2009b; Han et al, 2010). We found that STAT5IEC KO mice developed severe ileitis (Fig 2A) with a higher ileal histo-pathology and ulceration scores (Fig 2B and C). These data suggest that STAT5 signalling in enterocytes is required for protection against intestinal injury and mucosal wound healing.
Figure 1. Depletion of STAT5 in the IECs predisposes mice to DSS-induced injury and delays intestinal mucosal wound healing.

STAT5IEC KO mice were treated with 3% DSS for 7 days, or for 5 days followed by 5 days of water, or for 7 days followed by 14 days of water.
A-F. Disease severity (A), histological score (B,C), weight gain (D), colon length (E) and histological scores during disease recovery (F) were evaluated. Ulcer area is designated by brackets, DC: distal colon, MC: middle colon. Results were expressed as the mean ± SEM (n = 10), *p < 0.01 versus DC in the STAT5IEC WT mice and #p < 0.01 versus MC in the STAT5IEC WT mice. Original magnification, ×100, bar = 100 µm.
G. The paraffin-embedded slides were from 9 healthy controls (CON) and 10 patients with CD. Phospho-tyrosine STAT5 (pSTAT5) was determined by IH in CON, inflamed crypt in CD (CD), and normal crypts in CD (N-CD). The results were quantitated as pSTAT5 positive colonic IECs per Crypt. Results were expressed as the mean ± SEM, original magnification, ×400, bar = 100 µm.
Figure 2. Depletion of STAT5 in enterocytes increases NSAID ileal injury.
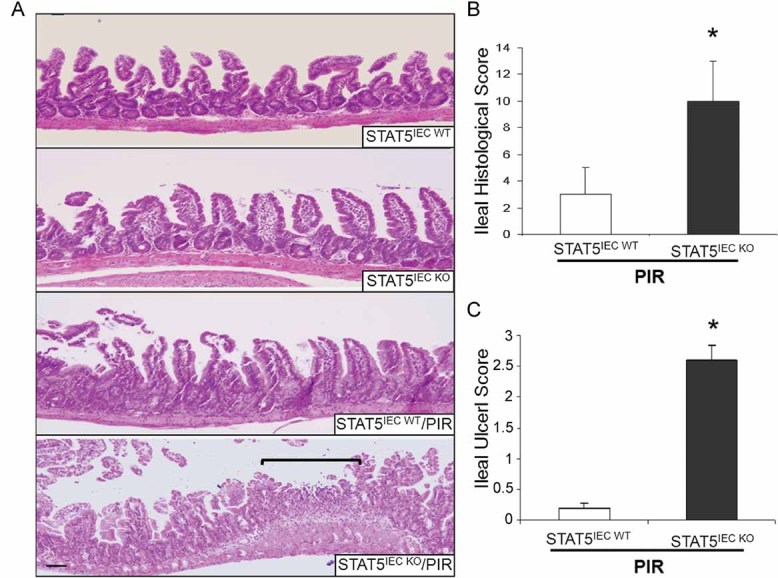
STAT5IEC WT and STAT5IEC KO mice received PIR in the chow for 2 weeks.
A-C. Disease severity (A), ileal histological score (B) and ileal ulceration score (C) were determined, ulcer area is designated by brackets, results were expressed as the mean ± SEM (n = 10), *p < 0.01 versus STAT5IEC WT/PIR. Original magnification, ×100, bar = 100 µm.
Loss of STAT5 signalling enhances intestinal mucosal adaptive immune response and delays mucosal repair during inflammation
We next measured intestinal immune cytokine levels and inflammatory cell infiltration. Real-time RT-PCR of colonic mucosa demonstrated that local mRNA levels of all examined cytokines, including TNF-α, IFN-γ, IL-6, IL-17 and IL-23, were comparable in STAT5IEC WT and STAT5IEC KO mice under basal conditions. Similarly, these cytokines were elevated in both genotypes following 7-days DSS acute injury, with the exception for reduced levels of IL-10 in STAT5IEC KO (Fig 3A). All tested cytokine levels remained significantly higher in STAT5IEC KO, however, as compared to STAT5IEC WT mice after a 5-day recovery period (Fig 3A). Importantly, isolated regulatory T cells (CD4+CD25+Foxp3+), activated CD4+ T cells (CD4+CD44hiCD62Llo) and macrophages (CD4−CD11C−CD11b+F4/80+) from mesenteric lymph nodes (MLN) were also significantly higher in STAT5IEC KO mice in the recovery group than the recovering STAT5IEC WT mice, as measured by flow cytometry (FACS) (Fig 3B, Supporting information Fig S3A). Consistently, colonic mucosal F4/80+ macrophages and Foxp3+ regulatory T cells were markedly increased in STAT5IEC KO mice in the recovery group compared to the recovering STAT5IEC WT mice, as shown by immunohistochemistry staining (IH) (Supporting information Fig S3B and C). Severe intestinal mucosal inflammation was characterized by a larger MLN size (Fig 3C, Supporting information Fig S3D) and a significantly higher amount of bacterial translocation to the MLN in STAT5IEC KO mice relative to STAT5IEC WT mice after a 5-day recovery (Fig 3D). Therefore, loss of IEC STAT5 deregulates the protective immune response to gut injury, significantly delaying the intestinal mucosal repair. Together, these data indicate that enterocyte STAT5 is required for control of intestinal mucosal immune response and mucosal wound repair.
Figure 3. Loss of STAT5 signalling exaggerates intestinal mucosal adaptive immune response.

Mice were treated with 3% DSS for 5 days followed by 5 days of water.
A. Colonic mucosal RNA was extracted; cytokine levels were quantitated by qPCR.
B. Regulatory T cells, activated T cells and macrophages were determined in MLNs using FACS. Results were expressed as the mean ± SEM (n = 10). *p < 0.01 versus STAT5IEC WT/DSS, ∧p < 0.01 versus STAT5IEC WT/DSS + water recovery, #p < 0.01 versus STAT5IEC WT and STAT5IEC KO/DSS.
C,D. MLNs were weighed (C), and bacterial translocation in the isolated MLNs (D) was determined in STAT5IEC WT and STAT5IEC KO mice by colony-forming units (CFU). Results were expressed as the mean ± SEM (n = 10), *p < 0.05 versus STAT5IEC WT, #p < 0.05 versus STAT5 IEC WT/DSS, ∧p < 0.05 versus STAT5IEC WT/DSS + water recovery.
Depletion of IEC STAT5 enhances NF-κB activation in response to gut injury
NF-κB activation promotes chronic intestinal inflammation in both CD and UC (Han et al, 2006; Schreiber et al, 1998). Our data exhibited that nuclear levels of RelA/p65 remained at significantly higher levels in the colonic mucosa of STAT5IEC KO mice than in littermate controls either under basal conditions or during water recovery after the cessation of DSS (Fig 4A). In addition, colonic IκBα expression, an endogenous suppressor of NF-κB activity, was consistently and remarkably reduced under basal conditions or in the water recovery group in STAT5IEC KO mice compared to STAT5IEC WT mice (Fig 4B). Most importantly, our data exhibited that depletion of IEC STAT5 in mice increased basal NF-κB activity in IECs, as determined by the nuclear abundance and phosphorylation of RelA/p65 (Fig 4C and D). Together, these data suggest that there is an interaction between IEC STAT5 and NF-κB in the regulation of the intestinal immune response to injury. IEC STAT5 could suppress NF-κB nuclear activation by stabilizing IκBα to ameliorate mucosal inflammation (STAT5 → IκBα⊣ NF-κB).
