

Abstract
We previously identified a series of methylsulfonylnitrobenzoates (MSNB's) that block the interaction of the thyroid hormone receptor with its coactivators. MSNB's inhibits coactivator binding through irreversibly modifying cysteine 298 of thyroid hormone receptor (TR). Although MSNB's have better pharmacological features than our first generation inhibitors (β-aminoketones) they contain a potentially unstable ester linkage. Here we report the bioisosteric replacement of the ester linkage with a thiazole moiety, yielding sulfonylnitrophenylthiazoles (SNPT's). An array of SNPT's representing optimal side chains from the MSNB series was constructed using parallel chemistry and evaluated to test their antagonism of the TR-coactivator interaction. Selected active compounds were evaluated in secondary confirmatory assays including regulation of thyroid response element driven transcription in reporter constructs and native genes. In addition the selected SNPT's shown to be selective for TR relative to other nuclear hormone receptor (NR).
Introduction
The nuclear hormone receptors (NR) are transcription factors that are therapeutic targets for metabolic disease, immunology, reproductive health, and cancer.1-3 The NR superfamily includes the thyroid hormone receptors (TR), TRα and TRβ, that regulate development, growth, and metabolism.4, 5 Although the TR isoforms are widely expressed, they follow tissue specific patterns that vary with developmental stage.6, 7 The TR isoforms have distinct regulatory roles.8, 9
Thyroid hormone (T3) regulates transcriptional responses mediated by TR,9 which contains an amino terminal transcription activation domain (AF-1), a central DNA binding domain (DBD), and a carboxyl terminal ligand binding domain (LBD) that contains a T3-inducible coactivator binding domain, AF-2.10 TR usually functions as a heterodimer with the retinoid X receptor (RXR). At low levels of T3, TR binds corepressors using the AF-2 domain and suppresses basal transcription at thyroid)responsive elements (TREs). In response to increasing concentrations of T3, TR undergoes a conformational change, releasing corepressor proteins and binding coactivator proteins, thus activating gene transcription.11, 12 The dominant family of coactivators is the SRC's, which include SRC1 (NcoA1), SRC2 (GRIP1/TIF2), and SRC3 (AIB1/TRAM1/RAC3/ACTR).13 The SRC's include both nuclear receptor interaction (NID) and activation domains. The SRC's NID includes a variable number of a conserved NR box motif, containing the LXXLL sequence, that binds to the TR's AF-2 domain.14, 15 This interaction is mediated by a small, well defined binding pocket16 that makes the AF-2 domain an ideal target for developing inhibitors of TR-SRC interactions. Although a number of small molecule modulators of TR have been developed recently, including agonists such as GC-1,17-19 TRIAC,20 KB-141,21, 22and antagonists such as NH-3,23-25 most target the ligand binding pocket in the LBD.
We have previously reported a series β-aminoketones that disrupt the TR-coactivator interaction without affecting T3 binding.26-28 Unfortunately these compounds suffered from multiple liabilities in vivo thus requiring development of a new scaffold. The second generation TRβ-SRC2 inhibitors, methylsulfonylnitrobenzoates (1, MSNB's), were identified in a quantitative high throughput screen (qHTS).29 Both the β-aminoketones and MSNB's have a similar inhibition mechanism, irreversibly modifying Cys298 within the AF-2 domain of TR.30 However, the MSNB's have two major advantages for the development of TR-coactivator inhibitors for use in vivo. First, MSNB members are predicted to lack the cardiac activity exhibited by the β-aminoketones because they lack the basic tertiary amines that lead to ion channel binding. Second, the MSNB's are more stable than the β-aminoketones at physiological pH. The MSNB's have two distinct structural features: the methylsulfonyl group that acts as a leaving group and ester-linked acetamide group that appears to target the compound to the AF-2 domain.
Carboxylic esters are often metabolically unstable in vivo due to facile hydrolysis by esterases in multiple compartments and intrinsic chemical instability in the stomach. A common strategy to replace esters is to use heterocyclic bioisosteres with increased stability to degradation.31, 32 A structural analysis indicated that thiazole-linked MSNB's, called sulfonylnitrophenylthiazoles (SNPT), gave good alignments between the requisite aromatic and side chain groups of the MSNB's (Figure 1). For this reason, we modified the MSNB structure to produce SNPT's. Here we report an efficient method of parallel synthesis of SNPT's and their evaluation as thyroid hormone receptor-coactivator inhibitors.
Figure 1.

(A) Structural modification of MSNB's leading to SNPT's. (B) The translucent shape is the van der Waals surface of MSNB's and SNPT's. The colors of translucent represent electrostatics of both molecules; red (negative), blue (positive), and white (neutral). Overall there is good alignment between the MSNB's and the SNPT's thus indicating their theoretical viability as more stable bioisosteres.
Results and Discussion
Chemistry
Reagents and conditions
(a) H2O2, K2CO3, DMSO, 60 °C, 0.5 h; (b) Lawesson's reagent, 1,4-dioxane, 110 °, 2 h; (c) 2-chloro-2-ketoacetate 7, EtOH, reflux, 24-36 h; (d) NaSMe or RSH/K2CO3, THF, 50 °C, 18 h; (e) m-CPBA, DCM, 24-36 h, rt; (f) LiOH, H2O/THF, rt, 3-5 h; (g) amines, PyBOP, DIEA, DMF, rt, 24 h; (h) bromo-oxobutanoate 12, EtOH, reflux, 24 h.
