

Background: dG lesion derived from potent carcinogen benzo[a]pyrene causes mutations through DNA replication.
Results: Pol ζ and REV1 are essential to mutagenic, but not accurate, translesion DNA synthesis.
Conclusion: DNA synthesis across identical DNA damage can be catalyzed by a different set of polymerases.
Significance: The results have revealed an important role for DNA polymerases, pol ζ and REV1, in inducing mutations.
Keywords: DNA Damage Response, DNA Polymerase, DNA Repair, Mutagenesis Mechanisms, Protein-Protein Interactions, Y Family DNA Polymerases, Benzo[a]pyrene, Gene Knockout Cells, Pol zeta, Translesion DNA Synthesis
Abstract
The DNA synthesis across DNA lesions, termed translesion synthesis (TLS), is a complex process influenced by various factors. To investigate this process in mammalian cells, we examined TLS across a benzo[a]pyrene dihydrodiol epoxide-derived dG adduct (BPDE-dG) using a plasmid bearing a single BPDE-dG and genetically engineered mouse embryonic fibroblasts (MEFs). In wild-type MEFs, TLS was extremely miscoding (>90%) with G → T transversions being predominant. Knockout of the Rev1 gene decreased both the TLS efficiency and the miscoding frequency. Knockout of the Rev3L gene, coding for the catalytic subunit of pol ζ, caused even greater decreases in these two TLS parameters; almost all residual TLS were error-free. Thus, REV1 and pol ζ are critical to mutagenic, but not accurate, TLS across BPDE-dG. The introduction of human REV1 cDNA into Rev1−/− MEFs restored the mutagenic TLS, but a REV1 mutant lacking the C terminus did not. Yeast and mammalian three-hybrid assays revealed that the REV7 subunit of pol ζ mediated the interaction between REV3 and the REV1 C terminus. These results support the hypothesis that REV1 recruits pol ζ through the interaction with REV7. Our results also predict the existence of a minor REV1-independent pol ζ recruitment pathway. Finally, although mutagenic TLS across BPDE-dG largely depends on RAD18, experiments using Polk−/− Polh−/− Poli−/− triple-gene knockout MEFs unexpectedly revealed that another polymerase(s) could insert a nucleotide opposite BPDE-dG. This indicates that a non-Y family polymerase(s) can insert a nucleotide opposite BPDE-dG, but the subsequent extension from miscoding termini depends on REV1-polζ in a RAD18-dependent manner.
Introduction
The human genome constantly suffers from DNA damage induced by endogenous and exogenous sources, and the damage often blocks DNA synthesis catalyzed by replicative DNA polymerases unless it is removed before they reach damage sites. Upon blocking, a group of specialized DNA polymerases takes over DNA synthesis across a lesion, which is termed translesion DNA synthesis (TLS).5 TLS DNA polymerases can synthesize DNA across a damaged base often at a cost of mutations that mostly are targeted at the lesion site. Among 15 mammalian DNA polymerases (1), Y family DNA polymerases (2) play major roles in TLS. This family includes pol η, pol ι, pol κ, and REV1. These polymerases have in common a wide catalytic space that accommodates unusual base pairs (3, 4). Human pol η is the product of the gene responsible for xeroderma pigmentosum variant, an inherited disorder highly predisposed to skin cancer caused by sunlight exposure (5, 6). This polymerase conducts a very efficient and relatively accurate DNA synthesis across UV-induced cyclobutane pyrimidine dimers. Thus, pol η plays an important role in protecting cells from the deleterious effects of unrepaired cyclobutane pyrimidine dimers. Although pol ι has been reported to play a role in dealing with unrepaired oxidative DNA damage (7), the physiological substrates for pol ι, as well as pol κ, have not yet been established.
REV1, another member of the Y family, does not have a canonical DNA polymerase activity but instead shows a deoxycytidyl transferase activity (8, 9). This enzyme activity plays a role in TLS across certain classes of lesions (10, 11). REV1 also plays a noncatalytic role in TLS (12) and physically interacts with the other Y family polymerases (13, 14) and the REV7 subunit of pol ζ (15, 16). The biological significance of these interactions remains to be determined. Pol ζ, containing at least two subunits of REV3 and REV7, belongs to the B family DNA polymerases and also plays a very important role in TLS. REV3 has a catalytic activity, and REV7 is an accessory subunit whose function remains to be established. It is generally believed that this polymerase plays a role in extending a primer across from DNA lesions rather than in inserting a nucleotide opposite lesions (17, 18). REV1 and pol ζ are involved in the production of most spontaneous mutations in Saccharomyces cerevisiae (19) and DNA damage-induced mutations (20, 21). REV1 and pol ζ appear to function together during TLS, but the mechanism by which they cooperate has not yet been established.
The involvement of Y family polymerases in TLS is controlled by the monoubiquitination of proliferating cell nuclear antigen (PCNA), which is catalyzed by the RAD6-RAD18 ubiquitination complex (22, 23). This PCNA monoubiquitination is thought to allow the recruitment of Y family polymerases with one or two copies of ubiquitin-binding domain to stalled replication sites (24–26). Thus, most players involved in TLS and a major regulatory mechanism have been revealed. However, several important questions remain to be answered, including: (i) How is an appropriate polymerase(s) selected for each DNA lesion? (ii) What is the noncatalytic role for REV1? (iii) How does pol ζ have an access to a stalled site? (iv) How is a series of polymerase switches regulated during TLS?
Because pol η, pol κ, and pol ι could play a redundant role in inserting a nucleotide opposite various lesions, it is very informative to use Polk−/− Polh−/− Poli−/− triple (TKO), as well as double (DKO), gene knockout cells in TLS experiments. In this paper, we analyze a mechanism for TLS across a benzo[a]pyrene-derived dG adduct, using single- and multiple-gene knockout mouse embryonic fibroblasts (MEFs). Benzo[a]pyrene, a well known environmental carcinogen, is present in the smokes of tobacco and fossil fuel combustion. Its metabolite, benzo[a]pyrene dihydrodiol epoxide (BPDE), forms bulky DNA adducts mainly on dG. The results obtained in in vitro experiments by several groups have shown that human pol κ bypassed through BPDE-dG with inserting dC opposite the lesion, whereas human pol η preferentially inserted dA (27–29). On the other hand, human pol ι and yeast pol ζ inserted no nucleotide opposite this lesion (27–29). A study with siRNA-treated human cells (18) has suggested that pol κ or pol η engages in the insertion of a nucleotide opposite this lesion, and pol ζ catalyzes the extension from the inserted nucleotide. To elucidate the mammalian TLS mechanism for this adduct more precisely, we conducted site-specific experiments in genetically engineered MEFs. Our results have revealed that two distinct pathways can operate in the TLS across BPDE-dG: the miscoding TLS depends on REV1 and pol ζ, but the accurate TLS does not necessarily require these two proteins. This finding indicates that a nucleotide inserted opposite the lesion influences the subsequent extension step in multiple TLS pathways. Our results also have shown that the insertion of a nucleotide opposite BPDE-dG can be catalyzed relatively efficiently by non-Y family polymerase(s). Furthermore, the results of our biological and biochemical experiments strongly suggest that REV1 recruits pol ζ to a stalled replication site through the interaction with the REV7 subunit of pol ζ, and this recruitment mechanism plays an important role for pol ζ to catalyze TLS.