Figure 4. STAT5 in IECs regulates NF-κB activation in response to gut injury.
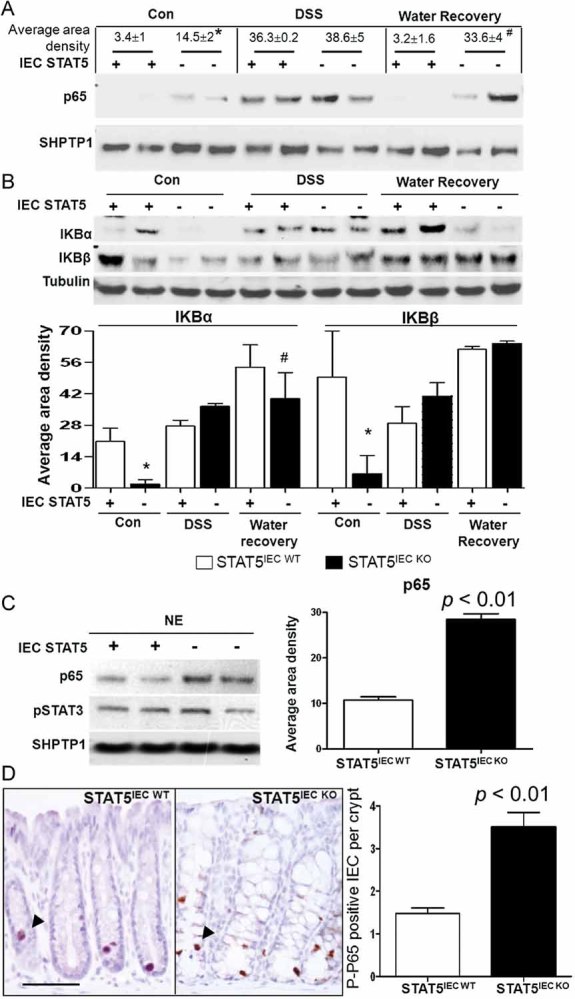
Mice were treated with 3% DSS for 5 days followed by 5 days of water. Colonic mucosa was extracted, and NE and CE recovered.
A,B. p65 nuclear abundance was determined in NE (A), and IκBα and Iκβ were detected in CE (B). Signal intensity was determined by densitometry, results were expressed as the mean ± SEM (n = 5). *p < 0.05 versus STAT5IEC WT mice, #p < 0.05 versus STAT5IEC WT mice in the water recovery group.
C. IECs were isolated from STAT5IEC WT and STAT5IEC KO mice, p65 nuclear abundances were determined by WB.
D. Colon sections were immunostained with phosphoserine p65 (P-p65) as indicated by arrows. Results were expressed as average positive cells per crypt, n = 5, bar = 100 µm.
Depletion of STAT5 in IECs upregulates MLCK activation, disrupts zonula occluden (ZO) assembly and leads to increased TJ permeability
Our data demonstrated that STAT5IEC KO mice exhibited a disrupted intestinal barrier under basal conditions, characterized by increased para-cellular permeability in both ileum and colon (Fig 5A). STAT5IEC KO mice also exhibited an increased focal colonic IEC apoptosis, as measured by IH analysis of cleaved Caspase-3, and this was accompanied by a slight increase in proliferation of IECs (Supporting information Fig S4A and B). However, low levels of IEC apoptosis that are highly localized are insufficient to cause global and persistent TJ barrier dysfunction, leading to IBD (Clayburgh et al, 2005; Turner, 2009). To further understand the molecular basis by which STAT5 signalling mediates the regulation of IEC barrier function, we thus studied long MLCK mRNA levels in intestinal mucosa and phosphorylation of the myosin light chain (pMLC) in IECs, as these factors have been implicated as key regulators of TJ permeability (Turner et al, 1997). Both quantitative PCR and immunofluorescence (IF) staining showed that MLCK RNA and pMLC were upregulated in STAT5 deficient IECs (Fig 5B and C). Protein and RNA were then isolated from IECs of STAT5IEC WT and STAT5IEC KO mice using either EDTA-based or LCM to determine pMLC levels and TJ proteins (TJPs) expression. The ratio of pMLC and MLC was increased in STAT5 deficient IECs (Fig 5D). Immunoblot and quantitative PCR also demonstrated that ZO-1, -2 and -3, but not other TJP components, had significantly reduced protein and mRNA levels in STAT5-deficient ileal and colonic IECs compared to control IECs (Fig 5D and E and Supporting information Fig S4A). Consistently, IF staining confirmed that among the various components of TJPs, the distributions of ZO-1, ZO-2 and -3 were disrupted in IECs in STAT5IEC KO mice (Fig 5F). MLCK has been demonstrated to directly interact with ZO-1 actin binding region (ABR), MLCK-dependent barrier regulation is associated with ZO-1 stabilization at the TJ (Marchiando et al, 2010; Yu et al, 2010). We found elevated levels of pMLC and reduced ZO abundance in STAT5-deficient IECs compared to STAT5IEC WT IECs (Fig 5D and Supporting information Fig S5A), indicating that STAT5 in enterocytes may stabilize ZO assembly under basal conditions, possibly acting on MLCK activity.
Figure 5. Depletion of STAT5 in IECs upregulates MLCK activation and disrupts ZO assembly, leading to increased TJ permeability.

- Colonic and ileal paracellular permeability was determined with everted gut sac in STAT5IEC WT and STAT5IEC KO mice.
- Colonic and ileal mucosal RNA was extracted; long MLCK mRNA levels were quantitated by qPCR. Results were expressed as the mean ± SEM (n = 10). *p < 0.05 versus STAT5IEC WT mice.
- Colonic and ileal frozen sections were immunostained with pMLC (red). Negs are negative controls, n = 5.
- Ileal IECs were isolated from STAT5IEC WT and STAT5IEC KO mice, pMLC and MLC, ZO-1, -2 and -3, JAM-A, claudin-2 and occludin were identified with WB. Signal intensity was determined by densitometry. MLCK activation was expressed as the ratio of pMLC and MLC. n = 5.
- Ileal IECs were isolated using LCM, ZO-1, -2 and -3 mRNA levels were determined by qPCR, *p < 0.05 versus STAT5IEC WT mice, n = 10.
- Ileal frozen sections were, respectively, immunostained with ZO-1 (green) or ZO-3 (red), n = 5. Insets are the confocal images. Original magnification, ×100, bar = 50 µm.