Chemistry
The bioisostere hypothesis was tested by constructing a compound array containing all of the variations of the warhead and side chains that were reasonably potent in the MSNB background, with replacement of the ester by one of two thiazole linkages. This allowed any synergistic interactions affecting potency to adjust to the new core. The SNPT array was constructed using a parallel chemistry method with three diversification steps (Figure 2). First, six α-haloketones were employed to selectively give either the 4- or 5-carboxamide's (x variation). Second, three thiols were used to provide sulfonyl group diversity (y variation). Finally, 24 amines were employed as the third building block (z variation). The amine series were chosen to systematically vary the size, electrostatics, and hydrophobicity at this position. All of three sets of building blocks selected for this compound array are shown in Figure 3.
Figure 2.

Plan for the construction of SNPT compound array.
Figure 3.

Building Blocks (R1-R3/R4).
The synthetic route to the SNPT's is depicted in Scheme 1. Commercially available chloronitrobenzonitrile 4 was converted to benzamide 5 through oxidation with hydrogen peroxide. Benzamide 5 was then converted to thiobenzamide 6 with high yield by treatment with Lawesson's reagent. Intermediate 6 was treated with various 2-chloro-oxoacetates 7 and 3-bromo-oxoacetate 12 to give the arrays of 5-carboxyesters 8 and 4-carboxyesters 13{x}, respectively. Next, compound arrays 8 and 13 were reacted with three thiolates (sodium methanethiolate, n-butanethiol/K2CO3, and benzylthiol/K2CO3), yielding sulfide compound arrays 9 {x,y} and 14{x,y}, respectively. Oxidation with m-CPBA gave the sulfonyl compound sets 10 and 15, followed by hydrolysis to give 5-carboxylic acid SNPT array 11{x,y} and 4-carboxylic acid SNPT array 16{x,y}, respectively. This chemistry performed cleanly enough to allow going from starting material 4 to the penultimate step without column purification, with all intermediates being purified by crystallization. In the last step carboxylic acids arrays 11 and 16 were treated with 24 amines, PyBOP, and DIEA at room temperature to give the final SNPT arrays (2{x,y,z} and 3{x,y,z}). The final products were purified using flash chromatography followed by automated preparative HPLC. The identity of all compounds was established using NMR and MS. The purity was confirmed by LC/MS/UV/ELSD/CLND. The yield and purity of the SNPT's is available in Supporting Information.
Scheme 1.

Synthesis of SNPT analogs
Biology
All of the SNPT's were evaluated to test their TR-coactivator (COA) antagonism using a previously reported fluorescence polarization (FP) in replicate dose-response experiments using TRβ-LBD and Texas Red-labeled SRC2-2 peptide (Tx-SRC2-2).26, 34 Compounds were serially diluted in 10 3-fold steps from a 10000 μM DMSO stock. The resulting concentration series of each SNPT were transferred to the assay wells using hydrodynamic pins with a final concentration of 0.1% DMSO. All assays were run in triplicate and the entire experiment replicated twice, for a total of 6 replicates; the data are reported as average values across all assays as IC50 value with 95% confidential range (Supporting Information). Fifty-two out of 291 SNPT's tested showed detectable inhibitory activity (EC50 < 60 μM). Among the active analogs, 19 had IC50 values below 10 NM (Table 1).
Table 1.
Summary of pharmacological properties of SNPTs.
| No | Structure | TRβ-SRC2-2 Inhibition, IC50 (μM, FP assay)a | TRE Response Inhibition (%, at 5 μM)a | Permeabilityb x10-6 cm/s | Solubilityb (μM) | HepG2 Cytotoxicity, EC50 (μM)b |
|---|---|---|---|---|---|---|
| 1 (MSNB) |
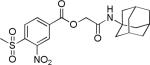
|
4.1±0.2 | 6.2±5.0 | 167±44 | 2.9±0.8 | 16.4±0.4 |
| 2{4,1,5} |
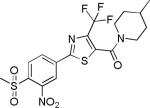
|
0.31±0.17 | 8.1±0.6 | 1322±159 | 6.4±0.3 | >27 |
| 2{4,1,4} |
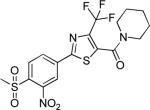
|
0.66±0.57 | 2.3±2.7 | 791±101 | 1.6±0.9 | >27 |
| 2{1,1,4} |
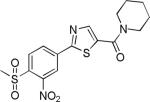
|
1.3±0.6 | 11.9±6.2 | 287±17 | 5.0±0.7 | >27 |
| 2{5,1,4} |
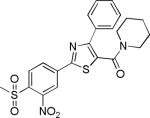
|
1.4±0.3 | no inhibition | 971±210 | 2.3±0.8 | >27 |
| 2{3,1,4} |
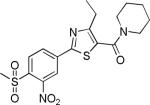
|
1.6±0.9 | 11.6±1.3 | 657±43 | 50.1±1.1 | 11.2 |
| 2{1,1,1} |
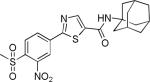
|
1.7±0.9 | no inhibition | 379±39 | 0.7±0.1 | >27 |
| 2{2,1,4} |
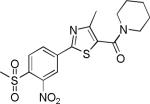
|
1.8±0.6 | 9.8±6.0 | 280±42 | 18.0±0.9 | >27 |
| 2{4,1,2} |
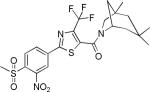
|
2.1±1.6 | 15.4±5.2 | 8±1 | 1.0±0.8 | >27 |
| 2{3,1,2} |
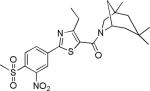
|
2.4±1.1 | 42.1±5.9 | 949±195 | 2.8±0.5 | 10.1 |
