

Abstract
The field of pancreatic stem and progenitor cell biology has been hampered by a lack of in vitro functional and quantitative assays that allow for the analysis of the single cell. Analyses of single progenitors are of critical importance because they provide definitive ways to unequivocally demonstrate the lineage potential of individual progenitors. Although methods have been devised to generate "pancreatospheres" in suspension culture from single cells, several limitations exist. First, it is time-consuming to perform single cell deposition for a large number of cells, which in turn commands large volumes of culture media and space. Second, numeration of the resulting pancreatospheres is labor-intensive, especially when the frequency of the pancreatosphere-initiating progenitors is low. Third, the pancreatosphere assay is not an efficient method to allow both the proliferation and differentiation of pancreatic progenitors in the same culture well, restricting the usefulness of the assay.
To overcome these limitations, a semi-solid media based colony assay for pancreatic progenitors has been developed and is presented in this report. This method takes advantage of an existing concept from the hematopoietic colony assay, in which methylcellulose is used to provide viscosity to the media, allowing the progenitor cells to stay in three-dimensional space as they undergo proliferation as well as differentiation. To enrich insulin-expressing colony-forming progenitors from a heterogeneous population, we utilized cells that express neurogenin (Ngn) 3, a pancreatic endocrine progenitor cell marker. Murine embryonic stem (ES) cell-derived Ngn3 expressing cells tagged with the enhanced green fluorescent protein reporter were sorted and as many as 25,000 cells per well were plated into low-attachment 24-well culture dishes. Each well contained 500 μL of semi-solid media with the following major components: methylcellulose, Matrigel, nicotinamide, exendin-4, activin βB, and conditioned media collected from murine ES cell-derived pancreatic-like cells. After 8 to 12 days of culture, insulin-expressing colonies with distinctive morphology were formed and could be further analyzed for pancreatic gene expression using quantitative RT-PCR and immunoflourescent staining to determine the lineage composition of each colony.
In summary, our colony assay allows easy detection and quantification of functional progenitors within a heterogeneous population of cells. In addition, the semi-solid media format allows uniform presentation of extracellular matrix components and growth factors to cells, enabling progenitors to proliferate and differentiate in vitro. This colony assay provides unique opportunities for mechanistic studies of pancreatic progenitor cells at the single cell level.
Keywords: Developmental Biology, Issue 57, Pancreas, insulin-expressing cells, embryonic stem cells, colony assay, progenitor cells, 3-dimensional culture, semi-solid media, Matrigel, methylcellulose
Protocol
1. Differentiation of murine ES cells to pancreatic-like cells in vitro to generate conditioned media (CM) for colony assay
Procedures for murine ES cell maintenance, embryoid body (EB) formation in suspension culture, and attachment culture for further differentiation have been previously described1-3 and are not the focus of this report. A schematic presentation of the steps involved in our differentiation protocol is shown in Figure 1.
In order to obtain high-quality conditioned media, it is important to first generate 6-day-old EBs in which at least 30-40% of them are with diameters of at least 150 μm. The larger EBs should contain dark spots under the light microscope, suggesting enhanced differentiation (Figure 2). Selection of high quality fetal calf serum (FCS) is critical to generate such EBs. High quality FCS lots are selected based on their ability to support the differentiation of EBs towards insulin 1, glucagon and amylase 2A gene expression in the later stages (day 18-20) as detected by RT-PCR analysis. Furthermore, when day-6 EBs are transferred from suspension culture to attachment culture (approximately 80 - 100 EBs per well in 6 well plates) it is important to gently rotate the plates to gather the day-6 EBs into the center of the wells. For reasons unknown, maneuvers that enhance cell-cell contacts result in better development of pancreatic-like cells from EBs in the subsequent attachment culture stage.
On culture day 13, replace the attachment culture media (DMEM/F-12 (1:1), 15% knockout serum replacement, 2 mM L-glutamine, 50 U/mL penicillin, 50 μg/mL streptomycin) with fresh attachment culture media containing10 mM nicotinamide, 0.1 nM exendin-4, and 10 ng/mL human recombinant activin ßB (all concentrations are final).
