

Abstract
Objective
Platelet-derived growth factor (PDGF) and its receptor (PDGFR) promote fibrosis in scleroderma (SSc) dermal fibroblasts, which produce excessive reactive oxygen species (ROS). PDGFR is phosphorylated upon PDGF stimulation, and dephosphorylated by protein tyrosine phosphatases (PTPs), including PTP1B. In this study we determine whether the thiol-sensitive PTP1B is affected by ROS, thus enhancing PDGFR phosphorylation (p-PDGFR) and collagen I (Col I) synthesis. The effect of a thiol antioxidant, n-acetylcysteine (NAC), was also investigated.
Methods
Fibroblasts were isolated from skin. A phosphate release assay was used for PTP1B activity.
Results
ROS and Col I were significantly higher in SSc fibroblasts, accompanied by significantly lower amounts of free thiols compared to normal fibroblasts. After PDGF stimulation, not only were the PDGFR and ERK1/2 phosphorylated to a greater extent, but the ability to produce PTP1B was also hampered in SSc fibroblasts. PTP1B activity was significantly inactivated in SSc fibroblasts, which resulted from cysteine oxidation by higher levels of ROS, since oxidation of multiple PTPs, including PTP1B, was observed. Decreased PTP1B expression in normal fibroblasts led to increased Col I. NAC restored the low PTP1B activity, improved the profile of p-PDGFR, decreased the numbers of tyrosine-phosphorylated proteins and Col I, and scavenged ROS in SSc fibroblasts.
Conclusion
We introduce a new mechanism by which ROS promote a profibrotic phenotype in SSc fibroblasts through oxidative inactivation of PTP1B leading to pronounced PDGFR activation. Our study also provides a novel molecular mechanism by which NAC therapy may act on ROS and PTP1B to benefit SSc patients.
Scleroderma (SSc) is characterized by Raynaud’s phenomenon, proliferative vascular lesions, and fibrosis of the skin and internal organs. The exact mechanism of tissue scarring is still unknown, however, the abnormal phenotype has been suggested to be due to excessive production of extracellular matrixes, such as collagen I (Col I), by fibroblasts (1). These cells proliferate rapidly, stain positive for α-smooth muscle actin, and therefore possess a myofibroblast phenotype (2).
Platelet-derived growth factor (PDGF) and its receptor (PDGFR) have been suggested as key elements in fibrosis. Increased PDGF activity was observed in plasma-derived serum from patients with progressive SSc (3). Immunohistological study revealed increased expression of PDGF and PDGFR in SSc skin biopsies (4, 5). Cytokines such as transforming growth factor β (TGFβ) and interleukin-1α stimulate PDGFR expression and PDGF production in SSc fibroblasts, and appear to be critical for promoting fibrosis in SSc fibroblasts (6, 7). PDGF stimulates the Ras- ERK1/2- reactive oxygen species (ROS) signaling pathway and results in stimulation of Col gene expression (8). All these studies point to the importance of PDGFR signaling in the development of skin lesions in SSc.
Reactive oxygen species such as superoxide (O2•−) act as messengers to maintain cellular functions through oxidation-reduction reaction (redox) signaling (9). However, when the production of ROS exceeds the cellular antioxidant capacity and disturbs the balance of the normal redox state, this can lead to oxidative damage of proteins, lipids, and DNA. Increased oxidative damage by ROS has been suggested in the pathogenesis of SSc (8, 10–14). The imbalance of the redox state can cause endothelial dysfunction and injury (15), fibroblast proliferation (16), increased Col synthesis (11), and favoring autoantibody production through a Th2 profile (17); all major characteristics of SSc. Free radical generators such as bleomycin can result in skin fibrosis similar to SSc (18), and in fact, bleomycin injected into mouse skin serves as an animal model of SSc (19). Recently an animal model for SSc has been established by injecting different forms of ROS subcutaneously (20). N-acetylcysteine (NAC), a thiol antioxidant, decreases O2•− production in vitro (11), and shows promising results in alleviating SSc symptoms (14, 21–23). NAC is also used as a treatment for idiopathic pulmonary fibrosis (24). These studies imply that there can be a thiol-oxidation mechanism involved in SSc oxidative stress, since NAC not only acts as a free radical scavenger (25), but also acts specifically on protein thiols to support glutathione synthesis (26) and generate free sulfhydryl groups (27).
The protein tyrosine phosphatase (PTP) family is a critical regulator of a variety of cellular signaling pathways including the PDGFR. These PTPs act by dephosphorylating the activation signals elicited by protein tyrosine kinases. PTPs are characterized by their signature motif, HC(X)5R, which contains a cysteine (Cys) residue that is essential for their catalytic activity. The low pKa of this Cys residue allows it to function as a nucleophile, but also makes it susceptible to oxidation. Several studies have shown that the Cys residue is oxidized in the presence of ROS for various PTPs (28, 29). These observations imply that the increased ROS in SSc can oxidize PTPs that are involved in the PDGFR signaling pathway.
In this study, we examined the effect of excessive oxidative stress on PTP1B, a phosphatase that has been shown to dephosphorylate the PDGFR (30, 31). The effect of the thiol antioxidant, NAC, on cellular ROS production, PDGFR phosphorylation, tyrosine-phosphorylated proteins, and PTP1B activity was also investigated.
