

Abstract
1-Nitropyrene (1-NP), a mutagen and potential carcinogen, is the most abundant nitro polyaromatic hydrocarbon in diesel exhaust, which reacts with DNA to form predominantly N-(deoxyguanosin-8-yl)-1-aminopyrene (dGAP). If not repaired, this DNA lesion is presumably bypassed in vivo by any of human Y-family DNA polymerases kappa (hPolκ), iota (hPolτ), eta (hPolη), and Rev1 (hRev1). Our running start assays demonstrated that each of these enzymes was indeed capable of traversing a site-specifically placed dGAP on a synthetic DNA template but hRev1 was stopped after lesion bypass. The time required to bypass 50% of the dGAP sites (t50bypass ) encountered by hPolη, hPolκ and hPolτ was determined to be 2.5 s, 4.1 s, and 106.5 s, respectively. The efficiency order of catalyzing translesion synthesis of dGAP (hPolη > hPolκ > hPolτ >> hRev1) is the same as the order for these human Y-family enzymes to elongate undamaged DNA. Although hPolη bypassed dGAP efficiently, replication by both hPolκ and hPolτ was strongly stalled at the lesion site and at a site immediately downstream from dGAP. By employing pre-steady state kinetic methods, a kinetic basis was established for polymerase pausing at these DNA template sites. Besides efficiency of bypass, the fidelity of those low-fidelity polymerases at these pause sites was also significantly decreased. Thus, if the translesion DNA synthesis of dGAP in vivo is catalyzed by a human Y-family DNA polymerase, e.g. hPolη, the process is certainly mutagenic.
INTRODUCTION
Both normal metabolic processes and environmental factors constantly damage cellular DNA and produce 1,000 to 1,000,000 DNA lesions per cell per day. If these DNA lesions are not repaired by cellular DNA repair machinery, they will stall replicative DNA polymerases and replication machinery.1-4 To rescue stalled DNA replication, cellular DNA repair machinery temporarily switches to a Y-family DNA polymerase that primarily functions in the bypass of DNA lesions in vivo. Out of the 16 identified human DNA polymerases, four enzymes belong to the Y-family: DNA polymerases eta (hPolη), kappa (hPolκ), iota (hPolτ), and Rev1 (hRev1). These enzymes have been shown to catalyze both error-free and error-prone translesion DNA synthesis (TLS) in vitro and in vivo.5-7 For example, hPolη catalyzes error-free bypass of cis-syn cyclobutane thymidine dimer (cis-syn TT)8, 9 and its inactivation by mutations leads to Xeroderma Pigmentosum Variant (XPV) disease that increases incidence of sunlight-induced skin cancer.8, 10 In addition, hPolη has been shown in vitro to bypass other lesions including apurinic/apyrimidinic (abasic) sites,11-13 N-(deoxyguanosin-8-yl)-N-acetyl-2-aminofluorene (AAF-dG),13 and N2-deoxyguanosine adducts of benzo[a]pyrene (BPDE-dG).12 hPolκ, a homolog of the well-studied Y-family member Sulfolobus solfataricus DNA Polymerase IV (Dpo4), is capable of bypassing abasic sites,11 AAF-dG, and BPDE-dG,14 and efficiently elongating mispaired primer termini.15 hPolτ has been shown to bypass abasic sites,11 cis-syn TT,16 and AAF-dG.17 hRev1 preferentially incorporates dCTP opposite any template base18 and DNA lesions including abasic sites11 and BPDE-dG.19 These biochemical studies demonstrate that there is a significant overlap in lesion bypass abilities of the four human Y-family members. At present, it is unclear which human Y-family enzyme bypasses which DNA lesion(s) in vivo.
Despite growing concerns about the effects of air pollution on human genomic stability, there are no comprehensive studies of the bypass of air pollutant-induced DNA adducts catalyzed by human Y-family DNA polymerases. The metabolites of 1-nitropyrene (1-NP), a product of incomplete gasoline combustion and one of the most abundant polycyclic aromatic hydrocarbons (PAH),20-22 react with DNA to primarily form N-(deoxyguanosin-8-yl)-1-aminopyrene (dGAP, Supplementary Figure 1).20 Because 1-NP is a potent mutagen and carcinogen in rodents,23 it is classified as a class 2B carcinogen.24, 25 Moreover, the dGAP lesion is shown to be mutagenic in bacterial and mammalian cells.24, 25 Thus, it is interesting to identify which human Y-family DNA polymerase(s) bypass(es) dGAP in vitro and in vivo. Previously, we have shown that the kinetics of nucleotide incorporation opposite dGAP and the subsequent extension of the dGAP bypass product catalyzed by Dpo4 are significantly altered.26 A minimum kinetic mechanism for the dGAP bypass has also been established via pre-steady-state kinetic methods.26 Here, we employed these methods and kinetically assessed the dGAP bypass abilities of human Y-family DNA polymerases in order to shed light on which of these enzymes are better suited to bypass dGAP in vivo.
MATERIAL AND METHODS
Materials
Reagents were purchased from the following companies: OptiKinase from USB Corporation, [γ−32P]ATP from MP Biochemicals, and dNTPs from GE Healthcare. hPolη, hRev1, and hPolκ were expressed and purified as previously described.11 hPolτ was expressed and purified as previously described11 with one modification: the GST-tag was not removed in order to increase the protein stability of hPolτ.
DNA Substrates
The DNA template 26-mer-dGAP (Table 1) was synthesized and purified as previously described.26 Other DNA oligomers listed in Table 1 were purchased from Integrated DNA Technologies. The radiolabeling of the primers and the annealing of a primer and a template were performed as described previously.26, 27
Table 1.
DNA Substrates for dGAP.
| Primers | |
| 17-mer | 5′-AACGACGGCCAGTGAAT-3′ |
| 20-mer | 5′-AACGACGGCCAGTGAATTCG-3′ |
| 21-mer | 5′-AACGACGGCCAGTGAATTCGC-3′ |
|
| |
| Templates | |
| 26-mer | 3′-TTGCTGCCGGTCACTTAAGCGCGCCC-5′ |
| a26-mer-dGAP | 3′-TTGCTGCCGGTCACTTAAGCGCGCCC-5′ |
|
| |
| DNA Trap | |
| D-1 (21/41-mer) | 5′-CGCAGCCGTCCAACCAACTCA-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTAGCAGCTAGGTTACGGCAGG-5′ | |
G designates the 1-AP adduct on C8 position of dG (dGAP).
Buffers
All pre-steady-state kinetic assays were performed in reaction buffer R (50 mM HEPES, pH 7.5 at 37 °C, 5 mM MgCl2, 50 mM NaCl, 0.1 mM EDTA, 5 mM DTT, 10 % glycerol, and 0.1 mg/ml BSA). All electrophoresis mobility shift assays (EMSA) were performed in buffer S (50 mM Tris-Cl, pH 7.5 at 23 °C, 5 mM MgCl2, 50 mM NaCl, 5 mM DTT, 10 % glycerol, and 0.1 mg/ml BSA). All reported concentrations were final after mixing solutions.
Running Start Assays
The running start assay was performed similarly as described previously.11, 26, 28-30 Briefly, a preincubated solution of 5′-[32P]-labeled DNA (100 nM) and a human Y-family enzyme (1 μM) in buffer R was rapidly mixed with a solution containing all four dNTPs (200 μM each) at 37 °C via a rapid chemical-quench flow apparatus (KinTek). The reactions were quenched with 0.37 M EDTA after various times, and the reaction products were analyzed by denaturing polyacrylamide gel electrophoresis (PAGE, 20 % polyacrylamide, 8 M urea).
Quantitative analysis of the running start assays was performed by determining the relative lesion bypass efficiencies (dGAP bypass%) as a function of reaction time. For each time point t, dGAP bypass% was calculated as the ratio of the bypass events to the encounter events (Equation 1):
| (1) |
where the total dGAP bypass events (B) was calculated from the concentration of all intermediate products with sizes greater than or equal to the 21-mer, and the total dGAP “encounter” events (E) equaled the summation of the 20-mer concentration and the total dGAP bypass events (B). To quantitatively define the dGAP bypass efficiency, t50bypass was defined as the time required to bypass 50% of the total dGAP lesions encountered.
Electrophoresis Mobility Shift Assays
hPolη (10-400 nM), hPolκ (65-950 nM), or hRev1 (15-550 nM) was titrated onto a solution containing 5′-[32P]-labeled DNA (10 nM) in buffer S at 23 °C. Native PAGE was used to separate the binary complex E•DNA from free DNA. After quantitation using a Typhoon Trio (GE Healthcare), the concentration of the binary complex was plotted as a function of the enzyme concentration. The data were fit to Equation 2 using Kaleidagraph (Synergy software):
| (2) |
where Eo and Do are the initial enzyme and DNA concentrations, respectively, and Kd, DNA is the equilibrium dissociation constant for E•DNA at 23 °C.