| 2{4,1,16} |
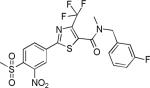
|
2.8±1.8 | 26.0±2.0 | 271±79 | 1.2±0.9 | >27 |
| 2{4,1,5} |
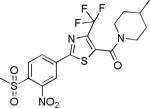
|
3.3±1.3 | 5.8±7.1 | 702±34 | 38.8±1.1 | >27 |
| 2{4,1,14} |
|
3.3±1.4 | 17.9±4.7 | 1499±536 | 1.2±0.7 | >27 |
| 2{4,1,15} |
|
3.4±2.5 | 24.7±3.3 | 221±21 | 0.8±0.1 | >27 |
| 2{4,1,11} |
|
4.2±2.9 | 33.4±1.4 | 84±17 | 0.2±0.5 | >27 |
| 2{4,1,9} |
|
5.2±2.7 | 17.5±6.8 | 43±6 | 0.4±0.4 | >27 |
| 2{2,1,2} |
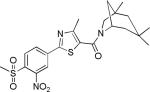
|
7.1±1.9 | 42.1±5.6 | 1416±41 | 5.7±1.0 | >27 |
| 2{2,1,5} |
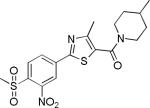
|
7.4±3.4 | 8.1±5.2 | 912±205 | 6.7±0.4 | >27 |
| 3{6,1,13} |
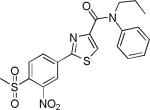
|
8.2±2.0 | 14.6±2.8 | 1619±260 | 2.4±0.1 | >27 |
| 2{1,1,16} |
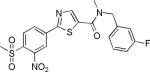
|
8.5±4.4 | 26.7±2.6 | 1031±288 | 2.4±0.4 | >27 |
The IC50 values are for the inhibition of coregulatory peptide binding (SRC2-2) to the TR-LBD using a fluorescence polarization assay. The EC50 values are for cellular proliferation inhibition (HepG2) using the measurement of total ATP content with the CellTiter-Glo® (Promega) method. T3-responsed TRE-luciferase inhibitory activity was evaluated in HEK293 cells transfected with both CMV-TRβ and a DR4-TRE driven luciferase expression vector. The data were normalized to co-transfected constitutive Renilla luciferase activity. Solubility was measured using the Millipore method at pH 7.4 in PBS. Permeability was measured using the parallel artificial membrane permeation assay (PAMPA) at pH 7.4. Compounds are ordered by potency of TRβ and SRC2-2 inhibition.
Values are the mean of two independent experiments in triplicate.
Values are the mean of a single triplicate experiment.
Comparing potency trends between classes of substituent at R1, R2, and R3/R4 allowed an initial analysis of structure activity relationships, (Figure 4). In general, 5-carboxamide-SNPTs 2{x,y,z} gave better inhibitory potency compared to 4-carboxamide-SNPTs 3{x,y,z}. Among the 4-carboxamide-SNPTs 3{x,y,z} the most active compound was compound 3{6,1,13} with 8 μM IC50 value. Among the 5- carboxamide-SNPTs 2{x,y,z}, a wide range of substituents were tolerated at R1, but the phenyl substitutions 2{5,y,z} were mostly inactive, with the exception of 2{5,1,4} (IC50 = 1 μM). The coactivator- binding site on TR is shallow, thus it is likely that large phenyl groups are not favorable for binding. Remarkably CF3 substituted compounds 2{4,y,z} showed slightly better TR-SRC2 antagonism (Figure 4A). Two compounds in this subseries, 2{4,1,5} and 2{4,1,4}, showed the most potent activity (IC50 values of 0.3 NM and 0.6 μM, respectively).
Figure 4.

Analysis of SNPT's evaluation based on substitution position and class.
The potency was significantly less tolerant of variation in R2 with most n-butyl- and benzyl-SNPTs exhibiting no inhibitory activity regardless of substitution on R1 and R3/R4. However, 55 out of 136 methyl-SNPTs (R2=Me) gave detectable inhibition with potency depending on the substitution pattern at R1 and R3/R4 (Figure 4B). Only two compounds from the n-butyl and benzyl-SNPT subseries showed any activity (17 μM for 2{2,2,4}, 20 μM for 2{2,3,4}, 20 μM for 2{3,2,4}, and 19 μM for 2{3,3,4}in Supporting Information).
Finally, the binding pocket seems to be fairly tolerant of a wide range of substituents at R3, with both aliphatic amines and non-aliphatic amines (anilines, benzylamines, and piperazines) giving reasonable potency (Figure 4C). It is apparent that small amines are more favorable than large amines. For instance, the piperidine (z=4) and 4-methylpiperidine (z=5) substituted series showed generally good inhibitory activity while those series containing larger amines such as anilines, benzylamines, and phenylpiperazines did not. However, activity is not simply described by sterics as hydrophilic-aliphatic amines such as morpholines (z=3) and 1-methylpiperazine (z=6) showed weak or no inhibitory activity. This indicates that both electrostatic and hydrophobic interactions are important in this portion of the binding site. These results strongly support our previous finding that hydrophilic atoms on the amide group of the MSNB series and on side chain of β-aminoketone series were not favorable in this portion of the binding pocket.28, 30
MSNB's inhibit the TR-coactivator interaction through alkylation of the Cys 298 residue located on the AF-2 cleft by nucleophilic replacement with methylsulfonyl group.30 Our results indicate that a bulky sulfonyl group hinders nucleophilic attack by this cysteine residue. In addition, larger alkyl groups appear not to fit within the relatively shallow and small binding pocket on TRβ surface thus blocking reaction.