Materials and Methods
Cell culture
Both normal (NL) and SSc dermal fibroblasts were kindly provided by Dr. Sergio Jimenez from Thomas Jefferson University (32). SSc cells were obtained from the forearm of patients with diffuse SSc, ages range from 32–57 years old. They were maintained in RPMI with 10% fetal bovine serum (FBS), penicillin, and streptomycin. The cells were switched to RPMI with 0.1% FBS for 24 hr before they were stimulated with PDGF-BB (30 ng/ml, R&D Systems, Minneapolis, MN). Passages between 6–12 were used.
Superoxide detection
Two days before the experiment, NL and SSc dermal fibroblasts were switched to RPMI with 0.1% FBS to avoid the effect of FBS on O2•− production. When needed,20 mM NAC (Sigma-Aldrich, St. Louis, MO) was added to the cell culture media. Cellular O2•− was measured using dihydroethidium (DHE, Invitrogen, Carlsbad, CA). The nuclei were stained using 4',6-diamidino-2-phenylindole (DAPI, Invitrogen, Carlsbad, CA). Fluorescence was detected using an Olympus FV-500 confocal microscope and photographs were taken at 400x.
mRNA extraction and qRT-PCR
Total RNA was isolated using RNAeasy miniRNA isolation kits (Qiagen, Valencia, CA). cDNA was prepared using Verso cDNA synthesis kits (Thermo Scientific, Rockford, IL). Quantative PCR was performed using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen, Carlsbad, CA), with specific primers for human PTP1B, Col I, and β-actin. All samples were run in duplicate using a Mastercycler ep realplex thermal cycler (Eppendorf, Hauppauge, NY) and analyzed using Eppendorf software.
Detection of PDGFR phosphorylation
Both NL and SSc dermal fibroblasts were incubated with or without 20 mM NAC overnight and stimulated with PDGF. Cell lysates were obtained and equal amount of lysate proteins was incubated with immobilized mouse anti-human phospho- tyrosine monoclonal antibody (Cell Signaling Technology, Danvers, MA) overnight at 4°C. Rabbit anti-human PDGFRβ antibody (Cell Signaling Technology, Danvers, MA) was used to probe for phosphorylated PDGFR after SDS-PAGE and Western blotting. The immunoprecipitated tyrosine- phosphorylated proteins were detected using mouse anti- human phospho- tyrosine antibody (Cell Signaling Technology, Danvers, MA).
Immunofluorescence
Cells grown in 8-well chambers were fixed and blocked before probing with mouse anti-human Col I monoclonal antibodies (Abcam, Cambridge, MA). They were subsequently incubated with Alexa Fluoro donkey anti-mouse antibodies (Invitrogen, Carlsbad, CA). The nuclei were stained using DAPI.
Western blotting and immunoprecipitation
Equal amounts of cell lysate were loaded onto polyacrylamide gels and separated by SDS-PAGE. The proteins were then transferred onto nitrocellulose membranes via Western blotting. The blots were probed with either PTP1B (Abcam, Cambridge, MA), phosphorylated ERK1/2, total ERK1/2 (both from Cell Signaling Technologies, Danvers, MA), oxidized PTPs (R&D Systems, Minneapolis, MN), or β-actin (Sigma-Aldrich, St. Louis, MO). The blots were scanned and densitometric analysis was carried out using UN-SCAN-IT software (Silk Scientific, Orem, UT). Immunoprecipitation was carried out incubating equal amount of protein from NL and SSc dermal fibroblasts with human anti-rabbit PTP1B antibody (Novus Biologicals, Littleton, CO, 20 μg/mg protein). Rabbit IgG (Thermo Scientific, Rockford, IL) was incorporated as a negative control. The proteins captured by the protein A/G beads were eluted and subjected to SDS-PAGE and Western blotting. The blot was then probed with mouse anti-human oxidized PTP antibody and rabbit anti-human PTP1B antibody.
PTP1B activity assay
PTP1B activity assay was carried out using the PTP1B DuoSet IC kit from R&D Systems (Minneapolis, MN). Mouse anti-human PTP1B antibodies that capture both active and inactive PTP1B were immobilized. After washing away unbound proteins, a synthetic phosphopeptide substrate was added that was dephosphorylated by active phosphatases to generate free phosphate and unphosphorylated peptide. The free phosphate was detected by a sensitive dye-binding assay using malachite green and molybdic acid. By calculating the rate of phosphate release, the activity of the phosphatase was determined.
PTP1B knock-down studies
Normal dermal fibroblasts were plated in 6 well plates and allowed to grow to approximately 60% confluence. They were then transfected with either 50 nM control siRNA (Invitrogen, Carlsbad, CA) or PTP1B siRNA (Santa Cruz Biotechnology) for 72 hours in RPMI containing 10% FBS. Both PTP1B and Col I levels were then quantified by qPCR.
Analysis of free thiol content
Free sulfhydryl groups on proteins were determined by using 5,5’-dithiobis (2-nitrobenzoic acid) (DTNB). Briefly, cell lysate was mixed with Tris-EDTA buffer (Tris-base 0.25 M, EDTA 20 mM, pH 8.2). The absorbance of the mixture was measured at 412 nm. DTNB in methanol was then added, incubated for 15 min, and the absorbance was measured again at 412 nm. After subtracting the absorbance obtained before adding DTNB, the net absorbance was used to calculate the thiol content in the samples using a molar extinction coefficient of 13600 M−1cm−1 at 412 nm and normalized to the level of total cellular protein with a final unit of μM/mg protein.
