

Abstract
The activity of ATP-sensitive potassium (KATP) channels is governed by the concentration of intracellular ATP and ADP and is thus responsive to the metabolic status of the cell. Phosphorylation of KATP channels by protein kinase A (PKA) or protein kinase C (PKC) results in the modulation of channel activity and is particularly important in regulating smooth muscle tone. At the molecular level the smooth muscle channel is composed of a sulfonylurea subunit (SUR2B) and a pore-forming subunit Kir6.1 and/or Kir6.2. Previously, Kir6.1/SUR2B channels have been shown to be inhibited by PKC, and Kir6.2/SUR2B channels have been shown to be activated or have no response to PKC. In this study we have examined the modulation of channel complexes formed of the inward rectifier subunit, Kir6.2, and the sulfonylurea subunit, SUR2B. Using a combination of biochemical and electrophysiological techniques we show that this complex can be inhibited by protein kinase C in a Ca2+-dependent manner and that this inhibition is likely to be as a result of internalization. We identify a residue in the distal C terminus of Kir6.2 (Ser-372) whose phosphorylation leads to down-regulation of the channel complex. This inhibitory effect is distinct from activation which is seen with low levels of channel activity.
Keywords: ABC Transporter, Calcium, Ion Channels, Potassium Channels, Protein Kinase C (PKC), ATP-sensitive K+ Channel
Introduction
ATP-sensitive potassium (KATP)2 channels couple cell metabolism to membrane potential in many cell types and control insulin release, vascular smooth muscle tone, and excitability in neurons and muscle (1). Inhibition by ATP and activation by nucleotide diphosphates allow the metabolic state of the cell to control membrane potential and cell excitability. Additionally, activation of KATP channels as a result of metabolic stress such as ischemia and hypoxia has been shown to protect muscle, heart, and brain (2).
KATP channels are composed of an octomeric complex consisting of four pore-forming subunits of the Kir6.x subfamily of inwardly rectifying potassium channels (Kir6.1 or Kir6.2) and four regulatory sulfonylurea receptor subunits (SUR1, SUR2A, or SUR2B), belonging to the ATP-binding cassette superfamily of proteins (1, 3, 4). The assembly of one pore-forming subunit (Kir6.1 or Kir6.2) with a particular SUR generates currents with distinct nucleotide sensitivities and pharmacological properties (4, 5). For example, the pancreatic β-cell KATP channel is composed of Kir6.2 and SUR1 (6, 7), Kir6.1 and SUR2B are thought to form the vascular smooth muscle KATP channel (8–10), Kir6.2 and SUR2B are present in nonvascular smooth muscle and portal vein (11–15), and Kir6.2 and SUR2A comprise the cardiac KATP channel (16). Characteristically, the vascular smooth muscle KATP channel has a lower single-channel conductance and has an absolute dependence on nucleotide diphosphates for activity (8). In contrast, in portal vein and colonic smooth muscle cells the single-channel conductance and nucleotide regulation are more compatible with a channel complex containing Kir6.2 (11, 15, 17).
Regulation of proteins by intracellular signals through protein kinase-mediated phosphorylation is an important mechanism by which the activity of many ion channels, including KATP channels, can be modulated (18, 19). Phosphorylation of ion channels is most commonly catalyzed by protein kinase A (PKA) and protein kinase C (PKC), where phosphorylation of serine or threonine residues leads to an alteration in channel properties by modifying kinetics and/or the number of channels at the membrane (20). Regulation of KATP channels in the vasculature by protein kinases is of particular importance as vasoconstrictors, such as angiotensin II, for example, modulate channel activity by activating PKC (21). A number of studies have shown that the KATP channel composed of Kir6.1/SUR2B is inhibited by PKC (11, 22–24). A short motif containing specific sites for PKC phosphorylation has been found on the pore-forming Kir6.1 subunit (23). There are two main subfamilies of PKC enzymes regulated by Gq/11-coupled receptors: conventional and novel. Conventional PKCs require Ca2+ and diacylglycerol to function, whereas novel PKCs require diacylglycerol but are Ca2+-independent. Several groups have demonstrated the role of a Ca2+-independent isoform of PKC, PKCϵ, in the regulation of Kir6.1 (22, 25, 26). PKC has also been shown to regulate KATP channels containing the Kir6.2 subunit (24, 27–30). Interestingly, PKC has a dual effect on Kir6.2, both up-regulating and down-regulating channel activity by phosphorylating threonine 180 (29). In addition, trafficking studies have revealed that PKC initiates internalization of the channel complex leading to decreased channel activity (28, 30). To date Thr-180 is the only specific PKC site to be identified on the Kir6.2 subunit, and the mechanism behind its phosphorylation by PKC has not been investigated. In this study, we identify a new site for PKC-mediated phosphorylation leading to channel inhibition and additionally show Ca2+ dependence of PKC phosphorylation at this site.
EXPERIMENTAL PROCEDURES
Molecular Biology and Cell Culture
Single-point mutations were introduced into the mouse Kir6.2 clone and the maltose-binding protein (MBP)-Kir6.2C vector by PCR using the Stratagene XL Mutagenesis kit according to the manufacturer's instructions. Human embryonic kidney (HEK) 293 cells stably transfected with Kir6.1/SUR2B and Kir6.2/SUR2B were maintained in G418 and Zeocin selective media as described previously (9, 31). Transfection of Kir6.2ΔC26 or the mutant Kir6.2 constructs with SUR2B was carried out using FuGENE HD (Roche Diagnostics) as per the manufacturer's instructions. GFP (100 ng) was co-transfected to enable transfection success and efficiency to be assessed, and cells were patched 48 h after transfection.
MBP Fusion Proteins and in Vitro Phosphorylation Assays
Expression and purification of the wild-type and mutant MBP-Kir6.2C were carried out as described previously (22). The protein was judged to be approximately 90% pure based on SDS-PAGE analysis. In vitro phosphorylation assays using the wild-type and mutant MBP-Kir6.2C were carried out using the method described previously (22, 32). Briefly, 1 mg of protein was bound to 100 μl of amylose resin (New England Biolabs). After subsequent washes with a HEPES wash buffer the amylose-bound protein was incubated in a mix containing [γ-32P]ATP (2 μCi) in the presence or absence of the catalytic subunit of PKC (20 ng to give a final concentration of 8 nm; Calbiochem) for 2 h at 37 °C. The sample was washed a further five times to remove excess radioactivity before subjecting it to SDS-PAGE (10%) and autoradiography. Determination of the band intensity was carried out using ImageJ (National Institutes of Health), and statistical analysis using an unpaired Student's t test was carried out using Prism v4 (GraphPad).
Phosphopeptide Mapping of PKC-phosphorylated Residues
The MBP-Kir6.2C protein was phosphorylated as above with the following alterations. [γ-32P]ATP was omitted from the assay, and the following wash buffer was used (50 mm HEPES, pH 7.4, 1 mm EDTA, 10% glycerol, 5 mm sodium pyrophosphate, 50 mm sodium fluoride, 1 mm sodium vanadate, 10 mm glycerol phosphate, 1 mm DTT, 1% Triton X-100). After incubation with PKC and further washing the sample was resuspended in 2 × SDS loading buffer before denaturation for 4 min at 85 °C. Samples were loaded onto a 10% Novex gel (Invitrogen) and subsequently stained with Coomassie Blue. The relevant bands were excised and subjected to phosphopeptide mapping at the Fingerprints Proteomics Facility, University of Dundee.
In Cell Western Assay
The In Cell Western assay was carried out as described by Meimaridou et al. (33). In brief, Chinese hamster ovary-K1 (CHO-K1) cells were seeded into 24-well dishes and transfected with Kir6.2-HA/SUR2B using Turbofect (Fermentas). After 48 h, cells were washed with PBS and incubated with DMSO, 1 μm PdBu, or 1 μm 4α-phorbol for 30 min at 37 °C. Cells were then fixed with 3.7% formaldehyde for 20 min before half of the wells were permeabilized with 0.1% Triton X-100. The other wells were left unpermeabilized to measure the amount of protein expressed at the cell surface. Cells were blocked with PBS + 5% nonfat milk solution for 1.5 h at room temperature followed by overnight incubation (4 °C) with anti-HA antibody (1:500 dilution; Sigma) in blocking solution. Cells were washed with PBS before incubation with IRDye 800CW goat anti-mouse secondary antibody for 1 h. After washing with PBS the fluorescent signal was measured using a LI-COR Odyssey plate reader. Data are presented as cell surface protein over total protein, normalized to DMSO (control).