On culture day 16, collect the media from step 1.1 and filter it through a 0.2 μm polyethersulfone membrane. From this point forward the collected media will be referred to as conditioned media (CM). Aliquot the CM into 15 mL Falcon tubes and store it at -80°C until just prior to semi-solid culture (see Section 3 below).
2. Obtaining pancreatic-like progenitors in single cell suspension using a fluorescence activated cell sorter
A knock-in murine ES cell line,4 designated as Ngn3-EGFP, where the EGFP reporter gene replaced one allele of the Ngn3 locus was differentiated as previously described.2 Ngn3 is a basic helix-loop-helix (bHLH) containing transcription factor expressed by pancreatic endocrine progenitors during development.5 Day-16 cultures were normally sorted to obtain differentiated Ngn3-EGFP+ and Ngn3-EGFP- cells.1,2
- Dissociation of differentiated cells into a single cell suspension.
- Incubate day-16 cultures with 0.25% trypsin-EDTA (3 min, 37°C) to dislodge the cells from culture wells.
- Add 10% FCS to stop trypsin activity.
- Wash cells with magnesium and calcium-free phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (BSA), and centrifuge at 300 x g for 5 min.
- Remove supernatant and incubate the cell clumps with 4 mg/mL collagenase B and 2000 U/mL deoxyribonuclease (DNase) I (30 min, 37°C) to produce a single cell suspension. Use a 1000 μL pipette to disrupt the cell pellets every 10 min to hasten the dissociation process.
- Wash cells with PBS containing 0.1% BSA, and centrifuge at 300 x g for 5 min.
- Remove supernatant and resuspend the cells in PBS containing 0.1% BSA.
- Preparation of cells for sorting.
- Add 2 μL DNase I (1 MU/ mL) for every mL cell suspension to prevent cell reaggregation during sorting.
- Pass the single cell suspension through a 40 to 70 μm mesh to remove large cell aggregates.
- Add DAPI (1 μg/mL final concentration) to the single cell suspension.
- Acquire flow cytometry data on a cell sorter. A MoFlo MLS (Beckman Coulter) cell sorter was used for this report.
- Gating of cell populations for sorting. Ngn3-EGFP+ and Ngn3-EGFP- cells are gated first with forward scatter vs. side scatter (Figure 3A), followed by DAPI vs. side scatter (Figure 3B), pulse width vs. side scatter (Figure 3C), and finally FL1 vs. auto-fluorescence (Figure 3D). The final sort gate of Ngn3-EGFP+ cells is intentionally moved to the right (Figure 3E; arrow) in order to limit contamination of Ngn3-EGFP- cells during sorting. For negative control gating, either day-6 EBs from Ngn3-EGFP ES line or the same stage, day-16 culture from a non-transgenic line, such as TL1 ES cells (Figure 3D, left panel), can be used.
Sort Ngn3-EGFP+ and Ngn3-EGFP- cells.
3. Plating sorted cells to semi-solid culture
- Matrigel preparation.
- Aliquot the Matrigel to 1 mL per tube and store at -20°C before use.
- Frozen aliquots should be placed at 4°C overnight or on ice for 3 hours to thaw just prior to use. Thawed aliquots must remain on ice during semi-solid culture preparation to avoid resolidification.
- Methylcellulose solution (3.3%) preparation. Methylcellulose powder cannot be autoclaved. In order to obtain bacteria-free methylcellulose solution, use clean weighing boats and autoclaved stir bars, beakers, flasks and other hardware during preparation process.
- One day before the methylcellulose solution preparation, store 200 mL of sterile double distilled water at 4°C. Also, prepare 500 mL of 2x DMEM/F12 media (prepare by dissolving powdered DMEM/ F-12 in doubled distilled water) containing 100 U/mL penicillin and 100 μg/mL streptomycin and store at 4°C.
- On the day of the methylcellulose preparation, add 500 mL of sterile double distilled water to a 1000 mL glass beaker with a piece of aluminum foil on top. Boil for at least 30 min to rid the water of air bubbles.