Active Site Titration Assays
A preincubated solution of 5′-[32P]-labeled DNA (10-450 nM) and hPolτ (27.5 nM, UV-based concentration) in buffer R was reacted with correct dNTP at saturating concentrations at 37 °C for various time intervals prior to be quenched by EDTA (0.37 M). The reaction products were resolved and quantitated as described above. The burst reaction amplitude (A) was plotted as a function of DNA concentrations and the data were then fit to Equation 3 to determine Kd, DNA.
| (3) |
Nucleotide Incorporation Efficiency and Fidelity Measurements
Single-turnover kinetic assays were employed to obtain the kp (maximum dNTP incorporation rate) and Kd, dNTP (equilibrium dissociation constant for dNTP from E•DNA•dNTP) for single nucleotide incorporation as described previously.26, 31 Briefly, a preincubated solution of 5′-[32P]-labeled DNA (20 nM) and hPolη (130 nM) or hPolτ (130 nM) in buffer R was mixed with increasing concentrations of an incoming dNTP at 37 °C. For hPolκ and hRev1, a preincubated solution of 5′-[32P]-labeled DNA (30 nM) and hPolκ (300 nM) or hRev1 (120 nM) in buffer R was mixed with increasing concentrations of an incoming dNTP. The reactions were terminated after various times by the addition of 0.37 M EDTA. Reaction products were resolved and quantitated as described above. The time course of product formation at each dNTP concentration was fit to Equation 4:
| (4) |
where kobs is the observed reaction rate constant. Next, the plot of the kobs values as a function of dNTP concentrations was fit to Equation 5:
| (5) |
From this plot, the dNTP incorporation efficiency (kp/Kd, dNTP) was also calculated for each enzyme. The dNTP incorporation fidelity was determined using Equation 6:
| (6) |
DNA Trap Assays
A preincubated solution of hPolη (130 nM) and 5′-[32P]-labeled DNA (20 nM) in buffer R was rapidly mixed with a solution of 5 μM unlabeled DNA trap D-1 (Table 1) and a correct dNTP at a saturating concentration in buffer R for various times before the reactions were quenched with 0.37 M EDTA. Reaction products were resolved and quantitated as described above. The plot of the product concentration as a function of reaction time was fit to Equation 7:
| (7) |
where Eo is the active hPolη concentration, A1 and A2 the reaction amplitudes of the first and second phases, respectively, and k1 and k2 the rate constants of the first and second phases, respectively.
RESULTS
Bypass of dGAP catalyzed by human Y-family DNA polymerases
To determine if each of the four human Y-family DNA polymerases was able to bypass dGAP (Supplementary Figure 1), we performed running start assays (Figure 1) with both a control DNA substrate 17-mer/26-mer and a damaged DNA substrate 17-mer/26-mer-dGAP containing a site-specifically placed dGAP on template 26-mer-dGAP (Table 1). Notably, hPolκ synthesized much more 25-mer than 26-mer with 17-mer/26-mer after long reaction times while an intermediate product 24-mer significantly accumulated between 6 s to 60 s (Figure 1C). Because hPolκ is known to create −1 frameshift mutations11, 14, 32, 33 and the template 26-mer contains a dC-rich 5′-terminus (Table 1), the 25-mer was likely the true full-length product synthesized by hPolκ after hPolκ slipped once within the dC-rich region. The strong early accumulation of 24-mer in Figure 1C supports this possibility. If the possibility is proven to be true, the small amount of 26-mer was likely generated by hPolκ via a single blunt-end addition onto the 25-mer as we have previously observed with Dpo4.34 Interestingly, the blunt-end addition phenomenon was more significant with hPolη (Figure 1A). This enzyme has been found previously to catalyze blunt-end additions onto other DNA substrates.13, 35, 36 With the control template 26-mer, hPolη, hPolκ, and hPolτ were able to synthesize full-length products after 1 s, 6 s, and 60 s, respectively (Figures 1A, 1C, and 1E). In stark contrast, hRev1 only incorporated five nucleotides and did not synthesize full-length products even after 12 hrs (Figure 1G). The reaction was not carried out beyond 12 hrs was due to both enzyme stability and in vivo relevance. Interestingly, with the damaged template 26-mer-dGAP, hPolη was not significantly stalled by dGAP and synthesized the full-length products (26-mer and 27-mer) after 3 s (Figure 1B). In comparison, hPolκ and hPolτ needed 60 s and 7,200 s, respectively, to achieve the same task (Figures 1D and 1F). Thus, the presence of a single dGAP slowed hPolκ and hPolτ by 10- and 120-fold, respectively. In addition, there was a significant accumulation of intermediate products 20-mer and 21-mer with both hPolκ and hPolτ, indicating that these enzymes encountered significant difficulty in bypassing the lesion and carrying out the subsequent extension steps (Figures 1D and 1F). As expected, hRev1, a dCTP polymerase, did not synthesize full-length products with 26-mer-dGAP (Figure 1H). The longest product after 12 hrs was a small of 21-mer, suggesting that hRev1 was able to incorporate a nucleotide opposite dGAP but could not extend the DNA lesion bypass product.
Figure 1.

Running start assays at 37 °C. A preincubated solution of 100 nM 5′-[32P]-labeled 17-mer/26-mer (A, C, E and G) or 17-mer/26-mer-dGAP (B, D, F and H) and 1 μM hPolη (A and B), hPolκ (C and D), hPolτ (E and F), or hRev1 (G and H) was rapidly mixed with a solution of four dNTPs (200 μM each) for various times before being quenched with 0.37 M EDTA. Sizes of important products are denoted and the 21st position marks the location of dGAP from the 3′-terminus of the DNA template.
We further analyzed these running start assays by determining both dGAP bypass% using Equation 1 (Material and Methods) and bypass efficiency t50bypass for each enzyme.11 A plot of dGAP bypass% versus reaction time for each enzyme (Supplementary Figure 2) yielded the corresponding t50bypass value (Table 2), which was defined as the time required for each enzyme to bypass 50% of the total dGAP sites encountered. Similar analysis was also performed with control DNA and yielded t50 values for each enzyme to traverse the corresponding dG site (Table 2). These t50bypass values (Table 2) indicate that hPolη bypassed dGAP encountered and bypassed 50% of dGAP sites 1.6- and 42.6-fold faster than hPolκ and hPolτ, respectively. When comparing the t50bypass and t50 values for each enzyme, hPolη, hPolκ, and hPolτ became 3.1-, 2.7-, and 7.1-fold slower, respectively, in the presence than in the absence of dGAP. Thus, dGAP had only a slight inhibitory effect on the overall DNA polymerase activity of hPolη and hPolκ but significantly slowed DNA synthesis catalyzed by hPolτ. The varying effects of dGAP on these three Y-family enzymes suggest that each enzyme may utilize a unique mechanism for dGAP bypass.
Table 2.
dGAP bypass efficiencies of human Y-family DNA polymerases.
Calculated as the time required to bypass 50% of the dGAP sites.
Calculated as the time required to bypass 50% of dG, or 20-mer.
Effects of dGAP on DNA binding affinities
The accumulation of the intermediate 21-mer during dGAP bypass (Figure 1) suggests that the presence of the bulky 1-AP adduct could have weakened DNA binding to human Y-family DNA polymerases. To evaluate this possibility, we estimated the binding affinity of human Y-family enzymes to 20-mer/26-mer, 20-mer/26-mer-dGAP, 21-mer/26-mer, 21-mer/26-mer-dGAP (Table 1) via gel electrophoresis mobility shift assays (EMSA). An example of gel image for EMSA using hPolη and 20-mer/26-mer-dGAP is shown in Supplementary Figure 3. The Kd, DNA values for the binding of hPolη to other DNA substrates were measured similarly and are listed in Table 3. Interestingly, the calculated ratios in Table 3 demonstrate that the presence of dGAP increased the binding affinity of DNA to hPolη. The structures of Dpo4 with BPDE-dG37 and yeast Polη with AAF-dG38 suggest that the hydrophobic 1-AP adduct in complexes hPolη•20-mer/26-mer-dGAP and hPolη•21-mer/26-mer-dGAP likely interacted with the amino acid residues in the Little Finger domain of hPolη.
Table 3.
Binding affinity of hPolη, hPolτ, and hRev1 to normal and damaged DNA at room temperature.
| Enzyme | DNA Substrate | With Adducta (nM) |
Without Adductb (nM) |
Affinity Ratioc |
|---|---|---|---|---|
| hPolη | 20-mer/26-mer | 7.9 ± 0.3 | 23 ± 2 | 3.0 |
| 21-mer/26-mer | 8.8 ± 0.5 | 26 ± 1 | 2.9 | |
| hPolτd | 20-mer/26-mer | 64 ± 8 | 56 ± 12 | 0.9 |
| 21-mer/26-mer | ND | 43 ± 4 | ND | |
| hRev1 | 20-mer/26-mer | 85 ± 5 | 118 ± 7 | 1.4 |
| 21-mer/26-mer | ND | ND | ND |
With adduct refers to 26-mer-dGAP.