Cell Surface Biotinylation
Surface expression of Kir6.2 in the presence and absence of PdBu was investigated using the Pierce Cell Surface Protein Isolation kit (Thermo Scientific). In brief, CHO-K1 cells were seeded into 10-cm dishes and transfected with Kir6.2-HA or Kir6.2-HA-S372A/SUR2B, and enhanced GFP for analysis of transfection efficiency. 48 h after transfection cells were washed twice with ice-cold PBS supplemented with 1 mm CaCl2 and 1 mm MgCl2. Cells were then incubated with 1 μm PdBu or DMSO for 30 min at 37 °C. Following this, cells were incubated with PBS containing EZ-Link Sulfo-NHS-SS-Biotin for 2 h at 4 °C. Cells were again washed with PBS and the reaction quenched with Pierce Quenching Solution as supplied. Cells were harvested and resuspended in TDET-S buffer (1% Triton X-100, 0.4% deoxycholic acid, 5 mm EDTA, 25 mm Tris, pH 7.4, 150 mm NaCl, 10% glycerol) containing protease inhibitors (Complete Mini EDTA-free; Roche Applied Science) and phosphatase inhibitors (PhosStop; Roche Applied Science). The resuspended cells were sonicated (5 × 1 s) and left for 30 min on ice with periodic vortexing every 5 min. The lysate was obtained by centrifuging at 10,000 × g for 2 min, and this was then incubated with avidin overnight at 4 °C to allow the biotinylated protein to bind. Following several washes with TDET-S buffer, labeled protein was eluted into 2 × SDS loading buffer + DTT at 95 °C for 5 min. Western blotting was used to detect labeled protein using a rat monoclonal anti-HA antibody (Roche Applied Science) or a rabbit anti-calnexin antibody (Santa Cruz Biotechnology) and the ECL detection kit (GE Healthcare).
Rubidium Flux
An 86Rb+ assay was used to determine the amount of flux passing through KATP channels in response to various drugs as described previously (34, 35). HEK293 cells stably expressing either Kir6.1/SUR2B or Kir6.2/SUR2B were used. The cells (in 6-well dishes) were incubated for 24 h with 86RbCl (0.037 MBq/ml) before being washed three times with HBS assay medium (10 mm HEPES, pH 7.4, 10 mm glucose, 130 mm NaCl, 7 mm KCl, 2 mm CaCl2, and 1 mm MgCl2). Cells were then preincubated with 2 ml of HBS ± PKC inhibitors, 1 μm staurosporine or 3 μm GF109203X, or the calcium chelator, 20 nm BAPTA-AM for 15 min at 25 °C. PdBu or 4α-phorbol was then added to each well to give a final concentration of 1 μm (concentration commonly used to activate PKC), and the cells were incubated for 5 min at 37 °C before channel activators/inhibitors were added as follows (DMSO, 10 μm pinacidil, 10 μm pinacidil + 10 μm glibenclamide). After 15 min further incubation at 25 °C, the supernatant was aspirated into vials, and the cells were lysed using HBS + 2% Triton X-100 solution and collected. All vials were assayed for 86Rb content by measurement of Cherenkov radiation in a liquid scintillation counter (TriCarb, Packard 2000CA). Efflux was expressed as a percentage relative to the total amount of radioactivity incorporated. One-way ANOVA followed by a Bonferroni test was used to determine statistical significance as indicated in the text.
Electrophysiology
Whole cell patch clamp recordings were performed as described previously (22). Capacitance transients and series resistance in whole cell recordings were compensated electronically by using amplifier circuitry (Axopatch 200B). Data were filtered at 1 kHz using the filter provided with the Axopatch 200B (four-pole Bessel) and sampled at 5 kHz using a Digidata 1440 (Axon Instruments). Currents were acquired and analyzed using pClamp 10 (Axon Instruments). The intracellular (pipette) solution contained 140 mm KCl, 1.2 mm MgCl2, 1 mm CaCl2, 10 mm EGTA, and 5 mm HEPES, 1 mm MgATP, and 0.5 mm NaUDP, pH 7.2, using KOH. The free Ca2+ concentration was estimated using Webmaxc standard software. An additional 5.57 mm CaCl2 was added to the standard pipette solution to give a higher free intracellular Ca2+ concentration of ∼300 nm. The bath solution contained 140 mm KCl, 2.6 mm CaCl2, 1.2 mm MgCl2 and 5 mm HEPES, pH 7.4. For inside-out recordings the bath and pipette solutions contained 140 mm KCl, 1.4 mm MgCl2, 1 mm EGTA, 10 mm HEPES, pH 7.2. Pipette resistances were between 2 and 4 megohms for whole cell recordings and 5 and 8 megohms for single-channel recordings. All chemicals were obtained from Sigma-Aldrich. Agents were applied to the bath using a gravity-driven perfusion system.
Data Analysis
To analyze the rate of current inhibition, time course data were fitted using the following equation (in Origin 6),
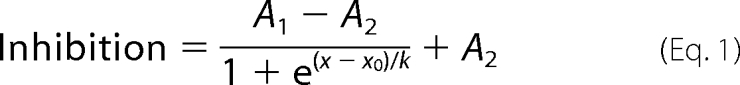 |
where x is the time at any given point, x0 is the time at no inhibition, and k is the time constant. A1 is the initial Y value, and A2 is the final Y value. Statistical analysis was carried out using one-way ANOVA with a Bonferroni post hoc test or a paired Student's t test as appropriate. Statistical significance is indicated in the figure legends. Data are presented as mean ± S.E. Where appropriate, current density is presented normalized to the control.
RESULTS
Activation of PKC by PdBu Inhibits 86Rb+ Flux through Kir6.2/SUR2B Channels and Shows Ca2+ Dependence
It is well established that PKC regulates the activity of Kir6.1/SUR2B currents. However, the same is not true for the KATP channel composed of Kir6.2/SUR2B. To investigate this further, a 86Rb+ (as a K+ surrogate) flux assay was used to measure flux in response to a PKC activator, PdBu, and various PKC inhibitors (staurosporine and the more selective GF109203X). HEK293 cells stably expressing either Kir6.1 or Kir6.2 and SUR2B were used. 10 μm pinacidil failed to activate flux in the presence of 1 μm PdBu (DMSO, 15.75 ± 1.04%; pinacidil, 34.21 ± 1.08%; PdBu + pinacidil, 15.52 ± 0.87%; PdBu + pinacidil + glibenclamide, 17.86 ± 0.85%, n = 9, p < 0.01 compared with pinacidil) in the Kir6.1/SURB cell line (second panel, Fig. 1A). Interestingly, this was also the case with the Kir6.2/SUR2B line (DMSO, 37.84 ± 4.11%; pinacidil, 71.44 ± 2.15%; PdBu + pinacidil, 47.19 ± 2.63%; PdBu + pinacidil + glibenclamide, 23.77 ± 5.51%, n = 9, p < 0.01 compared with pinacidil) (Fig. 1B). The inactive phorbol ester 4α-phorbol was used to rule out nonspecific phorbol effects. Activation of flux by pinacidil through both Kir6.1/SUR2B and Kir6.2/SUR2B was unaffected by 1 μm 4α-phorbol. To ascertain whether the PdBu-induced inhibition of pinacidil-activated flux is due to PKC, we used PKC inhibitors in the presence of PdBu. Pinacidil-induced flux through both Kir6.1/SUR2B and Kir6.2/SUR2B was restored to pre-PdBu levels in the presence of the PKC inhibitors, 1 μm staurosporine and 3 μm GF109203X, suggesting that inhibition of flux through both channels was due to activation of PKC (Fig. 1). Additionally, we tested whether there was a Ca2+ dependence of the PKC effect on the flux through either channel. To this end, the cells were preincubated in 20 nm BAPTA-AM, a Ca2+ chelator. In the case of Kir6.1/SUR2B, the presence of BAPTA-AM had no effect on the inhibition of flux by PdBu (BAPTA-AM + PdBu, 17.81 ± 0.63%; BAPTA-AM + PdBu + pinacidil, 19.03 ± 0.44%, n = 9, p > 0.05, Fig. 1A). However, in cells expressing Kir6.2/SUR2B, BAPTA-AM prevented the inhibition previously seen in the presence of PdBu (BAPTA-AM + PdBu, 38.51 ± 6.14%; BAPTA-AM + PdBu + pinacidil, 78.17 ± 2.76%, n = 9, p > 0.01, Fig. 1B), suggesting that in the case of Kir6.2/SUR2B the PKC effect is Ca2+-dependent.
FIGURE 1.
PKC-mediated inhibition of 86Rb+ efflux through Kir6.2/SUR2B is Ca2+-dependent. Mean 86Rb+ efflux data from HEK293 cells stably transfected with Kir6.1/SUR2B (A) or Kir6.2/SUR2B (B) are shown. Transfected cells were preincubated with HEPES-buffered saline ± PKC inhibitors, 1 μm staurosporine (stau), or 3 μm GF109203X (GFX), or the calcium chelator, 20 nm BAPTA-AM (B-AM) for 15 min prior to a 5-min incubation with 1 μm PdBu or 4α-phorbol (4αP) at 37 °C before channel activators/inhibitors were added as follows (DMSO, 10 μm pinacidil, 10 μm pinacidil + 10 μm glibenclamide). Mean 86Rb+ flux was calculated as percentage efflux of initial 86Rb+ content. Data are shown as mean ± S.E. (error bars), n = 9. **, p < 0.01; ***, p < 0.001 when compared with PdBu.