Statistical analysis
Results were expressed as mean ± S.E. To determine the differences between the groups, Student’s t-test were performed. P-values of less than 0.05 with two-tailed analysis were considered statistically significant.
Results
Superoxide production in dermal fibroblasts
As shown in Figure 1, our results indicated that O2•− was significantly higher in SSc dermal fibroblasts compared to NL cells. In the presence of NAC, a thiol antioxidant, the level of O2•− was significantly reduced in SSc dermal fibroblasts.
Figure 1.

Production of O2•− in NL and SSc dermal fibroblasts by fluorescence microscopy. NL (top panels) and SSc dermal fibroblasts (bottom panels) were treated with DHE, a O2•− trap that releases red fluorescence when it encounters O2•− . Nuclei were counterstained with DAPI. Staining was detected using a fluorescence microscope and images were taken at 400x. A significant amount of O2•− was detected in SSc fibroblasts compared to NL (left panels) while in the presence of NAC, O2•− in SSc fibroblasts decreased (right panels). These representative images were from 3 NL and 3 SSc lines.
Thiol content in NL and SSc dermal fibroblasts
In NL dermal fibroblasts, the total free thiol content was 464 ± 18 μM/mg protein, while in SSc dermal fibroblasts, it was significantly lower at 344 ± 15 μM/mg protein (mean ± SE, n=3, p<0.01). These results suggest that a portion of the protein thiols in SSc dermal fibroblasts was modified into oxidation products that do not react with DTNB. This also confirms the O2•− stainingin Figure 1 and suggests that there is increased oxidative stress in SSc dermal fibroblasts compared to NL cells.
Phosphorylation of PDGFR after PDGF stimulation
As shown in Figure 2A, in NL dermal fibroblasts, PDGF-stimulated PDGFR phosphorylation reached maximum at 10 min (p<0.05 vs. non-stimulated [NS]) and returned back to baseline at 1 hr. In contrast, PDGFR phosphorylation maximized at 10 min (p<0.05 vs. NS) and remained phosphorylated at 1 hr (p<0.05 vs. NS) in SSc dermal fibroblasts. At 30 min, the extent of phosphorylated PDGFR in SSc dermal fibroblasts was significantly higher than that in NL cells (p<0.05). These results suggest that phosphatases that dephosphorylate PDGFR, such as PTP1B, might be deficient or inactivated in SSc dermal fibroblasts. To determine whether oxidative stress plays a role in the prolonged PDGFR phosphorylation seen in SSc dermal fibroblasts, NAC was added to cell culture media and tyrosine phosphorylated proteins were immunoprecipitated. As shown in Figure 2B, in the presence of NAC, PDGFR phosphorylation occurred at 10 and 30 min for NL dermal fibroblasts. A similar pattern was observed for SSc dermal fibroblasts, with less intensity at 30 min compared to NL cells. Comparing the time courses of PDGFR phosphorylation in the presence or absence of NAC (Figure 2A and 2B), addition of NAC significantly decreased the extent of PDGFR phosphorylation in both NL and SSc dermal fibroblasts. After PDGF stimulation for 10 and 30 min, the PDGFR is activated through dimerization as indicated in Figure 2A and B.
Figure 2.

PDGF-stimulated PDGFR and protein phosphorylation in NL and SSc dermal fibroblasts. A. In NL cells, PDGF significantly stimulated PDGFR phosphorylation (p-PDGFR) at 10 and 30 min (p<0.05 vs. NL not-stimulated [NS]). In SSc fibroblasts, p-PDGFR increased significantly until 1 hr (p<0.05 vs. SSc NS). At 30min, the level of p-PDGFR in SSc dermal fibroblasts was significantly higher than in NL cells. B. In the presence of NAC, p-PDGFR occurred at 10 and 30 min in both NL and SSc dermal fibroblasts. PDGF significantly stimulated p-PDGFR at 10 and 30 min in SSc dermal fibroblasts (p<0.05 vs. SSc NS). The intensity of p-PDGFR was significantly lower in the presence of NAC in both NL and SSc dermal fibroblasts. C. In the absence of NAC, multiple proteins were phosphorylated in both NL and SSc dermal fibroblasts. At 1 hour phosphorylated proteins were still visible. PDGF induced more phosphorylated proteins in SSc than in NL cells. D. In the presence of NAC, PDGF stimulated protein tyrosine phosphorylation in both NL and SSc dermal fibroblasts, but with less intensity compared to those in Figure 2C. Results are expressed as mean ± S.E. and p<0.05 is considered significant. n≥3 NL subjects and patients.
Protein tyrosine phosphorylation after PDGF stimulation
To examine the effect of NAC on other tyrosine phosphorylated proteins, the immunoprecipitated samples in Figure 2A and 2B were subjected to Western blotting and probed with mouse anti-human protein tyrosine phosphorylation antibodies. The 25 and 50 kDa bands are the light and heavy chains of IgG. The tyrosine phosphorylated protein patterns in the absence of NAC (Figure 2C) showed that, PDGF induced phosphorylation on multiple proteins in both NL and SSc dermal fibroblasts. Most of these proteins were still visible at 1 hr, and more proteins were phosphorylated in SSc dermal fibroblasts compared to those in NL cells. In the presence of NAC (Figure 2D), fewer proteins were phosphorylated by PDGF in both NL and SSc dermal fibroblasts, consistent with the results observed with PDGFR phosphorylation. These results suggest that in addition to the PDGFR pathway, many more proteins were phosphorylated in SSc dermal fibroblasts, and NAC not only affects PDGFR, but also many other proteins. Since NAC decreases cellular ROS, these results indicate that the prolonged phosphorylation of PDGFR in SSc dermal fibroblasts might be due to oxidative inactivation of multiple thiol-sensitive phosphatases that dephosphorylate PDGFR. And since PTPs tend to have multiple substrates, the increased phosphorylated proteins seen here can also be a result of inactivation of a certain PTP.