- Place a large sized stir bar (at least 3 inches in length) into a 2000 mL glass flask.
- Measure 300 mL of boiling water and transfer it to the flask.
- Place the flask on a stir plate (without heating). Slowly stir the hot water to avoid generating air bubbles.
- Weigh 33 grams of methylcellulose powder and very slowly add it to the hot water. Hot water is necessary to wet the methylcellulose powder efficiently and aid the subsequent dissolution in cold temperature.
- Continue to stir the powder until the temperature is reduced to approximately 40-45°C.
- Add the 200 mL cold sterile double distilled water to the cooling methylcellulose mixture. Immediately after, you should start to see the solution turn transparent and viscous, indicating that methylcellulose is dissolving into the aqueous phase. Hand swirl the flask to ensure the solution is mixed thoroughly.
- Add the 500 mL cold 2x DMEM/F12 media. Continue to swirl by hand to mix.
- Place the flask on a stir plate in a cold room, and slowly stir the methylcellulose solution for 24-48 hours.
- Aliquot the solution into 125 mL per storage bottle, and store at -20°C. A sample should be dispensed into a sterile petridish and placed in 37°C incubator to test sterility. Thawed methylcellulose solution can be kept in refrigerator for up to 2 months.
Conditioned media preparation. Thaw CM (see section 1) just prior to use. Keep the CM on ice throughout semi-solid culture preparation.
Prepare the following growth factors: 1.0 M nicotinamide, 0.1 μM exendin-4, 10 μg/mL recombinant human activin βB, and 10 μg/mL VEGF-A. Store the nicotinamide and exendin-4 at -20°C and activin βB and VEGF-A at -80°C, and thaw them immediately before use. All semi-solid culture media components should be kept on ice during plating. It should be noted that addition of VEGF-A was later found not necessary for colony formation.2
Fetal calf serum preparation. FCS should be heat inactivated before use. Incubate the FCS for 30 minutes at 56°C in a water bath to inactivate complement proteins. If the initial FCS stock bottle holds more than 500 mL aliquot it into 125 mL vials. The increased surface area will ensure even and thorough heating of the entire volume. Allow the bottles to cool at room temperature for at least 1 hour before storing at -20°C. Thawed FCS is 0.2μm filtered before use.
- Mix culture components with sorted cells. Keep the cells and all semi-solid media components on ice during preparation at all times to prevent solidification of Matrigel. All supplies that come in contact with Matrigel including, pipette tips, syringes and needles, should be cold. Place these supplies at -20°C at least half an hour before use.
- Determine the quantity of cells to be seeded per well in a 24-well plate. Up to 25,000 cells can be plated per 24-well. One could perform a titration of cell density in initial experiments to determine the optimal seeding cell number.
- Add the culture components to a 5 mL polystyrene tube with a snap cap in the sequence described in Table 1 (note that the cells are added last to avoid premature stimulation). Add the 3.3 % methylcellulose to the polystyrene tubes first and the sorted cells last. Note that the 3.3 % methylcellulose solution needs to be drawn up and added to the polystyrene tubes with a 16 ½ gauge needle and an adequately marked graduated syringe. In general, in order to correctly measure the volume of viscous solutions, such as the 3.3 % methylcellulose solution and the final culture mixture, it is important to aspirate some solution in the syringe first, and then immediately push the solution out to expel the air. The 3.3 % methylcellulose solution may be warmed to room temperature if it is too difficult to draw out. However, ensure it is cooled on ice after it is dispensed into the polystyrene tubes and before the addition of Matrigel.
- Plate the semi-solid media containing sorted cells.
- After ensuring the caps of the polystyrene tubes are firmly in place, shake the tubes by hands vigorously and thoroughly. Air bubbles will be generated during this process.
- Place the tubes on ice for 5 min or until the small air bubbles surface.
- Use a 1mL syringe with an 16 ½ or 18 ½ -gauge needle to aspirate the contents. Follow the advice in 3.6.2 to draw accurate volume of viscous solution without air in syringes.