PdBu Inhibits Whole Cell Kir6.2/SUR2B Currents with High Intracellular Ca2+ Concentrations
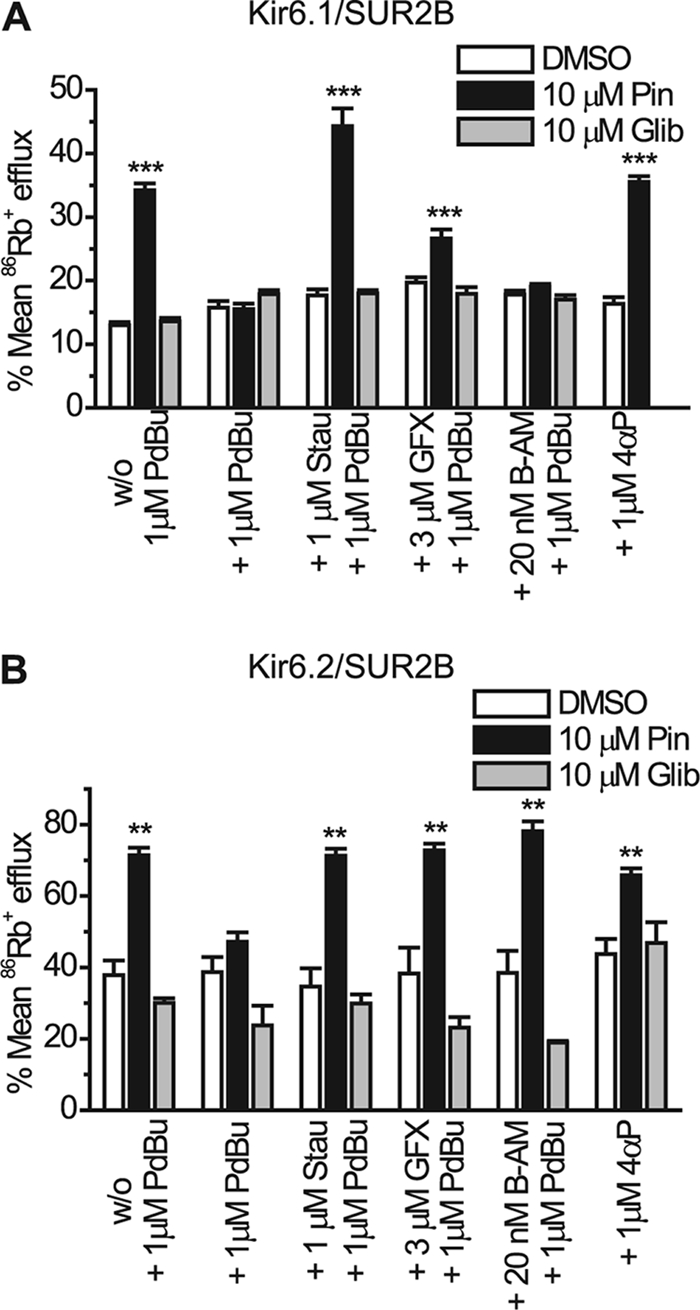
In our previous studies we failed to see the inhibition of the Kir6.2/SUR2B channel current upon PKC activation using the whole cell configuration of the patch clamp (22). However, because the flux data described above showed that PKC does inhibit Kir6.2/SUR2B, possibly via a Ca2+-dependent mechanism, we investigated whether the lack of PKC inhibition seen previously may have been due to the low free Ca2+ concentration present in the pipette. The standard whole cell pipette solution normally used for such recordings had a free intracellular Ca2+ concentration of ∼20 nm. To test the Ca2+ dependence of the PKC effect on Kir6.2/SUR2B we subjected stably transfected HEK 293 cells to whole cell patch clamping using concentrations of free intracellular Ca2+ in the pipette ranging from 20 to 300 nm. Current was elicited by a repetitive 1-s voltage ramp from −100 mV to +100 mV every 15 s from a holding potential of 0 mV. Whole cell currents evoked through Kir6.2/SUR2B channels by 10 μm pinacidil at −50 mV (−193.48 ± 6.15 pA/pF, n = 5) were not significantly affected by the application of 1 μm PdBu (−188.23 ± 12.05 pA/pF, n = 5, p > 0.05) when the intracellular Ca2+ concentration was buffered to 20 nm (Fig. 2, A and E). 10 μm glibenclamide (−20.47 ± 12.71 pA/pF, n = 5) completely inhibited the pinacidil-evoked current. Increasing the free intracellular Ca2+ to 50 nm did not lead to a significant PdBu-induced inhibition of KATP currents (pinacidil, −160.5 pA/pF ± 41.21; PdBu, −156.06 ± 43.7 pA/pF, n = 6, p < 0.05, Fig. 2, B and E). In contrast, when the intracellular solution was buffered to ∼100 nm free Ca2+, PdBu significantly inhibited the pinacidil-evoked current (pinacidil, −240.07 ± 17.62 pA/pF; PdBu, −152.8 ± 12.24 pA/pF, n = 5, p < 0.01) through Kir6.2/SUR2B channels (Fig. 2, C and E). The PdBu-induced inhibition was not reversed upon washing with pinacidil (wash, −147.57 ± 12.38 pA/pF, n = 5, p > 0.05 compared with PdBu). Glibenclamide further reduced the current to control levels (control, −44.02 ± 3.66 pA/pF; glibenclamide, −59.64 ± 3.22, n = 5, p > 0.05 compared with control). When the concentration of free Ca2+ was further increased to ∼300 nm, PdBu inhibited the current (pinacidil, −117.23 pA/pF ± 29.8; PdBu, −37.61 ± 15.83 pA/pF, n = 12, p < 0.001) through Kir6.2/SUR2B channels (Fig. 2, D and E). Glibenclamide further reduced the current to control levels (control, −18.32 ± 9.14 pA/pF; glibenclamide, −19.49 ± 5.50, n = 12, p > 0.05 compared with control). PdBu inhibited ∼50% (50.46 ± 10.89%, n = 12) of the glibenclamide-sensitive current in high intracellular Ca2+ (300 nm) compared with <1% (0.27 ± 1.28%, n = 5) inhibition by PdBu when the Ca2+ concentration was low (20 nm) (Fig. 2F). To confirm that the inhibition observed with PdBu was due to PKC as suggested by the flux data, we incubated cells containing Kir6.2/SUR2B channels with 1 μm staurosporine, a protein kinase inhibitor, and subjected them to whole cell patch clamp. Fig. 2, G and H, shows that in the presence of staurosporine, 1 μm PdBu does not prevent pinacidil from activating currents, nor does it inhibit the current following maximal activation by pinacidil (1 μm staurosporine, −32.17 ± 11.86 pA/pF; 1 μm PdBu, −37.75 ± 9.35 pA/pF; 10 μm pinacidil, −132.39 ± 38.82 pA/pF; 10 μm glibenclamide, −26.45 ± 12.86 pA/pF, n = 3). The inactive phorbol ester 4α-phorbol (in the presence of high intracellular Ca2+) did not significantly modulate Kir6.2/SUR2B current (10 μm pinacidil, −444.27 ± 51.74 pA/pF; 1 μm 4α-phorbol, −465.52 ± 42.63 pA/pF, n = 4, p > 0.05). 10 μm glibenclamide (−42.08 ± 10.26, n = 4) completely inhibited the pinacidil-activated current (Fig. 3). This indicates that a nonspecific effect of phorbol ester treatment was not responsible for the inhibition of the current in the presence of PdBu.
FIGURE 2.

PKC activation by PdBu is Ca2+-dependent and inhibits whole cell Kir6.2/SUR2B currents. Whole cell currents recorded from HEK293 cells stably transfected with Kir6.2/SUR2B are shown. For time course experiments, currents were evoked using a series of ramps (1 s every 15 s from −100 mV to +100 mV), and drugs were superfused as indicated by the solid bars. Current-voltage (I-V) relationships were constructed by a series of voltage steps (20-mV increments) from −100 mV to +100 mV from a holding potential of 0 mV. Representative time course data (i) (at −50 mV) and (ii) I-V relationship show the effects of 1 μm PdBu on KATP currents recorded in 20 nm (A), 50 nm (B), 100 nm (C), and 300 nm (D) free intracellular Ca2+ in the pipette. E, mean whole cell patch data were taken at −50 mV in 20 (n = 5), 50 (n = 6), 100 (n = 5), and 300 (n = 8) nm free intracellular Ca2+ and normalized to the control (mean ± S.E. (error bars), **, p < 0.01; ***, p < 0.001 compared with pinacidil). F, PdBu-induced inhibition of Kir6.2/SUR2B currents in 20, 50, 100, and 300 nm free intracellular Ca2+ (mean ± S.E., ***, p < 0.001) was compared. G, representative time course experiment (i) and I-V relationship (ii) in the presence of 1 μm staurosporine (stau) with 300 nm free intracellular Ca2+ in the pipette is shown. H, mean whole cell data in the presence of staurosporine at −50 mV with 300 nm free intracellular Ca2+ were normalized to the control (mean ± S.E., n = 8, *, p < 0.05 compared with control).
FIGURE 3.