EXPERIMENTAL PROCEDURES
Cell Lines
Rev1−/− MEF (30), Rad18−/− cells (31), and the introduction of human REV1 cDNA to Rev1−/− MEF (30) were described before. A Rev3L−/− MEF line was generously provided by Dr. Richard D. Wood (University of Texas M. D. Anderson Cancer Center). The Polk−/− Polh−/− Poli−/− TKO MEF was generated as follows: Polk−/− mice (32) were mated with 129-strain mice carrying a nonsense mutation in the Poli gene (33) to obtain Polk−/− Poli−/− DKO mice. The DKO mice were mated with Polh−/− mice (34) to generate TKO mice. TKO MEFs were established as spontaneously arisen immortalized cells. Immortalized Polh−/− Poli−/− DKO MEFs were obtained similarly by a cross of single-gene knockout mice and spontaneous immortalization (34). Their genotypes were confirmed by genomic PCR (supplemental Fig. S1). The UV sensitivity of various gene knockout MEFs is shown in supplemental Fig. S2.
Construction of Gapped Site-specifically Modified Plasmid
The synthesis of oligonucleotides containing (+)-trans-anti-BPDE-dG was reported previously (35). The 5′ end of a modified 15-mer, 5′-TCCTCGTGBCCTCTC, where B represents BPDE-dG, was ligated to a 14-mer, 5′-CCATCTCCTCCATC, and its 3′ end was elongated by five nucleotides using Klenow enzyme, following the annealing to a complementary scaffold 54-mer, 5′-AGGTAGAGAGGTCACGAGGAGATGGAGGAGATGGA20. The resulting 34-mer, 5′-CCATCTCCTCCATCTCCTCGTGBCCTCTCTACCT, was purified by electrophoresis in a denaturing 20% (w/v) polyacrylamide gel and then annealed to its complementary uracil-containing 34-mer, 5′-TTCCAGGUAGAGAUCUCACUAGGAGAUGGAGGAG. This annealing resulted in the formation of four-nucleotide overhangs on both ends and three base mismatches opposite and adjacent to the adduct (see Fig. 1B). Annealed oligonucleotides were incorporated into pMTEX4 (Ref. 30; see Fig. 1A) by ligating to BsaI and BsmBI sites of the vector. Closed circular DNA was isolated by ultracentrifugation in a cesium chloride-ethidium bromide continuous gradient. To make a gap opposite BPDE-dG, 500 ng of a modified construct was incubated, just prior to transfection, with 1 unit of uracil-DNA glycosylase (New England Biolabs) for 30 min at 37 °C, followed by treatment with 10 units of apurinic/apyrimidinic endonuclease I (New England Biolabs) for 30 min at 37 °C (see Fig. 1B). These treatments made BPDE-dG resistant to nucleotide excision repair.
FIGURE 1.

Preparation of BPDE-dG-bearing plasmid and oligonucleotide probes used for analysis. A, vector-employed, BPDE-dG insertion site (open circle) is located between BsaI and BsmBI. Py, mouse polyoma virus; ori, replication origin; amp, ampicillin resistance gene; blaS, blasticidin S resistance gene. B, preparation of gapped plasmid, B with gray shading represents BPDE-dG. Note three mismatches at 5′-BCC/5′-UCU. A, C, G, and T probes determine a TLS event targeting BPDE-dG, whereas L and R probes determine the presence of the inserted modified oligonucleotide.
TLS Experiments in MEFs
The cells were cultured under 5% (v/v) CO2 at 37 °C in Dulbecco's modified Eagle's medium supplemented with fetal bovine serum (10%, v/v), penicillin (100 units/ml), and streptomycin (100 μg/ml). The cells (1 × 106) were seeded in a 25-cm2 flask and cultured overnight, after which they were transfected overnight with 500 ng of a freshly prepared, gapped construct together with 250 ng of internal control plasmid, pMTKm, by the FuGENE 6 transfection reagent (Roche Applied Science). pMTKm was constructed by replacing the blasticidin S and ampicillin resistance genes in pMTEX4 with the kanamycin resistance gene. The following day, the cells were detached by treating with trypsin-EDTA, transferred to a 150-cm2 flask, and cultured for 3 days.
Analysis of TLS Events
Progeny plasmids were recovered from cells by the method of Hirt (36) and analyzed for translesional events. The recovered plasmids were treated with DpnI (4 units) and BglII (20 units) for 1 h to remove unreplicated input DNA and progeny plasmids derived from the residual complementary strand, respectively. NEB10-β electrocompetent Escherichia coli (araD139, Δ(ara,leu)7697, fhuA, lacX74, galK16, galE15, mcrA, f80d(lacZΔM15), recA1, relA1, endA1, nupG, rpsL, rph, spoT1, Δ(mrr-hsdRMS-mcrBC)) (New England Biolabs) was transformed with progeny plasmids and plated on YT(1×) agar plates containing ampicillin (100 μg/ml) and blasticidin S (50 μg/ml) or kanamycin (50 μg/ml) alone. Because the adduct incorporation site is located very close to the blasticidin S resistance gene (see Fig. 1A), transformants carrying progeny plasmid with deletions around the adduct site will not grow on a blasticidin S-containing plate and are excluded from the analysis. E. coli transformants with the internal control plasmid, pMTKm, will grow on plates containing kamamycin. The ratios of the number of ampicillin/blasticidin S-resistant colonies (TLS products) to the number of kanamycin-resistant colonies (internal control) were determined for each MEF line, and a relative TLS efficiency was determined. E. coli colonies were picked up individually and analyzed for a sequence of the adducted region by oligonucleotide hybridization using probes shown in Fig. 1B. Probes L and R were used to confirm the presence of the oligonucleotide insert and to detect untargeted mutations and small deletions around the adduct site. These mutants were also excluded from the analysis. Probes A, C, G, and T detected targeted base substitutions. Examples of oligonucleotide hybridization are presented in supplemental Fig. S3. DNA sequencing was conducted when any of these four probes did not hybridize.
Yeast Three-hybrid Assay
The cDNA of the human REV3 gene was generously provided by Dr. P. E. M. Gibbs with the permission of Dr. C. W. Lawrence (University of Rochester). The cDNAs of human REV1 and REV7 genes were purchased from OriGene Technologies (Rockville, MD). cDNAs were cloned in frame into the multiple cloning sites of pBridge and pGADT7 vectors (Clontech) by standard molecular biology techniques. pBridge has two cloning sites: constitutive site I and Met25 promoter-regulated inducible site II. The expression of a gene cloned into the site II is repressed in a medium containing 1 mm methionine. When PCR was employed, corresponding sequences were confirmed by DNA sequencing. The principle of yeast three-hybrid assay has been depicted in Fig. 3A. S. cerevisiae AH109 (MATa, trp1-901, leu2-3, 112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2::GAL1UAS-GAL1TATA-HIS3, GAL2UAS-GAL2TATA-ADE2,URA3::MEL1UAS-MEL1TATA-lacZ, MEL1) (Clontech) was transformed with pBridge and pGADT7 constructs by the Yeastmaker yeast transformation system 2 (Clontech). Yeast was plated on a synthetic dextrose/−Leu/−Trp agar plate and incubated at 28 °C for 48 h. Well isolated colonies were picked up and suspended in a 0.9% (w/v) NaCl solution, and a 10-μl aliquot was spotted on two-nutrient (−Leu/−Trp), four-nutrient (−Ade/−His/−Leu/−Trp), and five-nutrient (−Ade/−His/−Leu/−Trp/−Met) dropout synthetic dextrose agar plates. The plates were incubated at 28 °C for 48–72 h, and the interaction between proteins was judged based on the growth of yeast on the nutrients dropout synthetic dextrose plates (see Fig. 3B).