Next, the SNPT compound array was tested for the compounds’ ability to inhibit T3-mediated transcription of a luciferase reporter gene assay. This was done using a single concentration of inhibitor (5 μM) and the data was summarized in Table 1. Strikingly, biochemically highly potent compounds 2{4,1,5} and 2{4,1,4} had weak transcriptional inhibitory activity at 5 μM concentration (8.1 and 2.3% inhibition, respectively). In addition 2{5,1,4} and 2{1,1,1}, which exhibited good biochemical inhibitory activity (1.4 and 1.7 μM IC50), completely failed to inhibit T3-response luciferase expression. Instead biochemically moderately active compounds 2{3,1,2} and 2{2,1,2} significantly inhibited T3-response luciferase activity..
To further validate transcriptional inhibition of T3-driven genes by SNPT compounds, we performed RT-PCR experiments using two well-accepted thyroid-responsive genes: PEPCK and MMP11, which are known to be T3-responsive in HepG2 cells (Figure 5).30, 35 Cells were co-treated with T3 (100 nM) and compound (10 μM). Controls included NH3, a ligand antagonist of T3, and a representative MSNB (1), a known T3 antagonist. mRNA was isolated, and real-time PCR experiments were carried out on the diluted cDNA prepared from each mRNA sample. Both genes were inhibited by SNPT's, with efficacy matching that of the ligand antagonist NH-3. Compound 2{3,1,2} had slightly better inhibitory potency for both genes in comparison to other inhibitors. This data provide support that SNPT analogs effectively block TR-mediated gene transcription at native response elements in live cells.
Figure 5.

Regulation of native T3-controlled genes in HepG2 cells by the treatment with SNPTs. The cells were exposed to compounds at a fixed concentration in the presence of T3 for 24 h. RT-PCR was carried out to determine transcription levels of the PEPCK (Panel A) and MMP11 (Panel B) genes. The ΔΔCt method was used to calculate fold induction of expression. Error bars represent the standard errors of two independent experiments in performed in triplicate. *, P < 0.05, **, P < 0.01, *** P < 0.005.
Solubility and permeability of the compounds in the SNPT array were evaluated to elucidate likely relationships between biochemical assay and cellular assays. Compound solubility was determined in PBS buffer containing 1% DMSO, reflecting the conditions of the biochemical assays. SNPTs generally possessed relatively poor solubility (0.4-39 μM), but most of the potent compounds showed reasonable solubility, being freely soluble at concentrations well above their biochemical potency. Permeability of compounds was measured using a PAMPA at pH 7.4. All of the compounds showed acceptable to good permeability (> 40 × 10-6 cm/s) except 2{4,1,2} (8 × 10-6 cm/s). No correlation was observed between cellular activity and solubility or permeability.
We also measured the cytotoxicity of selected inhibitors in HepG2, a hepatocellular carcinoma derived line. Most of SNPTs showed no cytotoxic effects in the concentration range being evaluated (EC50 > 27 μM). Only three compounds, 2{3,1,4}, 2{3,1,2}, and 2{2,1,1}, showed weak cytotoxicity potency (11.2, 10.1, and 6.4 μM, respectively)
The association of TRβ with SRC2-2 is ligand dependent.36 Previously, we reported that MSNB's could not block the binding of T3 to TRβ.30 In order to confirm the suspected mechanism of action, we tested the antagonism of T3 binding by SNPT's using a ([125I]-T3) competition assay.37 None of SNPT's blocked T3 binding at concentrations up to 60 μM (data not shown). Thus, SNPT's do not appear to act as T3 competitive antagonists.
We examined the NR specificity of the SNPT series by testing the following NR-coactivator interactions in FP assays: TRα with SRC2-2 and peroxisome proliferator-activated receptor gamma (PPARγ) with vitamin D receptor interacting protein-2 (DRIP-2). The coactivators were selected on the basis of previously published work mapping the preferred interaction partners.38, 39 Additionally, PPARγ was chosen because it contains a reactive cysteine in the ligand binding pocket that can be alkylated by electrophilic reagents and might potentially give poor selectivity. A set of 27 compounds was surveyed (Figure 6). In general, most the compounds were selective for the TR's but exhibited similar antagonistic potency towards both TRα and TRβ. However, 2{4,1,4}, 2{4,1,2}, 2{4,1,3} and 2{4,1,15}, which possess a 4-CF3 at the 4-position, are more potent against TRα by at least 5-fold. Compounds 2{3,1,1}, 2{2,1,1}, and 2{1,1,2} were more potent against TRβ by 4- to 5-fold although they are only modestly potent (IC50's ranging from 11-15 μM). This set of compounds had almost no effect on PPARγ.
Figure 6.

Biochemical selectivity of SNPT analogs in inhibiting coregulator binding to other NR family members. Compounds are ordered by potency against TRβ. The coactivator and NR interactions tested were: SRC2-2 with TRα, and DRIP-2 with PPARγ. All values are the mean of two independent fluorescence polarization experiments, each carried out in triplicate.
In considering the aggregate of all the data, the best-balanced two compounds are 2{2,1,2} and 2{3,1,2}. These two compounds showed both good thyroid hormone receptor coactivator interaction inhibitory potency and reasonable cellular toxicity. In addition these molecules significantly inhibited T3- mediated target gene expression in both luciferase based transcription assay and real-time PCR experiments. While their physiochemical properties are not ideal, they are both Class II compounds40 with high permeability and low solubility. Reasonable formulation strategies exist for handling Class II compounds.41 Further optimization will likely focus on careful studies of the amine substitution pattern where the SAR is apparently subtle.