Without adduct refers to 26-mer.
Calculated as (Kd, DNA)Normal/(Kd, DNA)Damaged.
Values were determined using active site titration assays.
ND denoted ‘not determined’.
All given errors were derived from data fitting.
When EMSA was performed for each of the other human Y-family enzymes, we were only able to estimate the Kd, DNA values for hRev1 (Table 3). There was no obvious mobility shift with hPolκ and hPolτ during EMSA, likely due to their weak binding to DNA. For hPolτ, active site titration assays (Materials and Methods) were employed to estimate the Kd, DNA values (Table 3). These values were similar to a previously reported value of 44 ± 7 nM with an undamaged DNA substrate.39 We did not measure the Kd, DNA of hPolτ•21-mer/26-mer-dGAP because catalysis (see below) was slower than the equilibration of the enzyme and DNA (E + DNA ↔ E•DNA). For hPolκ, it is not proper to use active site titration assays to estimate the Kd, DNA values because only 15% of recombinant hPolκ has been previously found to be active in such an assay.40 With undamaged DNA, the Kd, DNA of a similar hPolκ construct used in this paper (Materials and Methods) has been estimated to be 96 ± 21 nM at 25 °C by using a fluorescence anisotropy assay.41 Based on the above published and measured Kd, DNA values as well as calculated affinity ratios in Table 3, we concluded that hPolη bound to both damaged and undamaged DNA with the highest affinity and the impact of the 1-AP adduct on DNA binding to human Y-family DNA polymerases was not significant enough to account for the strong polymerase pausing during TLS shown in Figure 1. As we have discovered previously with other DNA lesions,26, 29, 42 it is likely that nucleotide incorporation kinetics was dramatically influenced by dGAP.
Effect of dGAP on the kinetics of nucleotide incorporation
hPolη
To determine the effect of dGAP on nucleotide incorporation efficiency at or near the lesion site, we performed single nucleotide incorporation assays with each human Y-family DNA polymerase under single-turnover conditions. For example, a preincubated solution of 5′-[32P]-labeled DNA 20-mer/26-mer-dGAP (20 nM) and hPolη (130 nM) at 37 °C was rapidly mixed with varying dCTP concentrations for various reaction times before being quenched with EDTA (0.37 M). The reaction mixtures were analyzed by denaturing PAGE and quantitated by using a Typhoon Trio (Materials and Methods). Subsequently, the product concentrations were plotted against reaction times and each time course was fit to Equation 4 to obtain an observed reaction rate constant (kobs) (Figure 2A). The plot of the kobs values versus dCTP concentrations was then fit to Equation 5 to obtain an apparent maximum dCTP incorporation rate constant (kp) of 8.1 ± 0.2 s−1 and an apparent equilibrium dissociation constant (Kd, dNTP) of 287 ± 24 μM opposite dGAP (Figure 2B). The efficiency (kp/Kd, dNTP) of correct dCTP incorporation opposite dGAP was then calculated to be 2.8×10−2 μM−1s−1 (Table 4).
Figure 2.

Kinetics of correct dCTP incorporation onto 20-mer/26-mer-dGAP catalyzed by hPolη under single-turnover conditions. (A) A preincubated solution of hPolη (130 nM) and 5′-[32P]-labeled 20-mer/26-mer-dGAP (20 nM) was rapidly mixed with increasing concentrations of dCTP (25 μM, •; 50 μM, ■; 100 μM, △; 200 μM, ▲; 400 μM, ○; 800 μM, ◆; 1200 μM, □) for various time intervals at 37 °C. Each time course was fit to Equation 4 to obtain a kobs for a specific dCTP concentration. (B) The plot of kobs values against dCTP concentrations was fit to Equation 5 to yield a Kd, dCTP of 287 ± 24 μM and a kp of 8.1 ± 0.2 s−1.
Table 4.
Kinetic parameters of nucleotide incorporation onto damaged DNA catalyzed by hPolη.
| dNTP |
Kd, dNTP (μM) |
kp (s−1) |
kp/Kd, dNTP (μM−1s−1) |
Efficiency Ratioa,b |
Fidelityc | Fidelity Ratiob,d |
Probabilitye (%) |
|---|---|---|---|---|---|---|---|
| Template dGAP (20/26mer-dGAP) | |||||||
| dCTP | 287 ± 24 | 8.1 ± 0.2 | 2.8×10−2 | 20 | - | - | 77 |
| dATP | 634 ± 66 | (8.7 ± 0.4)×10−1 | 1.4×10−3 | 1.9 | 4.8×10−2 | 10 | 3.9 |
| dGTP | 65 ± 7 | (5.2 ± 0.2)×10−2 | 8.1×10−4 | 1.4 | 2.8×10−2 | 14 | 2.2 |
| dTTP | 492 ± 67 | 3.0 ± 0.2 | 6.0×10−3 | 1.1 | 1.8×10−1 | 15 | 17 |
| Template dC (21/26mer-dGAP) | |||||||
| dGTP | 125 ± 18 | 2.2 ± 0.1 | 1.8×10−2 | 48 | - | - | 22 |
| dATP | 335 ± 34 | (3.3 ± 0.1)×10−1 | 1.0×10−3 | 14 | 5.3×10−2 | 3.1 | 1.2 |
| dCTP | 119 ± 14 | 7.3 ± 0.2 | 6.1×10−2 | 0.3 | 7.7×10−1 | 37 | 75 |
| dTTP | 632 ± 71 | (9.5 ± 0.5)×10−1 | 1.5×10−3 | 3.1 | 7.7×10−2 | 14 | 1.8 |
Calculated as (kp/Kd, dNTP)normal/(kp/Kd, dNTP)damaged.
The values are from Supplementary Table 1 using control 26mer DNA template.
Calculated as (kp/Kd, dNTP)incorrect/[(kp/Kd, dNTP)correct + (kp/Kd, dNTP)incorrect].
Calculated as Fidelitydamaged/Fidelitynormal.
Calculated as ((kp/Kd, dNTP)damaged/[Σ(kp/Kd, dNTP)damaged])×100.
All given errors were derived from data fitting.
To determine if nucleotide incorporation efficiency and fidelity were affected within the vicinity of the bulky 1-AP adduct, we performed similar single-turnover kinetic assays for each correct or incorrect nucleotide with control or damaged DNA substrates and the resulting kinetic parameters are listed in Table 4 and Supplementary Table 1. In the presence of either 20-mer/26-mer or 21-mer/26-mer (Table 1), hPolη incorporated correct nucleotides with 50-500 fold higher kp/Kd, dNTP values than incorrect nucleotides, which are mainly derived from the kp differences (Supplementary Table 1). Relative to corresponding control DNA substrates, hPolη incorporated correct nucleotides onto 20-mer/26-mer-dGAP and 21-mer/26-mer-dGAP with 6-18 fold lower kp values and 3-fold higher Kd, dNTP values, leading to 20-48 fold lower kp/Kd, dNTP values (Table 4). The large decreases in nucleotide incorporation efficiency with the damaged DNA appear to be contradictory to the conclusion of weak polymerase pausing as revealed by Figure 1B. However, the overall polymerase efficiency at the lesion site and the immediate downstream site only dropped by 7-16 fold if the 3-fold tighter DNA binding affinity (Table 3) was taken into consideration. Moreover, the relative high kp values (2-8 s−1) for correct nucleotide incorporation further diminished intermediate product accumulation near the lesion site in Figure 1B. Interestingly, the nucleotide incorporation fidelity of hPolη was 16-fold lower on average with the damaged DNA substrates than undamaged ones and thus, significant percentages of incorporated nucleotides opposite dGAP and the downstream template base dC were incorrect (Table 4). Remarkably, hPolη misincorporated dCTP onto 21-mer/26-mer-dGAP with 3-fold higher kp and kp/Kd, dNTP values than it incorporated correct dGTP. Taken together, hPolη was highly error-prone although it catalyzed efficient TLS of dGAP.
hPolκ
The kinetic parameters for correct and incorrect nucleotide incorporations with control (Supplementary Table 2) and damaged (Table 5) DNA substrates were determined under similar single-turnover kinetic conditions as described for hPolη (Supplementary Figure 4). Like hPolη, hPolκ incorporated correct dNTPs onto damaged DNA with decreased efficiency than onto undamaged DNA. Notably, the decrease is less dramatic during the dGAP bypass step (5.7-fold) than during the extension step (208-fold) and hPolκ was 123-fold less efficient during the later step than during the former step. These efficiency differences and the low kp value (2.3×10−3 s−1) for correct dGTP incorporation onto 21-mer/26-mer-dGAP contributed to the strong accumulation of the intermediate product 21-mer (Figure 1D).
Table 5.