Phosphorylation of ERK1/2 after PDGF stimulation
PDGF-stimulated ERK1/2 phosphorylation reached maximum at 45 min and returned to baseline at 4 hr in NL dermal fibroblasts (Figure 3A). In contrast, significant phosphorylation of ERK1/2 was observed at 10 min after PDGF stimulation in SSc dermal fibroblasts, and persisted until 4 hr. At 4 hr, the extent of ERK1/2 phosphorylation was significantly different between NL and SSc dermal fibroblasts (p<0.05). These results agree with what was observed in Figure 2A, in that the enhanced and prolonged PDGFR phosphorylation in SSc dermal fibroblasts leads to enhanced and prolonged activation of its downstream signaling pathway, ERK1/2, while in NL cells, the profile of PDGFR phosphorylation parallels the pattern of ERK1/2 phosphorylation, which is shorter and less pronounced. Although the basal level of p-ERK1/2 increased in both NL and SSc cells (p>0.05 vs. corresponding NS in Figure 3A) in the presence of NAC (Figure 3B), PDGF stimulation did not result in further significant ERK1/2 phosphorylation in both NL and SSc fibroblasts (p>0.05). The increased basal level of p-ERK1/2 in the presence of NAC might be due to its effect on negative regulators of the MAPK pathway. These results also suggest that the beneficial effect of NAC on Col I production might in part be due to some other factors, such as affecting the expression of matrix metalloproteinases. Taken together, the PDGFR and its downstream signaling pathway were activated to a greater extent in SSc dermal fibroblasts, while NAC, a thiol antioxidant, altered the extent of PDGF stimulation in both NL and SSc dermal fibroblasts.
Figure 3.

ERK1/2 phosphorylation patterns and Col I in NL and SSc dermal fibroblasts. A. In NL cells, PDGF significantly stimulated ERK1/2 phosphorylation (p-ERK1/2) at 45 min and 2 hr (p<0.05 vs. NL not-stimulated [NS]). In SSc fibroblasts, p-ERK1/2 increased significantly from 10 min until 4 hr (p<0.05 vs. SSc NS). At 4 hr, the level of p-ERK1/2 in SSc dermal fibroblasts was significantly higher than in NL cells. B. In the presence of NAC, although the basal level of p-ERK1/2 increased slightly in both NL and SSc cells (p>0.05), PDGF stimulation did not result in significant p-ERK1/2. C. Col I mRNA levels were significantly higher in SSc fibroblasts compared to NLs. NAC significantly decreased Col I mRNA levels in SSc cells, but had no significant effect in NL dermal fibroblasts. D. A significant amount of Col I was detected in SSc fibroblasts compared to NL. In the presence of NAC, Col I in SSc fibroblasts decreased. These representative images were from 3 NL and SSc lines.
Expression of Col I in NL and SSc dermal fibroblasts
At basal level and 10 min after PDGF stimulation, Col I mRNA was significantly higher in SSc dermal fibroblasts compared to NL (p<0.05, Figure 3C). After 4 hr of PDGF stimulation, Col I mRNA significantly decreased compared to NS. NAC significantly decreased Col I mRNA levels in SSc dermal fibroblasts at several time points after PDGF incubation, while it had no significant effect in NL cells. At the protein level, there was more Col I staining in SSc dermal fibroblasts compared to NL cells (Figure 3D). In the presence of NAC, the expression of Col I in SSc dermal fibroblasts decreased to similar levels that were observed in NL cells. These results indicate the final response of PDGFR activation. In SSc dermal fibroblasts, enhanced PDGFR activation leads to increased ERK1/2 phosphorylation, and therefore more Col I synthesis. By acting on scavenging ROS and deactivating the PDGFR pathway, NAC decreased Col I synthesis in SSc dermal fibroblasts.
Expression of PTP1B in NL and SSc dermal fibroblasts
As shown in Figure 4A, in the presence of PDGF, the mRNA levels of PTP1B in NL dermal fibroblasts increased slightly at 2 and 4 hrs, while the expression in SSc dermal fibroblasts remained the same. When stimulated with PDGF, PTP1B protein expression increased gradually and was maximal at 45 min and 2 hr (p<0.05 vs. NS), and there was no difference at 4 hr compared to NS in NL dermal fibroblasts (Figure 4B). However, in SSc dermal fibroblasts, similar to the mRNA results in Figure 4A, the protein expression of PTP1B remained the same when stimulated with PDGF. At basal state, there was no difference in PTP1B expression in NL and SSc dermal fibroblasts (NS groups in Figure 4B; 0.46 ± 0.10 in NL vs. 0.49 ± 0.14 in SSc dermal fibroblasts). In addition, the level of PTP1B in SSc dermal fibroblasts was significantly lower than that in NL cells at 45 min (p<0.05). Based on these results, it appears that at basal level, the same amount of PTP1B is present in NL and SSc dermal fibroblasts. However, when stimulated with PDGF, PTP1B increases in NL cells, but not in SSc dermal fibroblasts.
Figure 4.