FIGURE 3.

REV7-mediated interaction between REV1 and REV3 in yeast. A, principle of the assay. GAL4 BD-fused bait protein (REV3) and nontagged REV7 are expressed from pBridge. The expression of REV7 from the promoter, PMet25, is repressed in the presence of methionine in medium, and hence the gene (REV7) cloned in the cloning site II is not expressed. It becomes active in the absence of methionine and expresses the gene. The GAL4 AD-fused prey protein (REV1) is expressed constitutively from pGADT7. When three gene products form a heterotrimer (panel a), a marker gene is expressed. The absence of a mediator protein (REV7) (panel b) or overexpression of a mediator protein (panel c) causes a failure of the formation of a heterotrimer, and a marker gene is not expressed. B, formation of heterotrimer as judged by the growth of a yeast host on selective media. The numbers correspond to those in Table 2, showing the combination of genes tested. Panels I, II, and III are two-nutrient (Leu and Trp), four-nutrient (Leu, Trp, Ade, and His), and five-nutrient (Leu, Trp, Ade, His, and Met) dropout media, respectively.
Mammalian Three-hybrid Assay
The Matchmaker Mammalian Assay Kit 2 (Clontech) was used. Instead of pVP16 (activation domain vector), we employed pIRESneo2 (Clontech) to express two proteins from one vector simultaneously. The C terminus corresponding to the last 111 amino acids (1141–1251) of human REV1 cDNA fused in frame to the VP16 activation domain (AD) was cloned into the multiple cloning site of pIRESneo2. The full-length human REV7 cDNA replaced the G418 resistance gene. The XbaI-SalI fragment (681 base pairs, amino acid 1776–2003) of human REV3 was cloned into pM (a vector with GAL4 DNA-binding domain) in frame. This fragment codes for a protein region interacting with REV7. Each construct was confirmed by DNA sequencing. Experiments were conducted according to the manufacturer's protocol, using COS7 cells as a host and pG5SEAP as a gene activation assay vector. Protein-protein interaction was determined by measuring alkaline phosphatase secreted into a culture medium, using a Great EscAPe SEAP chemiluminescence detection kit (Clontech). The principle of this assay is similar to that of the yeast three-hybrid assay (see Fig. 3A) except that the expression of REV7 is not inducible, but constitutive, in this assay.
RESULTS
Fidelity of TLS across BPDE-dG Adduct
In our previous site-specific studies (30, 37), we used covalently closed, circular double-stranded plasmid containing a single lesion and three consecutive base mismatches at the adducted region. The advantage of the use of this construct is to allow us to study TLS taking place at a replication fork. A disadvantage is that a lesion could be subject to DNA repair, which undermines the quantitative determination of TLS efficiency. BPDE base adducts are known to be good substrates for nucleotide excision repair, and all of our MEFs are proficient in this repair. To obviate this problem, we inserted BPDE-dG in a gapped, single-stranded region of a plasmid (Fig. 1B; refer to “Experimental Procedures” for details). In this construct, TLS likely takes place during gap-filling reaction.
First, the fidelity of TLS was determined in wild-type MEFs (Fig. 2A and supplemental Table S1). This analysis revealed a high frequency (>90%) of site-specific miscoding events consisting of BPDE-dG G* → T (73%), G* → A (12%), G* → C (1%), and others (5%). Others included mutations at the 5′ and 3′ nearest neighboring bases and multiple mutations together with targeted mutations. Thus, a frequency of correct TLS, G* → G events generated by the insertion of correct dC opposite BPDE-dG accounted for only 9%. This coding spectrum was highly reproducible: refer to the coding events observed in wild-type MEFs in Fig. 2 (A and C). Supplemental Tables S1 and S2 provide detailed numbers from which Fig. 2 was generated.
FIGURE 2.

Relative efficiency and targeted coding specificity of translesion DNA synthesis across BPDE-dG (A and C) and H-ϵdC (B). A and B, TKO, Polk−/− Polh−/− Poli−/− triple-gene knockout MEF. k/i, Polk−/− Poli−/− double-gene knockout MEF; h/i, Polh−/− Poli−/− double-gene knockout MEF. A value for wild-type MEF is set to 100%. Coding specificity is color-coded. Red, BPDE-dG (G*) or H-ϵdC (C*) → T; blue, G* or C* → A; purple, G* or C* → C; green, G* or C* → G; black, others. Detailed numbers, %, and statistics are reported in supplemental Tables S1 and S2.
Relatively Efficient TLS in Absence of Three Y Family Polymerases of Pol κ, Pol η, and Pol ι
TLS consists of two steps: insertion of a nucleotide opposite a lesion and extension from this newly formed terminus. It is widely thought that pol η, pol κ, and pol ι are involved in the insertion step with occasional participation in the subsequent extension step. To examine whether these polymerases play critical roles during TLS across the site-specific BPDE-dG, we employed pol κ/pol η/pol ι-deficient TKO MEFs. The inactivation of these polymerases is expected to result in a marked defect in TLS if any other polymerases cannot catalyze nucleotide insertion opposite this lesion. Unexpectedly, the efficiency and coding specificity of TLS were not greatly affected in TKO MEFs (Fig. 2A and supplemental Table S1). Similar results were obtained in pol κ/pol ι- and pol η/pol ι-deficient DKO MEFs, although modest changes were observed in coding specificity (supplemental Table S1). These results imply that a non-Y family polymerase(s) can insert a nucleotide, largely incorrect nucleotides (dA and dT), opposite BPDE-dG in the absence of those three Y family polymerases.
We also examined TLS across heptanone-etheno dC (H-ϵdC), using the same gapped DNA construct. In wild-type MEFs, H-ϵdC was highly miscoding by directing the insertion of dTMP or dAMP (Fig. 2B and supplemental Table S1) as previously reported (30, 37). In TKO MEFs, TLS efficiency dropped to 21% of that in wild-type MEFs, indicating that the TLS across H-ϵdC depends on pol η, pol κ, and/or pol ι. Two TKO MEF lines (2091 and 2095) separately established from littermates were more sensitive to UV irradiation than pol κ-, pol η-, and pol κ/pol ι-deficient MEFs (supplemental Fig. S2). These results indicate that TKO MEFs are indeed deficient in pol η, pol κ, and pol ι, thus supporting the above interpretation that the insertion of a nucleotide opposite BPDE-dG can be catalyzed relatively efficiently by a non-Y family polymerase(s) in the absence of the three Y family polymerases.