Knockdown of stat5 in IEC monolayers increases MLC phosphorylation and disrupts TJ, leading to hyperpermeability in IEC monolayer
To confirm our in vivo studies, we grew HT-29 IEC monolayers on Transwell filters and used RNA interference (RNAi) to achieve approximately 90% knockdown of STAT5 expression (Fig 6B). We observed that paracellular permeability in 17-day post-confluent HT-29 cell monolayers, assessed by the apical-to-basolateral flux of FITC-dextran 4 kDa (FD4), was markedly increased by knocking down STAT5 expression. Transepithelial electrical resistance (TEER) was reduced in STAT5 knockdown monolayers (Fig 6A). This reduction was accompanied by an elevation in pMLC levels as well as a reduction in the levels of ZO-1, -2 and -3, but not in the levels of other membrane-associated TJPs (Fig 6B). Of note, knockdown of STAT5 did not alter cleaved Caspase-3 or the expression of the pro-apoptotic factor Bax in well-differentiated IEC monolayers (Fig 6B). To further confirm these findings, we utilized IF to analyse TJ distribution in HT-29 monolayers grown on the Transwell inserts. We found that distribution of ZO-1 and -3 along cell boundaries was disrupted by STAT5 knockdown (Supporting information Fig S5B). The distribution of all other TJP components remained unaffected by STAT5 knockdown. Accordingly, these findings demonstrate that STAT5 signalling plays a non-redundant role in ZO expression and assembly; our data is consistent with a model that STAT5 may regulate TJ permeability by modulating MLCK activity to stabilize ZO assembly under basal conditions (STAT5⊣ MLCK → ZOs).
Figure 6. Knockdown of STAT5 in IEC monolayers increases TJ permeability.
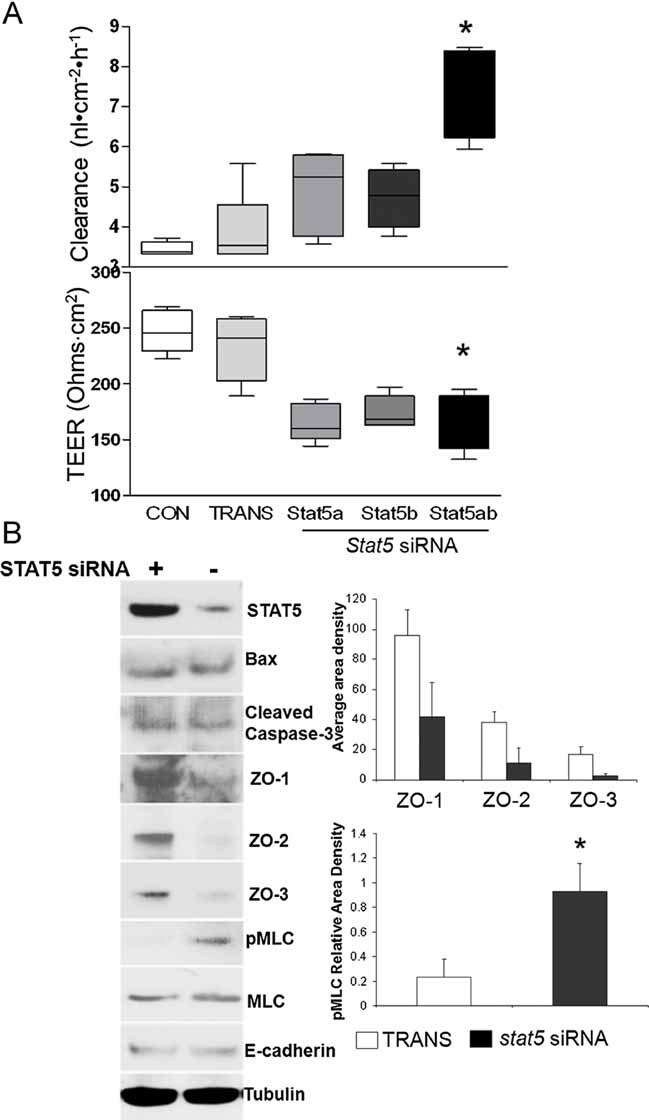
- HT-29 IEC monolayers, grown on transwell filters, were interfered with stat5a, stat5b or stat5a and b (stat5) siRNA. Paracellular permeability was assessed as apical-to-basolateral flux of FITC dextran (FD-4) clearance and TEER. Results were expressed as the mean ± SEM (n = 7). TRANS: transfection reagent only. *p < 0.01 versus CON and TRANS.
- Total proteins (TP) were extracted from post-confluent HT-29 cell monolayer, ZO-1, -2 and -3, STAT5, cleaved Caspase-3, E-cadherin, pMLC and MLC were measured by WB (n = 5). Signal intensity was determined by densitometry. *p < 0.01 versus TRANS.
STAT5 knockdown amplifies proinflammatory cytokine-induced hyperpermeability in IEC monolayer
NF-κB activation is sufficient to upregulate transcription of MLCK from the long MLCK promoter, and some evidences suggest that TJPs (TJPs; occludin, claudin-1 and ZO-1) are internalized through an NF-κB-dependent pathway (Graham et al, 2006; Tang et al, 2010). We thus ask whether there is a transcriptional regulation between NF-κB and STAT5 activation in MLCK-mediated IEC barrier function that impacts intestinal immune response to mucosal inflammation. To mimic in vivo inflammatory signalling, we combined IFN-γ (10 ng/ml) with TNF-α (10 ng/ml) to basolaterally stimulate the HT-29 monolayers. Cytokine-induced monolayer hyperpermeability was significantly enhanced by STAT5 knockdown compared to cytokine-treated controls (Fig 7A). We then demonstrated that NF-κB activity and pMLC levels were consistently increased in stat5 knockdown HT-29 IEC cell monolayers under baseline conditions, and were enhanced by IFN-γ and TNF-α co-administration in the presence of stat5 RNAi (Fig 7B and C). Conversely, IκBα, but not IκBβ, was reduced in STAT5 knockdown HT-29 IEC monolayers. This reduction was potentiated by IFN-γ and TNF-α co-administration in the presence of stat5 RNAi (Fig 7D). We next measured NF-κB and MLCK promoter activity by Electrophoretic Mobility Shift Assay (EMSA). Consistently, we found that NF-κB-mediated transcription as well as DNA binding activity on the NF-κB binding element from the MLCK promoter was promoted in IEC monolayers by stat5 RNAi, and was further enhanced in the cytokine-stimulated monolayers with STAT5 knockdown (Fig 7E and F). Taken together, STAT5 modulates NF-κB DNA binding in the MLCK promoter to suppress TNF-α-induced MLCK transcription; whereas STAT5 deficiency in IECs increases NF-κB-activated MLCK to induce a persistent TJ barrier dysfunction, resulting in the impairment of intestinal mucosal wound healing in response to gut injury.
Figure 7. STAT5 Knockdown amplifies pro-inflammatory cytokine-induced hyperpermeability in IEC monolayer.

HT-29 IEC monolayers on transwell filters, with and without stat5 RNAi, were exposed to IFN-γ (10 ng/ml) induction for 18 h and then TNF-α (10 ng/ml) stimulation for 12 h.
A. Paracellular permeability in HT-29 cell monolayers was assessed as FD4 clearance by the apical-to-basolateral flux of FD4 and TEER. The inset indicated STAT5 knockdown in the monolayers. Results were expressed as the mean ± SEM (n = 6).
B-D. HT-29 monolayer was interfered with stat5 siRNA, p65 nuclear abundance (B), pMLC levels in TP (C) and cytosolic IκB α and β levels (D) were assessed in the presence and absence of IFN-γ and TNF-α co-stimulation. Signal intensity was determined by densitometry, results were expressed as the mean ± SEM (n = 5).
E,F. DNA binding activity in the NF-κB promoter activity (E) or MLCK promoter containing κB binding sites (F) was analysed with nuclear extracts by EMSAs. Cold competition assays were performed by adding unlabeled oligonucleotides in 100-fold molar excess; NF-κB identity was confirmed by specific p65 antibody (2 µg) as indicated, and the arrow represented the shifted bands. The representative result is shown from five repeated experiments.