Expression and enzymatic activity of PTP1B in NL and SSc dermal fibroblasts. A. PDGF-stimulated mRNA expression of PTP1B in NL and SSc dermal fibroblasts was not significantly different. B. At the protein level, PDGF stimulated PTP1B production was significantly higher at 45 min and 2 hr (p<0.05 vs. NS) in NL cells. However, in SSc cells, PDGF incubation did not alter PTP1B expression, and at 45 min there was a significant difference between NL and SSc (p<0.05). C. In SSc dermal fibroblasts, the PTP1B activity was significantly lower compared to NL (p<0.05). In the presence of NAC, the activity in SSc cells was restored. D. When PTP1B was significantly knocked down in NL dermal fibroblasts, Col I mRNA significantly increased (p<0.05), mimicking what was observed in SSc dermal fibroblasts. Results are expressed as mean ± S.E. and p<0.05 is considered significant. n≥4 NL subjects and patients.
Enzyme activity of PTP1B in NL and SSc dermal fibroblasts
PTP1B activity was significantly lower in SSc dermal fibroblasts compared to NL cells (20.1 ± 2.7 and 11.9 ± 1.3 nmoles in NL and SSc dermal fibroblasts, p<0.05, Figure 4C). In contrast, in the presence of the antioxidant NAC, PTP1B activity was restored in SSc dermal fibroblasts (16.2 ± 0.4 and 19.2 ± 2.1 nmoles in NL and SSc dermal fibroblasts, respectively). Therefore in the presence of excess O2•− in SSc dermal fibroblasts, the PTP1B activity is inactivated; while O2•− is eliminated by NAC, the activity of PTP1B is restored. These results imply that the increased oxidative stress in SSc dermal fibroblasts inactivates PTP1B, resulting in increased PDGFR phosphorylation, ERK1/2 activation, and increased Col I synthesis. The presence of NAC not only eliminates O2•− in these cells, but also restores PTP1B activity, therefore decreasing the extent of PDGFR phosphorylation and its downstream events.
PTP1B expression and Col I production
To elucidate the direct involvement of PTP1B in Col I production, we knocked down PTP1B expression in NL dermal fibroblasts and assessed the expression of Col I. As shown in Figure 4D, PTP1B expression was significantly knocked down by siRNA treatment (p<0.05). Under this condition, Col I expression increased approximately 40% (p<0.05). These results suggest PTP1B, possibly through the PDGFR pathway, controls Col I production in dermal fibroblasts. This further supports the notion that it plays a significant role in skin fibrosis.
Oxidation of PTPs in NL and SSc dermal fibroblasts
To examine whether PTPs are oxidized in dermal fibroblasts, oxidized PTPs were probed using antibodies that recognizes the oxidized Cys residue at their active site. The oxidized PTP profile of 3 NL subjects and 5 SSc patients is shown in Figure 5A. In NL subjects, 2 bands were observed, though subject 3 had stronger bands and one was located at a higher molecular weight. In SSc dermal fibroblasts, at least 3 PTPs were oxidized, with stronger intensities compared to NL cells. The profile for each SSc patient appears slightly different, with consistent oxidation occurring at 50 kD, which could be PTP1B. We then pulled down PTP1B from cell lysates and probed with the same antibody again, there was approximately 2 fold more oxidized PTP1B in SSc dermal fibroblasts compared to NLs (n=3, p<0.05, Figure 5B). These results suggest that the inactivation of PTP1B is due to oxidation of its active site.
Figure 5.

Oxidation of PTPs in NL and SSc dermal fibroblasts. Oxidized PTPs in NL and SSc dermal fibroblasts were visualized using mouse anti-oxidized human PTP antibody after Western blotting. Equal amounts of protein were subjected to immunoprecipitation using rabbit anti-human PTP1B antibody. The blot was then probed with mouse anti-oxidized human PTP antibody. A. Dermal fibroblasts from 3 NL subjects and 5 SSc patients were used for this study. Overall, more PTPs were oxidized in SSc dermal fibroblasts compared to NLs. In NL cells, 2 bands were observed in all subjects though at different molecular weight. In SSc dermal fibroblasts, at least 3 PTPs were oxidized. The 50 kD protein, which we postulate as PTP1B, was oxidized in all SSc dermal fibroblasts. B. We confirmed that the 50 kD protein was indeed PTP1B by immunoprecipitation. There was significantly more oxidized PTP1B in SSc dermal fibroblasts compared to NL. Results are expressed as mean ± S.E. and p<0.05 is considered significant. n ≥ 3 NL subjects and patients.
Discussion
In this study, we provide evidence that PTP1B is inactivated by ROS in SSc dermal fibroblasts, leading to prolonged phosphorylation of the PDGFR. As summarized in Figure 6, we show that ROS are elevated in SSc dermal fibroblasts compared to NL cells. The increased oxidative stress leads to a significant decrease in the total protein thiol groups in SSc dermal fibroblasts, as well as oxidation at the Cys active site of PTP1B. Oxidation of PTPs leads to inactivation of their phosphatase activity, resulting in the prolonged phosphorylation profile of PDGFR and ERK1/2 and thus increased Col I expression in SSc dermal fibroblasts. In addition, we showed a direct relationship between PTP1B expression and Col I synthesis by knocking down PTP1B in NL fibroblasts. The inclusion of NAC, a thiol antioxidant, reconfirms the impact of oxidative stress on PTP1B, as lower PTP1B activity in SSc dermal fibroblasts is observed with high levels of O2•−, and when the O2•− is eliminated by NAC, not only is the PTP1B activity restored, but the PDGFR and its downstream events, including Col I expression, become similar to those in NL cells. Our results suggest a new mechanism by which oxidative stress promotes a fibrotic phenotype in SSc dermal fibroblasts through oxidative inactivation of phosphatases leading to pronounced PDGFR activation.