Deficiency in Rev1 or Rev3 Gene Greatly Affects TLS
In contrast to the result in TKO MEFs, inactivation of REV1 or pol ζ (REV3) greatly affected not only the efficiency but also the coding specificity of TLS past BPDE-dG (Fig. 2C and supplemental Table S2). The inactivation of the Rev1 gene reduced TLS efficiency to 41% of that in wild-type MEFs and significantly increased the frequency of correct G* → G events from 7 to 48% with a concomitant decrease in the frequency of G* → T transversions from 74 to 32% (supplemental Table S2). This increase in the frequency of correct TLS events in REV1-deficient MEFs excludes the possibility of REV1 serving as a major dCMP inserter. The knockout of the Rev3L gene, which codes for the catalytic subunit of pol ζ, caused similar but more drastic changes: the TLS efficiency decreased to 13% of that in wild-type MEFs and the correct (G* → G) TLS events accounted for almost all TLS (94%). The effects of the inactivation of the Rev1 and Rev3L genes on the TLS events were very reproducible, and similar effects of their inactivation on TLS were also observed with a different 5′ neighboring base (5′-TG*C instead of 5′-GG*C) (data not shown). These results indicate that both pol ζ and REV1 are involved in the major mutagenic TLS across BPDE-dG and that they are not critical to the minor accurate TLS pathway. Pol ζ and REV1 contribute to the mutagenic pathway through the catalytic and noncatalytic functions, respectively. Our results also indicate that REV1 is not completely epistatic to pol ζ. The greater effects of the inactivation of the Rev3L gene have also been observed with the bypass of H-ϵdC.6 These findings imply that pol ζ has a minor REV1-independent pathway to access replication-blocked sites.
The REV1 C-terminal Region Is Critical for Pol ζ-dependent TLS
The above results support the idea that REV1 and pol ζ are involved in the same mutagenic TLS pathway for BPDE-dG and that the dCMP transferase activity of REV1 does not play a role in this TLS. Because the accessory subunit of pol ζ, REV7, is known to interact with a C-terminal region of REV1 (15), it seems likely that this interaction is important to the collaboration of REV1 with pol ζ. To investigate the biological significance of this interaction, we expressed human REV1 protein (1251 amino acids) and its mutant lacking the C-terminal 111 amino acids, hREV1(1–1140), in Rev1−/− MEFs (refer to supplemental Fig. S4 for their expressions) and determined TLS events. As shown above, the major coding events are G* → T and G* → G in wild-type and Rev1−/− MEFs, respectively (Fig. 2C and supplemental Table S2). We focused on these coding events to investigate the effects of the expression of exogenous hREV1 in the complementation experiments. The expression of the intact REV1(1–1251), although it did not fully complement the REV1 deficiency, caused a decrease in the frequency of correct TLS from 55 to 31% and hence the increase in mutagenic TLS from 45 to 69% (Table 1). This increase was mainly caused by that in G* → T transversions (from 26 to 49%), and G* → T transversions became predominant among the coding specificities. On the other hand, the expression of the deletion mutant failed to reverse the coding specificity, and correct TLS remained dominant: 64%. The higher frequencies of correct G* → G TLS events in both complemented Rev1−/− MEFs, especially those complemented with hREV1(1–1251) (wild type:hREV1(1–1251) = 9%:31%), might result from the dCMP transferase activity of ectopically overexpressed REV1, unlike the endogenous level of REV1. (Note that the dCMP transferase domain is located upstream of the deletion and intact in the deletion mutant. This mutant REV1 partially complemented the REV1 deficiency in the cytotoxicity test of 4-nitroquinoline-N-oxide (supplemental Fig. S5), suggesting the contribution of the dCMP transferase activity to the partial recovery of its resistance to this DNA-damaging agent.) The biological results indicate that the REV1 C-terminal 111-amino acid region is important for the noncatalytic function of REV1. Although an experiment using a REV1 point mutant that is specifically defective in interacting with REV7 should be conducted before reaching a conclusion, our results suggest that the REV7-mediated formation of the REV1-pol ζ complex is critical to the pol ζ-catalyzed extension reaction.
TABLE 1.
Complementation of Rev1−/− MEF with intact and C-terminally deleted hREV1for TLS coding events
| DNA lesion | Rev1−/− MEF complemented with: | Total effective no. sequenced | Correct TLS |
Incorrect TLS |
|||
|---|---|---|---|---|---|---|---|
| G* → G | G* → T | G* → A | G* → C | Others | |||
| BPDE-dG (G*) | Wild type | 96 (100%)a | 9 (9%)b | 65 (68%)b | 13 (14%) | 3 (3%) | 6 (6%) |
| Rev1−/− | 96 (100%) | 53 (55%)b | 25 (26%)b | 14 (15%) | 1 (1%) | 3 (3%) | |
| hREV1 (1–1251) | 177 (100%)a | 55 (31%)b | 86 (49%)b | 25 (14%) | 4 (2%) | 7 (4%) | |
| hREV1 (1–1140) | 186 (100%) | 119 (64%)b | 44 (24%)b | 19 (10%) | 1 (0.5%) | 3 (2%) | |
a The distribution of coding events are statistically significant at p < 0.001 when compared with that in Rev1−/− cells, and they are highlighted with bold type.
b p < 0.001 when compared with the corresponding event in Rev1−/− cells. The significant numbers are highlighted with bold type.
Formation of Ternary Complex of Human REV1, REV7, and REV3 Proteins
The above results suggest a vital role for the interaction between REV1 and REV7 in recruiting pol ζ to a replication-blocked site. To examine this possibility, we conducted yeast and mammalian three-hybrid assays. We employed pBridge and pGADT7 vectors to investigate the ternary interaction among the human REV1, REV3, and REV7 proteins in yeast cells (Fig. 3). Because REV1, when cloned in various DNA-binding domain (BD) vectors, was found to show positive signals with empty AD vectors (such as pGADT7), the REV1 full-length cDNA and its fragments were cloned into pGADT7 to fuse in frame to the GAL4 activation domain. Fragments of REV3 cDNA were cloned into the cloning site I of pBridge to form the GAL4 DNA-binding domain fusion protein. The REV7 cDNA was cloned into the cloning site II that was negatively controlled by methionine (refer to Fig. 3A for the principle of this assay). As shown previously (15), the interaction between REV7 and REV1 and that between REV7 and the C-terminal half (amino acid 1776–3130) of REV3 were readily detected (No. 1 and No. 2 of Table 2). A positive signal for interaction between REV3(1776–3130) and REV1 was detected in the presence of REV7 cDNA (No. 5) but not in its absence (No. 3 and No. 4). Interestingly, such a positive signal was observed under the uninduced conditions (i.e. in the presence of 1 mm methionine in the medium) but not the induced conditions (i.e. in the absence of methionine) (No. 6). The presence of the Met25 promoter at the cloning site II was necessary for the ternary complex formation (No. 7). Refer to Fig. 3B for the growth of yeast on selective media. Thus, the regulation of REV7 expression by the Met25 promoter appears to be leaky, and a small amount of REV7 may be optimal for the formation of the ternary complex of REV1, REV3(1776–3130), and REV7 proteins. Therefore, the ternary complex seems to be formed on a delicate balance among the REV proteins, and overexpression of REV7 causes preferential formations of REV7-REV1 and REV7-REV3(1776–3130) binary complexes, thereby inhibiting the formation of the REV1-REV7-REV3(1776–3130) ternary complex (Fig. 3A). The experiments using various fragments of REV3(1776–3130) and REV1 identified regions responsible for the interaction (No. 9 to No. 22): the C-terminal region (1141–1251) of REV1 and the central region (1776–2003) of REV3 containing the REV7-interacting sequence were necessary and sufficient for the interaction. These results clearly show that REV7 mediates interaction between REV3(1776–3130) and REV1.