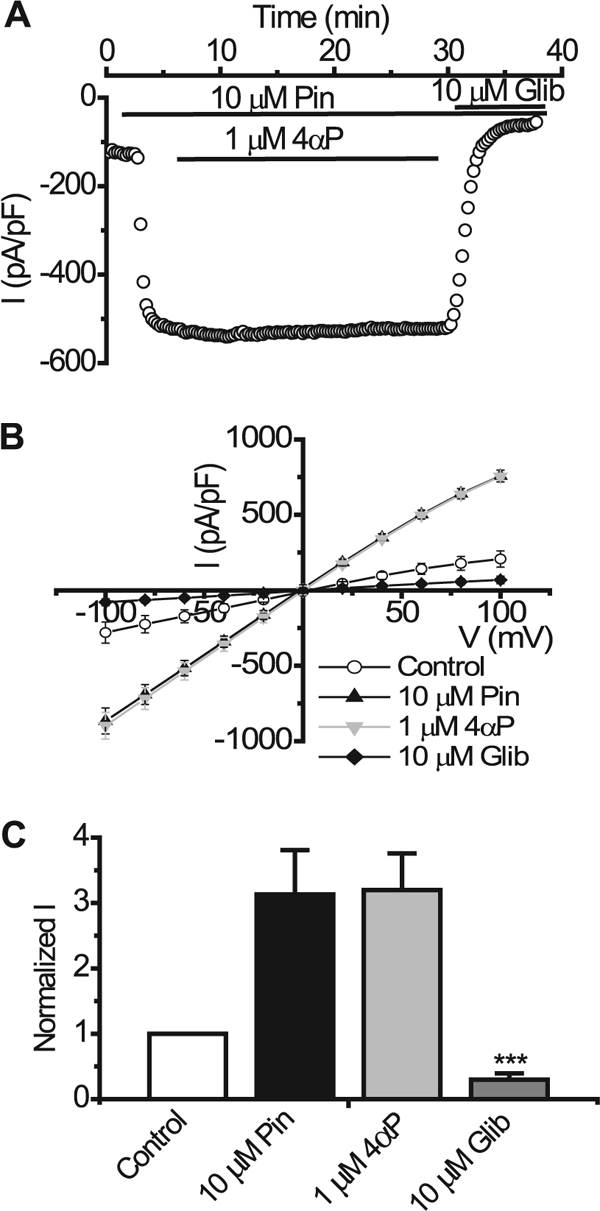
Inactive phorbol ester, 4α-phorbol, does not inhibit pinacidil-evoked currents through Kir6.2/SUR2B channels. A, whole cell recording from HEK293 cell stably transfected with Kir6.2/SUR2B. Currents were evoked using a series of 1-s voltage ramps from −100 mV to +100 mV from a holding potential of 0 mV every 15 s in 300 nm free intracellular Ca2+. KATP current was activated by superfusing the patch with 10 μm pinacidil (Pin), 1 μm 4α-phorbol (4αP), and 10 μm glibenclamide (Glib) were superfused in the presence of pinacidil as indicated by the solid bars. B, mean I-V relationship. Currents were elicited by a series of voltage steps from −100 to +100 mV in 20-mV increments. C, normalized mean currents at −50 mV in the presence and absence (Control) of 1 μm 4α-phorbol (mean ± S.E. (error bars), n = 4, ***, p < 0.001 compared with pinacidil).
Kir6.2ΔC26 Truncation Mutant
We next investigated the location of the potential site of action of PKC on Kir6.2 by using a truncated mutant of Kir6.2 where the last 26 amino acids have been deleted. This mutant is functional with or without SUR (36). In this case, we transfected Kir6.2ΔC26 with SUR2B to allow better expression and also to allow regulation by the KATP channel opener pinacidil and blocker glibenclamide. Fig. 4 shows that application of 1 μm PdBu, with a pipette solution containing a high (300 nm) intracellular Ca2+ concentration, did not significantly modulate the pinacidil-activated current (pinacidil, −268.4 ± 51.39 pA/pF; PdBu, −257.02 ± 47.14 pA, pF, n = 6, p > 0.05). Subsequent application of 10 μm glibenclamide (−42.01 ± 2.98 pA/pF) inhibited the pinacidil-evoked current to base line (−36.06 ± 14.86 pA/pF). The lack of an effect on this mutant by PdBu suggests that the site for PKC phosphorylation lies somewhere in the last 26 amino acids of the C terminus of Kir6.2.
FIGURE 4.
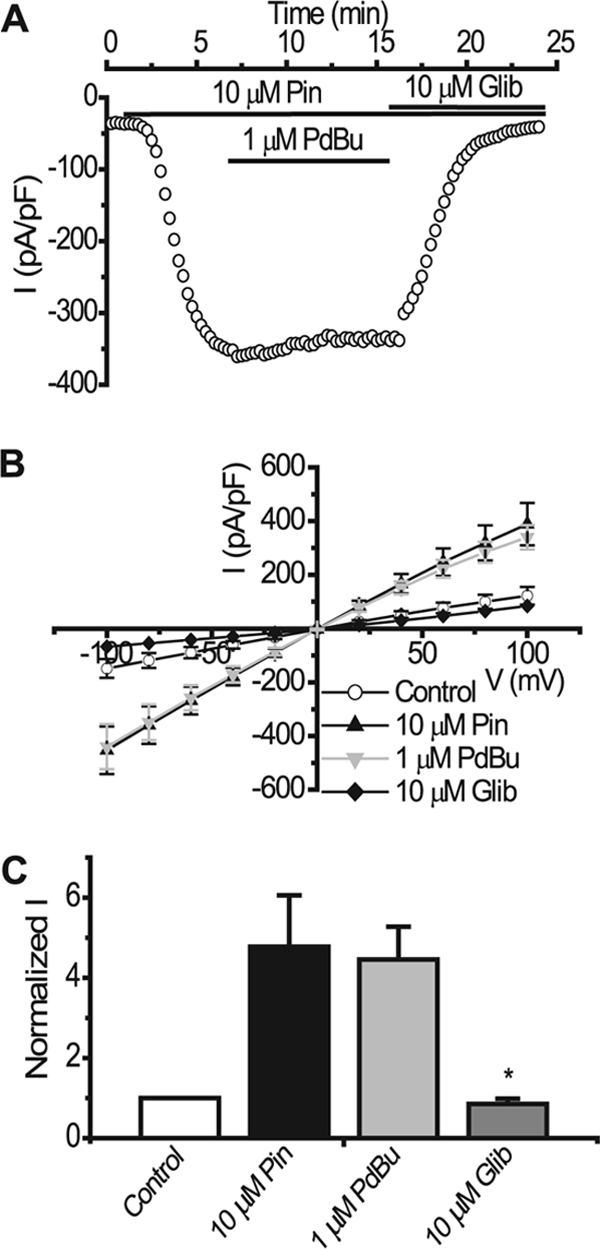
PdBu does not inhibit pinacidil-activated Kir6.2ΔC26/SUR2B currents. A, representative time course trace showing no PdBu effect on pinacidil-activated current in HEK293 cells expressing Kir6.2ΔC26/SUR2B at −50 mV. Drugs were applied as indicated by the solid bars. B, mean I-V relationship. Currents were elicited by a series of voltage steps from −100 to +100 mV in 20-mV increments. C, normalized mean data from experiments like the one in A (mean ± S.E. (error bars), n = 10, *, p < 0.05 compared with pinacidil).
Serine at Position 372 in the C Terminus of Kir6.2 Is a Site for PKC-mediated Phosphorylation
To elucidate the exact site for PKC phosphorylation on Kir6.2, the C-terminal region of Kir6.2 (amino acid residues 185–390) was fused to a MBP (MBP-Kir6.2C) and in vitro phosphorylated by PKC. The phosphorylated proteins were then sent for phosphopeptide mapping (Fingerprints Proteomics Facility, University of Dundee). The proteomic data revealed several potential sites and one residue in particular, a serine at position 372 of Kir6.2 was found to be modified by a phosphate group and therefore a potential phosphorylation site for PKC (Fig. 5A).
FIGURE 5.

Ser-372 is phosphorylated in vitro by PKC. A, amino acid sequence of the C terminus (185–390) of the mouse Kir6.2 subunit. Fusion of this sequence to MBP and subsequent phosphopeptide mapping showed that the Ser-372 (highlighted) residue is modified by a phosphorylation group. The residues removed by the ΔC26 truncation are underlined. Bi, Coomassie-stained 10% SDS-PAGE of MBP-Kir6.2C and the MBP-Kir6.2C S372A mutant in the presence (+) or absence (−) of PKC. Bii, corresponding autoradiograph after 65 h. Marker sizes in kDa are as indicated in the figures. C, graph showing difference in the band intensities between the MBP-Kir6.2C and the MBP-Kir6.2C-S372A mutant in the presence of PKC. Error bars, S.E.; n = 4, *, p < 0.05.
To determine the effect of the proposed PKC phosphorylation of the Ser-372 residue, it was mutated to an alanine residue within the MBP-Kir6.2C protein. Both the wild-type and the S372A proteins were then subjected to in vitro phosphorylation using [γ-32P]ATP as the phosphate substrate. In the presence of PKC, MBP-Kir6.2C is phosphorylated (Fig. 5Bii). MBP-Kir6.2C Ser-372 also shows some phosphorylation; however, this is considerably less than that of the wild-type protein (Fig. 5Bii). The amount of protein loaded on the gel is similar after staining with Coomassie Blue (Fig. 5Bi), indicating that the reduction in phosphorylation is most likely due to the presence of the S372A mutation. Densitometric analysis of the intensity of the autoradiography bands clearly shows a significant reduction of ∼75% in the amount of phosphorylation of the mutant protein compared with the wild type (Fig. 5C). It would therefore appear that the Ser-372 residue is one of the main phosphorylation sites in the Kir6.2 C terminus.