Depletion of long MLCK restores intestinal barrier dysfunction in STAT5IEC KO mice, and facilitates the intestinal mucosal wound healing in response to gut injury
To test whether MLCK mediates IEC barrier dysfunction in STAT5IEC KO, we crossed MLCKKO mice with our STAT5 flox mice, and then depleted IEC STAT5 by crossing these mice with villin-cre mice (MLCK/STAT5IEC KO). We found that depletion of long MLCK reduced increased IEC para-cellular permeability in STAT5IEC KO mice (Fig 8A), suggesting that epithelial MLCK mediates the STAT5 regulation of TJ permeability and STAT5 suppresses MLCK-dependent barrier dysfunction under basal conditions. We next tested the role of IEC STAT5 activation in the protection of epithelial MLCK-induced mucosal injury. To test this, we used CA-MLCK Tg and MLCKKO mice. During recovery from DSS-induced colonic injury, CA-MLCK Tg mice exhibited slower wound healing and repair of intestinal mucosa than littermate controls as characterized by a significantly higher histological score (Fig 8B and C), more severe rectal bleeding, slower weight gain, shorter colon length, softer stools, and worse diarrhea than littermate controls. MLCKKO mice were apparently protected against DSS-induced colonic injury either in DSS acute injury or during recovery (Fig 8B and C). Most importantly, STAT5 activation in IECs was abrogated in CA-MLCK Tg mice following DSS exposure and water recovery as compared to littermate controls, whereas knockout of long MLCK apparently restored the activation of STAT5 in IECs in response to DSS injury (Fig 8D). Taken together, these findings suggest that IEC STAT5 is required for control of the intestinal mucosal immune response and promotion of mucosal wound repair.
Figure 8. Depletion of long MLCK restores intestinal barrier dysfunction in STAT5IEC KO mice, and facilitates the intestinal mucosal wound healing in response to gut injury.
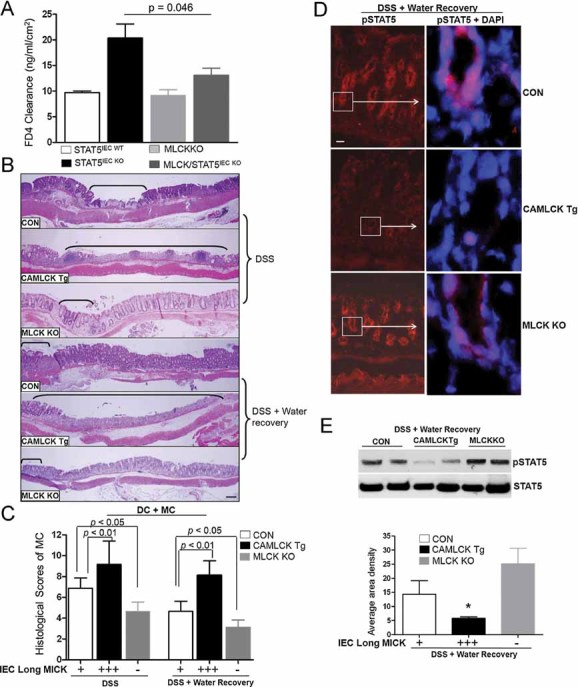
A. Ileal paracellular permeability was determined with everted gut sac in MLCK/STAT5IEC KO mice, results were expressed as the mean ± SEM (n = 7). CA-MLCK, MLCK KO and littermate control mice (CON) were treated with 3% DSS for either 7 days or for 5 days followed by 5 days of water.
B-D. Disease severity (B) and histological scores (C) were evaluated. Ulcer area is designated by brackets, results were expressed as the mean ± SEM, n = 10. (D) pSTAT5 was determined by IF in CA-MLCK Tg, MLCKKO and littermate control mice exposure to 5 days DSS followed by 5 days of water, n = 7.
E. IECs were isolated from CA-MLCK Tg, MLCKKO and littermate control mice and NE extracted. pSTAT5 nuclear abundance was determined by WB in NE. Signal intensity was determined by densitometry, results were expressed as the mean ± SEM, n = 5. *p < 0.01 versus controls.
Knockdown of RelA/p65 in IEC monolayers prevents hyperpermeability induced by interfering with stat5 expression
To further demonstrate that STAT5 regulates NF-κB activity in IECs, further modulates MLC phosphorylation, we performed a simultaneous knockdown of STAT5 and RelA/p65 in HT-29 monolayers. We observed that paracellular permeability was increased in STAT5 knockdown monolayers, but not in HT-29 monolayers following RelA/p65 knock down (Fig 9A). Monolayer permeability induced by STAT5 knockdown was further potentiated by IFN-γ (10 ng/ml) and TNF-α (10 ng/ml) co-administration, while RelA/p65 knockdown promoted resistance of proinflammatory cytokine-induced hyperpermeability in monolayers (Fig 9A). Surprisingly, STAT5 and RelA/p65 double knockdown rescued the hyperpermeability that was induced by pro-inflammatory cytokines in single STAT5 knockdown monolayers (Fig 9A). Thus, the reduction of NF-κB may prevent hyperpermeability caused by the loss of STAT5 in IEC monolayers. Furthermore, we consistently found that STAT5 knockdown increased the levels of pMLC and potentiated cytokine-induced pMLC, which was restored by concurrent RelA/p65 knockdown (Fig 9B). Knockdown of STAT5 and basolateral stimulation with cytokines, however, did not increase the expression of the pro-apoptotic protein Bax (Fig 9B). Taken together, STAT5 in enterocytes may negatively regulate NF-κB activation of MLCK and facilitates the protection of intestinal hyperpermeability induced by pro-inflammatory cytokines (STAT5⊣ NF-κB → MLCK; Fig 9C).
Figure 9. Knockdown of RelA/p65 in IEC monolayers rescues hyperpermeability induced by interfering with stat5 expression.

- HT-29 IEC monolayers, grown on Transwell filters, were interfered with RelA/p65 and/or stat5a and b (stat5) siRNA and exposed to IFN-γ (10 ng/ml) induction for 18 h and then stimulation with TNF-α (10 ng/ml) for 12 h. Para-cellular permeability in monolayers was assessed as FD-4 clearance and TEER.
- Total proteins (TP) were extracted, P-p65, p65, pMLC, MLC and Bax were determined by WB. Results were expressed as the mean ± SEM (n = 6). ∧p < 0.01 versus Controls (transfection reagent only), *p < 0.05 versus cytokine-treated controls, &p < 0.05 versus STAT5 knockdown monolayer. #p < 0.01 versus cytokine-treated STAT5 knockdown monolayer.
- STAT5 signalling in the IEC barrier function and mucosal injury. STAT5⊣ MLCK → ZOs feedback mechanism stabilizes IEC TJ barrier function under basal conditions (1); STAT5⊣ NFκB → MLCK negative feedback loop regulates IEC barrier function under inflammatory conditions (2) and mucosal immune responses to gut injury (3). MA, membrane-associated TJPs.