TABLE 2.
Analysis for ternary complex formation of REV proteins by yeast three-hybrid assay
| No. | pBridge |
pGADT7 | Interactiona | |
|---|---|---|---|---|
| Constitutive site I | Inducible site II | |||
| 1 | REV7 | REV1 | + | |
| 2 | REV7 | REV3 (1776–3130) | + | |
| 3 | REV3 (1776–3130) | (b) | REV1 | − |
| 4 | REV3 (1776–3130) | REV1 | − | |
| 5 | REV3 (1776–3130) | (REV7)b | REV1 | + |
| 6 | REV3 (1776–3130) | REV7 | REV1 | − |
| 7 | REV3 (1776–3130) | (ΔPMet25, REV7)c | REV1 | − |
| 8 | REV3 (1776–3130) | ΔPMet25, REV7 | REV1 | − |
| 9 | REV3 (1776–3130) | (REV7) | − | |
| 10 | REV3 (1776–3130) | REV7 | − | |
| 11 | REV3 (1776–3130) | (REV7) | REV1 (1141–1251) | + |
| 12 | REV3 (1776–3130) | REV7 | REV1 (1141–1251) | − |
| 13 | REV3 (1776–3130) | (REV7) | REV1 (1–1140) | − |
| 14 | REV3 (1776–3130) | REV7 | REV1 (1–1140) | − |
| 15 | REV3 (1776–3130) | REV1 (1141–1251) | − | |
| 16 | REV3 (1776–3130) | (ΔPMet25, REV7) | REV1 (1141–1251) | − |
| 17 | REV3 (1776–2003) | (REV7) | REV1 | + |
| 18 | REV3 (1776–2003) | (REV7) | REV1 (1141–1251) | + |
| 19 | REV3 (2004–3130) | (REV7) | REV1 | − |
| 20 | REV3 (2004–3130) | (REV7) | REV1 (1141–1251) | − |
| 21 | REV3 (1776–2003) | REV1 | − | |
| 22 | REV3 (1776–2003) | REV1 (1141–1251) | − | |
a Determined by the growth on a plate with a four- or five-nutrient dropout medium (refer to Fig. 3B).
b REV7 was induced by removing methionine from four-nutrient dropout medium. Parentheses indicate no induction.
c Deletion of Met25 promoter.
To test for the interaction in mammalian cells, we devised a mammalian three-hybrid assay by employing pIRESneo2 instead of pVP16 used in the Matchmaker mammalian assay kit 2 (Clontech). A fusion of the VP16 activation domain with REV1(1141–1251) and untagged REV7 was transferred to and expressed from pIRESneo2, and the GAL4 DNA-binding domain fused to REV3(1776–2003) was expressed from another vector pM. These two plasmid derivatives were introduced into COS7 cells, together with the reporter plasmid pG5SEAP that contains the alkaline phosphatase gene under the control of a GAL4-responsible element. Interaction between the REV1 and REV3 fragments was monitored periodically by measuring the activity of alkaline phosphatase secreted into a culture medium. The results showed positive interaction between the REV3 and REV1 fragments only under the co-expression of REV7 (Fig. 4). We repeated such experiments several times under different conditions and obtained the same results. The results of this assay confirmed the phenomenon observed in the yeast three-hybrid assay: REV7 mediates the interaction between REV3 and REV1.
FIGURE 4.
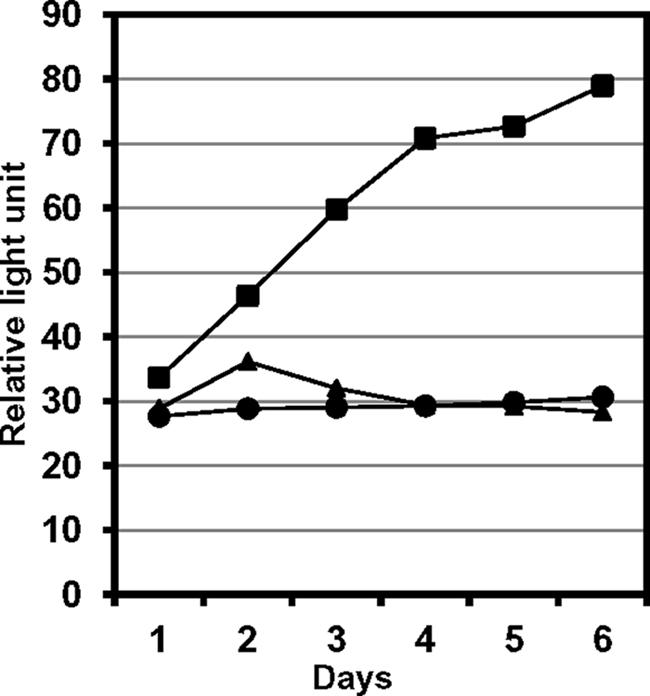
REV7-mediated interaction between REV1 and REV3 in mammalian cells. The following genes were expressed in the monkey kidney cell line, COS7. Square, GAL4 BD-REV3(1776–2003) fusion, VP16-AD-REV1(1141–1251) fusion, and full-length REV7; circle, BD-REV3(1776–2003) and AD-REV1(1141–1251); triangle, BD-REV3(1776–2003), AD and REV7. Culture media were collected from day 1 through day 6 following transfection and assayed for BD-REV3(1776–2003):AD-REV1(1141–1251) interaction by a Great EscAPe SEAP chemiluminescence detection kit (Clontech). Similar results were obtained in several experiments conducted under different conditions. Refer to the legend to Fig. 3 for the principle of assay and “Experimental Procedures” for details.
TLS in Rad18−/− MEF
We studied the effect of RAD18 inactivation on the TLS across BPDE-dG. RAD18 is an E3 ubiquitin ligase and functions together with RAD6 to monoubiquitinate PCNA upon DNA damage (22, 23). Consistent with the current model that monoubiquitinated PCNA contributes to recruiting TLS polymerases to a replication-stalled site, all of the mammalian Y family polymerases contain one or two copies of ubiquitin-binding domain (38). In Rad18−/− MEFs, the TLS efficiency decreased to 26% of that in wild-type MEFs (Fig. 2C and supplemental Table S2), which was much lower than in TKO MEFs. Because Y family polymerases are not absolutely essential for the nucleotide insertion opposite BPDE-dG (see above), we ascribe this decrease to the impaired extension across from BPDE-dG. The increase from 7 to 29% in the frequency of G* → G correct TLS events observed in Rad18−/− MEFs is reminiscent of that in the Rev1−/− MEFs (supplemental Table S2). Thus, the effect of RAD18 inactivation on the TLS appears to be mainly caused by the lack of participation of REV1.