Kinetic parameters of nucleotide incorporation onto damaged DNA catalyzed by hPolκ.
| dNTP |
Kd, dNTP (μM) |
kp (s−1) |
kp/Kd, dNTP (μM−1s−1) |
Efficiency Ratioa,b |
Fidelityc | Fidelity Ratiob,d |
Probabilitye (%) |
|---|---|---|---|---|---|---|---|
| Template dGAP (20-mer/26-mer-dGAP) | |||||||
| dCTP | 267 ± 40 | 1.7 ± 0.1 | 6.5×10−3 | 5.7 | - | - | 71 |
| dATP | 497 ± 98 | (7.0 ± 0.6)×10−1 | 1.4×10−3 | 0.4 | 1.8×10−1 | 11 | 15 |
| dGTP | 346 ± 80 | (3.4 ± 0.3)×10−1 | 9.9×10−4 | 1.1 | 1.3×10−1 | 4.5 | 11 |
| dTTP | 726 ± 237 | (2.4 ± 0.4)×10−1 | 3.2×10−4 | 2.5 | 4.7×10−2 | 2.2 | 3.5 |
| Template dC – pause site (21-mer/26-mer-dGAP) | |||||||
| dGTP | 43 ± 7 | (2.3 ± 0.1)×10−3 | 5.3×10−5 | 208 | - | - | 60 |
| dATP | 203 ± 36 | (5.5 ± 0.3)×10−3 | 2.7×10−5 | 1.5 | 3.4×10−1 | 94 | 31 |
| dCTP | 693 ± 45 | (4.1 ± 0.1)×10−3 | 5.9×10−6 | 12 | 1.0×10−1 | 17 | 6.7 |
| dTTP | 468 ± 90 | (12 ± 1)×10−4 | 2.5×10−6 | 16 | 4.5×10−2 | 13 | 2.8 |
Calculated as (kp/Kd, dNTP)normal/(kp/Kd, dNTP)damaged.
The values are from Supplementary Table 2 using control 26mer DNA template.
Calculated as (kp/Kd, dNTP)incorrect/[(kp/Kd, dNTP)correct + (kp/Kd, dNTP)incorrect].
Calculated as Fidelitydamaged/Fidelitynormal.
Calculated as ((kp/Kd, dNTP)damaged/[Σ(kp/Kd, dNTP)damaged])×100.
All given errors were derived from data fitting.
With control DNA substrates, the nucleotide incorporation fidelity of hPolκ was calculated to be in the range of 10−3 to 10−2 (Supplementary Table 2). The low nucleotide incorporation fidelity of hPolκ was further worsened by 2-94 folds and was in the range of 10−2 to 10−1 with the damaged DNA substrates (Table 5). The probability for hPolκ to incorporate incorrect nucleotides during TLS of dGAP was estimated to be as high as 30% (Table 5).
hPolτ
Like hPolη and hPolκ, the kinetic parameters for nucleotide incorporations with undamaged (Supplementary Table 3) and damaged (Table 6) DNA substrates were determined by employing single-turnover kinetic assays (Supplementary Figure 5). Since misincorporation of either dATP or dCTP onto 21-mer/26-mer-dGAP catalyzed by hPolτ was not detected even after eight hours, the corresponding kinetic parameters with either 21-mer/26-mer-dGAP or 21-mer/26-mer were not determined. Like hPolκ, hPolτ incorporated correct nucleotides onto damaged DNA with lower efficiency than onto undamaged DNA and the impact was determined to be 7- and 4,545-fold with 20-mer/26-mer-dGAP and 21-mer/26-mer-dGAP, respectively (Table 6). Accordingly, 21-mer/26-mer-dGAP was extended by hPolτ with more than 1,000-fold lower kp and kp/Kd, dNTP values than 20-mer/26-mer-dGAP. These kinetic differences led to the strong accumulation of the intermediate product 21-mer in Figure 1F.
Table 6.
Kinetic parameters of nucleotide incorporation onto damaged DNA catalyzed by hPolτ.
| dNTP |
Kd, dNTP (μM) |
kp (s−1) |
kp/Kd, dNTP (μM−1s−1) |
Efficiency Ratioa,b |
Fidelityc | Fidelity Ratiob,d |
Probabilitye (%) |
|---|---|---|---|---|---|---|---|
| Template dGAP (20-mer/26-mer-dGAP) | |||||||
| dCTP | 380 ± 66 | (8.8 ± 0.6)×10−2 | 2.3×10−4 | 7.0 | - | - | 97 |
| dATP | 338 ± 22 | (10.3 ± 0.2)×10−4 | 3.0×10−6 | 5.0 | 1.3×10−2 | 1.4 | 1.3 |
| dGTP | 252 ± 29 | (5.3 ± 0.2)×10−4 | 2.1×10−6 | 7.6 | 9.0×10−3 | 0.9 | 0.9 |
| dTTP | 762 ± 60 | (13.1 ± 0.5)×10−4 | 1.7×10−6 | 118 | 7.3×10−3 | 0.1 | 0.7 |
| Template dC – pause site (21-mer/26-mer-dGAP) | |||||||
| dGTP | 264 ± 76 | (5.9 ± 0.6)×10−5 | 2.2×10−7 | 4.5×103 | - | - | 47 |
| dATP | ND | ND | - | - | - | - | - |
| dCTP | ND | ND | - | - | - | - | - |
| dTTP | 573 ± 73 | (14.1 ± 0.8)×10−5 | 2.5×10−7 | 680 | 5.3×10−1 | 5.5 | 53 |
Calculated as (kp/Kd, dNTP)normal/(kp/Kd, dNTP)damaged.
The values are from Supplementary Table 3 using control 26mer DNA template.
Calculated as (kp/Kd, dNTP)incorrect/[(kp/Kd, dNTP)correct + (kp/Kd, dNTP)incorrect].
Calculated as Fidelitydamaged/Fidelitynormal.
Calculated as ((kp/Kd, dNTP)damaged/[Σ(kp/Kd, dNTP)damaged])×100.
ND denoted ‘not determined’.
All given errors were derived from data fitting.
With 20-mer/26-mer-dGAP, all three incorrect nucleotides were incorporated onto 20-mer/26-mer-dGAP by hPolτ with comparable low efficiencies and probabilities (Table 6). In comparison, incorrect dTTP was incorporated onto 21-mer/26-mer-dGAP with similar efficiency and probability to those of correct dGTP (Table 6). These kinetic data suggest that hPolτ was more error-prone during the subsequent extension step than during dGAP bypass.
hRev1
Consistent with Figure 1H, hRev1 could not incorporate any nucleotide onto 21-mer/26-mer-dGAP after three hours. Thus, we only determined the kinetic parameters for nucleotide incorporations onto 20-mer/26-mer-dGAP (Table 7) and the corresponding control substrate 20-mer/26-mer (Supplementary Table 4) under single turnover conditions (Supplementary Figure 6). Like the other three human Y-family enzymes, hRev1 incorporated dCTP opposite dGAP with 22-fold lower efficiency than opposite undamaged dG, which was predominantly contributed by the kp difference. In addition, all three incorrect dNTPs were also incorporated with lower efficiency when the template base dG was damaged, leading to a higher fidelity with 20-mer/26-mer-dGAP. Thus, hRev1 was slightly more error-free but catalytically slower as it bypassed dGAP.
Table 7.
Kinetic parameters of nucleotide incorporation onto damaged DNA catalyzed by hRev1.
| dNTP |
Kd, dNTP (μM) |
kp (s−1) |
kp/Kd, dNTP (μM−1s−1) |
Efficiency Ratioa,b |
Fidelityc | Fideltiy Ratioc |
Probabilitye (%) |
|---|---|---|---|---|---|---|---|
| Template dGAP (20-mer/26-mer-dGAP) | |||||||
| dCTP | 5.2 ± 0.9 | (3.3 ± 0.2)×10−2 | 6.5×10−3 | 22 | - | - | 99.8 |
| dTTP | 78 ± 18 | (2.6 ± 0.3×10−4 | 3.4×10−6 | 7.6×103 | 5.2×10−4 | 3.0×10−3 | 0.1 |
| dATP | 103 ± 25 | (4.4 ± 0.4×10−4 | 4.2×10−6 | 17 | 6.5×10−41.3 | 1.3 | 0.1 |
| dGTP | 143 ± 34 | (4.2 ± 0.3)×10−4 | 3.0×10−6 | 1.8×103 | 4.6×10−4 | 0.1 | ~0 |
Calculated as (kp/Kd, dNTP)normal/(kp/Kd, dNTP)damaged.
The values are from Supplementary Table 4 using control 26mer DNA template.
Calculated as (kp/Kd, dNTP)incorrect/[(kp/Kd, dNTP)correct + (kp/Kd, dNTP)incorrect].
Calculated as Fidelitydamaged/Fidelitynormal.
Calculated as ((kp/Kd, dNTP)damaged/[Σ(kp/Kd, dNTP)damaged])×100.