To investigate whether this site has functional significance, we mutated Ser-372 in Kir6.2 either to an alanine (Kir6.2-S372A) or a glutamate (Kir6.2-S372E). These mutants were transiently transfected along with SUR2B into HEK293 cells and subjected to whole-cell patch clamp using high (∼300 nm) intracellular Ca2+. Kir6.2-S372A/SUR2B currents were activated ∼2-fold by 10 μm pinacidil (at −50 mV: control, −48.74 ± 10.28 pA/pF; pinacidil, −89.94 ± 13.92 pA/pF, n = 10, p < 0.01), application of 1 μm PdBu (−91.16 ± 13.72 pA/pF, p > 0.05 compared with pinacidil) failed to reduce this pinacidil-activated current even after a prolonged 10-min application (Fig. 6, A and B). 10 μm glibenclamide (−38.34 ± 9.11 pA/pF) fully reversed the effects of pinacidil. The phosphomimetic mutant Kir6.2-S372E/SUR2B (Fig. 6, C and D) was also activated by pinacidil (control, −36.62 ± 10.37 pA/pF; pinacidil, −63.27 ± 10.76, n = 8, p < 0.001 compared with control) but to a lesser extent, and 1 μm PdBu had no effect on this current (−63.77 ± 10.53 pA/pF, n = 8, p > 0.05). Fig. 6E shows that the inhibition of the mutant channels is negligible compared with the WT Kir6.2/SUR2B channel (WT, 50.46 ± 10.89%; Kir6.2-S372A/SUR2B, 2.42 ± 2.8%; Kir6.2-S372E/SUR2B, 1.35 ± 3.56%). Comparison of the current density and relative activation by pinacidil of the two Ser-372 mutants (Fig. 6F) revealed that S372E (−36.62 ± 10.37 pA/pF) had lower basal current density than S372A (−48.74 ± 10.28 pA/pF) and that pinacidil activated the S372E mutant (−63.27 ± 10.76 pA/pF) to a lesser extent than the S372A mutant (−89.95 ± 13.92 pA/pF). Taken together, these data suggest that PKC likely phosphorylates Ser-372 to regulate the Kir6.2 channel.
FIGURE 6.

Kir6.2-S372A/SUR2B and Kir6. 2-S372E/SUR2B currents are not inhibited by PdBu. A, representative time course trace recorded from a HEK293 cell expressing Kir6.2-S372A/SUR2B. Currents were evoked using a series of repetitive voltage steps from −100 to +100 mV. Drugs were superfused where indicated by the solid bars. B, summary of the mean currents recorded at −50 mV from cells expressing Kir6.2-S372A/SUR2B in the presence of the indicated drugs (mean ± S.E. (error bars), n = 10, **, p < 0.01 compared with pinacidil). C, typical time course trace from a cell expressing Kir6.2-S372E/SUR2B. D, normalized mean currents from Kir6.2-S372E/SUR2B channels in the presence of the indicated drugs (mean ± S.E., n = 8, *, p < 0.05). E, comparison of the percentage inhibition in the presence of 1 μm PdBu of Kir6.2/SUR2B (n = 4), Kir6.2-S372A/SUR2B (n = 10) and Kir6.2-S372E/SUR2B (n = 8), respectively (***, p < 0.001). F, summary of the mean current density of Kir6.2-S372A/SUR2B and Kir6.2-S372E/SUR2B at two time points and in the presence of 10 μm pinacidil.
PdBu Reduces Surface Expression of Kir6.2
To investigate whether phosphorylation of Kir6.2 also affects the amount of protein at the cell surface we used an In Cell Western assay. We used CHO-K1 cells because we have found in HEK293 cells that there is some leak, and “control” intracellular proteins are often labeled by the apparent surface treatment. This assay uses an anti-HA antibody to look at cell surface expression of proteins (Fig. 7A). In the presence of 1 μm PdBu, there was a significant reduction in surface expression of Kir6.2 (68% reduced compared with control, p < 0.01, Fig. 7, A and B). When the inactive phorbol, 4α-phorbol, was added, a small reduction in surface expression occurred (13%, not significant, Fig. 7, A and B), indicating that PdBu-induced activation of PKC reduces the amount of channel present at the surface. This was further confirmed using cell surface biotinylation (Fig. 7, C and D). Kir6.2 can be detected in both the total protein and in the cell lysates in the presence and absence of PdBu (Fig. 7C). In the absence of PdBu treatment, Kir6.2 was visible in the eluted sample, suggesting that it is present at the cell surface (Fig. 7C). In the presence of PdBu, there was a clear reduction in the amount of protein detected in the eluted sample, signifying that the protein is no longer at the cell surface (Fig. 7C). When the Kir6.2 S372A mutant was investigated, no apparent change was visible between the surface-labeled protein in the presence and absence of PdBu (Fig. 7D). Calnexin was used as a control intracellular protein and was only found in the cell lysate samples and not in the surface-labeled samples (Fig. 7E).
FIGURE 7.

PdBu reduces surface expression of Kir6.2. A, representative 24-well plate showing visualization of cell surface expression using the In Cell Western Assay. The left side of the plate shows the surface labeling of nonpermeabilized cells, and the right side indicates labeling of the total protein in the Triton X-100-permeabilized cells. B, bar chart showing the percentage of Kir6.2 surface expression in the presence of 1 μm PdBu or 1 μm 4α-phorbol (4αP). Error bars, S.E. **, p < 0.01 compared with control. C, Western blot using the anti-HA antibody to show the changes in Kir6.2-HA at the surface of CHO-K1 cells in the presence or absence of PdBu. T, total protein; L, cell lysate; E, protein eluted from avidin. D, Western blot using the anti-HA antibody to determine surface expression of Kir6.2 S372A in the absence (−) and presence (+) of PdBu. E, Western blot using anti-calnexin antibody to detect the intracellular control protein calnexin. Marker sizes are shown in kDa.
Dual Action of PKC on Kir6.2
Previous studies have shown a possible dual effect of PKC on the Kir6.2 subunit. To investigate this, we superfused whole cell basal Kir6.2/SUR2B currents with 1 μm PdBu (without prior pinacidil activation). Fig. 8, A and E, shows that in 300 nm free intracellular Ca2+, the basal current (−18.32 ± 9.14 pA/pF) was increased in the presence of 1 μm PdBu (−43.57 ± 17.5 pA/pF, n = 5, p > 0.05) and subsequently inhibited by 10 μm glibenclamide (−19.49 ± 5.5 pA/pF), suggesting as shown previously (29) that PKC may have a dual role in the regulation of Kir6.2. Similar experiments with Kir6.2ΔC26/SUR2B (Fig. 8, B and F) showed that low basal current levels (−36.06 ± 14.86 pA/pF) were significantly increased in the presence of 1 μm PdBu (−168.37 ± 32.73 pA/pF, n = 10, p < 0.05) and inhibited by glibenclamide (−57.21 ± 22.13 pA/pF, p > 0.05 compared with control). Addition of 1 μm PdBu also increased basal currents through Kir6.2-S372A/SUR2B mutant channels to an extent similar to that seen with Kir6.2ΔC26/SUR2B (Fig. 8, C and F: control, −98.19 ± 12.74 pA/pF; PdBu, −260.36 ± 35.82 pA/pF, p < 0.01), suggesting that PKC action on Ser-372 is not responsible for up-regulation of the Kir6.2 current. The PdBu-induced activation of Kir6.2/SUR2B was not Ca2+-dependent, as when the free intracellular Ca2+ concentration was reduced to 100 nm a substantial increase was seen on PdBu application (Fig. 8, A and E: control, −78.06 ± 29.22 pA/pF; PdBu, −162.66 ± 33.18 pA/pF, p < 0.01). This increase was not reversed on wash-off (wash, −164.43 ± 30.01). A further reduction in intracellular Ca2+ to 20 nm did not have any effect on PdBu-induced activation of Kir6.2/SUR2B current (Fig. 8, A and E: control, −62.3 ± 7.26 pA/pF; PdBu, −176.28 ± 13.20 pA/pF, p < 0.01). Activation of basal current by PdBu was greater in the lower Ca2+ concentrations (Fig. 8E). In 20 and 100 nm Ca2+ basal currents increased by 3–4-fold compared with ∼2-fold increase with 300 nm Ca2+ (p < 0.05). To show that the current activation by PdBu is mediated directly through reconstituted KATP channels, we measured the response to PdBu and glibenclamide of the basal current in mock-transfected HEK293 cells (Fig. 8, D and F). 1 μm PdBu (−10.26 ± 2.09 pA/pF) and subsequent application of 10 μm glibenclamide (−9.86 ± 2.65 pA/pF) had no effect on basal currents (−10.82 ± 2.7 pA/pF, n = 4, p > 0.05), thus confirming that the PdBu-induced increase in current is due to the activation of KATP channels via PKC. Comparison of the rates of activation of basal and inhibition of pinacidil-activated Kir6.2/SUR2B current in 300 nm intracellular Ca2+ by PdBu showed that activation was ∼5-fold faster than inhibition (Fig. 8F, mean time constants for inhibition and activation, τ (min), were 4.89 ± 1.36; and 1.12 ± 0.37 respectively, p < 0.05). The rate of activation of both the Kir6.2ΔC26/SUR2B (τ (min), 0.69 ± 0.15) and Kir6.2-S372A/SUR2B (τ (min), 0.96 ± 0.3) currents by PdBu were also significantly quicker than the rate of inhibition of the WT channel (Fig. 8G, p < 0.05).