Conclusion
This paper describes the structural modification of the previously reported MSNB series of thyroid receptor antagonists using a bioisosteric approach to remove a potentially labile ester group linking the two critical pharmacophore elements for the inhibitors. In order to identify the best combination of linker and optimal versions of each portion of the molecule, the candidate array of inhibitors (SNPT's) was constructed using a parallel chemistry method with three diversification steps. Antagonism of SNPT's toward TR-coactivator binding was evaluated, revealing the most potent biochemical inhibitors of this interaction reported to date. Among 291 SNPTs analogs tested, sixty compounds inhibited the interaction between TRβ and SRC2-2 peptide. The cellular activity of SNPT analogs was explored using a TRE responsive luciferase reporter gene assay, demonstrating that a number of compounds were potent inhibitors of T3 induction of this element. This antagonism of TR-mediated T3 signaling was confirmed on native response elements using RT-PCR. Interestingly, moderately active compounds in the FP assay significantly inhibited in the cellular level, whereas biochemically highly potent compounds showed weak inhibitory activity. While a number of potent cellular antagonists were identified, they tended not to be among the most potent from the biochemical assay, which is attributed to overly high reactivity of some of the compound array members leading to poor selectivity. The best two compounds 2{2,1,2} and 2{3,1,2} showed moderate inhibitory activity in biochemical assay, but exhibited stronger inhibitory activity in cellular level. We successfully changed the metabolically unfavorable ester group of MSNB's to a thiazole group without any loss of activity in both biochemical assay and cellular assays. This result indicated that the SNPTs can be used as new tools for use in the further TR biology studies. Currently we are studying their activity in in vivo models.
Experimental Section
Chemistry
All materials were obtained from commercial suppliers and used without further purification. All solvents used were dried using an aluminum oxide column. Thin-layer chromatography was performed on pre-coated silica gel 60 F254 plates. Purification of intermediates was carried out by normal phase column chromatography (SP1 [Biotage], Silica gel 230-400 mesh). Chromatographic separation was performed using a UPLC-MS (BEH C18 1.7 μ, 2.1 × 50 mm column, Waters Corp.). Data were acquired using Masslynx v.4.1 and analyzed using the Openlynx software suite. The flow was then split to an evaporative light scattering detector (ELSD) and SQ mass spectrometer. The total flow rate was 1.0 mL/min and gradient program started at 90% A (0.1% formic acid in H2O), changed to 95 % B (0.1% formic acid in ACN), then to 90% A. The mass spectrometer was operated in positive-ion mode with electrospray ionization. NMR spectra were recorded on a Bruker 400 MHz and NMR peaks were assigned by MestReNova (5.2.2). Identity of all final compounds was confirmed by proton NMR and by mass spectrometry. The purity of all final compounds was assessed using LC/MS/UV/ELSD with the purity (>95%) being assigned as the average determined by UV/ELSD (see Supporting Information for details).
4-Chloro-3-nitrobenzamide (5)
To a solution of 4-chloro-3-nitrobenznitrile 4 (30g, 164 mmol) and potassium carbonate (27.3 g, 197 mmol) in DMSO (400 mL) were cautiously added hydrogen peroxide (27.9 mL, 30% aqueous solution). The reaction mixture was heated for 15 min at 60 °C, then cooled to room temperature. The reaction mixture was poured to ice water to afford precipitate, then the precipitate was washed with water to give the desired product 5 (25 g, 76%). 1H NMR (400 MHz, DMSO) δ 8.52 (d, J = 2.1 Hz, 1H), 8.27 (d, J = 24.5 Hz, 1H), 8.16 (dd, J = 8.4, 2.1 Hz, 1H), 7.92 – 7.87 (m, 1H), 7.78 (s, 1H); 13C NMR (101 MHz, DMSO) δ 164.81, 147.31, 134.37, 132.52, 131.90, 127.85, 124.65.
4-Chloro-3-nitrobenzothioamide (6)
To a solution of compound 5 (25 g, 125 mmol) in 1,4-dioxane (300 mL) was added Lawesson's reagent (25.2 g, 62.3 mmol). The reaction mixture was stirred for 2 h at 110 °C. The reaction mixture was concentrated in vacuo, then diluted with ethyl acetate, and washed with water. The organic solution was concentrated and the resulting solid product crystallized from dichloromethane to afford a yellow solid product (25 g, 93%). 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 2.2 Hz, 1H), 8.04 (dd, J = 8.4, 2.2 Hz, 1H), 7.66 (br, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.19 (br, 1H); 13C NMR (101 MHz, CDCl3) δ 198.17, 138.54, 132.14, 131.31, 130.50, 123.56.
Ethyl 2-(4-chloro-3-nitrophenyl)-4-methylthiazole-5-carboxylate (8{2})
To a solution of compound 6 (3 g, 13.9 mmol) in EtOH (40 mL) was added ethyl 2-chloro-3-oxobutanoate (2.9 mL, 20.8 mmol). The reaction mixture was stirred for overnight at 110 °C, and then cooled down to rt. The resulting precipitate was isolated by filtration and washed with MeOH to give pure white solid product (3.5 g, 77%). 1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 2.1 Hz, 1H), 8.07 (dd, J = 8.4, 2.1 Hz, 1H), 7.66 – 7.56 (m, 1H), 4.38 (q, J = 7.1 Hz, 2H), 2.79 (s, 3H), 1.40 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 165.26, 161.77, 161.40, 148.40, 132.88, 132.63, 130.53, 129.00, 123.54, 123.44, 61.65, 17.48, 14.33.