- Slowly dispense 500 uL of media into each 24-well. Use the low attachment 24-well plates only. Add the media directly to the center of the well. The semi-solid media will automatically spread out in the wells. For each experimental sample, quadruplicated wells are routinely used in order to obtain statistically significant results. Only the 4 innermost columns of a horizontally oriented 24-well plate are used for culture. Fill the remaining wells with sterile water to aid humidification of the cell-containing culture.
- Incubate the cells in 37°C and 5% CO2 air.
- Delayed addition of growth factors. It is possible to add growth factors after the initiation of the culture. Such maneuvers may be used to address the survival effects of certain growth factors. Growth factor solutions up to 100 μL per well may be dripped via a 1 mL syringe with a 26 G 3/8 needle and allowed to diffuse into the semi-solid media.
4. Observing colony formation under the light microscope
Cells should be observed immediately after plating to ensure that single cells are randomly and evenly distributed throughout out the wells. If most of the cells or the resulting colonies are distributed at the margin of the wells, this will indicate that dispensing the semi-solid media into the wells was too forceful (see section 3.7.4). Due to the meniscus effect colony density at the margins of the culture wells may be higher compared to those residing in the center.
Colony number quantification. Due to the 3-dimensional feature of the media, the colonies will appear at different focal points. Thus, an adjustment of the focus for each colony may be required during observation by light microscopy. Attach a grid under the 24-well when counting in order to avoid registering the same colony twice. A grid may be obtained by cutting the wall of a NUNC petridish (Cat. No.169558). A 10x objective lens on a light microscope should be used to observe and count the colonies.
5. Representative Results:
Previous experiments had shown that starting with 2.5 x 105 murine R1 ES cells (in 50 mL media total) one could expect to generate approximately 5000 appropriate sized day-6 EBs. The Ngn3-EGFP ES cells were less efficient; 4 times more cells were required to generate similar-sized EBs by day 6. Because 25 day-6 EBs contain an average of 1.88 ± 0.67 x 105 cells,1 approximately 37.6 x 106 cells can be obtained from 5000 EBs. During attachment culture the cells are expected to expand 2.72 ± 0.32 fold.1 Thus, 5000 day-6 EBs will yield approximately 100 x 106 day-16 cells before sorting. After gating, the Ngn3-EGFP+ cells will make up approximately 10 % of the total day-16 cell population (Protocol Text Section 2.2.5 and Figure 3).
Example of the photomicrographs of insulin-expressing colonies derived from murine ES cells is shown in Figure 4. These colonies have a distinctive morphology; small (~60-100 μm) round colonies with individual cells in the colonies that are small, dark, and not light reflective. In our previous publication,2 real-time RT-PCR and double immunofluorescent staining of individually hand-picked colonies revealed expression of insulin, C-peptide and glucagon. These results demonstrate the ability of single Ngn3-EGFP+ cells to differentiate into cells containing at least two islet hormones simultaneously, resembling primitive first-wave like endocrine cells.
The colony assay for dispersed single cells enables the study for those progenitors that have the capacity to initiate proliferation and differentiation in vitro (Figure 5). Thus, this is an assay that can measure the intrinsic function of individual progenitor cells. The progenitors that have the capability of becoming insulin-expressing colonies are termed insulin-expressing colony forming units (ICFUs) (Figure 5). Consistent to the idea that Ngn3 marks endocrine progenitor cells in the developing pancreas,5 Ngn3-EGFP+, but not Ngn3-EGFP- cells from mES cell-derived populations are enriched for the ICFUs.2 However, the frequency of these progenitors is still low; approximately 0.1 % to 0.6 % of the total Ngn3-EGFP+ population isolated from day 16 culture are ICFUs.2 Regardless, a clear difference in colony-forming efficiency is expected, with Ngn3-EGFP+ cells the highest, followed by the presorted cells, and then the Ngn3-EGFP- cells.