FIGURE 8.

PdBu activates basal Kir6.2 currents. A–D, representative whole cell recordings showing the effect of PdBu on Kir6.2/SUR2B (A), Kir6.2ΔC26/SUR2B (B), Kir6.2-S372A/SUR2B (C), and mock-transfected cells and their basal currents (D). Currents were evoked using a series of 1-s voltage ramps from −100 mV to +100 mV from a holding potential of 0 mV. Recordings were carried out using 300 nm free intracellular Ca2+ unless indicated otherwise. E, mean data showing PdBu-induced activation of basal Kir6.2/SUR2B current in 20, 100, and 300 nm free intracellular Ca2+ concentrations (mean ± S.E. (error bars), n = 6, 4, and 6 cells respectively, *, p < 0.05). F, normalized mean data comparing the effect of PdBu on Kir6.2/SUR2B, Kir6.2ΔC26/SUR2B, Kir6.2-S372A/SUR2B and mock-transfected HEK293 cell basal current at −50 mV (mean ± S.E., n = 6, 5, 4, and 4 cells, respectively). G, comparison of the rate of PdBu-induced inhibition of Kir6.2/SUR2B current with the rates of PdBu-induced increase of Kir6.2/SUR2B, Kir6.2ΔC26/SUR2B, and Kir6.2-S372A/SUR2B basal current (mean ± S.E., n = 6, 5, and 4, respectively, *, p < 0.05).
Phosphomimetic of Ser-372 Does Not Alter ATP Sensitivity of Kir6.2/SUR2B
Earlier studies have shown that phosphorylation of Kir6.2 alters the sensitivity of the channel to ATP (29, 37). To investigate whether there is a correlation between ATP sensitivity and potential PKC phosphorylation of Ser-372, we tested the relative ATP sensitivity of the WT (Kir6.2/SUR2B) channel and the phosphomimetic S372E mutant channel using the inside-out patch clamp configuration (Fig. 9, A and B). Under these experimental conditions the patches contained large numbers of single channels, and a mean current was measured to assess channel activity. Both channels exhibited similar levels of sensitivity to varying concentrations of internal ATP, with IC50 values of ∼57 μm for WT and 55 μm for the S372E mutant. The S372E mutation had no effect on the Hill coefficient for the ATP dose-response curve of Kir6.2 (1.52 and 1.5, respectively) (Fig. 9C). The data suggest that in the case of Ser-372, a phosphomimetic of PKC phosphorylation does not change ATP sensitivity of the Kir6.2/SUR2B KATP channel.
FIGURE 9.

Phosphomimetic mutation S372E does not alter the ATP sensitivity of the Kir6.2/SUR2B channel. A and B, representative currents from inside-out patches containing Kir6.2/SUR2B (A) and Kir6.2-S372E/SUR2B (B) channels. The excised patches were held at −50 mV in symmetrical 140 mm K+ solution and subsequently exposed to different intracellular concentrations of ATP. C, relative ATP sensitivity of Kir6.2/SUR2B and Kir6.2-S372E/SUR2B. Data were grouped from four to eight inside-out patches and fitted to the following equation: Relative current = 1/(1 + ([ATP]/IC50)k) where relative current is the current relative to the current measured in the absence of ATP and k is the Hill coefficient.
DISCUSSION
Activation of KATP channels in the sarcolemma and mitochondria protects against the metabolic insult of ischemia in a number of tissues but particularly the heart (2, 38–40). Furthermore, KATP channel function can be regulated by cell surface receptors and by intracellular signaling molecules such as PKC (21, 41). PKC has been shown to act on several isoforms of KATP channels in different ways (11, 22–24, 28, 29). In particular, the sites and mechanisms responsible for PKC regulation of the predominant vascular smooth muscle channel subtype, Kir6.1/SUR2B, are well defined (11, 22, 23, 42, 43). PKC activation leads to inhibition of the channel, and this effect is thought to be a result of direct phosphorylation of Kir6.1 (22–24). The regulation of KATP channels containing the Kir6.2 subunit is not as clear. PKC has been shown to both activate and inhibit the Kir6.2/SUR1 and Kir6.2/SUR2A channels depending on the intracellular ATP concentration (29). When expressed with SUR2B, the Kir6.2 channel is activated by PKC or shows no response, depending on the recording conditions (22–24). Single-channel inside-out patch recordings have shown that PKC has a dual action on KATP channels containing the Kir6.2 subunit (co-expressed with either SUR1 or SUR2A), activating when ATP is high and inhibiting when ATP is low (29). When Kir6.2 is co-expressed with SUR2B, inside-out patch recordings in relatively high ATP (0.5 mm) showed that PKC activation with PdBu activates the channel (24). Conversely, we and others have shown, using whole cell patch clamp studies, that PKC has no effect on recombinant Kir6.2/SUR2B currents (22, 23).
Our 86Rb+ efflux studies on Kir6.1/SUR2B and Kir6.2/SUR2B showed that pinacidil-induced efflux through both channels was reduced in the presence of PdBu, and this effect was reversed by the PKC inhibitors staurosporine and GF109203X (Fig. 1). The apparent inhibition of flux through Kir6.2/SUR2B was surprising in our hands, as we had not observed this effect in our previous whole cell patch clamp experiments (22). Interestingly, when cells expressing Kir6.2/SUR2B channels were incubated with a Ca2+ chelator, PdBu failed to inhibit the pinacidil-induced efflux, suggesting that the PKC effect on this isoform of the KATP channel is Ca2+-dependent. Kir6.1/SUR2B, on the other hand, was still inhibited by PdBu in the presence of the chelator, agreeing with the consensus that a novel Ca2+-independent PKC phosphorylates this channel (22, 26, 43). These data led us to hypothesize whether there may be a Ca2+-dependent component to the regulation of Kir6.2 by PKC. The lack of an effect by PdBu in whole cell patch clamp experiments may well have been due to a low intracellular free Ca2+ concentration (∼20 nm) in the pipette (22, 23). In fact, we found this to be the case, there was no effect with PdBu with low intracellular Ca2+ (∼20 nm and ∼50 nm); however, upon increasing the free Ca2+ concentration (∼100 or 300 nm) in the pipette, application of PdBu led to ∼40–50% inhibition of the glibenclamide-sensitive current. Previous studies have used at least 100 nm free Ca2+ to study Ca2+-dependent PKC effects on ion channels (44, 45). Our data imply that the PdBu-induced inhibition of Kir6.2/SUR2B requires a typical resting level of 100 nm free Ca2+ or higher. Although we have not investigated the identity of the specific PKC isoform responsible for inhibition of Kir6.2-containing KATP channels in this study, several Ca2+-dependent PKC isoforms have been shown to be expressed in vascular smooth muscle and the heart, including PKCs α, β, and γ (43, 46, 47). Furthermore, angiotensin II-evoked inhibition of inwardly rectifying K+ channels in rabbit coronary arterial cells has been shown to be mediated by PKCα (48). Recent studies have shown a clear translocation of PKCα from the cytosol to the membrane following stimulation with a phorbol ester in aortic and arterial smooth muscle cells (43, 49). More specifically for our studies, the Ca2+-dependent isoforms PKCα, βI, and βII show a strong endogenous presence in HEK293 cells (44). The inhibition of Kir6.2/SUR2B current as a result of PKC activation was slow, taking ∼20 min to reach steady state at room temperature, suggesting that the inhibition we see with PdBu is likely to be due to PKC-induced internalization rather than a direct effect on plasma membrane channel activity and reduction in open probability. Indeed, our biochemical studies using cell surface expression assays (Fig. 7) show significantly (∼68%) reduced cell surface expression of Kir6.2 in the presence of the PKC activator PdBu. This would concur with recent studies implicating PKC activation in the stimulation of endocytosis of KATP channels containing the Kir6.2 subunit (28, 30). The inhibitory effect was also irreversible within the time course of our experiments. However, this is not surprising as it is known that phorbol ester activation of PKC promotes an irreversible and active membrane-bound complex of the kinase (50).
PKC phosphorylation of Kir6.2 has been shown by a number of groups; however, to date only one specific phosphorylation target (Thr-180) has been elucidated (24, 28–30). It has been shown that this site was not responsible for the PKC-mediated down-regulation of Kir6.2 (28). Phosphopeptide mapping identified the amino acid residue Ser-372 as a target for PKC phosphorylation, and this was confirmed using in vitro phosphorylation assays. There was ∼75% reduction in the phosphorylation of MBP-Kir6.2C-S372A compared with the wild-type protein. Additionally, there was no clear reduction in the cell surface expression of the Kir6.2-S372A protein in the presence of PdBu (Fig. 7D). Furthermore, our whole cell patch clamp data strongly suggest that Ser-372 is a likely site for down-regulation of the channel. In support of this, the Kir6.2-S372A/S372E and Kir6.2ΔC26 (Ser-372 lies within the last 26 amino acids of the C-terminal mutant) channels were not inhibited by PKC activation. The Kir6.2-S372E mutant showed typically lower basal expression and a smaller response to pinacidil, an effect expected as this mutant is a phosphomimetic. Given the precedent in the literature (28, 30), it is likely that PKC promotes internalization through endocytosis of the Kir6.2 subunit; however, the phosphorylation site is adjacent to the RKR motif known to influence forward trafficking of the KATP channel complex (1). Thus, it is also a possibility that forward trafficking is inhibited and these are topics for future investigation.