Ethyl 4-methyl-2-(4-(methylthio)-3-nitrophenyl)thiazole-5-carboxylate (9{2,1})
To a solution of compound 8{2} (0.5g, 1.5 mmol) in THF (5 mL) was added sodium methane thiolate (0.13 g, 1.8 mmol). The reaction mixture stirred for 1 hr at rt. The resulting precipitate was filtered and washed with THF and ethyl acetate to afford a yellow solid product (0.5g, 97%).
1H NMR (400 MHz, CDCl3) δ 8.83 (d, J = 2.0 Hz, 1H), 8.16 (dd, J = 8.5, 2.0 Hz, 1H), 7.45 (d, J = 8.6 Hz, 1H), 4.37 (q, J = 7.1 Hz, 2H), 2.79 (s, 3H), 2.56 (s, 3H), 1.40 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 166.45, 161.97, 161.33, 145.54, 142.27, 130.91, 129.48, 126.22, 124.11, 122.61, 61.51, 17.53, 16.16, 14.36.
Ethyl 4-methyl-2-(4-(methylsulfonyl)-3-nitrophenyl)thiazole-5-carboxylate (10{2,1})
To a solution of compound 9{2,1} (0.27 g, 0.8 mmol) in DCM was added m-chloroperoxybenzoic acid (0.49 g, 2.0 mmol, < 70% maximum activity). The reaction mixture was stirred for 6 h at rt and washed with water and sat. sodium bicarbonate. The organic solution was dried over MgSO4 and concentrated in vaccuo. The crude product was purified by recrystallization with DCM to give a white solid product (0.27 g, 91%).
10{2,1}1H NMR (400 MHz, CDCl3) δ 8.39 (s, 1H), 8.24 – 8.18 (m, 2H), 4.32 (q, J = 7.1 Hz, 2H), 3.40 (s, 3H), 2.74 (s, 3H), 1.34 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 163.91, 161.77, 161.52, 149.71, 139.21, 134.99, 132.37, 129.87, 124.90, 122.70, 61.87, 45.18, 17.50, 14.32.
4-Methyl-2-(4-(methylsulfonyl)-3-nitrophenyl)oxazole-5-carboxylic acid (11{1,1})
To a solution of compound 10{1,1} (40 mg, 0.11 mmol) in H2O/THF (4 mL, 1:3 v/v) was added lithium hydroxide (3.2 mg, 0.14 mmol). The reaction mixture was stirred for 3 h at rt, neutralized by 1 N HCl to pH 5-6, resulting in formation of solid precipitate. The solid product was washed with water to give pure white solid product (20 mg, 54%).
11{1,1} 1H NMR (400 MHz, DMSO) δ 8.67 (d, J = 1.8 Hz, 1H), 8.58 (s, 1H), 8.55 (dd, J = 8.3, 1.8 Hz, 1H), 8.28 – 8.25 (m, 1H), 3.54 (s, 3H); 13C NMR (101 MHz, DMSO) δ 167.26, 161.67, 149.08, 148.86, 138.18, 133.88, 133.17, 132.34, 130.46, 122.44, 44.43.
Amides (2{x,y,z} and 3{x,y,z})
To a solution of carboxylic acid (50 mmol) in DMF (0.25 mL) in a 48 position Mettler Toledo XT reaction block was added PyBOP (50 mmol, 0.2 mL of 0.3 M solution in DMF) and TEA (75 mmol, 0.05 mL of 1.5 M solution in DMF) followed by the appropriate amine (55 mmol, 0.55 ml of 1 M solution in DMF ). The reactions were stirred at rt for 24 h and concentrated using a GeneVac HT)4. The crude product mixtures were dissolved in EtOAc (1 mL), filtered through a silica) packed column, and washed with EtOAc (2 × 3 mL). The organic solutions were concentrated using a GeneVac HT)4 and dissolved in DMSO (1 mL). The crude products were purified by automated HPLC/MS. NMR and MS of all final compounds are available in Supporting Information.
Supplementary Material
Acknowledgement
This work was supported by NIH grant DK58080, the American Lebanese Syrian Associated Charities (ALSAC), and St. Jude Children's Research Hospital (SJCRH).
Abbreviations
- MSNB
Methylsulfonylnitrobenzoate
- SNPT
sulfonylnitrophenylthiazole
- NR
nuclear hormone receptor
- TR
thyroid hormone receptor
- RXR
Retinoid X receptor
- PPAR
peroxisome proliferator)activated receptor
- AF
transcription activation
- NID
Nuclear receptor interaction domain
- LBD
ligand binding domain
- TRE
thyroid) responsive element
- T3
triiodothyronine
- SRC
steroid receptor coactivator
- COA
coactivator
- PAMPA
parallel artificial membrane permeability assay
- FP
fluorescence polarization
- PDK
pyruvate dehydrogenase kinase
- PEPCK
phosphoenolpyruvate carboxykinase
- MMP11
matrix metallopeptidase 11
Footnotes
Compound evaluation. All biological and pharmacological methods have been previously published and followed the established procedures. For details, see the Supporting Information.