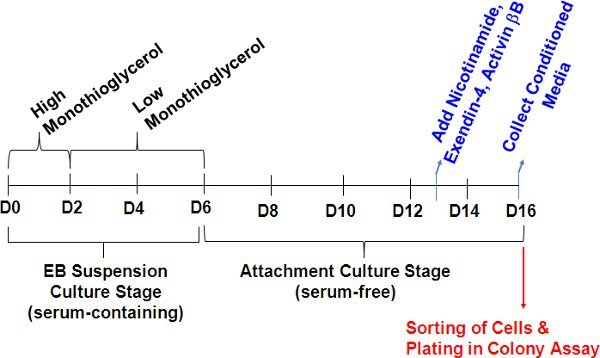 Figure 1. Timeline of in vitro murine ES cells differentiation to pancreatic-like cells. For conditioned media (CM) generation, murine ES cells are grown in suspension with 15 % FCS for the first six days with decreasing doses of monothioglycerol (6.0 x 10-3 M for the first two days and 6.0 x 10-4 M for the following four days) to generate EBs. Day-6 EBs (80-100 per well) are then transferred to attachment 6-well dishes containing 15 % knockout serum replacement. 5% FCS may be added between days 6-10 of culture to hasten the attachment of EBs. Nicotinamide, exendin-4, and human recombinant activin βB are then added to the media on culture day 13 (see section 1.1). Media is collected on culture day 16 and at this point has become conditioned media. Day 16 cells are then sorted and plated into semi-solid media for colony assay.
Figure 1. Timeline of in vitro murine ES cells differentiation to pancreatic-like cells. For conditioned media (CM) generation, murine ES cells are grown in suspension with 15 % FCS for the first six days with decreasing doses of monothioglycerol (6.0 x 10-3 M for the first two days and 6.0 x 10-4 M for the following four days) to generate EBs. Day-6 EBs (80-100 per well) are then transferred to attachment 6-well dishes containing 15 % knockout serum replacement. 5% FCS may be added between days 6-10 of culture to hasten the attachment of EBs. Nicotinamide, exendin-4, and human recombinant activin βB are then added to the media on culture day 13 (see section 1.1). Media is collected on culture day 16 and at this point has become conditioned media. Day 16 cells are then sorted and plated into semi-solid media for colony assay.
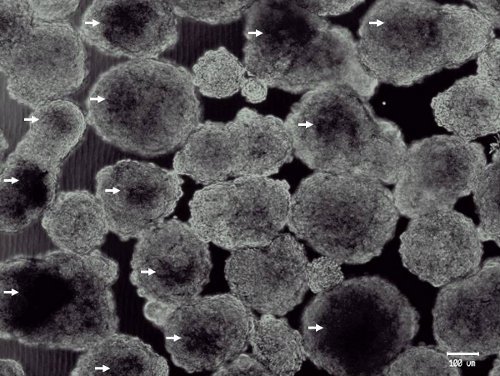 Figure 2. Representative photomicrograph of large day-6 embryoid bodies derived from murine embryonic stem cells. Ideal day-6 EBs are at least 150 μm in diameter and have darkened centers (arrows). Expression of early pancreatic progenitor markers (such as Pdx-1 and Sox9) is enhanced in these EBs (data not shown).3
Figure 2. Representative photomicrograph of large day-6 embryoid bodies derived from murine embryonic stem cells. Ideal day-6 EBs are at least 150 μm in diameter and have darkened centers (arrows). Expression of early pancreatic progenitor markers (such as Pdx-1 and Sox9) is enhanced in these EBs (data not shown).3
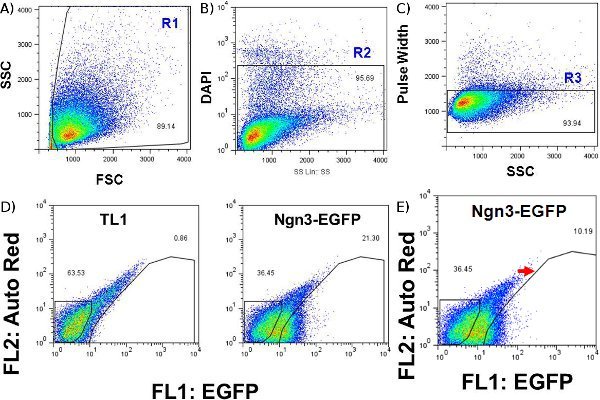 Figure 3. Sorting gates for mES cell-derived day-16 Ngn3-EGFP expressing cells. (A) Forward and side scatter gates, indicative of cell size and granularity respectively, eliminate non-cellular debris. (B) DAPI is used to identify dead cells, which will be stained positive when excited at 405-413 nm and emission detected with a 450/40 filter. (C) Pulse width, the width of the electronic forward scatter pulse, reveals doublets, which should be eliminated before being passed through the fluorescence activated cell sorter. (D) Gating of EGFP (channel FL1) versus autofluorescence using day-16 cells from a control parental cell line such as TL1 (left panel) illuminates true fluorescence and thus the Ngn3-EGFP+ cell population (right panel). (E) Red arrow shows manual right-shift of EGFP+ gate just before sorting to increase separation of false positive cells. Events in both (D) and (E) are gated based on regions (R1 - R3) shown in (A-C).