Our observations that Kir6.2ΔC26/SUR2B and in particular Kir6.2-S372A/SUR2B basal currents were activated by PKC support another PKC phosphorylation site such as Thr-180; and given the fairly quick increase in current, this is most likely to be due to an increase in channel activity. Interestingly, the potentiating effect on Kir6.2 does not appear to be Ca2+-dependent, as even at low Ca2+ we see a substantial increase in current (Fig. 8); however, this does not completely rule out the possibility of a conventional PKC also acting on Kir6.2 to increase current through it. Patch clamp studies of guinea pig ventricular myocytes suggest that this could be true. KATP channels in these cells, thought to contain Kir6.2 as the pore-forming subunit, have been shown to be regulated by both conventional and novel isoforms of PKC (51). Rainbow et al. have also shown that two isoforms of PKC (PKCα and PKCϵ) regulate voltage-gated K+ channels in rat mesenteric arteries depending on which receptor pathway is activated (45). Our data imply that the activation and inhibition of current through Kir6.2 by PKC may be similarly isoform-specific. Previous studies have shown that phosphorylation of Thr-180 leads to a decrease in ATP sensitivity of Kir6.2 (29, 37). In our hands, the relative ATP sensitivity of the WT channel and the phosphomimetic Kir6.2-S372E mutant channel did not change, suggesting that by implication phosphorylation of Ser-372 does not affect ATP binding to Kir6.2. Thr-180 is thought to line the hydrophobic pocket where the adenine ring of ATP sits explaining why modification of this residue reduces ATP sensitivity (52). Conversely, Ser-372 is at the distal C terminus of Kir6.2 and has not been shown to be involved in ATP binding. The Ser-372 residue has also been implicated as a site for PKA phosphorylation (27). However, it is not uncommon for both PKA and PKC to act on the same site on a protein (53–55). Indeed, on the Kir6.1 subunit one site (Ser-385) has been implicated as a target for both PKC and PKA (23, 56). PKA and PKC phosphorylation have opposing effects on KATP channels in smooth muscle, with the former leading to activation and vasodilation and the latter inhibition and subsequent vasoconstriction (21, 25, 57). Thus, other factors must be at play to give context to the net signaling response, and for example these might be additional phosphorylation sites on SUR2B or other proteins in the complex and/or particular intracellular nucleotide or Ca2+ conditions.
In this study, using protein biochemistry and electrophysiology, we have shown a relatively slow inhibition of Kir6.2/SUR2B by PKC and shown that this is Ca2+-dependent consistent with the action of a conventional PKC. In addition, we have identified a novel site (Ser-372) for Ca2+-dependent PKC phosphorylation on the Kir6.2 subunit.
This work was supported by the British Heart Foundation.
- KATP
- ATP-sensitive K+ channel
- DMSO
- dimethyl sulfoxide
- Kir
- inwardly rectifying potassium channel
- MBP
- maltose-binding protein
- pA
- picoampere
- pF
- picofarad
- PdBu
- phorbol 12,13-dibutyrate
- 4α-phorbol
- 4α-phorbol dideconate
- SUR
- sulfonylurea receptor.
REFERENCES
- 1. Rodrigo G. C., Standen N. B. (2005) ATP-sensitive potassium channels. Curr. Pharm. Des. 11, 1915–1940 [DOI] [PubMed] [Google Scholar]
- 2. Yellon D. M., Downey J. M. (2003) Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol. Rev. 83, 1113–1151 [DOI] [PubMed] [Google Scholar]
- 3. Babenko A. P., Aguilar-Bryan L., Bryan J. (1998) A view of SUR/Kir6.x, KATP channels. Annu. Rev. Physiol. 60, 667–687 [DOI] [PubMed] [Google Scholar]
- 4. Seino S. (1999) ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu. Rev. Physiol. 61, 337–362 [DOI] [PubMed] [Google Scholar]
- 5. Tucker S. J., Gribble F. M., Proks P., Trapp S., Ryder T. J., Haug T., Reimann F., Ashcroft F. M. (1998) Molecular determinants of KATP channel inhibition by ATP. EMBO J. 17, 3290–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aguilar-Bryan L., Nichols C. G., Wechsler S. W., Clement J. P., 4th, Boyd A. E., 3rd, González G., Herrera-Sosa H., Nguy K., Bryan J., Nelson D. A. (1995) Cloning of the β cell high affinity sulfonylurea receptor: a regulator of insulin secretion. Science 268, 423–426 [DOI] [PubMed] [Google Scholar]
- 7. Inagaki N., Gonoi T., Clement J. P., 4th, Namba N., Inazawa J., Gonzalez G., Aguilar-Bryan L., Seino S., Bryan J. (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270, 1166–1170 [DOI] [PubMed] [Google Scholar]
- 8. Beech D. J., Zhang H., Nakao K., Bolton T. B. (1993) K channel activation by nucleotide diphosphates and its inhibition by glibenclamide in vascular smooth muscle cells. Br. J. Pharmacol. 110, 573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cui Y., Giblin J. P., Clapp L. H., Tinker A. (2001) A mechanism for ATP-sensitive potassium channel diversity: functional coassembly of two pore-forming subunits. Proc. Natl. Acad. Sci. U.S.A. 98, 729–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamada M., Isomoto S., Matsumoto S., Kondo C., Shindo T., Horio Y., Kurachi Y. (1997) Sulfonylurea receptor 2B and Kir6.1 form a sulfonylurea-sensitive but ATP-insensitive K+ channel. J. Physiol. 499, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cole W. C., Malcolm T., Walsh M. P., Light P. E. (2000) Inhibition by protein kinase C of the KNDP subtype of vascular smooth muscle ATP-sensitive potassium channel. Circ. Res. 87, 112–117 [DOI] [PubMed] [Google Scholar]
- 12. Gopalakrishnan M., Whiteaker K. L., Molinari E. J., Davis-Taber R., Scott V. E., Shieh C. C., Buckner S. A., Milicic I., Cain J. C., Postl S., Sullivan J. P., Brioni J. D. (1999) Characterization of the ATP-sensitive potassium channels (KATP) expressed in guinea pig bladder smooth muscle cells. J. Pharmacol. Exp. Ther. 289, 551–558 [PubMed] [Google Scholar]
- 13. Inoue I., Nagase H., Kishi K., Higuti T. (1991) ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 352, 244–247 [DOI] [PubMed] [Google Scholar]
- 14. Isomoto S., Kondo C., Yamada M., Matsumoto S., Higashiguchi O., Horio Y., Matsuzawa Y., Kurachi Y. (1996) A novel sulfonylurea receptor forms with BIR (Kir6.2), a smooth muscle type ATP-sensitive K+ channel. J. Biol. Chem. 271, 24321–24324 [DOI] [PubMed] [Google Scholar]
- 15. Koh S. D., Bradley K. K., Rae M. G., Keef K. D., Horowitz B., Sanders K. M. (1998) Basal activation of ATP-sensitive potassium channels in murine colonic smooth muscle cell. Biophys. J. 75, 1793–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alekseev A. E., Hodgson D. M., Karger A. B., Park S., Zingman L. V., Terzic A. (2005) ATP-sensitive K+ channel channel/enzyme multimer: metabolic gating in the heart. J. Mol. Cell. Cardiol. 38, 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang H. L., Bolton T. B. (1996) Two types of ATP-sensitive potassium channels in rat portal vein smooth muscle cells. Br. J. Pharmacol. 118, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Levitan I. B. (1994) Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu. Rev. Physiol. 56, 193–212 [DOI] [PubMed] [Google Scholar]
- 19. Light P. (1996) Regulation of ATP-sensitive potassium channels by phosphorylation. Biochim. Biophys. Acta 1286, 65–73 [DOI] [PubMed] [Google Scholar]
- 20. Jonas E. A., Kaczmarek L. K. (1996) Regulation of potassium channels by protein kinases. Curr. Opin. Neurobiol. 6, 318–323 [DOI] [PubMed] [Google Scholar]
- 21. Quayle J. M., Nelson M. T., Standen N. B. (1997) ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol. Rev. 77, 1165–1232 [DOI] [PubMed] [Google Scholar]
- 22. Quinn K. V., Cui Y., Giblin J. P., Clapp L. H., Tinker A. (2003) Do anionic phospholipids serve as cofactors or second messengers for the regulation of activity of cloned ATP-sensitive K+ channels? Circ. Res. 93, 646–655 [DOI] [PubMed] [Google Scholar]