DISCUSSION
Global depletion of STAT5 predisposes mice to experimental colitis (Han et al, 2009a), however, the phenotype could be caused by the defective STAT5 signalling in IEC compartment and/or sub-epithelial laminar propria (Yao et al, 2006). We have recently shown that STAT5 in enterocytes is required for GM-CSF regulation of homeostatic responses to gut injury (Han et al, 2010). Thus, in this study, we investigated whether enterocyte STAT5 directly regulates intestinal homeostatic responses to injury. We demonstrated that an IEC-specific stat5 deletion increases the susceptibility of these animals to DSS-induced colitis as well as to NSAID-induced ileal injury in mice (Fig 1B); STAT5IEC KO mice with colitis exhibited delayed intestinal mucosal wound healing (Fig 1C), as characterized by significantly higher levels of colonic mucosal inflammatory cells, Th1 and Th17 cytokines and NF-κB activation in STAT5IEC KO mice as compared to STAT5IEC WT mice after a 5-day recovery period. Consistently, the extended time of DSS treatment and recovery resulted in the worse mucosal injury and poorer healing in STAT5IEC KO mice (Fig 1F). These extra time points excluded the possibility that it is a particular kinetic of recovery which could affect the mucosal physiology and wound healing in response to gut injury. Accordingly, enterocyte STAT5 is required for intestinal mucosal wound healing in response to gut injury.
TNF-α, IFN-γ and IL-1β induce NF-κB activation in the well-differentiated IECs, leading to a progressive increase in MLCK protein expression. This process has been postulated to be an important mechanism that contributes to intestinal inflammation (Al-Sadi et al, 2009; Turner, 2009). Epithelial-specific IκBα mutant transgenic mice exhibit attenuated T cell-induced diarrhea and abrogated increased paracellular permeability (Tang et al, 2010). These studies suggest that aberrant activation of NF-κB in IECs controls the expression of many inflammatory cytokines involved in the pathogenesis of IBD. In normal mammary gland development and differentiation, NF-κB regulates the milk protein gene β-casein by negatively interfering with STAT5 tyrosine phosphorylation (Geymayer & Doppler, 2000). Conversely, we reported that STAT5 signalling antagonize NF-κB binding activity to regulate epithelial survival (Han et al, 2006; Han et al, 2009a). Our current studies showed that depletion of STAT5 in IECs resulted in reduction of IκBα cytosolic abundance, upregulation of NF-κB nuclear activation and delayed mucosal wound healing. Interestingly, consistent with the recent report (Smyth et al, 2011), we found that combination of IFN-γ and TNF-α increased STAT5 nuclear abundance in HT-29 monolayers (Supporting information Fig S5C). We think that simultaneous up-regulation of STAT5 also reflects a feed-back response by IEC monolayer during injury; in other words, the up-regulated STAT5 plays a protective role in the monolayer TJ disintegration induced by cytokine-mediated NFκB and MLCK activation. Together, these data support an interactive regulation between the STAT5 and NF-κB pathways, and this interaction may impact IEC barrier function and regulate mucosal wound repair (Fig 9C).
The disrupted IEC barrier has been proposed to play a critical role in initiating disease through broadly activating mucosal immune responses and accelerating the onset and severity of immune-mediated colitis (Blair et al, 2006; Su et al, 2009), for instance, genetic ablation of IL-10 increases small bowel para-cellular permeability, leading to the spontaneous colitis in mice (Madsen et al, 1999). Conversely, preservation of the integrity of the IEC barrier may promote intestinal mucosal healing in IBD (Dignass, 2001). In our studies, STAT5IEC KO mice exhibited IEC barrier dysfunction at baseline, which was characterized by significantly elevated paracellular permeability, focal colonic IEC apoptosis, and slightly increased proliferation; however, knockdown of STAT5 in well-differentiated IEC monolayers did not increase apoptosis under basal conditions. In patients with active IBD, increased IEC apoptosis, erosions and ulcer-type lesions contribute to focal barrier defects. In contrast, morphological breakage and loss of TJ strands are associated with the global barrier defects seen in active IBD (Schulzke et al, 2009; Zeissig et al, 2007). Ultrastructural analyses confirmed that the bulk of the transmucosal transport occurred across TJ rather than via gross epithelial defects, such as ulcers (Machen & Erlij, 1972; Shen et al, 2010). Therefore, the IEC apoptosis in STAT5IEC KO mice may be secondary to up-regulated proinflammatory cytokines caused by IEC TJ barrier disruption, infiltrated luminal bacteria, and sequentially activated adaptive immune reaction.
Furthermore, our data demonstrated that NF-κB activation, MLCK mRNA levels and phosphorylation of MLC were upregulated, and that TJ assembly was disrupted in STAT5IEC KO mice. Among the various components of TJ, ZO-1, -2 and -3 exhibited reduced protein and RNA levels as well as a disrupted distribution. Knockdown of STAT5 activated NF-κB, elevated pMLC levels and reduced ZO abundance, causing monolayer hyperpermeability. ZO-1, -2 and -3 have been shown to localize to the adherens junction (AJ) with cadherin and catenins prior to the localization of other membrane-associated TJPs, such as occludin (Mitic & Anderson, 1998). ZO proteins regulate both AJ and TJ function by directly binding to F-actin (Yamazaki et al, 2008). Depletion of both ZO-1 and -2 in the EpH4 mammary epithelial cell line completely abrogates TJ assembly and genetic deletion of ZO genes results in embryonic lethality (Xu et al, 2008). More importantly, MLCK may regulate TJ barrier function by directly mediating ZO-1 exchange and anchoring (Yu et al, 2010), and MLC phosphorylation caused ZO-1 redistribution, which can be reversed by MLCK inhibition (Shen & Turner, 2006). Thus, ZO proteins may play a key role in directing the regulation of TJ assembly. We speculate that STAT5 controls TJ integrity by regulating MLCK-dependent ZO assembly or by direct interaction with ZO proteins; however, our data do not reveal a direct interaction between TJ assembly and STAT5 activity. Taken together, these findings show that STAT5 signalling plays a non-redundant role in ZO expression and assembly.
A constitutively active truncated MLCK (tMLCK)-mediated MLC phosphorylation is sufficient to trigger regulation of TJ barrier function and ZO-1 redistribution in IEC monolayers (Shen et al, 2006). We found that long form MLCK expression as well as pMLC was elevated in STAT5 deficient and knockdown IEC monolayers under basal conditions, and STAT5 regulated MLCK promoter activity. Conversely, depletion of long MLCK reduced increased IEC para-cellular permeability in STAT5IEC KO mice. Thus, we speculate that STAT5 controls TJ assembly under basal conditions, partially by regulating MLCK-dependent ZO assembly (STAT5⊣ MLCK → ZOs). We found that, during water recovery, CA-MLCK Tg mice exhibited slower wound healing and repair of intestinal mucosa than littermate controls. Conversely, MLCK KO mice were resistant to DSS-induced mucosal injury. Importantly, STAT5 activation in IECs was abrogated in CA-MLCK Tg mice and restored in long form MLCK KO mice following DSS exposure compared to littermate controls. Therefore, the impaired STAT5 signalling in IECs delayed intestinal epithelial wound healing, and IEC STAT5 signalling is required for repairing mucosal wound induced by loss of IEC barrier. However, our data also suggest that other factors could contribute to the maintenance of increased IEC TJ permeability during mucosal inflammation. The on-going studies with an extension of mouse genotypes will further determine the other factors involved in the intestinal phenotypes due to loss of epithelial STAT5.