DISCUSSION
Our results have shown that at least two distinct TLS pathways can operate on BPDE-dG in MEFs: REV1 and pol ζ are required for the predominant incorrect TLS, but the three Y family polymerases (pol η, pol κ, and pol ι) are not essential. In contrast, neither REV1 nor pol ζ is critical to the minor correct TLS, although they may participate in this TLS in wild-type MEFs. We also have shown that REV1 recruits pol ζ through the interaction with its REV7 subunit. Although this is the major recruitment mechanism of pol ζ, a REV1-independent minor pathway also exists.
TLS across BPDE-dG
TLS is an important mechanism for protecting cells from lethal effects of unrepaired DNA damage. Recent studies have revealed the general TLS mechanism as outlined in the introduction. However, TLS is heterogeneous even for an identical adduct because the stereochemistry and conformation of DNA adducts, the surrounding DNA sequence context, and DNA polymerases involved greatly influence the outcome of TLS. We previously reported that frequencies of base pair substitution mutations targeted chemically defined BPDE-dG adducts were greatly influenced by a sequence context (35), suggesting that a conformational difference of the adduct influences nucleotide insertion. Thus, the much higher miscoding frequency (>90%) observed in our study compared with that (<14%) reported by Livneh and co-workers (18) is likely due to a difference in the sequence context employed. Another major difference between the results of the two studies is that in polymerases involved in TLS: Livneh and co-workers have concluded that pol κ and pol ζ contribute to the error-free TLS across BPDE-dG by inserting dCMP opposite the lesion and extending from this terminus, respectively. Our results indicate that pol ζ is critical to the mutagenic, but not accurate, TLS (Fig. 2C and supplemental Table S2). Pol κ is a good candidate for catalyzing this accurate TLS in pol ζ-deficient MEFs because this polymerase was shown to catalyze accurate TLS of BPDE-dG in vitro (27–29). We cannot obtain a definite answer to this possibility at present because of the lack of pol κ-pol ζ DKO MEFs. The reason for this discrepancy is not clear at present, but the efficiency of extension even from the correct dC terminus might be influenced by a surrounding sequence.
Our results obtained in TKO MEFs also indicate that none of pol η, pol ι, and pol κ is critically required for the TLS across BPDE-dG in the current sequence context (Fig. 2A and supplemental Table S1). The results suggest that non-Y family polymerase(s) can catalyze nucleotide insertion opposite BPDE-dG, but the primer extension greatly depends on pol ζ. Two possibilities are envisaged regarding a mechanism for the nucleotide insertion opposite BPDE-dG (Fig. 5). The first possibility is that a replicative polymerase (pol δ or pol ϵ), which first encounters the lesion, adds a nucleotide to the primer terminus before it dissociates from a template. Narita et al. (39) have recently shown that although human pol δ mostly stops synthesis at the end of a template, the enzyme also adds one or more nucleotides (mostly one dAMP) to the terminus of a fully elongated daughter strand. When pol δ or pol ϵ adds a nucleotide opposite BPDE-dG prior to dissociation, pol ζ should be essential for the extension from incorrect termini. On the other hand, when pol δ or pol ϵ adds dCMP, this terminus could also be extended by a polymerase such as pol κ as verified by in vitro extension studies (27–29). A second scenario is that another non-Y family polymerase catalyzes nucleotide insertion opposite BPDE-dG in the absence of pol η, polκ, and pol ι when a replicative polymerase dissociates without adding any nucleotide upon the block of DNA synthesis by the adduct. However, in wild-type MEFs, it is possible that pol κ inserts dCMP and pol η and/or pol ι insert(s) a miscoding nucleotide opposite BPDE-dG, as shown in in vitro studies (28), and then pol ζ extends from the new terminus.
FIGURE 5.

Model for TLS across BPDE-dG and recruitment of pol ζ to a replication-stalled site. Following the insertion of a nucleotide (N) opposite BPDE-dG (red triangle), extension from this newly created terminus requires pol ζ when N is incorrect A, T, or G. On the other hand, when N is correct, C (the extension) does not necessarily depend on pol ζ. Pol κ might conduct this extension. Pol ζ is recruited by REV1 through the interaction between REV1 and REV7, a subunit of pol ζ (thick red arrow). Then REV1 may interact with monoubiquitinated PCNA at its ubiquitin-binding motifs to bring pol ζ to a stalled site. This is the major pol ζ recruitment pathway. A REV1-independent minor pathway (thin red arrow) also exists, but its mechanism is unclear. U, 1, 3, and 7 represent monoubiquitin, REV1, REV3 and REV7, respectively.
Role for REV1 in TLS
Among the TLS-specialized DNA polymerases, REV1 plays a unique role in TLS by serving as a protein platform (40) although its deoxycytidyl transferase activity plays a role for TLS past some lesions (10, 11). It is generally thought that REV1 and pol ζ function together during TLS. However, the mechanism by which they cooperate is still unclear in mammalian cells. Our results show that REV7 mediates the interaction between REV3 and REV1 and that the C terminus of REV1 and the REV7-interacting region of REV3 are involved in this interaction. These results of protein-protein interaction among REV proteins agree with those reported by Hara et al. (41). In their report, however, the biological significance of this interaction has not yet been established because they used, in a toxicity test, a mutant REV7 that did not interact with either REV3 or REV1, resulting in the lack of the formation of even pol ζ. We show that the REV1 C terminus is important to the pol ζ-catalyzed error-prone TLS (Table 1). The same region of REV1 interacts with the other Y family polymerases (13, 14), but its biological significance is unclear at present. The importance of the C terminus of REV1 protein to DNA damage tolerance has been reported in yeast (42) and chicken DT40 cells (26). Our results, together with those of Hara et al., support the hypothesis that REV1 guides pol ζ to a stalled site (Fig. 5). However, when we compare the effects of the Rev3L and Rev1 gene knockouts on TLS across BPDE-dG (Fig. 2C and supplemental Table S2) and H-ϵdC,6 we have consistently observed that the knockout of the Rev3L gene induces a greater effect than does that of the Rev1 gene, suggesting the presence of a REV1-independent minor pathway for pol ζ recruitment.