Figure 6.

Summary of our work. In NL dermal fibroblasts, adequate amounts of ROS perform their role in redox signaling and are not harmful in oxidizing cellular proteins. When PDGF binds to the PDGFR, the receptor is phosphorylated and downstream signaling pathways such as ERK1/2 are activated, leading to a response such as collagen I production. PTP1B is active and controls the extent of phosphorylation on PDGFR. In SSc dermal fibroblasts, the increased production of ROS inactivates PTP1B through oxidation of the Cys residue in its active site. When PDGF binds to its receptor, due to PTP1B inactivation, prolonged PDGFR phosphorylation results in an increased downstream response. In the presence of the thiol antioxidant NAC, ROS decrease, the oxidized Cys residue on PTP1B is reduced, the activity is restored, and the response returns to baseline. P: tyrosine phosphorylation; -SH: free thiols; -SOxH: oxidized thiols; ROS: reactive oxygen species; NAC: n-acetylcysteine;
 : oxidation;
: oxidation;
 : less oxidation;
: less oxidation;
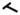 : dephosphorylation;
: dephosphorylation;
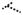 : unable to dephosphorylate
: unable to dephosphorylate
Our findings of increased ROS production in SSc agree with previous reports (8, 11, 12, 14, 33). The source of O2•− is believed to be NADPH oxidase. When SSc dermal fibroblasts were treated with diphenylene iodonium, a NADPH oxidase inhibitor, O2•− diminished (11). In addition, increased expression of the NADPH oxidase subunits was observed in monocytes and dermal fibroblasts isolated from SSc patients (11, 12). As protein thiols are very sensitive to ROS and can be oxidized, we measured cellular free thiols as another marker for oxidative stress. We showed that the free thiol content in SSc dermal fibroblasts is significantly lower than that in NL cells, again suggesting the existence of oxidative stress in SSc. It has been shown that total plasma thiols in SSc patients were significantly lower compared to NL subjects (34, 35).
In this study we hypothesize that the prolonged phosphorylation of the PDGFR in SSc fibroblasts is a result of decreased expression of or ROS-inactivated PTPs. We cannot rule out the role of protein kinases in driving PDGFR phosphorylation, as studies have emphasized the importance of these enzymes in the pathogenesis of SSc (36). However, it appears that protein tyrosine kinases are not direct targets of ROS. In addition, protein tyrosine phosphorylation is controlled through the coordinated actions of tyrosine kinases and PTPs. These proteins not only have housekeeping functions, but also are highly regulated and play crucial roles in many cell functions and in different diseases. A recent study showed that the expression of protein phosphatase 2A was significantly lower in SSc fibroblasts (37). The authors suggested that this led to constitutive activation of the TGFβ pathway and enhanced ERK1/2 phosphorylation, which was also seen in our study.
We examined the expression and function of PTP1B, which has been reported to be a negative regulator of receptor tyrosine kinases such as PDGFR (30, 31). The association of PTP1B and PDGFR is further supported by a study using PTP1B knockout mice (38). In addition to the PDGFR pathway, PTP1B appears to be a negative regulator in both insulin and leptin signaling pathways (39, 40). Epidermal growth factor receptor (41) and tyrosine-phosphorylated proteins such as STAT5 (42) have also been shown to be targets of PTP1B. Although the expression of PTP1B is similar in NL and SSc dermal fibroblasts (NS in Figure 4B), the activity of PTP1B in SSc fibroblasts is significantly lower, suggesting that only a portion of the expressed PTP1B is active in SSc fibroblasts. We further showed that the inactivation is due to oxidation of its active site. Oxidation of PTPs is not rare. In fact, reversible and transient oxidation of PTPs is regarded as a redox regulatory mechanism for receptor tyrosine kinases to control their phosphorylation state (43). However, when a constant and large amount of ROS is present, prolonged inactivation of PTP activity may occur. Indeed, it has been shown that in human cancer cells, increased ROS led to production of sulfenic, sulfinic, and sulfonic acids at the active site of PTP1B, resulting in its inactivation (44). Similar to their results, we showed in this study that increased ROS in SSc dermal fibroblasts not only inactivates PTP1B, but also oxidizes at least 3 PTPs, including PTP1B. The decreased antioxidant capacity in SSc (45) may also exacerbate the inactivation.
The reason for the increased expression of PTP1B in NL cells after PDGF stimulation requires further investigation. Since protein synthesis requires a longer time, we postulate that the increase in PTP1B might be due to release of PTP1B from the phosphorylated or sumoylated pool, since the antibody we used only recognizes the free PTP1B. This might be a defense mechanism in NL cells to counteract the activation of PDGFR. However this mechanism is lost in SSc dermal fibroblasts, as the expression of PTP1B did not change after PDGF treatment.