All given errors were derived from data fitting.
Biphasic kinetics of nucleotide incorporation catalyzed by hPolη
Previously, we have shown that correct nucleotide incorporation opposite dGAP and the downstream template base dC catalyzed by Dpo4 in the presence of a DNA trap follows biphasic kinetics and that the amplitudes of both the fast and slow phases are higher during the dGAP bypass step.26 To explore if human Y-family DNA polymerases also follow similar biphasic kinetics during dGAP bypass in the presence of a DNA trap, hPolη was chosen as a model enzyme because it bound DNA tightly (Table 3), a condition for the DNA trap assay.26, 29, 42-45 Here, we performed single dGTP incorporation onto either 21-mer/26-mer or 21-mer/26-mer-dGAP in the presence of an unlabeled DNA trap which was in large excess over 5′-radiolabeled DNA. First, we determined the effectiveness of 5 μM D-1 (Table 1) as a DNA trap. This trap was effective to prevent correct dGTP incorporation onto radiolabeled 21-mer/26-mer (20 nM) by hPolη within 33 s (Supplementary Figure 7). Next, we conducted the DNA trap assay for correct dGTP incorporation onto either 21-mer/26-mer or 21-mer/26-mer-dGAP (Material and Methods). The concentration of products was plotted as a function of reaction time (Figure 3) and the plot was fit to Equation 6 to yield kinetic parameters listed in Supplementary Table 5. Unlike Dpo4,26 hPolη followed biphasic kinetics with both control and damaged DNA substrates. With 21-mer/26-mer, the amplitude of the fast kinetic phase (A1 = 39%) is smaller than that of the slow kinetic phase (A2 = 47%) while the former (k1 = 72.2 s−1) occurs with a higher reaction rate constant than the latter (k2 = 4.4 s−1). Interestingly, the sum of the fast and slow reaction rate constants (A1k1 + A2k2 = 30 s−1) for 21-mer/26-mer is close to the kp value (39 s−1, Supplementary Table 1) determined by using the single-turnover kinetic assay. For dGTP incorporation onto 21-mer/26-mer-dGAP, both A1 and k1 were larger than corresponding A2 and k2 (Supplementary Table 5). The sum of reaction rate constants for 21-mer/26-mer-dGAP (A1k1 + A2k2 = 1.5 s−1) is also approximately equal to the kp value in Table 4 (2.2 s−1). Similar relationship has been previously observed with other lesions and Dpo4.26, 29, 42-45 This is not surprising because both kp and the sum (A1k1 + A2k2) reflect the rate constant of nucleotide incorporation during single binding event between an enzyme and DNA. Furthermore, all four biphasic kinetic parameters with 21-mer/26-mer-dGAP are smaller than the counterparts with 21-mer/26-mer, suggesting that the presence of dGAP negatively affected the biphasic kinetics of nucleotide incorporation in the presence of a DNA trap.
Figure 3.
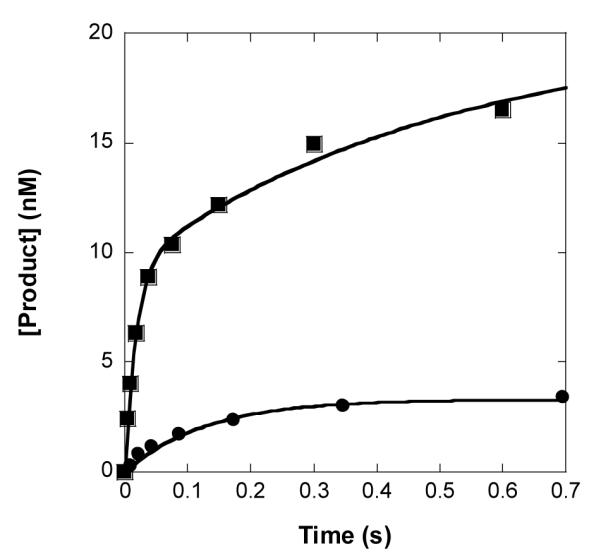
Biphasic kinetics of correct dGTP incorporation onto 21-mer/26-mers catalyzed by hPolη in the presence of a DNA trap. A preincubated solution of hPolη (130 nM) and 5′-[32P]-labeled 21-mer/26-mer (20 nM, ■) or 21-mer/26-mer-dGAP (20 nM, •) was mixed with the solution of dGTP (0.8 mM, ■; 1.2 mM, •) and a DNA trap D-1 21/41-mer (5 μM, Table 1) for various time intervals. The plot of the product concentrations against reaction times was fit to Equation 7. For 21-mer/26-mer, the fast phase had a reaction amplitude of 7.8 ± 0.3 nM and a reaction rate constant of 72 ± 5 s−1 while the slow phase had a reaction amplitude of 9.5 ± 0.3 nM and a reaction rate constant of 4.4 ± 0.5 s−1. For 21-mer/26-mer-dGAP, the fast phase had a reaction amplitude of 2.9 ± 0.1 nM and a reaction rate constant of 11 ± 1 s−1 while the slow phase had a reaction amplitude of 1.1 ± 0.2 nM and a reaction rate constant of 0.20 ± 0.02 s−1.
DISCUSSION
In this paper, we have compared the abilities of four human Y-family DNA polymerases to catalyze TLS of dGAP in the same reaction conditions. All of them except hRev1 bypassed a single site-specifically placed lesion dGAP and synthesized full-length products with different efficiency (Figure 1, Table 2). There was more accumulation of the intermediate product 21-mer than 20-mer synthesized by hPolκ (Figure 1D) and hPolτ (Figure 1F). This suggests that for these enzymes, the dGAP bypass step was less problematic than the subsequent extension step. Similar strong polymerase pausing immediately downstream from a bulky DNA lesion site has also been observed for the bypass of dGAP 26 and AAF-dG46 catalyzed by Dpo4 and for the bypass of BPDE-dG47 and AAF-dG catalyzed by yeast Polη.38 To understand the polymerization patterns in Figure 1, it is necessary to establish a kinetic basis for how each human Y-family DNA polymerase catalyzes TLS of dGAP.
Comparison of efficiency of human Y-family DNA polymerases with undamaged DNA
For efficient DNA synthesis, a DNA polymerase must bind to DNA tightly and incorporate nucleotides with high substrate specificity (kp/Kd, dNTP). Since the Kd, DNA values do not change significantly within the temperature range of 23 to 37 °C, we ranked the DNA-binding affinity order of human Y-family enzymes as hPolη > hPolτ > hPolκ ≈ hRev1 based on the Kd, DNA values (Table 3) and the affinity of undamaged DNA to hPolκ (96 ± 21 nM) measured by fluorescence anisotropy at 25 °C.41 Notably, the Kd, DNA values of human Y-family DNA polymerases were within 5-fold and are not the major factor to differentiate their relative polymerization efficiency. In contrast, the kp/Kd, dNTP values for correct nucleotide incorporation onto undamaged DNA by these enzymes varied by several orders of magnitude (Supplementary Tables 1-4) and likely governed the polymerization efficiency order. For example, the order of kp/Kd, dNTP values of dCTP incorporation opposite dG in 20-mer/26-mer was as followed: hPolη (0.56 μM−1s−1) > hRev1 (0.14 μM−1s−1) > hPolκ (0.037 μM−1s−1) > hPolτ (0.0016 μM−1s−1). Similarly, the kp/Kd, dGTP values for correct dGTP incorporation onto 21-mer/26-mer were ranked as hPolη (0.84 μM− 1s−1) > hPolκ (0.011 μM−1s−1) > hPolτ (0.0016 μM−1s−1). Although we did not measure the kp/Kd, dNTP value for dGTP incorporation onto 21-mer/26-mer by hRev1, our previous work has shown that this deoxycytidyl transferase preferentially incorporates dCTP, rather than a correct incoming nucleotide, opposite dC, dA, or dT with 103-104 fold lower kp/Kd, dNTP than incorporating dCTP opposite dG.18 Accordingly, the kp/Kd, dNTP values for dGTP incorporation onto 21-mer/26-mer follow the order of hPolη > hPolκ > hPolτ > hRev1. Although we did not determine the kp/Kd, dNTP values for correct dNTP incorporations at other positions along template 26-mer, the kp/Kd, dNTP order was expected to follow hPolη > hRev1 > hPolκ > hPolτ at each of the two template dG positions and hPolη > hPolκ > hPolτ > hRev1 at each of the other five template positions. Taken together, the overall efficiency for the elongation of primer 17-mer to 26-mer along the template 26-mer is expected to follow the order of hPolη > hPolκ > hPolτ >> hRev1. Interestingly, this prediction was consistent with the speed order of elongating 17-mer/26-mer to full-length products by human Y-family enzymes: hPolη (1 s) > hPolκ (6 s) > hPolτ (60 s) >> hRev1 (> 12 hrs) (Figure 1).