Supporting Information
Experimental procedures, tabulated activity data, and characterization data for intermediates and final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.He J, Cheng Q, Xie W. Minireview: Nuclear receptor-controlled steroid hormone synthesis and metabolism. Mol Endocrinol. 2010;24:11–21. doi: 10.1210/me.2009-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulman IG. Nuclear receptors as drug targets for metabolic disease. Adv Drug Deliv Rev. 2010;62:1307–1315. doi: 10.1016/j.addr.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun G, Shi Y. Nuclear receptors in stem cells and their therapeutic potential. Adv Drug Deliv Rev. 2010;62:1299–1306. doi: 10.1016/j.addr.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kress E, Samarut J, Plateroti M. Thyroid hormones and the control of cell proliferation or cell differentiation: paradox or duality? Mol Cell Endocrinol. 2009;313:36–49. doi: 10.1016/j.mce.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 6.Oppenheimer JH, Schwartz HL. Molecular basis of thyroid hormone-dependent brain development. Endocr Rev. 1997;18:462–475. doi: 10.1210/edrv.18.4.0309. [DOI] [PubMed] [Google Scholar]
- 7.Yaoita Y, Brown DD. A correlation of thyroid hormone receptor gene expression with amphibian metamorphosis. Genes Dev. 1990;4:1917–1924. doi: 10.1101/gad.4.11.1917. [DOI] [PubMed] [Google Scholar]
- 8.Brent GA. Tissue-specific actions of thyroid hormone: insights from animal models. Rev Endocr Metab Disord. 2000;1:27–33. doi: 10.1023/a:1010056202122. [DOI] [PubMed] [Google Scholar]
- 9.Harvey CB, Williams GR. Mechanism of thyroid hormone action. Thyroid. 2002;12:441–446. doi: 10.1089/105072502760143791. [DOI] [PubMed] [Google Scholar]
- 10.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alonso M, Goodwin C, Liao X, Ortiga-Carvalho T, Machado DS, Wondisford FE, Refetoff S, Weiss RE. In vivo interaction of steroid receptor coactivator (SRC)-1 and the activation function-2 domain of the thyroid hormone receptor (TR) beta in TRbeta E457A knock-in and SRC-1 knockout mice. Endocrinology. 2009;150:3927–3934. doi: 10.1210/en.2009-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul BD, Buchholz DR, Fu L, Shi YB. SRC-p300 coactivator complex is required for thyroid hormone-induced amphibian metamorphosis. J Biol Chem. 2007;282:7472–7481. doi: 10.1074/jbc.M607589200. [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol. 2003;17:1681–1692. doi: 10.1210/me.2003-0116. [DOI] [PubMed] [Google Scholar]
- 14.Savkur RS, Burris TP. The coactivator LXXLL nuclear receptor recognition motif. J Pept Res. 2004;63:207–212. doi: 10.1111/j.1399-3011.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- 15.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng W, Ribeiro RC, Wagner RL, Nguyen H, Apriletti JW, Fletterick RJ, Baxter JD, Kushner PJ, West BL. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science. 1998;280:1747–1749. doi: 10.1126/science.280.5370.1747. [DOI] [PubMed] [Google Scholar]
- 17.Yuan C, Lin JZ, Sieglaff DH, Ayers SD, Denoto-Reynolds F, Baxter JD, Webb P. Identical Gene Regulation Patterns of T3 and Selective Thyroid Hormone Receptor Modulator GC-1. Endocrinology. 2012;153:501–511. doi: 10.1210/en.2011-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grover GJ, Egan DM, Sleph PG, Beehler BC, Chiellini G, Nguyen NH, Baxter JD, Scanlan TS. Effects of the thyroid hormone receptor agonist GC-1 on metabolic rate and cholesterol in rats and primates: selective actions relative to 3,5,3'-triiodo-L-thyronine. Endocrinology. 2004;145:1656–1661. doi: 10.1210/en.2003-0973. [DOI] [PubMed] [Google Scholar]
- 19.Baxter JD, Webb P, Grover G, Scanlan TS. Selective activation of thyroid hormone signaling pathways by GC-1: a new approach to controlling cholesterol and body weight. Trends Endocrinol Metab. 2004;15:154–157. doi: 10.1016/j.tem.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Messier N, Langlois MF. Triac regulation of transcription is T(3) receptor isoform- and response element-specific. Mol Cell Endocrinol. 2000;165:57–66. doi: 10.1016/s0303-7207(00)00266-5. [DOI] [PubMed] [Google Scholar]
- 21.Bryzgalova G, Effendic S, Khan A, Rehnmark S, Barbounis P, Boulet J, Dong G, Singh R, Shapses S, Malm J, Webb P, Baxter JD, Grover GJ. Anti-obesity, anti-diabetic, and lipid lowering effects of the thyroid receptor beta subtype selective agonist KB-141. J Steroid Biochem Mol Biol. 2008;111:262–267. doi: 10.1016/j.jsbmb.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 22.Garg N, Li YL, Garcia Collazo AM, Litten C, Ryono DE, Zhang M, Caringal Y, Brigance RP, Meng W, Washburn WN, Agback P, Mellstrom K, Rehnmark S, Rahimi-Ghadim M, Norin T, Grynfarb M, Sandberg J, Grover G, Malm J. Thyroid receptor ligands. Part 8: Thyromimetics derived from N-acylated-alpha-amino acid derivatives displaying modulated pharmacological selectivity compared with KB-141. Bioorg Med Chem Lett. 2007;17:4131–4134. doi: 10.1016/j.bmcl.2007.05.049. [DOI] [PubMed] [Google Scholar]