Since oxidative stress in SSc dermal fibroblasts was accompanied by significantly lower free thiol content, we hypothesize that NAC, a thiol antioxidant (25–27) that showed beneficial effects in SSc (11, 14, 20–23), will benefit the cells by decreasing cellular ROS and replenishing free cellular thiols. Indeed we showed that NAC not only decreased cellular O2•−, it also restored PTP1B activity along with improvement of the PDGFR phosphorylation and Col I profile. Our results agree with a previous report showing that NAC prevented serum- induced PDGFR phosphorylation and its downstream signaling pathways by scavenging O2•− produced by NADPH oxidase (46). Another report showed that in cultured vascular smooth muscle cells, NAC reduced PDGFR phosphorylation and increased PTP activity (47).
PTP1B is not the only phosphatase that regulates the phosphorylation state of PDGFR. A number of thiol-sensitive phosphatases, such as SHP-2 (30) and density-enhanced phosphatase-1 (DEP-1) (48), have been shown to dephosphorylate the PDGFR. Preliminary results show that the expression of SHP-2 seems to differ in SSc fibroblasts compared to NL cells when cells are treated with PDGF. In addition, in nonstimulated SSc fibroblasts, the expression of DEP-1 appears to be higher (data not shown). Whether the phosphatase activities of DEP-1 and SHP-2 are affected by ROS needs further investigation. If indeed DEP-1 and SHP-2 are inactivated, the antioxidative effect of NAC on PDGFR phosphorylation and protein tyrosine phosphorylation (Figure 2) may thus be a result of restoring the activities of a combination of phosphatases. Nonetheless, we showed that at least one phosphatase, PTP1B, is inactive when excessive O2•− is present in SSc dermal fibroblasts, and its activity is restored when NAC is present, with a simultaneous change in the PDGFR phosphorylation pattern.
In conclusion, our findings provide evidence that the increased oxidative stress in SSc fibroblasts oxidizes PTP1B and renders it inactivated, and therefore amplifies the PDGFR signaling pathway. We not only introduce a new mechanism for the role of oxidative stress in the pathogenesis of fibrosis in SSc, but also provided a novel molecular pathway by which NAC therapy may act on ROS and PTP1B to benefit SSc patients.
Acknowledgments
This work was supported by the National Institute of Health (AR48267 to A.E.K.), the Office of Research and Development, Medical Research Service, Department of Veterans Affairs, the Frederick G. L. Huetwell and William D. Robinson, MD, Professorship in Rheumatology, Scleroderma Research Foundation, NIH Center for Translational Science Activities grant UL1-RR-024986, the clinical research unit at the University of Michigan, American College of Rheumatology Research and Education Foundation grant (Academic Reentry Award to K.P), the Linda Dolce Scleroderma Research Fund, and by the Marvin and Betty Danto and the Jonathan and Lisa Rye Endowments for Scleroderma Research at the University of Michigan.
References
- 1.LeRoy EC. Increased collagen synthesis by scleroderma skin fibroblasts in vitro: a possible defect in the regulation or activation of the scleroderma fibroblast. J Clin Invest. 1974;54:880–9. doi: 10.1172/JCI107827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jelaska A, Korn JH. Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. 2000;43:2230–9. doi: 10.1002/1529-0131(200010)43:10<2230::AID-ANR10>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 3.Pandolfi A, Florita M, Altomare G, Pigatto P, Donati MB, Poggi A. Increased plasma levels of platelet-derived growth factor activity in patients with progressive systemic sclerosis. Proc Soc Exp Biol Med. 1989;191:1–4. doi: 10.3181/00379727-191-42880. [DOI] [PubMed] [Google Scholar]
- 4.Gay S, Jones RE, Jr, Huang GQ, Gay RE. Immunohistologic demonstration of platelet-derived growth factor (PDGF) and sis-oncogene expression in scleroderma. J Invest Dermatol. 1989;92:301–3. doi: 10.1111/1523-1747.ep12276895. [DOI] [PubMed] [Google Scholar]
- 5.Klareskog L, Gustafsson R, Scheynius A, Hallgren R. Increased expression of platelet-derived growth factor type B receptors in the skin of patients with systemic sclerosis. Arthritis Rheum. 1990;33:1534–41. doi: 10.1002/art.1780331011. [DOI] [PubMed] [Google Scholar]
- 6.Yamakage A, Kikuchi K, Smith EA, LeRoy EC, Trojanowska M. Selective upregulation of platelet-derived growth factor alpha receptors by transforming growth factor beta in scleroderma fibroblasts. J Exp Med. 1992;175:1227–34. doi: 10.1084/jem.175.5.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawaguchi Y, Hara M, Wright TM. Endogenous IL-1alpha from systemic sclerosis fibroblasts induces IL-6 and PDGF-A. J Clin Invest. 1999;103:1253–60. doi: 10.1172/JCI4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Svegliati S, Cancello R, Sambo P, Luchetti M, Paroncini P, Orlandini G, et al. Platelet-derived growth factor and reactive oxygen species (ROS) regulate Ras protein levels in primary human fibroblasts via ERK1/2. Amplification of ROS and Ras in systemic sclerosis fibroblasts. J Biol Chem. 2005;280:36474–82. doi: 10.1074/jbc.M502851200. [DOI] [PubMed] [Google Scholar]