Comparison of the TLS of dGAP efficiency catalyzed by human Y-family DNA polymerases
When human Y-family enzymes elongated 17-mer along 26-mer-dGAP (Figure 1) the most troubled steps were the dGAP bypass step and the subsequent extension step. Opposite dGAP, dCTP was incorporated onto 20-mer/26-mer-dGAP with the following efficiency order of hPolη (2.8×10−2 μM−1s−1) > hRev1 = hPolκ (6.5×10−3 μM−1s−1) > hPolτ (2.3×10−4 μM−1s−1) (Tables 4-6), which is consistent with the t50bypass order of hPolη (2.5 s) < hPolκ (4.1 s) < hPolτ (107 s) (Table 2). During the subsequent extension step, dGTP was incorporated opposite dC in 21-mer/26-mer-dGAP with the kp/Kd, dNTP order of hPolη (1.8×10−2 μM−1s−1 > hPolκ (5.3×10−5 μM−1s−1) > hPolτ (2.2×10−7 μM−1s−1). Since hRev1 did not elongate 21-mer/26-mer-dGAP after several hours, its kp/Kd, dNTP value should be much lower than that of hPolτ. Thus, the order of nucleotide incorporation efficiency during the extension step is hPolη > hPolκ > hPolτ >> hRev1. On the basis of these kp/Kd, dNTP values and the small impact of dGAP on the Kd, DNA values (Results), we predicted that the overall efficiency of TLS of dGAP and possibly the elongation of 17-mer/26-mer-dGAP to full-length products follow the order of hPolη > hPolκ > hPolτ >> hRev1. Consistently, this order matched the speed order for creating full-length product 26-mer along template 26-mer-dGAP (Figure 1). Thus, hPolη is the most efficient human Y-family DNA polymerase to catalyze TLS of dGAP. Interestingly, the same conclusion has been reached by us for the TLS of an abasic site.11
A kinetic basis for polymerase pausing during TLS of dGAP
In the presence of dGAP, the accumulation of the intermediate products 20-mer and 21-mer was more obvious with hPolκ (Figure 1D) and hPolτ (Figure 1F) than with hPolη (Figure 1B). These data indicated that these former enzymes had different extents of difficulty in either binding damaged DNA or incorporating nucleotides opposite both dGAP and the downstream template base dC on template 26-mer-dGAP. Table 3 displays the small impact of dGAP on the Kd, DNA values of human Y-family DNA polymerases. In contrast, the kp/Kd, dNTP values were drastically affected by the presence of dGAP (Tables 4-6). For example, hPolη, hPolκ, and hPolτ were 20-, 6-, and 208-fold, respectively, less efficient when incorporating dCTP opposite dGAP than opposite dG in undamaged DNA. During the subsequent extension step, the embedded dGAP affected the kp/Kd, dNTP values of hPolη, hPolκ, and hPolτ by 48-, 208-, and 4,565-fold, respectively. Thus, the large decrease in correct nucleotide incorporation efficiency caused by dGAP was the main reason why these human Y-family enzymes paused during TLS of dGAP (Figure 1). Specifically, the decreases in kp/Kd, dNTP values were contributed by 2-4 fold higher Kd, dNTP values and up to 1,000-fold lower kp values. In addition, the larger negative effect of dGAP on the kp/Kd, dNTP values of hPolκ and hPolτ than on those of hPolη explained why both hPolκ and hPolτ paused more strongly than hPolη at the 20th and 21st template positions (Figure 1). The 3-fold higher binding affinity of hPolη to damaged DNA than to undamaged DNA (Table 3) and relatively high kp values (2 to 8 s−1) for correct nucleotide incorporations opposite both dGAP and downstream dC (Table 4) further lessened hPolη pausing at these sites.
Clearly, the intermediate product 21-mer accumulated more than 20-mer with both hPolκ (Figure 1D) and hPolτ (Figure 1F). This observation suggested that the subsequent extension step was affected by dGAP more than the DNA lesion bypass step with these enzymes. Consistently, the kp/Kd, dNTP value for correct nucleotide incorporation was 123-fold lower during the extension step than during the lesion bypass step with hPolκ (Table 5) while the difference was 1,045-fold lower with hPolτ (Table 6). Additionally, these efficiency differences were mainly contributed by ~1,000-fold decrease in kp. Thus, the conversion from 21-mer to 22-mer was less efficient and much slower than the conversion from 20-mer to 21-mer, leading to the stronger accumulation of 21-mer in Figures 1D and 1F.
Biphasic kinetics of nucleotide incorporation with hPolη
Surprisingly, we observed a fast phase (A1 = 39%, k1 = 72.2 s−1) and a slow phase (A2 = 47%, k2 = 4.4 s−1) during correct nucleotide incorporation onto undamaged 21-mer/26-mer catalyzed by hPolη in the presence of a DNA trap (Figure 3). In contrast, only the fast phase has been observed during nucleotide incorporation onto the same undamaged substrate catalyzed by Dpo4.26 At present, it is unclear what contributed to the slow phase observed with hPolη. Mechanistically, it is possible that 39% of the binary complex hPolη•DNA were in a productive conformation (E•DNAnP) while 47% of the complex were in a slightly non-productive manner (E•DNAnN). The latter was isomerized to become the former at a rate of 4.4 s−1. Since the total reaction amplitude (A1 + A2) of 86% is close to the calculated percentage of the binary complex (83%) based on the Kd, DNA value (Table 3), the remaining 14% of 21-mer/26-mer must not be bound by hPolη and were free in solution. For the unprecedented biphasic kinetics of nucleotide incorporation onto normal DNA, we proposed a mechanism shown in Figure 4A.
Figure 4.

Proposed kinetic mechanisms for nucleotide incorporation opposite undamaged dG (A) and damaged dGAP (B) catalyzed by hPolη. A1, fast phase amplitude; k1, fast phase reaction rate constant; A2, slow phase amplitude; Do, the initial concentration of DNA; k2, slow phase reaction rate constant; E, DNA polymerase; DNAn, DNA substrate; E•DNADn, dead-end binary complex; E•DNANn, nonproductive binary complex; E•DNAPn, productive binary complex; E•DNANn•dNTP, nonproductive ternary complex; E•DNAPn•dNTP, productive ternary complex; DNAn+1, extended DNA product by a base; PPi, pyrophosphate.
Like Dpo4,26 biphasic kinetics of correct dNTP incorporation onto 21-mer/26-mer-dGAP was observed with hPolη (Figure 3). In comparison to what was observed with 21-mer/26-mer, the total reaction amplitude of the fast (14%) and slow (5.3%) phases with 21-mer/26-mer-dGAP was only 19.3%, much less than the calculated reaction amplitude (93%) based on the Kd, DNA value in Table 3. Besides the decrease in reaction amplitudes in both phases, the values of k1 and k2 were also reduced by the presence of the bulky 1-AP adduct (Supplementary Table 5). Based on the above calculation, 7% of 21-mer/26-mer-dGAP was free in solution and 73.7% were in a dead-end conformation (E•DNAnD) which was not elongated within the observation time window in Figure 3. The physical nature of E•DNAnD is unclear at present. It is likely that hPolη bound to DNA in a catalytically incompetent conformation. It is also possible that some hPolη molecules bound to the blunt-end of 21-mer/26-mer-dGAP, rather than the staggered end, as suggested by the formation of 27-mer through a blunt-end addition (Figure 1B). On the basis of the above information, we proposed a kinetic mechanism for TLS of dGAP shown in Figure 4B, which has been previously applied to other DNA lesions and polymerases.26, 29, 42, 44 There are a few indirect structural evidences to support this mechanism. Previously, the NMR structure of a damaged 11-mer duplex reveals that the hydrophobic 1-AP adduct of an embedded dGAP:dC base pair is intercalated onto the DNA helix between adjacent Watson-Crick base pairs with the sugar of the modified dG possessing a syn glycosidic torsion angle, and the bases of the dGAP:dC base pair are displaced onto the major groove.48 If this conformation of dGAP is preserved within the active site of hPolη, the bulky 1-AP adduct would occupy the position of an incoming dNTP. Such a binary complex would not be elongated without undergoing dramatic structural changes and likely represents E•DNAnD. Consistently, a crystal structure of the binary complex of yeast Polη and DNA containing an AAF-dG38 shows that the AAF adduct stacks above the last primer-template base pair as proposed for E•DNAnD. The other crystal structure of the same binary complex illustrates that the enzyme rotates along DNA and allows the DNA adduct to partially block the active site. If 1-AP is in the quasi-intercalative conformation as AAF in the corresponding binary crystal structure, the binary complex hPolη•20-mer/26-mer-dGAP requires subtle to mild structural changes before elongation and thereby exists in the conformation of E•DNAnN. When 1-AP is completely rotated out of the DNA duplex, hPolη•21-mer/26-mer-dGAP will be in the conformation of E•DNAnP and be rapidly elongated after nucleotide binding.