- 23. Shi Y., Cui N., Shi W., Jiang C. (2008) A short motif in Kir6.1 consisting of four phosphorylation repeats underlies the vascular KATP channel inhibition by protein kinase C. J. Biol. Chem. 283, 2488–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thorneloe K. S., Maruyama Y., Malcolm A. T., Light P. E., Walsh M. P., Cole W. C. (2002) Protein kinase C modulation of recombinant ATP-sensitive K+ channels composed of Kir6.1 and/or Kir6.2 expressed with SUR2B. J. Physiol. 541, 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayabuchi Y., Davies N. W., Standen N. B. (2001) Angiotensin II inhibits rat arterial KATP channels by inhibiting steady-state protein kinase A activity and activating protein kinase Cϵ. J. Physiol. 530, 193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jun J. Y., Kong I. D., Koh S. D., Wang X. Y., Perrino B. A., Ward S. M., Sanders K. M. (2001) Regulation of ATP-sensitive K+ channels by protein kinase C in murine colonic myocytes. Am. J. Physiol. Cell Physiol. 281, C857–864 [DOI] [PubMed] [Google Scholar]
- 27. Béguin P., Nagashima K., Nishimura M., Gonoi T., Seino S. (1999) PKA-mediated phosphorylation of the human KATP channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J. 18, 4722–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu K., Huang C. S., Jan Y. N., Jan L. Y. (2003) ATP-sensitive potassium channel traffic regulation by adenosine and protein kinase C. Neuron 38, 417–432 [DOI] [PubMed] [Google Scholar]
- 29. Light P. E., Bladen C., Winkfein R. J., Walsh M. P., French R. J. (2000) Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc. Natl. Acad. Sci. U.S.A. 97, 9058–9063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Manna P. T., Smith A. J., Taneja T. K., Howell G. J., Lippiat J. D., Sivaprasadarao A. (2010) Constitutive endocytic recycling and protein kinase C-mediated lysosomal degradation control KATP channel surface density. J. Biol. Chem. 285, 5963–5973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Giblin J. P., Quinn K., Tinker A. (2002) The cytoplasmic C terminus of the sulfonylurea receptor is important for KATP channel function but is not key for complex assembly or trafficking. Eur. J. Biochem. 269, 5303–5313 [DOI] [PubMed] [Google Scholar]
- 32. Thomas A. M., Brown S. G., Leaney J. L., Tinker A. (2006) Differential phosphoinositide binding to components of the G protein-gated K+ channel. J. Membr. Biol. 211, 43–53 [DOI] [PubMed] [Google Scholar]
- 33. Meimaridou E., Gooljar S. B., Ramnarace N., Anthonypillai L., Clark A. J., Chapple J. P. (2011) The cytosolic chaperone Hsc70 promotes traffic to the cell surface of intracellular retained melanocortin-4 receptor mutants. Mol. Endocrinol. 25, 1650–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farzaneh T., Tinker A. (2008) Differences in the mechanism of metabolic regulation of ATP-sensitive K+ channels containing Kir6.1 and Kir6.2 subunits. Cardiovasc. Res. 79, 621–631 [DOI] [PubMed] [Google Scholar]
- 35. Muzyamba M., Farzaneh T., Behe P., Thomas A., Christesen H. B., Brusgaard K., Hussain K., Tinker A. (2007) Complex ABCC8 DNA variations in congenital hyperinsulinism: lessons from functional studies. Clin. Endocrinol. 67, 115–124 [DOI] [PubMed] [Google Scholar]
- 36. Tucker S. J., Gribble F. M., Zhao C., Trapp S., Ashcroft F. M. (1997) Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulfonylurea receptor. Nature 387, 179–183 [DOI] [PubMed] [Google Scholar]
- 37. Light P. E., Sabir A. A., Allen B. G., Walsh M. P., French R. J. (1996) Protein kinase C-induced changes in the stoichiometry of ATP binding activate cardiac ATP-sensitive K+ channels: a possible mechanistic link to ischemic preconditioning. Circ. Res. 79, 399–406 [DOI] [PubMed] [Google Scholar]
- 38. Garlid K. D., Paucek P., Yarov-Yarovoy V., Murray H. N., Darbenzio R. B., D'Alonzo A. J., Lodge N. J., Smith M. A., Grover G. J. (1997) Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels: possible mechanism of cardioprotection. Circ. Res. 81, 1072–1082 [DOI] [PubMed] [Google Scholar]
- 39. Miki T., Suzuki M., Shibasaki T., Uemura H., Sato T., Yamaguchi K., Koseki H., Iwanaga T., Nakaya H., Seino S. (2002) Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat. Med. 8, 466–472 [DOI] [PubMed] [Google Scholar]
- 40. Yamada M. (2010) Mitochondrial ATP-sensitive K+ channels, protectors of the heart. J. Physiol. 588, 283–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Flagg T. P., Enkvetchakul D., Koster J. C., Nichols C. G. (2010) Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol. Rev. 90, 799–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiao J., Garg V., Yang B., Elton T. S., Hu K. (2008) Protein kinase Cϵ induces caveolin-dependent internalization of vascular adenosine 5′-triphosphate-sensitive K+ channels. Hypertension 52, 499–506 [DOI] [PubMed] [Google Scholar]
- 43. Sampson L. J., Davies L. M., Barrett-Jolley R., Standen N. B., Dart C. (2007) Angiotensin II-activated protein kinase C targets caveolae to inhibit aortic ATP-sensitive potassium channels. Cardiovasc. Res. 76, 61–70 [DOI] [PubMed] [Google Scholar]
- 44. Leaney J. L., Dekker L. V., Tinker A. (2001) Regulation of a G protein-gated inwardly rectifying K+ channel by a Ca2+-independent protein kinase C. J. Physiol. 534, 367–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rainbow R. D., Norman R. I., Everitt D. E., Brignell J. L., Davies N. W., Standen N. B. (2009) Endothelin-I and angiotensin II inhibit arterial voltage-gated K+ channels through different protein kinase C isoenzymes. Cardiovasc. Res. 83, 493–500 [DOI] [PubMed] [Google Scholar]
- 46. Damron D. S., Nadim H. S., Hong S. J., Darvish A., Murray P. A. (1998) Intracellular translocation of PKC isoforms in canine pulmonary artery smooth muscle cells by ANG II. Am. J. Physiol. Lung Cell. Mol. Physiol. 274, L278–288 [DOI] [PubMed] [Google Scholar]
- 47. Ohanian V., Ohanian J., Shaw L., Scarth S., Parker P. J., Heagerty A. M. (1996) Identification of protein kinase C isoforms in rat mesenteric small arteries and their possible role in agonist-induced contraction. Circ. Res. 78, 806–812 [DOI] [PubMed] [Google Scholar]
- 48. Park W. S., Ko E. A., Han J., Kim N., Earm Y. E. (2005) Endothelin-1 acts via protein kinase C to block KATP channels in rabbit coronary and pulmonary arterial smooth muscle cells. J. Cardiovasc. Pharmacol. 45, 99–108 [DOI] [PubMed] [Google Scholar]
- 49. Nelson C. P., Willets J. M., Davies N. W., Challiss R. A., Standen N. B. (2008) Visualizing the temporal effects of vasoconstrictors on PKC translocation and Ca2+ signaling in single resistance arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 295, C1590–1601 [DOI] [PubMed] [Google Scholar]
- 50. Bazzi M. D., Nelsestuen G. L. (1989) Differences in the effects of phorbol esters and diacylglycerols on protein kinase C. Biochemistry 28, 9317–9323 [DOI] [PubMed] [Google Scholar]
- 51. Ito K., Sato T., Arita M. (2001) Protein kinase C isoform-dependent modulation of ATP-sensitive K+ channels during reoxygenation in guinea pig ventricular myocytes. J. Physiol. 532, 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Trapp S., Haider S., Jones P., Sansom M. S., Ashcroft F. M. (2003) Identification of residues contributing to the ATP binding site of Kir6.2. EMBO J. 22, 2903–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. French A. D., Fiori J. L., Camilli T. C., Leotlela P. D., O'Connell M. P., Frank B. P., Subaran S., Indig F. E., Taub D. D., Weeraratna A. T. (2009) PKC and PKA phosphorylation affect the subcellular localization of claudin-1 in melanoma cells. Int. J. Med. Sci. 6, 93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Safran A., Provenzano C., Sagi-Eisenberg R., Fuchs S. (1990) Phosphorylation of membrane-bound acetylcholine receptor by protein kinase C: characterization and subunit specificity. Biochemistry 29, 6730–6734 [DOI] [PubMed] [Google Scholar]
- 55. Safran A., Sagi-Eisenberg R., Neumann D., Fuchs S. (1987) Phosphorylation of the acetylcholine receptor by protein kinase C and identification of the phosphorylation site within the receptor δ subunit. J. Biol. Chem. 262, 10506–10510 [PubMed] [Google Scholar]
- 56. Quinn K. V., Giblin J. P., Tinker A. (2004) Multisite phosphorylation mechanism for protein kinase A activation of the smooth muscle ATP-sensitive K+ channel. Circ. Res. 94, 1359–1366 [DOI] [PubMed] [Google Scholar]
- 57. Quayle J. M., Standen N. B. (1994) KATP channels in vascular smooth muscle. Cardiovasc. Res. 28, 797–804 [DOI] [PubMed] [Google Scholar]