- 23.Shah V, Nguyen P, Nguyen NH, Togashi M, Scanlan TS, Baxter JD, Webb P. Complex actions of thyroid hormone receptor antagonist NH-3 on gene promoters in different cell lines. Mol Cell Endocrinol. 2008;296:69–77. doi: 10.1016/j.mce.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webb P, Nguyen NH, Chiellini G, Yoshihara HA, Cunha Lima ST, Apriletti JW, Ribeiro RC, Marimuthu A, West BL, Goede P, Mellstrom K, Nilsson S, Kushner PJ, Fletterick RJ, Scanlan TS, Baxter JD. Design of thyroid hormone receptor antagonists from first principles. J Steroid Biochem Mol Biol. 2002;83:59–73. doi: 10.1016/s0960-0760(02)00270-4. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen NH, Apriletti JW, Cunha Lima ST, Webb P, Baxter JD, Scanlan TS. Rational design and synthesis of a novel thyroid hormone antagonist that blocks coactivator recruitment. J Med Chem. 2002;45:3310–3320. doi: 10.1021/jm0201013. [DOI] [PubMed] [Google Scholar]
- 26.Arnold LA, Estebanez-Perpina E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J Biol Chem. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]
- 27.Arnold LA, Kosinski A, Estebanez-Perpina E, Fletterick RJ, Guy RK. Inhibitors of the interaction of a thyroid hormone receptor and coactivators: preliminary structure-activity relationships. J Med Chem. 2007;50:5269–5280. doi: 10.1021/jm070556y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang JY, Arnold LA, Zhu F, Kosinski A, Mangano TJ, Setola V, Roth BL, Guy RK. Improvement of pharmacological properties of irreversible thyroid receptor coactivator binding inhibitors. J Med Chem. 2009;52:3892–3901. doi: 10.1021/jm9002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson RL, Hwang JY, Arnold LA, Huang R, Wichterman J, Augustinaite I, Austin CP, Inglese J, Guy RK, Huang W. A quantitative high-throughput screen identifies novel inhibitors of the interaction of thyroid receptor beta with a peptide of steroid receptor coactivator 2. J Biomol Screen. 2011;16:618–627. doi: 10.1177/1087057111402199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang JY, Huang W, Arnold LA, Huang R, Attia RR, Connelly M, Wichterman J, Zhu F, Augustinaite I, Austin CP, Inglese J, Johnson RL, Guy RK. Methylsulfonylnitrobenzoates, a new class of irreversible inhibitors of the interaction of the thyroid hormone receptor and its obligate coactivators that functionally antagonizes thyroid hormone. J Biol Chem. 2011;286:11895–11908. doi: 10.1074/jbc.M110.200436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warmus JS, Flamme C, Zhang LY, Barrett S, Bridges A, Chen H, Gowan R, Kaufman M, Sebolt-Leopold J, Leopold W, Merriman R, Ohren J, Pavlovsky A, Przybranowski S, Tecle H, Valik H, Whitehead C, Zhang E. 2-Alkylamino- and alkoxy-substituted 2-amino-1,3,4-oxadiazoles-O-Alkyl benzohydroxamate esters replacements retain the desired inhibition and selectivity against MEK (MAP ERK kinase). Bioorg Med Chem Lett. 2008;18:6171–6174. doi: 10.1016/j.bmcl.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 32.Diana GD, Volkots DL, Nitz TJ, Bailey TR, Long MA, Vescio N, Aldous S, Pevear DC, Dutko FJ. Oxadiazoles as ester bioisosteric replacements in compounds related to disoxaril. Antirhinovirus activity. J Med Chem. 1994;37:2421–2436. doi: 10.1021/jm00041a022. [DOI] [PubMed] [Google Scholar]
- 33.The MOE program (Chemical Computing Group) was used.
- 34.Arnold LA, Estebanez-Perpina E, Togashi M, Shelat A, Ocasio CA, McReynolds AC, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. A high-throughput screening method to identify small molecule inhibitors of thyroid hormone receptor coactivator binding. Sci STKE. 2006;2006:pl3. doi: 10.1126/stke.3412006pl3. [DOI] [PubMed] [Google Scholar]
- 35.Sadana P, Hwang JY, Attia RR, Arnold LA, Neale G, Guy RK. Similarities and Differences between Two Modes of Antagonism of the Thyroid Hormone Receptor. ACS Chem Biol. 2011 doi: 10.1021/cb200092v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeyakumar M, Tanen MR, Bagchi MK. Analysis of the functional role of steroid receptor coactivator-1 in ligand-induced transactivation by thyroid hormone receptor. Mol Endocrinol. 1997;11:755–767. doi: 10.1210/mend.11.6.0003. [DOI] [PubMed] [Google Scholar]
- 37.Feau C, Arnold LA, Kosinski A, Guy RK. A high-throughput ligand competition binding assay for the androgen receptor and other nuclear receptors. J Biomol Screen. 2009;14:43–48. doi: 10.1177/1087057108326662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore JM, Galicia SJ, McReynolds AC, Nguyen NH, Scanlan TS, Guy RK. Quantitative proteomics of the thyroid hormone receptor-coregulator interactions. J Biol Chem. 2004;279:27584–27590. doi: 10.1074/jbc.M403453200. [DOI] [PubMed] [Google Scholar]
- 39.Feau C, Arnold LA, Kosinski A, Zhu F, Connelly M, Guy RK. Novel flufenamic acid analogues as inhibitors of androgen receptor mediated transcription. ACS Chem Biol. 2009;4:834–843. doi: 10.1021/cb900143a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 41.Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29:278–287. doi: 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.