PCNA Monoubiquitination and REV1 Recruitment
The knockout of the Rad18 gene greatly affected mutagenic TLS across BPDE-dG (Fig. 2C and supplemental Table S2), suggesting that the REV1/pol ζ-catalyzed TLS is under the regulation by this gene. RAD18, in a complex with RAD6, monoubiquitinates PCNA upon DNA damage, and therefore, REV1 containing two copies of ubiquitin-binding domain in its C-terminal region is likely to bind to monoubiquitinated PCNA to recruit pol ζ to a stalled replication fork (Fig. 5). Guo et al. (43) reported that mouse REV1, which does not contain a typical PCNA-interacting peptide sequence, binds to PCNA via the BRCT (carboxyl terminus of BRCA1 protein) domain located near the N terminus. However, de Groote et al. (44) recently argued against this assertion by showing the BRCT domain, together with a further N-terminal region, to bind to recessed, phosphorylated 5′ ends of double-stranded DNA. REV1 probably has a PCNA-binding site(s) in its C-terminal region (12, 46, 47). Furthermore, although Akagi et al. (48) have shown that the accumulation of human endogenous REV1 into locally UV-irradiated regions of nucleus depends on the interaction with pol η, Andersen et al. (45) have reported that the recruitment of the human endogenous REV1 is independent of the interaction with pol η. Our results obtained in TKO, DKO, and Rad18−/− MEFs also suggest that the recruitment of REV1 is independent of the interaction with pol η. There may be multiple pathways in the recruitment of REV1 as observed in the recruitment of pol ζ. Currently, it is still very controversial on the mechanism by which REV1 is recruited to a replication-blocked site, and further studies are necessary to clarify this intriguing question.
Supplementary Material
Acknowledgments
We thank Dr. Peter E. M. Gibbs (University of Rochester Medical Center) and Dr. Richard D. Wood (University of Texas M. D. Anderson Cancer Center) for the generous gifts of human REV3 cDNA and Rev3 gene knockout MEFs, respectively. We also thank Youngsil Jo (Stony Brook University) for technical assistance and Kenji Suzuki (Gakushuin University) for help completing the experiment shown in supplemental Fig. S2.
This work was supported, in whole or in part, by National Institutes of Health Grants CA076163 and ES018833 (to M. M.).

This article contains supplemental Tables S1 and S2 and Figs. S1–S5.
Anastasia Tsaalbi-Shtylik, Piya Temviriyanukul, Lieneke Uittenboogaard, Keiji Hashimoto, Richard Heideman, Johan Verspuy, Martin van der Valk, Raoul Kuiper, Daniella Salvatori, Willy Baarends, Heinz Jacobs, Masaaki Moriya, Jan Hoeijmakers, and Niels de Wind, manuscript in preparation.
- TLS
- translesion DNA synthesis
- pol
- DNA polymerase
- BPDE-dG
- benzo[a]pyrene dihydrodiol epoxide-derived dG adduct
- MEF
- mouse embryonic fibroblast
- TKO
- triple gene knockout
- DKO
- double gene knockout
- PCNA
- proliferating cell nuclear antigen
- AD
- activation domain
- BD
- DNA-binding domain
- BRCT
- carboxyl-terminus of BRCA1 protein.
REFERENCES
- 1. Lange S. S., Takata K., Wood R. D. (2011) DNA polymerases and cancer. Nat. Rev. Cancer 11, 96–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ohmori H., Friedberg E. C., Fuchs R. P., Goodman M. F., Hanaoka F., Hinkle D., Kunkel T. A., Lawrence C. W., Livneh Z., Nohmi T., Prakash L., Prakash S., Todo T., Walker G. C., Wang Z., Woodgate R. (2001) The Y-family of DNA polymerases. Mol. Cell 8, 7–8 [DOI] [PubMed] [Google Scholar]
- 3. Silverstein T. D., Johnson R. E., Jain R., Prakash L., Prakash S., Aggarwal A. K. (2010) Structural basis for the suppression of skin cancers by DNA polymerase ϵ. Nature 465, 1039–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biertümpfel C., Zhao Y., Kondo Y., Ramón-Maiques S., Gregory M., Lee J. Y., Masutani C., Lehmann A. R., Hanaoka F., Yang W. (2010) Structure and mechanism of human DNA polymerase ϵ. Nature 465, 1044–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Masutani C., Kusumoto R., Yamada A., Dohmae N., Yokoi M., Yuasa M., Araki M., Iwai S., Takio K., Hanaoka F. (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase ϵ. Nature 399, 700–704 [DOI] [PubMed] [Google Scholar]
- 6. Johnson R. E., Kondratick C. M., Prakash S., Prakash L. (1999) hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 285, 263–265 [DOI] [PubMed] [Google Scholar]
- 7. Petta T. B., Nakajima S., Zlatanou A., Despras E., Couve-Privat S., Ishchenko A., Sarasin A., Yasui A., Kannouche P. (2008) Human DNA polymerase ι protects cells against oxidative stress. EMBO J. 27, 2883–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nelson J. R., Lawrence C. W., Hinkle D. C. (1996) Deoxycytidyl transferase activity of yeast REV1 protein. Nature 382, 729–731 [DOI] [PubMed] [Google Scholar]
- 9. Lin W., Xin H., Zhang Y., Wu X., Yuan F., Wang Z. (1999) The human REV1 gene codes for a DNA template-dependent dCMP transferase. Nucleic Acids Res. 27, 4468–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou Y., Wang J., Zhang Y., Wang Z. (2010) The catalytic function of the Rev1 dCMP transferase is required in a lesion-specific manner for translesion synthesis and base damage-induced mutagenesis. Nucleic Acids Res. 38, 5036–5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wiltrout M. E., Walker G. C. (2011) The DNA polymerase activity of Saccharomyces cerevisiae Rev1 is biologically significant. Genetics 187, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ross A. L., Simpson L. J., Sale J. E. (2005) Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 33, 1280–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo C., Fischhaber P. L., Luk-Paszyc M. J., Masuda Y., Zhou J., Kamiya K., Kisker C., Friedberg E. C. (2003) Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 22, 6621–6630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ohashi E., Murakumo Y., Kanjo N., Akagi J., Masutani C., Hanaoka F., Ohmori H. (2004) Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells 9, 523–531 [DOI] [PubMed] [Google Scholar]
- 15. Murakumo Y., Ogura Y., Ishii H., Numata S., Ichihara M., Croce C. M., Fishel R., Takahashi M. (2001) Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J. Biol. Chem. 276, 35644–35651 [DOI] [PubMed] [Google Scholar]
- 16. Masuda Y., Ohmae M., Masuda K., Kamiya K. (2003) Structure and enzymatic properties of a stable complex of the human REV1 and REV7 proteins. J. Biol. Chem. 278, 12356–12360 [DOI] [PubMed] [Google Scholar]
- 17. Takata K., Wood R. D. (2009) Bypass specialists operate together. EMBO J. 28, 313–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shachar S., Ziv O., Avkin S., Adar S., Wittschieben J., Reissner T., Chaney S., Friedberg E. C., Wang Z., Carell T., Geacintov N., Livneh Z. (2009) Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 28, 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lawrence C. W. (2004) Cellular functions of DNA polymerase zeta and Rev1 protein. Adv. Protein Chem. 69, 167–203 [DOI] [PubMed] [Google Scholar]