Impact of dGAP on polymerase fidelity
The fidelity of nucleotide incorporation opposite dGAP (Tables 4-7) ranked as hRev1 (10−4) > hPolτ (10−3 to 10−2) > hPolη (10−2 to 10−1) = hPolκ (10−2 to 10−1). The calculated fidelity ratios implied that both hPolη and hPolκ were about 10-fold more error-prone when opposite dGAP than opposite undamaged dG. Interestingly, the bulky 1-AP adduct enhanced the fidelity of hRev1 significantly and hPolτ slightly during dGAP bypass. Such an impact of the 1-AP adduct on polymerase fidelity was more negatively significant during the subsequent extension step. Opposite 5′-dC from dGAP, hPolη incorporated incorrect dCTP with 3-fold higher efficiency than correct dGTP, hPolκ incorporated incorrect dATP with only 2-fold lower efficiency than correct dGTP, and hPolτ incorporated both correct dGTP and incorrect dTTP with almost equal kp/Kd, dNTP values, leading to lowered polymerase fidelity at this position (Tables 4-6). Taken together, our kinetic studies suggested that hPolη, hPolκ and hPolτ became more error-prone during TLS of dGAP than during normal DNA synthesis.
The pathways for the formation of DNA lesions in vivo and whether the DNA lesions stall cellular DNA replication machinery have not been thoroughly explored.49 So far, it is known that genetic markers, e.g. Umu gene, for mutagenesis activity in several cell lines are elevated in the presence of 1-NP or certain metabolites.21-25 These results suggest that the Y-family DNA polymerases are active in the presence of lesion dGAP. If so, the mutagenic bypass of dGAP catalyzed by these enzymes may lead to genetic mutations in vivo.25, 50-54
In summary, our kinetic data demonstrated that hPolη, hPolκ, and hPolτ were able to bypass dGAP and extend the lesion bypass product with dramatically different efficiency and fidelity values. In contrast, hRev1 was only able to bypass the bulky DNA lesion. A kinetic basis for polymerase pausing during error-prone TLS of dGAP was established.
Supplementary Material
ACKNOWLEDGMENTS
We sincerely thank David Beyer, Jessica Chadwick and Rebecca Frankel for their assistance in performing some of the single-turnover kinetic assays with hRev1 and hPolκ. We also thank Jason Fowler and Sean Newmister for purifying hRev1 and hPolκ.
FUNDING SUPPORT
This work was supported by National Science Foundation Career Award (MCB-0447899) to Z.S., National Science Foundation Grant (MCB-0960961) to Z.S., and National Institutes of Health Grant (ES009127) to A.K.B. and Z.S. S.M.S was supported by an American Heart Association Predoctoral Fellowship (Grant GRT00014861).
Footnotes
Abbreviations: AAF-dG, N-acetyl-2-aminofluorene adduct at the C8 position of deoxyguanosine; 1-AP, 1-aminopyrene; BPDE-dG, benzocpyrene 7,8-diol 9,10-epoxide-derived adduct at the N2 position of 2′-deoxyguanosine; BSA, bovine serum albumin; Dpo4, Sulfolobus solfataricus DNA polymerase IV; dGAP, N-(deoxyguanosin-8-yl)-1-aminopyrene; dNTP, 2′-deoxynucleoside 5′-triphosphate; TLS, translesion DNA synthesis.
SUPPORTING INFORMATION AVAILABLE The structure of dGAP, dGAP bypass% plot, the pre-steady state kinetic parameters of each enzyme, a gel image of EMSA and corresponding plot, and the gel image displaying the efficiency of the DNA trap assay are available as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Duvauchelle JB, Blanco L, Fuchs RP, Cordonnier AM. Human DNA polymerase mu (Pol mu) exhibits an unusual replication slippage ability at AAF lesion. Nucleic Acids Res. 2002;30:2061–2067. doi: 10.1093/nar/30.9.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kunkel TA. DNA replication fidelity. J. Biol. Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- (3).Freisinger E, Grollman AP, Miller H, Kisker C. Lesion (in)tolerance reveals insights into DNA replication fidelity. Embo J. 2004;23:1494–1505. doi: 10.1038/sj.emboj.7600158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gruz P, Shimizu M, Pisani FM, De Felice M, Kanke Y, Nohmi T. Processing of DNA lesions by archaeal DNA polymerases from Sulfolobus solfataricus. Nucleic Acids Res. 2003;31:4024–4030. doi: 10.1093/nar/gkg447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Boudsocq F, Ling H, Yang W, Woodgate R. Structure-based interpretation of missense mutations in Y-family DNA polymerases and their implications for polymerase function and lesion bypass. DNA Repair (Amst) 2002;1:343–358. doi: 10.1016/s1568-7864(02)00019-8. [DOI] [PubMed] [Google Scholar]
- (6).Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- (7).Lehmann AR. Replication of damaged DNA in mammalian cells: new solutions to an old problem. Mutat. Res. 2002;509:23–34. doi: 10.1016/s0027-5107(02)00227-0. [DOI] [PubMed] [Google Scholar]
- (8).Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- (9).Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999;18:3491–3501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- (11).Sherrer SM, Fiala KA, Fowler JD, Newmister SA, Pryor JM, Suo Z. Quantitative analysis of the efficiency and mutagenic spectra of abasic lesion bypass catalyzed by human Y-family DNA polymerases. Nucleic Acids Res. 2011;39:609–622. doi: 10.1093/nar/gkq719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang Y, Yuan F, Wu X, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z. Error-prone lesion bypass by human DNA polymerase eta. Nucleic Acids Res. 2000;28:4717–4724. doi: 10.1093/nar/28.23.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Masutani C, Kusumoto R, Iwai S, Hanaoka F. Mechanisms of accurate translesion synthesis by human polymerase eta. Embo J. 2000;19:3100–3109. doi: 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhang Y, Yuan F, Xin H, Wu X, Rajpal DK, Yang D, Wang Z. Human DNA polymerase kappa synthesizes DNA with extraordinarily low fidelity. Nucleic Acids Res. 2000;28:4147–4156. doi: 10.1093/nar/28.21.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Washington MT, Johnson RE, Prakash L, Prakash S. Human DINB1-encoded DNA polymerase kappa is a promiscuous extender of mispaired primer termini. Proc. Natl. Acad. Sci. U. S. A. 2002;99:1910–1914. doi: 10.1073/pnas.032594399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Haracska L, Johnson RE, Unk I, Phillips BB, Hurwitz J, Prakash L, Prakash S. Targeting of human DNA polymerase iota to the replication machinery via interaction with PCNA. Proc. Natl. Acad. Sci. U. S. A. 2001;98:14256–14261. doi: 10.1073/pnas.261560798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang Y, Yuan F, Wu X, Taylor JS, Wang Z. Response of human DNA polymerase iota to DNA lesions. Nucleic Acids Res. 2001;29:928–935. doi: 10.1093/nar/29.4.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Brown JA, Fowler JD, Suo Z. Kinetic basis of nucleotide selection employed by a protein template-dependent DNA polymerase. Biochemistry. 2010;49:5504–5510. doi: 10.1021/bi100433x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang Y, Wu X, Rechkoblit O, Geacintov NE, Taylor JS, Wang Z. Response of human REV1 to different DNA damage: preferential dCMP insertion opposite the lesion. Nucleic Acids Res. 2002;30:1630–1638. doi: 10.1093/nar/30.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Mitchelmore CL, Livingstone DR, Chipman JK. Conversion of 1-nitropyrene by Brown trout (Salmo trutta) and turbot (Scophtalamus maximus) to DNA adducts detected by 32P-postlabelling. Biomarkers. 1998;3:21–33. doi: 10.1080/135475098231345. [DOI] [PubMed] [Google Scholar]
- (21).Pohjola SK, Lappi M, Honkanen M, Rantanen L, Savela K. DNA binding of polycyclic aromatic hydrocarbons in a human bronchial epithelial cell line treated with diesel and gasoline particulate extracts and benzo[a]pyrene. Mutagenesis. 2003;18:429–438. doi: 10.1093/mutage/geg021. [DOI] [PubMed] [Google Scholar]
- (22).Pohjola SK, Savela K, Kuusimaki L, Kanno T, Kawanishi M, Weyand E. Polycyclic aromatic hydrocarbons of diesel and gasoline exhaust and DNA adduct detection in calf thymus DNA and lymphocyte DNA of workers exposed to diesel exhaust. Polycyclic Aromatic Compounds. 2004;24:451–465. [Google Scholar]