To definitively demonstrate that STAT5 regulates NF-κB activity in IECs, we simultaneously knocked down STAT5 and RelA/p65 in HT-29 IEC monolayers and stimulated IEC monolayers with the combination of IFN-γ and TNF-α (Wang et al, 2006). We observed that knockdown of RelA/p65 prevented the upregulated MLC phosphorylation and TJ hyperpermeability in IEC monolayers, which were induced by either STAT5 knockdown or proinflammatory cytokine administration. Most importantly, we found that loss of STAT5 enhanced the NF-κB and MLCK promoter activity in response to cytokine administration, and that STAT5 antagonizes NF-κB DNA binding in long MLCK promoter. Accordingly, IEC STAT5 may negatively regulate NF-κB activation of MLCK to modulate MLC phosphorylation (STAT5⊣ NFκB → MLCK), which protects IEC barrier function to promote IEC wound healing (Fig 9C).
Taken together, our findings suggest that a novel negative feedback mechanism (STAT5⊣ NFκB → MLCK) directly regulates intestinal epithelial TJ barrier function and immune responses to injury relevant to human IBD (Fig 9C). Our results demonstrate the regulation of epithelial STAT5 activity as a means to improve IEC barrier dysfunction and mucosal inflammation in IBD. Thus, these studies will provide a potential therapeutic target for treating intestinal barrier dysfunction and mucosal wound healing in IBD.
MATERIALS AND METHODS
Materials
All chemicals and antibodies were purchased from Sigma–Aldrich (St. Louis, MO) unless otherwise noted. Antibodies specific for MLC and phosphorylated MLC (serine 19) (pMLC), phospho-tyrosin694 specific STAT5 (pSTAT5), phospho-serine276 p65 (P-p65) antibodies were from Cell Signaling Technology (Danvers, MA). Antibodies specific for STAT5a and STAT5b were from Zymed Laboratories (South San Francisco, CA). Antibodies specific for STAT5, p65, p50, Bax and IκBα and β were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies specific for ZO-1, -2 and -3, occludin, JAM-A, claudin 1 and 2 were from Invitrogen (Carlsbad, CA). Another tyrosine (Y694/699) phosphorylation specific STAT5 antibody was from Upstate Biotechnology (Lake Placid, NY). CD4, CD25 and Foxp3 were from eBioscience (San Diego, CA). Antibodies specific for CD3, CD44, CD62L, CD11b and F4/80 were from BD Pharmingen (San Jose, CA). ON-TARGET plus SMARTpool for human stat5 a and b and RelA/p65 were purchased from DHARMACON (Lafayette, CO).
The paraffin-embedded colonic samples were collected from healthy controls and pediatric CD; the de-identified patient-based studies were approved by the CCHMC IRB (# 2009-1680).
The paper explained
PROBLEM
Dysregulated intestinal barrier function is a critical component of the pathogenesis of inflammatory bowel diseases (IBD). Recent genome-wide association studies (GWAS) have implicated that JAK − STAT signalling is involved in IBD pathogenesis. Mechanistic studies are now required to define functional effects of these pathways.
RESULTS
Our studies defined for the first time STAT5 target genes regulating TJ barrier function, and determined whether a novel negative feedback mechanism (STAT5⊣ NF-κB → MLCK) directly regulates IEC barrier function and response to injury relevant to human IBD.
IMPACT
Our studies are expected to reveal a novel mechanism of intestinal barrier dysfunction in IBD, providing a possible approach to promote intestinal mucosal wound healing. Targeting exploration of the STAT5⊣ NFκB → MLCK signalling pathways in regulation of IEC barrier function will enhance not only the understanding of mucosal innate immunity, but also provide a novel therapeutic target for treatment of epithelial barrier dysfunction in IBD.
Animal resources and maintenance
The animal study protocol has been approved by the CHRF Institutional Animal Care and Use Committee (IACUC). Breeding pairs for CA-MLCK Tg mice and MLCKKO mice have been obtained from Dr. Turner's laboratory at University of Chicago (Su et al, 2009). Breeding pairs of STAT5 floxed mice (STAT5F/F) were from Dr. Hennighausen's lab at NIDDK (Cui et al, 2004). C57BL/6 mice and Villin-Cre transgenic mice (B6.SJL-Tg) were ordered from Jackson Labs (Bar Harbor, MA). Deletion of STAT5 from intestinal epithelia was achieved by breeding STAT5F/F mice with villin-Cre transgenics. All mice used in these studies have been backcrossed with C57BL/6 for more than 10 generations and were re-genotyped with respect to STAT5 and Cre following necropsy. All studies were performed with littermate STAT5F/F mice designated as wild-type controls (STAT5IEC WT), and STAT5F/F, villin-Cre animals denoted as STAT5IEC KO. MLCKKO mice were crossed with our STAT5F/F mice, and then depleted IEC STAT5 by crossing these mice with villin-cre mice, these mice were denoted as MLCK/STAT5IEC KO. Mice were maintained in specific pathogen free (SPF) conditions in the Children's Hospital Research Foundation (CHRF) Animal Care Facility.
Animal model of colitis
Intestinal inflammation was induced by providing transgenic or control littermate mice with 3% dextran sulphate sodium (DSS) water (m.w. 36,000–50,000; MP Biomedicals) for either 7 days for acute studies or 5 days followed by 5 days of water for healing phase studies. Mice were sacrificed; the colon was removed, and dissected into three equal fragments: distal, middle and proximal colon. For each colon fragment, longitudinal cuts of tissue in which the three colon sections had clearly visible intestinal lumen were used in histological scoring with the observer blinded to genotype. Scoring parameters included quantitation of the area of middle and distal colon involved, edema, erosion/ulceration of the epithelial monolayer, crypt loss/damage and infiltration of immune cells into the mucosa. Total disease score was expressed as the mean of all combined scores per genotype (Steinbrecher et al, 2008).
Animal model of ileitis
To establish the animal model of ileitis, 5- or 6-week old male and female STAT5IEC KO and STAT5IEC WT mice were exposed to a NSAID, piroxicam (PIR), at 200 ppm in the chow for 2 weeks. Control mice continued on regular chow. Mice were then sacrificed following PIR exposure to determine effects upon the development of ileitis, and histology was scored as published (Han et al, 2010).
Measurement of paracellular intestinal permeability and bacterial translocation
Ileal and colonic paracellular permeability to the fluorescent tracer fluorescein isothiocyanate-dextran with a molecular mass of 4 kDa (FD-4) was determined using an everted gut sac method (Han et al, 2010). Fluorescence was measured using a fluorescence spectrophotometer (Biotek Instruments, VT) at an excitation wavelength of 492 and an emission wavelength of 515 nm. Permeability was expressed as the mucosal-to-serosal clearance of FD-4. Bacterial translocation to MLN was determined as described (Han et al, 2010).