Figure 3. Sorting gates for mES cell-derived day-16 Ngn3-EGFP expressing cells. (A) Forward and side scatter gates, indicative of cell size and granularity respectively, eliminate non-cellular debris. (B) DAPI is used to identify dead cells, which will be stained positive when excited at 405-413 nm and emission detected with a 450/40 filter. (C) Pulse width, the width of the electronic forward scatter pulse, reveals doublets, which should be eliminated before being passed through the fluorescence activated cell sorter. (D) Gating of EGFP (channel FL1) versus autofluorescence using day-16 cells from a control parental cell line such as TL1 (left panel) illuminates true fluorescence and thus the Ngn3-EGFP+ cell population (right panel). (E) Red arrow shows manual right-shift of EGFP+ gate just before sorting to increase separation of false positive cells. Events in both (D) and (E) are gated based on regions (R1 - R3) shown in (A-C).
 Figure 4. Representative photomicrographs of insulin-expressing colonies in semi-solid culture. Colonies are randomly distributed and appear as small dark, non-light reflective clusters. Individual colonies can later be assayed for gene and protein expression by RT-PCR and immunohistochemistry analyses, respectively (not shown; see video).
Figure 4. Representative photomicrographs of insulin-expressing colonies in semi-solid culture. Colonies are randomly distributed and appear as small dark, non-light reflective clusters. Individual colonies can later be assayed for gene and protein expression by RT-PCR and immunohistochemistry analyses, respectively (not shown; see video).
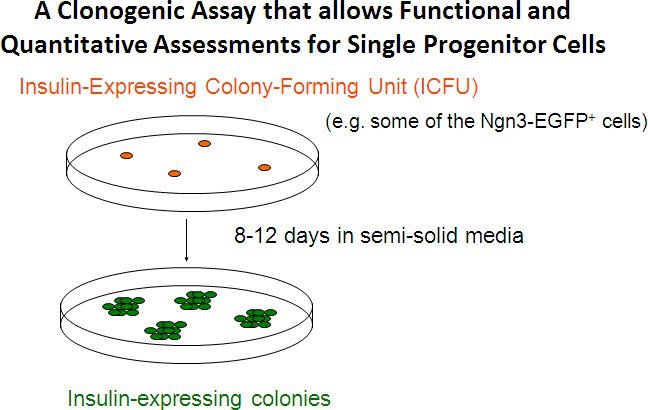 Figure 5. A colony assay that allows functional and quantitative assessments of single progenitor cells. Numeration of the resulting colonies is used to calculate the frequency of the progenitor cells among the total plated cells. The composition of cells within each colony is indicative of the lineage potential of the initiating progenitor. Using this assay, we found that Ngn3-EGFP+ cells derived from murine embryonic stem cells were enriched for insulin-expressing colony-forming units (ICFUs), progenitor cells that have the capability to differentiate into insulin-expressing colonies.2
Figure 5. A colony assay that allows functional and quantitative assessments of single progenitor cells. Numeration of the resulting colonies is used to calculate the frequency of the progenitor cells among the total plated cells. The composition of cells within each colony is indicative of the lineage potential of the initiating progenitor. Using this assay, we found that Ngn3-EGFP+ cells derived from murine embryonic stem cells were enriched for insulin-expressing colony-forming units (ICFUs), progenitor cells that have the capability to differentiate into insulin-expressing colonies.2
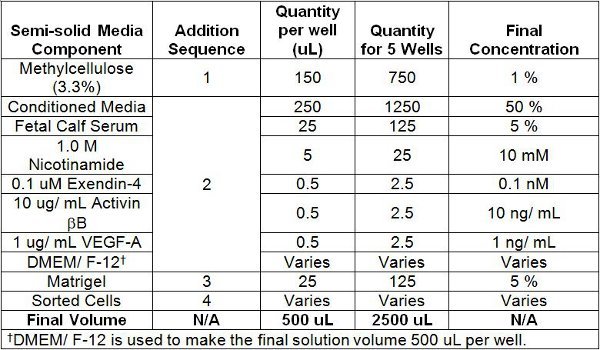 Table 1. Semi-solid Culture Media Components and Order of Addition.
Table 1. Semi-solid Culture Media Components and Order of Addition.
Discussion