- 20. Gibbs P. E., Wang X. D, Li Z., McManus T. P., McGregor W. G., Lawrence C. W., Maher V. M. (2000) The function of the human homolog of Saccharomyces cerevisiae REV1 is required for mutagenesis induced by UV light. Proc. Natl. Acad. Sci. U.S.A. 97, 4186–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jansen J. G., Tsaalbi-Shtylik A., Langerak P., Calléja F., Meijers C. M., Jacobs H., de Wind N. (2005) The BRCT domain of mammalian Rev1 is involved in regulating DNA translesion synthesis. Nucleic Acids Res. 33, 356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoege C., Pfander B., Moldovan G. L., Pyrowolakis G., Jentsch S. (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 [DOI] [PubMed] [Google Scholar]
- 23. Stelter P., Ulrich H. D. (2003) Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425, 188–191 [DOI] [PubMed] [Google Scholar]
- 24. Kannouche P. L., Wing J., Lehmann A. R. (2004) Interaction of human DNA polymerase ϵ with monoubiquitinated PCNA. A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 14, 491–500 [DOI] [PubMed] [Google Scholar]
- 25. Guo C., Tang T. S., Bienko M., Parker J. L., Bielen A. B., Sonoda E., Takeda S., Ulrich H. D., Dikic I., Friedberg E. C. (2006) Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol. Cell. Biol. 26, 8892–8900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Edmunds C. E., Simpson L. J., Sale J. E. (2008) PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell 30, 519–529 [DOI] [PubMed] [Google Scholar]
- 27. Suzuki N., Ohashi E., Kolbanovskiy A., Geacintov N. E., Grollman A. P., Ohmori H., Shibutani S. (2002) Translesion synthesis by human DNA polymerase κ on a DNA template containing a single stereoisomer of dG-(+)- or dG-(−)-anti-N2-BPDE (7,8-dihydroxy-anti-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene). Biochemistry 41, 6100–6106 [DOI] [PubMed] [Google Scholar]
- 28. Rechkoblit O., Zhang Y., Guo D., Wang Z., Amin S., Krzeminsky J., Louneva N., Geacintov N. E. (2002) trans-Lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J. Biol. Chem. 277, 30488–30494 [DOI] [PubMed] [Google Scholar]
- 29. Zhang Y., Wu X., Guo D., Rechkoblit O., Wang Z. (2002) Activities of human DNA polymerase κ in response to the major benzo[a]pyrene DNA adduct. Error-free lesion bypass and extension synthesis from opposite the lesion. DNA Repair 1, 559–569 [DOI] [PubMed] [Google Scholar]
- 30. Yang I. Y., Hashimoto K., de Wind N., Blair I. A., Moriya M. (2009) Two distinct translesion synthesis pathways across a lipid peroxidation-derived DNA adduct in mammalian cells. J. Biol. Chem. 284, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Watanabe K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., Yamaizumi M. (2004) Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 23, 3886–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ogi T., Shinkai Y., Tanaka K., Ohmori H. (2002) Polκ protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc. Natl. Acad. Sci. U.S.A. 99, 15548–15553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McDonald J. P., Frank E. G., Plosky B. S., Rogozin I. B., Masutani C., Hanaoka F., Woodgate R., Gearhart P. J. (2003) 129-derived strains of mice are deficient in DNA polymerase ι and have normal immunoglobulin hypermutation. J. Exp. Med. 198, 635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohkumo T., Kondo Y., Yokoi M., Tsukamoto T., Yamada A., Sugimoto T., Kanao R., Higashi Y., Kondoh H., Tatematsu M., Masutani C., Hanaoka F. (2006) UV-B radiation induces epithelial tumors in mice lacking DNA polymerase ϵ and mesenchymal tumors in mice deficient for DNA polymerase ι. Mol. Cell. Biol. 26, 7696–7706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moriya M., Spiegel S., Fernandes A., Amin S., Liu T., Geacintov N., Grollman A. P. (1996) Fidelity of translesional synthesis past benzo[a]pyrene diol epoxide-2′-deoxyguanosine DNA adducts. Marked effects of host cell, sequence context, and chirality. Biochemistry 35, 16646–16651 [DOI] [PubMed] [Google Scholar]
- 36. Hirt B. (1967) Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26, 365–369 [DOI] [PubMed] [Google Scholar]
- 37. Pollack M., Yang I. Y., Kim H. Y., Blair I. A., Moriya M. (2006) Translesion DNA synthesis across the heptanone–etheno-2′-deoxycytidine adduct in cells. Chem. Res. Toxicol. 19, 1074–1079 [DOI] [PubMed] [Google Scholar]
- 38. Bienko M., Green C. M., Crosetto N., Rudolf F., Zapart G., Coull B., Kannouche P., Wider G., Peter M., Lehmann A. R., Hofmann K., Dikic I. (2005) Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 310, 1821–1824 [DOI] [PubMed] [Google Scholar]
- 39. Narita T., Tsurimoto T., Yamamoto J., Nishihara K., Ogawa K., Ohashi E., Evans T., Iwai S., Takeda S., Hirota K. (2010) Human replicative DNA polymerase δ can bypass T-T (6–4) ultraviolet photoproducts on template strands. Genes Cells 15, 1228–1239 [DOI] [PubMed] [Google Scholar]
- 40. Nelson J. R., Gibbs P. E., Nowicka A. M., Hinkle D. C., Lawrence C. W. (2000) Evidence for a second function for Saccharomyces cerevisiae Rev1p. Mol. Microbiol. 37, 549–554 [DOI] [PubMed] [Google Scholar]
- 41. Hara K., Hashimoto H., Murakumo Y., Kobayashi S., Kogame T., Unzai S., Akashi S., Takeda S., Shimizu T., Sato M. (2010) Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase ζ and REV1. J. Biol. Chem. 285, 12299–12307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. D'Souza S., Waters L. S., Walker G. C. (2008) Novel conserved motifs in Rev1 C-terminus are required for mutagenic DNA damage tolerance. DNA Repair 7, 1455–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guo C., Sonoda E., Tang T. S., Parker J. L., Bielen A. B., Takeda S., Ulrich H. D., Friedberg E. C. (2006) REV1 protein interacts with PCNA. Significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell 23, 265–271 [DOI] [PubMed] [Google Scholar]
- 44. de Groote F. H., Jansen J. G., Masuda Y., Shah D. M., Kamiya K., de Wind N., Siegal G. (2011) The Rev1 translesion synthesis polymerase has multiple distinct DNA binding modes. DNA Repair 10, 915–925 [DOI] [PubMed] [Google Scholar]
- 45. Andersen P. L., Xu F., Ziola B., McGregor W. G., Xiao W. (2011) Sequential assembly of translesion DNA polymerases at UV-induced DNA damage sites. Mol. Biol. Cell 22, 2373–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sharma N. M., Kochenova O. V., Shcherbakova P. V. (2011) The non-canonical protein binding site at the monomer-monomer interface of yeast proliferating cell nuclear antigen (PCNA) regulates the Rev1-PCNA interaction and Polζ/Rev1-dependent translesion DNA synthesis. J. Biol. Chem. 286, 33557–33566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ohmori H., Hanafusa T., Ohashi E., Vaziri C. (2009) Separate roles of structured and unstructured regions of Y-family DNA polymerases. Adv. Protein Chem. Struct. Biol. 78, 99–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Akagi J., Masutani C., Kataoka Y., Kan T., Ohashi E., Mori T., Ohmori H., Hanaoka F. (2009) Interaction with DNA polymerase ϵ is required for nuclear accumulation of REV1 and suppression of spontaneous mutations in human cells. DNA Repair 8, 585–599 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.