Western blot (WB), Electrophoretic Mobility Shift Assay (EMSA), immunohistochemistry (IH) and immunofluorescence (IF)
Isolated colonic and ileal IECs, and mucosal tissue were saved. Total cellular protein (TP), cytosolic protein (CE) and nuclear protein (NE) extracts were prepared using cold RIPA buffer and the NE-PER kit per the manufacturers’ recommendations (Pierce, Rockford, IL). Expression of ZO-1, -2 and -3, occludin, claudin 1 and 2, junction adhesion molecule (JAM)-A, STAT5, MLC and pMLC abundance were detected in TP from the isolated IECs. The nuclear abundance of pSTAT5, pSTAT3 and p65 was detected in NE. IκBα and β were determined in CE. Band intensities were quantified as mean area density using ImageQuant (Molecular Dynamics, Sunnyvale, CA). pMLC was expressed as relative area density corrected by MLC bands intensity. One microgram NE was used for EMSAs. NF-κB DNA binding was performed using a duplex oligonucleotide probe containing the consensus NF-κB binding sequence (Han et al, 2009a; Ma et al, 2004). MLCK promoter activity was determined by EMSA, using a duplex oligonucleotide probe based on the downstream NFκB-binding element in the MLCK promoter region (Ye et al, 2006). DNA binding was detected according to LightShift chemiluminescent EMSA kit (Pierce, Rockford, IL) (Han et al, 2006). The sequences of the oligonucleotide probe are listed in the Supporting information Table. Frozen tissue sections from mouse ileum and colon (4 µm) were prefixed in paraformaldehyde. Tissue sections were labelled with ZO-1, -2 and -3, pMLC and pSTAT5 following FITC-conjugated or TRITC-conjugated goat anti-rabbit secondary antibodies, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were used for nuclear counterstaining. pSTAT5 (phosphotyrosine) and p65 (phosphoserine 276) were examined in paraffin embedded intestinal sections using VECTASTAIN Elite ABC system (Vector lab, Burlingame, CA). BrdU staining followed manufacturer's instructions (BrdU In Situ Detection Kit, BD Pharmingen) (Han et al, 2010). BrdU, cleaved Caspase-3, pSTAT5 and p65 positive cells were counted by a semi-quantitative method and expressed as average positive cells per crypt. Images were captured using a Zeiss microscope and Axioviewer image analysis software (Carl Zeiss Corp, Germany) (Han et al, 2009a).
Flow cytometry (FACS)
Fresh single cell suspensions were prepared from MLN. Cells were stained for surface markers anti-mouse CD11c, CD11b, F4/80, CD4, CD25, CD44, CD62L, CD3 or intracellular staining for anti-mouse Foxp3. The fluorescent signals were measured by FACS (LSR II, BD Biosciences). Data were analysed using FlowJo software (Tree Star, OR) (Han et al, 2009a).
Laser capture microdissection (LCM)
Briefly, approximately 200 crypts and adjacent surface epithelial cells from ileum were captured by a Veritas Microdissection System (Life Technologies, Carlsbad, CA); RNA was isolated with a PicoPure RNA Isolation kit (Arcturus) using published methods (Han et al, 2006; Han et al, 2011). The quality and concentration of RNA were measured by NanoDrop (Thermo Fisher). Total RNA (200 ng) was used to reversely transcribe to cDNA followed by a SYBR Green real-time PCR on the Mx4000 multiplex quantitative PCR instrument (Stratagene).
Real-time quantitative PCR (qPCR)
Total RNA was isolated from frozen tissue using RNeasy Mini Kit (QIAGEN, Valencia, CA) according to the manufacturer's protocol. Using specific gene primers (ZO-1, -2 and -3, occludin, and GAPDH), PCR reactions were performed with Brilliant II SYBR Green QPCR mix (Stratagene, La Jolla, CA) in the Mx3000p thermocycler (Stratagene). A relative amount for each gene examined was obtained from a standard curve generated by plotting the cycle threshold value against the concentration of a serially diluted RNA sample expressing the gene of interest. This amount was normalized to the level of β-actin or GAPDH RNA. The primer sequences are listed in the Supporting information Table followed by references.
RNA interference (RNAi) and IEC monolayer permeability assay (Han et al, 2003; Wang et al, 2005)
STAT5 a and b and RelA/p65 ON-TARGETplus SMARTpool transfection was performed with pre-confluent HT-29 cells (ATCC, passage 35–45) growing on Transwell inserts (Becton Dickinson, Bedford, MA). Groups with transfection reagents only (TRAN) were chosen as controls for RNAi. For subsequent experiments we used 17-day postconfluent HT-29 monolayers. The medium bathing the apical surface of the monolayers was replaced with 200 µl of DMEM complete medium containing FITC-Dextran (FD4, Sigma) at 25 mg/ml. The medium bathing the basolateral side of the monolayers was replaced with 500 µl of DMEM complete medium alone or DMEM supplemented with or without IFN-γ (10 ng/ml) induction for 18 h and then stimulation with TNF-α (10 ng/ml) for 12 h. Fluorescence in basolateral bathing medium was measured using a fluorescence spectrophotometer (Biotek Instruments, VT). The permeability of the monolayer was expressed as a clearance (C; nl cm−2 h−1). TEER was measured by E-VOM instrument (World Precision Instruments, Sarasota, FL). Results were expressed as Ohm cm2.
Statistical analysis
Results are presented as the mean ± SEM. Data were analysed using analysis of variance, two-tailed Student's t-test, and the Mann–Whitney test as appropriate (Prism, GraphPad, San Diego, CA). p-values ≤ 0.05 were considered significant.
Acknowledgments
We greatly appreciate Dr. Jerrold Turner at University of Chicago for revising our manuscript and providing outstanding technical assistance. We thank Drs. Simon Hogan, Marc Rothenberg, James Heubi and Mitchell Cohen at Cincinnati Children's Hospital Medical Center (CCHMC), and Dr. Mitchell Fink at University of California, Los Angeles for supporting this project; Dr. Lothar Hennighausen at NIDDK generously for providing STAT5F/F mice. This work was completed in part in NIH-funded Digestive Health Center (DHC) and Laser Mircodissection Core at CCHMC. Drs. Jun Li and Pranav Shivakumar at Dr. Jorge Bezerra's laboratory kindly provided us with pairs of primers and conditions for cytokine real time PCR. This work was supported by NIH Clinical and Translational Research Award KL2 RR026315 (XH), Cincinnati Children's Hospital Research Foundation Digestive Health Center (PHS Grant P30 DK078392), Crohns and Colitis Foundation of America (XH). RM was supported by grant SFB F28 from the Austrian Science Funds (FWF).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
XH conceived and designed the experiments; XH, SG, RZ, RM and JL performed the experiments; XH, RM, SG, LD, NS and KS analysed the data; LD, RM, RZ, KS and NS contributed reagents, materials and analysis tools; XH wrote the paper.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Al-Sadi R, Boivin M, Ma T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front Biosci. 2009;14:2765–2778. doi: 10.2741/3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayburgh DR, Rosen S, Witkowski ED, Wang F, Blair S, Dudek S, Garcia JG, Alverdy JC, Turner JR. A differentiation-dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. J Biol Chem. 2004;279:55506–55513. doi: 10.1074/jbc.M408822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignass AU. Mechanisms and modulation of intestinal epithelial repair. Inflamm Bowel Dis. 2001;7:68–77. doi: 10.1097/00054725-200102000-00014. [DOI] [PubMed] [Google Scholar]