Colony assays that allow quantitative and functional assessments of individual pancreatic progenitors have been limited so far, which greatly hampered the progress in the studies of pancreatic stem and progenitor cell biology. The essence of a colony assay is that it measures the intrinsic ability of progenitor cells, rather than solely their phenotype, allowing the functionality and lineage potentials of the progenitor cells to be observed. It is also a quantitative tool that can determine the number of progenitors in a given population. In the hematopoietic stem cell field, colony assays have played important roles in identification, as well as deciphering the growth factor requirements, interactions, and mechanisms governing the lineage commitment of these cells.6 In the field of pancreatic progenitor cell biology, pancreatic organ7,8 or bulk cultures9-13 have been devised, however, such assays do not address biological questions at the single cell level. Without single cell analyses, the existence of multi-potential pancreatic stem/progenitors cannot be proven unequivocally.14
Here we show that insulin-expressing colony-forming progenitors can be assayed using a three-dimensional, Matrigel-containing, semi-solid culture system. Although a suspension culture system15,16 has been developed which permits single adult pancreatic progenitors to proliferate and differentiate into "pancreatospheres", our colony assay provides additional advantages. First, our semi-solid culture media system allows plating of up to 25,000 sorted cells per 500 μL culture media per 24-well while maintaining the resulting colony formation in the linear range.2 In contrast, the suspension pancreatosphere assay requires the use of single cell deposition method from the cell sorter to allow single cell analyses, and thus a larger volume of culture media and incubator space are required. Therefore, our colony assay is relatively easy and cost-effective to conduct when large numbers of different cells need to be compared simultaneously for progenitor activities. Second, our semi-solid media allows uniform distribution of extracellular matrix components and growth factors, which allows both proliferation and differentiation to occur in the same well. In contrast, it is more difficult to provide such an environment, especially the matrix, to cells in suspension culture. We are aware that with our method it cannot be definitively proved that each colony is derived from a single cell, as two progenitors may be randomly positioned near each other and form one colony. However, this problem can be overcome by secondary analysis using single cell manipulation. Direct hand-picking of sorted single cells could definitively demonstrate the single cell origin of each colony.
In summary, the insulin-expressing colony-forming activity of progenitors in semi-solid media described herein provides unique opportunities for mechanistic studies of pancreatic progenitors at the single cell level. For example, in a prior report we found that addition of Matrigel, a crude mixture of extracellular matrix proteins, into the culture media is required for the formation of the insulin-expressing colonies from Ngn3-EGFP+ cells,2 suggesting the extracellular matrix is important for those progenitors. Using this colony assay, it will now be possible to further dissect and define specific matrix requirements for such activity. In addition to studying progenitors derived from mouse embryonic stem cells, we are currently investigating whether this in vitro assay system also supports formation of other distinctive colonies derived from the pancreas and liver of perinatal and adult mice, as well as from human cadaveric pancreatic tissue devoid of islets.