- (23).Hirose M, Lee MS, Wang CY, King CM. Induction of rat mammary gland tumors by 1-nitropyrene, a recently recognized environmental mutagen. Cancer Res. 1984;44:1158–1162. [PubMed] [Google Scholar]
- (24).Sabbioni G, Jones CR. Biomonitoring of arylamines and nitroarenes. Biomarkers. 2002;7:347–421. doi: 10.1080/13547500210147253. [DOI] [PubMed] [Google Scholar]
- (25).Malia SA, Vyas RR, Basu AK. Site-specific frame-shift mutagenesis by the 1-nitropyrene-DNA adduct N-(deoxyguanosin-8-y1)-1-aminopyrene located in the (CG)3 sequence: effects of SOS, proofreading, and mismatch repair. Biochemistry. 1996;35:4568–4577. doi: 10.1021/bi9525132. [DOI] [PubMed] [Google Scholar]
- (26).Sherrer SM, Brown JA, Pack LR, Jasti VP, Fowler JD, Basu AK, Suo Z. Mechanistic studies of the bypass of a bulky single-base lesion catalyzed by a Y-family DNA polymerase. J. Biol. Chem. 2009;284:6379–6388. doi: 10.1074/jbc.M808161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang L, Brown JA, Newmister SA, Suo Z. Polymerization fidelity of a replicative DNA polymerase from the hyperthermophilic archaeon Sulfolobus solfataricus P2. Biochemistry. 2009;48:7492–7501. doi: 10.1021/bi900532w. [DOI] [PubMed] [Google Scholar]
- (28).Fiala KA, Suo Z. Sloppy bypass of an abasic lesion catalyzed by a Y-family DNA polymerase. J. Biol. Chem. 2007;282:8199–8206. doi: 10.1074/jbc.M610719200. [DOI] [PubMed] [Google Scholar]
- (29).Brown JA, Newmister SA, Fiala KA, Suo Z. Mechanism of double-base lesion bypass catalyzed by a Y-family DNA polymerase. Nucleic Acids Res. 2008;36:3867–3878. doi: 10.1093/nar/gkn309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Brown JA, Pack LR, Fowler JD, Suo Z. Pre-steady-state kinetic analysis of the incorporation of anti-HIV nucleotide analogs catalyzed by human X- and Y-family DNA polymerases. Antimicrob. Agents Chemother. 2011;55:276–283. doi: 10.1128/AAC.01229-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Fiala KA, Suo Z. Pre-Steady-State Kinetic Studies of the Fidelity of Sulfolobus solfataricus P2 DNA Polymerase IV. Biochemistry. 2004;43:2106–2115. doi: 10.1021/bi0357457. [DOI] [PubMed] [Google Scholar]
- (32).Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Choi JY, Chowdhury G, Zang H, Angel KC, Vu CC, Peterson LA, Guengerich FP. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 2006;281:38244–38256. doi: 10.1074/jbc.M608369200. [DOI] [PubMed] [Google Scholar]
- (34).Fiala KA, Brown JA, Ling H, Kshetry AK, Zhang J, Taylor JS, Yang W, Suo Z. Mechanism of template-independent nucleotide incorporation catalyzed by a template-dependent DNA polymerase. J. Mol. Biol. 2007;365:590–602. doi: 10.1016/j.jmb.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).McCulloch SD, Kokoska RJ, Masutani C, Iwai S, Hanaoka F, Kunkel TA. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004;428:97–100. doi: 10.1038/nature02352. [DOI] [PubMed] [Google Scholar]
- (36).Johnson RE, Washington MT, Prakash S, Prakash L. Fidelity of human DNA polymerase eta. J. Biol. Chem. 2000;275:7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- (37).Bauer J, Xing G, Yagi H, Sayer JM, Jerina DM, Ling H. A structural gap in Dpo4 supports mutagenic bypass of a major benzo[a]pyrene dG adduct in DNA through template misalignment. Proc. Natl. Acad. Sci. U. S. A. 2007;104:14905–14910. doi: 10.1073/pnas.0700717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Schorr S, Schneider S, Lammens K, Hopfner KP, Carell T. Mechanism of replication blocking and bypass of Y-family polymerase {eta} by bulky acetylaminofluorene DNA adducts. Proc. Natl. Acad. Sci. U. S. A. 2010;107:20720–20725. doi: 10.1073/pnas.1008894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Washington MT, Johnson RE, Prakash L, Prakash S. Human DNA polymerase iota utilizes different nucleotide incorporation mechanisms dependent upon the template base. Mol. Cell Biol. 2004;24:936–943. doi: 10.1128/MCB.24.2.936-943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Carlson KD, Johnson RE, Prakash L, Prakash S, Washington MT. Human DNA polymerase kappa forms nonproductive complexes with matched primer termini but not with mismatched primer termini. Proc. Natl. Acad. Sci. U. S. A. 2006;103:15776–15781. doi: 10.1073/pnas.0605785103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lone S, Townson SA, Uljon SN, Johnson RE, Brahma A, Nair DT, Prakash S, Prakash L, Aggarwal AK. Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell. 2007;25:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- (42).Fiala KA, Hypes CD, Suo Z. Mechanism of abasic lesion bypass catalyzed by a Y-family DNA polymerase. J. Biol. Chem. 2007;282:8188–8198. doi: 10.1074/jbc.M610718200. [DOI] [PubMed] [Google Scholar]
- (43).Suo Z, Johnson KA. Effect of RNA secondary structure on the kinetics of DNA synthesis catalyzed by HIV-1 reverse transcriptase. Biochemistry. 1997;36:12459–12467. doi: 10.1021/bi971217h. [DOI] [PubMed] [Google Scholar]
- (44).Suo Z, Lippard SJ, Johnson KA. Single d(GpG)/cis-diammineplatinum(II) adduct-induced inhibition of DNA polymerization. Biochemistry. 1999;38:715–726. doi: 10.1021/bi981854n. [DOI] [PubMed] [Google Scholar]
- (45).Suo Z, Johnson KA. DNA secondary structure effects on DNA synthesis catalyzed by HIV-1 reverse transcriptase. J. Biol. Chem. 1998;273:27259–27267. doi: 10.1074/jbc.273.42.27259. [DOI] [PubMed] [Google Scholar]
- (46).Boudsocq F, Iwai S, Hanaoka F, Woodgate R. Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4): an archaeal DinB-like DNA polymerase with lesion-bypass properties akin to eukaryotic poleta. Nucleic Acids Res. 2001;29:4607–4616. doi: 10.1093/nar/29.22.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Choi JY, Guengerich FP. Adduct size limits efficient and error-free bypass across bulky N2-guanine DNA lesions by human DNA polymerase eta. J. Mol. Biol. 2005;352:72–90. doi: 10.1016/j.jmb.2005.06.079. [DOI] [PubMed] [Google Scholar]
- (48).Gu Z, Gorin A, Krishnasamy R, Hingerty BE, Basu AK, Broyde S, Patel DJ. Solution structure of the N-(deoxyguanosin-8-yl)-1-aminopyrene ([AP]dG) adduct opposite dA in a DNA duplex. Biochemistry. 1999;38:10843–10854. doi: 10.1021/bi9912138. [DOI] [PubMed] [Google Scholar]
- (49).Silvers KJ, Eddy EP, McCoy EC, Rosenkranz HS, Howard PC. Pathways for the mutagenesis of 1-nitropyrene and dinitropyrenes in the human hepatoma cell line HepG2. Environ. Health Perspect. 1994;102(Suppl 6):195–200. doi: 10.1289/ehp.94102s6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Watt DL, Utzat CD, Hilario P, Basu AK. Mutagenicity of the 1-nitropyrene-DNA adduct N-(deoxyguanosin-8-yl)-1-aminopyrene in mammalian cells. Chem. Res. Toxicol. 2007;20:1658–1664. doi: 10.1021/tx700131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Bacolod MD, Basu AK. Mutagenicity of a single 1-nitropyrene-DNA adduct N-(deoxyguanosin-8-yl)-1-aminopyrene in Escherichia coli located in a GGC sequence. Mutagenesis. 2001;16:461–465. doi: 10.1093/mutage/16.6.461. [DOI] [PubMed] [Google Scholar]
- (52).Bacolod MD, Krishnasamy R, Basu AK. Mutagenicity of the 1-nitropyrene-DNA adduct N-(deoxyguanosin-8-yl)-1-aminopyrene in Escherichia coli located in a nonrepetitive CGC sequence. Chem. Res. Toxicol. 2000;13:523–528. doi: 10.1021/tx000023r. [DOI] [PubMed] [Google Scholar]
- (53).Hilario P, Yan S, Hingerty BE, Broyde S, Basu AK. Comparative mutagenesis of the C8-guanine adducts of 1-nitropyrene and 1,6- and 1,8-dinitropyrene in a CpG repeat sequence. A slipped frameshift intermediate model for dinucleotide deletion. J. Biol. Chem. 2002;277:45068–45074. doi: 10.1074/jbc.M208103200. [DOI] [PubMed] [Google Scholar]
- (54).Malia SA, Basu AK. Mutagenic specificity of reductively activated 1-nitropyrene in Escherichia coli. Biochemistry. 1995;34:96–104. doi: 10.1021/bi00001a012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.