- Ferguson LR, Han DY, Fraser AG, Huebner C, Lam WJ, Morgan AR, Duan H, Karunasinghe N. Genetic factors in chronic inflammation: single nucleotide polymorphisms in the STAT-JAK pathway, susceptibility to DNA damage and Crohn's disease in a New Zealand population. Mutat Res. 2010;690:108–115. doi: 10.1016/j.mrfmmm.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Geymayer S, Doppler W. Activation of NF-kappaB p50/p65 is regulated in the developing mammary gland and inhibits STAT5-mediated beta-casein gene expression. Faseb J. 2000;14:1159–1170. doi: 10.1096/fasebj.14.9.1159. [DOI] [PubMed] [Google Scholar]
- Graham WV, Wang F, Clayburgh DR, Cheng JX, Yoon B, Wang Y, Lin A, Turner JR. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J Biol Chem. 2006;281:26205–26215. doi: 10.1074/jbc.M602164200. [DOI] [PubMed] [Google Scholar]
- Han X, Benight N, Osuntokun B, Loesch K, Frank SJ, Denson LA. Tumour necrosis factor alpha blockade induces an anti-inflammatory growth hormone signalling pathway in experimental colitis. Gut. 2007;56:73–81. doi: 10.1136/gut.2006.094490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Gilbert S, Groschwitz K, Hogan S, Jurickova I, Trapnell B, Samson C, Gully J. Loss of GM-CSF signalling in non-haematopoietic cells increases NSAID ileal injury. Gut. 2010;59:1066–1078. doi: 10.1136/gut.2009.203893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Mann E, Gilbert S, Guan Y, Steinbrecher KA, Montrose MH, Cohen MB. Loss of guanylyl cyclase C (GCC) signaling leads to dysfunctional intestinal barrier. PLos One. 2011;6:e1613. doi: 10.1371/journal.pone.0016139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Osuntokun B, Benight N, Loesch K, Frank SJ, Denson LA. Signal transducer and activator of transcription 5b promotes mucosal tolerance in pediatric Crohn's disease and murine colitis. Am J Pathol. 2006;169:1999–2013. doi: 10.2353/ajpath.2006.060186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Ren X, Jurickova I, Groschwitz K, Pasternak BA, Xu H, Wilson TA, Hogan SP, Denson LA. Regulation of intestinal barrier function by signal transducer and activator of transcription 5b. Gut. 2009a;58:49–58. doi: 10.1136/gut.2007.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Uchida K, Jurickova I, Koch D, Willson T, Samson C, Bonkowski E, Trauernicht A, Kim MO, Tomer G, et al. Granulocyte-macrophage colony-stimulating factor autoantibodies in murine ileitis and progressive ileal Crohn's disease. Gastroenterology. 2009b;136:1261–1263. doi: 10.1053/j.gastro.2008.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Uchiyama T, Sappington PL, Yaguchi A, Yang R, Fink MP, Delude RL. NAD+ ameliorates inflammation-induced epithelial barrier dysfunction in cultured enterocytes and mouse ileal mucosa. J Pharmacol Exp Ther. 2003;307:443–449. doi: 10.1124/jpet.103.056556. [DOI] [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22:711–721. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–885. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- Machen TE, Erlij D, Wooding FB. Permeable junctional complexes. The movement of lanthanum across rabbit gallbladder and intestine. J Cell Biol. 1972;54:302–312. doi: 10.1083/jcb.54.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen KL, Malfair D, Gray D, Doyle JS, Jewell LD, Fedorak RN. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis. 1999;5:262–270. doi: 10.1097/00054725-199911000-00004. [DOI] [PubMed] [Google Scholar]
- Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, II, Raleigh DR, Guan Y, Watson AJ, Montrose MH, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitic LL, Anderson JM. Molecular architecture of tight junctions. Annu Rev Physiol. 1998;60:121–142. doi: 10.1146/annurev.physiol.60.1.121. [DOI] [PubMed] [Google Scholar]
- Miyoshi K, Shillingford JM, Smith GH, Grimm SL, Wagner KU, Oka T, Rosen JM, Robinson GW, Hennighausen L. Signal transducer and activator of transcription (Stat) 5 controls the proliferation and differentiation of mammary alveolar epithelium. J Cell Biol. 2001;155:531–542. doi: 10.1083/jcb.200107065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, Huth M, Nikolaev A, Neufert C, Madison B, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- Neurath MF, Pettersson S, Meyer zum Buschenfelde KH, Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med. 1996;2:998–1004. doi: 10.1038/nm0996-998. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor kappa B inflammatory bowel disease. Gut. 1998;42:477–484. doi: 10.1136/gut.42.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulzke JD, Ploeger S, Amasheh M, Fromm A, Zeissig S, Troeger H, Richter J, Bojarski C, Schumann M, Fromm M. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci. 2009;1165:294–300. doi: 10.1111/j.1749-6632.2009.04062.x. [DOI] [PubMed] [Google Scholar]
- Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci. 2006;119:2095–2106. doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- Shen L, Turner JR. Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: tight junction dynamics exposed. Am J Physiol Gastrointest Liver Physiol. 2006;290:G577–G582. doi: 10.1152/ajpgi.00439.2005. [DOI] [PubMed] [Google Scholar]
- Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth D, Phan V, Wang A, McKay DM. Interferon-gamma-induced increases in intestinal epithelial macromolecular permeability requires the Src kinase Fyn. Lab Invest. 2011;91:764–777. doi: 10.1038/labinvest.2010.208. [DOI] [PubMed] [Google Scholar]
- Snow JW, Abraham N, Ma MC, Herndier BG, Pastuszak AW, Goldsmith MA. Loss of tolerance and autoimmunity affecting multiple organs in STAT5A/5B-deficient mice. J Immunol. 2003;171:5042–5050. doi: 10.4049/jimmunol.171.10.5042. [DOI] [PubMed] [Google Scholar]
- Steinbrecher KA, Harmel-Laws E, Sitcheran R, Baldwin AS. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol. 2008;180:2588–2599. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
- Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB, Abraham C, Turner JR. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology. 2009;136:551–563. doi: 10.1053/j.gastro.2008.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Clayburgh DR, Mittal N, Goretsky T, Dirisina R, Zhang Z, Kron M, Ivancic D, Katzman RB, Grimm G, et al. Epithelial NF-kappaB enhances transmucosal fluid movement by altering tight junction protein composition after T cell activation. Am J Pathol. 2010;176:158–167. doi: 10.2353/ajpath.2010.090548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, Abraham C, Turner JR. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology. 2006;131:1153–1163. doi: 10.1053/j.gastro.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kausalya PJ, Phua DC, Ali SM, Hossain Z, Hunziker W. Early embryonic lethality of mice lacking ZO-2, but Not ZO-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol Cell Biol. 2008;28:1669–1678. doi: 10.1128/MCB.00891-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Umeda K, Wada M, Nada S, Okada M, Tsukita S, Tsukita S. ZO-1- and ZO-2-dependent integration of myosin-2 to epithelial zonula adherens. Mol Biol Cell. 2008;19:3801–3811. doi: 10.1091/mbc.E08-04-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci USA. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2006;290:G496–G504. doi: 10.1152/ajpgi.00318.2005. [DOI] [PubMed] [Google Scholar]
- Yu D, Marchiando AM, Weber CR, Raleigh DR, Wang Y, Shen L, Turner JR. MLCK-dependent exchange and actin binding region-dependent anchoring of ZO-1 regulate tight junction barrier function. Proc Natl Acad Sci USA. 2010;107:8237–8241. doi: 10.1073/pnas.0908869107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.