Disclosures
We have nothing to disclose.
Acknowledgments
The authors would like to thank Dr. Klaus Kaestner for the gift of murine Ngn3-EGFP embryonic stem cell line, Mrs. Lucy Brown and the Analytical Cytometry Core for assistance in cell sorting. This work was funded in part by NIH grants R21DK069997 and R01DK081587 awarded to H.T.K. and from California Institute for Regenerative Medicine (CIRM) training grants awarded to M.W. and L.S.
References
- Ku HT. Committing embryonic stem cells to early endocrine pancreas in vitro. Stem cells. 2004;22:1205–1217. doi: 10.1634/stemcells.2004-0027. [DOI] [PubMed] [Google Scholar]
- Ku HT. Insulin-expressing colonies developed from murine embryonic stem cell-derived progenitors. Diabetes. 2007;56:921–929. doi: 10.2337/db06-0468. [DOI] [PubMed] [Google Scholar]
- Chen C. Characterization of an in vitro differentiation assay for pancreatic-like cell development from murine embryonic stem cells: detailed gene expression analysis. Assay. Drug. Dev. Technol. 2011;9:403–419. doi: 10.1089/adt.2010.0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Perreault N, Brestelli JE, Kaestner KH. Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes. 2002;16:1488–1497. doi: 10.1101/gad.985002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Ogawa M. Differentiation and proliferation of hematopoietic stem cells. Blood. 1993;81:2844–2853. [PubMed] [Google Scholar]
- Gittes GK, Galante PE, Hanahan D, Rutter WJ, Debase HT. Lineage-specific morphogenesis in the developing pancreas: role of mesenchymal factors. Development. 1996;122:439–447. doi: 10.1242/dev.122.2.439. [DOI] [PubMed] [Google Scholar]
- Miralles F, Battelino T, Czernichow P, Scharfmann R. TGF-beta plays a key role in morphogenesis of the pancreatic islets of Langerhans by controlling the activity of the matrix metalloproteinase MMP-2. J. Cell. Biol. 1998;143:827–836. doi: 10.1083/jcb.143.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir S. In vitro cultivation of human islets from expanded ductal tissue. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7999–8004. doi: 10.1073/pnas.97.14.7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R. Characterization of endocrine progenitor cells and critical factors for their differentiation in human adult pancreatic cell culture. Diabetes. 2003;52:2007–2015. doi: 10.2337/diabetes.52.8.2007. [DOI] [PubMed] [Google Scholar]
- Gao R, Ustinov J, Korsgren O, Otonkoski T. In vitro neogenesis of human islets reflects the plasticity of differentiated human pancreatic cells. Diabetologia. 2005;48:2296–2304. doi: 10.1007/s00125-005-1935-8. [DOI] [PubMed] [Google Scholar]
- Suarez-Pinzon WL, Lakey JR, Brand SJ, Rabinovitch A. Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet beta-cells from pancreatic duct cells and an increase in functional beta-cell mass. J. Clin. Endocrinol. Metab. 2005;90:3401–3409. doi: 10.1210/jc.2004-0761. [DOI] [PubMed] [Google Scholar]
- Hao E. Beta-cell differentiation from nonendocrine epithelial cells of the adult human pancreas. Nature Medicine. 2006;12:310–316. doi: 10.1038/nm1367. [DOI] [PubMed] [Google Scholar]
- Ku HT. Minireview: pancreatic progenitor cells--recent studies. Endocrinology. 2008;149:4312–4316. doi: 10.1210/en.2008-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaberg RM. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nature Biotechnology. 2004;22:1115–1124. doi: 10.1038/nbt1004. [DOI] [PubMed] [Google Scholar]
- Rovira M. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc. Natl. Acad. Sci. U. S. A. 2010;107:75–80. doi: 10.1073/pnas.0912589107. [DOI] [PMC free article] [PubMed] [Google Scholar]