- 9.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–30. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 10.Bruckdorfer KR, Hillary JB, Bunce T, Vancheeswaran R, Black CM. Increased susceptibility to oxidation of low-density lipoproteins isolated from patients with systemic sclerosis. Arthritis Rheum. 1995;38:1060–7. doi: 10.1002/art.1780380807. [DOI] [PubMed] [Google Scholar]
- 11.Sambo P, Baroni SS, Luchetti M, Paroncini P, Dusi S, Orlandini G, et al. Oxidative stress in scleroderma: maintenance of scleroderma fibroblast phenotype by the constitutive up-regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheum. 2001;44:2653–64. doi: 10.1002/1529-0131(200111)44:11<2653::aid-art445>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 12.Sambo P, Jannino L, Candela M, Salvi A, Donini M, Dusi S, et al. Monocytes of patients wiht systemic sclerosis (scleroderma spontaneously release in vitro increased amounts of superoxide anion. J Invest Dermatol. 1999;112:78–84. doi: 10.1046/j.1523-1747.1999.00476.x. [DOI] [PubMed] [Google Scholar]
- 13.Ogawa F, Shimizu K, Muroi E, Hara T, Hasegawa M, Takehara K, et al. Serum levels of 8-isoprostane, a marker of oxidative stress, are elevated in patients with systemic sclerosis. Rheumatology (Oxford) 2006;45:815–8. doi: 10.1093/rheumatology/kel012. [DOI] [PubMed] [Google Scholar]
- 14.Servettaz A, Guilpain P, Goulvestre C, Chereau C, Hercend C, Nicco C, et al. Radical oxygen species production induced by advanced oxidation protein products predicts clinical evolution and response to treatment in systemic sclerosis. Ann Rheum Dis. 2007;66:1202–9. doi: 10.1136/ard.2006.067504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blake DR, Winyard P, Scott DG, Brailsford S, Blann A, Lunec J. Endothelial cell cytotoxicity in inflammatory vascular diseases--the possible role of oxidised lipoproteins. Ann Rheum Dis. 1985;44:176–82. doi: 10.1136/ard.44.3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murrell GA, Francis MJ, Bromley L. Modulation of fibroblast proliferation by oxygen free radicals. Biochem J. 1990;265:659–65. doi: 10.1042/bj2650659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King MR, Ismail AS, Davis LS, Karp DR. Oxidative stress promotes polarization of human T cell differentiation toward a T helper 2 phenotype. J Immunol. 2006;176:2765–72. doi: 10.4049/jimmunol.176.5.2765. [DOI] [PubMed] [Google Scholar]
- 18.Finch WR, Rodnan GP, Buckingham RB, Prince RK, Winkelstein A. Bleomycin-induced scleroderma. J Rheumatol. 1980;7:651–9. [PubMed] [Google Scholar]
- 19.Yamamoto T, Takagawa S, Katayama I, Yamazaki K, Hamazaki Y, Shinkai H, et al. Animal model of sclerotic skin. I: Local injections of bleomycin induce sclerotic skin mimicking scleroderma. J Invest Dermatol. 1999;112:456–62. doi: 10.1046/j.1523-1747.1999.00528.x. [DOI] [PubMed] [Google Scholar]
- 20.Servettaz A, Goulvestre C, Kavian N, Nicco C, Guilpain P, Chereau C, et al. Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J Immunol. 2009;182:5855–64. doi: 10.4049/jimmunol.0803705. [DOI] [PubMed] [Google Scholar]
- 21.Sambo P, Amico D, Giacomelli R, Matucci-Cerinic M, Salsano F, Valentini G, et al. Intravenous N-acetylcysteine for treatment of Raynaud's phenomenon secondary to systemic sclerosis: a pilot study. J Rheumatol. 2001;28:2257–62. [PubMed] [Google Scholar]
- 22.Failli P, Palmieri L, D'Alfonso C, Giovannelli L, Generini S, Rosso AD, et al. Effect of N-acetyl-L-cysteine on peroxynitrite and superoxide anion production of lung alveolar macrophages in systemic sclerosis. Nitric Oxide. 2002;7:277–82. doi: 10.1016/s1089-8603(02)00120-9. [DOI] [PubMed] [Google Scholar]
- 23.Rosato E, Borghese F, Pisarri S, Salsano F. The treatment with N-acetylcysteine of Raynaud's phenomenon and ischemic ulcers therapy in sclerodermic patients: a prospective observational study of 50 patients. Clin Rheumatol. 2009;28:1379–84. doi: 10.1007/s10067-009-1251-7. [DOI] [PubMed] [Google Scholar]
- 24.Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2229–42. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- 25.Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med. 1989;6:593–7. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 26.Ceconi C, Curello S, Cargnoni A, Ferrari R, Albertini A, Visioli O. The role of glutathione status in the protection against ischaemic and reperfusion damage: effects of N-acetyl cysteine. J Mol Cell Cardiol. 1988;20:5–13. doi: 10.1016/s0022-2828(88)80174-3. [DOI] [PubMed] [Google Scholar]
- 27.Fung HL, Chong S, Kowaluk E, Hough K, Kakemi M. Mechanisms for the pharmacologic interaction of organic nitrates with thiols. Existence of an extracellular pathway for the reversal of nitrate vascular tolerance by N-acetylcysteine. J Pharmacol Exp Ther. 1988;245:524–30. [PubMed] [Google Scholar]
- 28.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–42. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 29.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–99. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 30.Klinghoffer RA, Kazlauskas A. Identification of a putative Syp substrate, the PDGF beta receptor. J Biol Chem. 1995;270:22208–17. doi: 10.1074/jbc.270.38.22208. [DOI] [PubMed] [Google Scholar]
- 31.Liu F, Chernoff J. Protein tyrosine phosphatase 1B interacts with and is tyrosine phosphorylated by the epidermal growth factor receptor. Biochem J. 1997;327:139–145. doi: 10.1042/bj3270139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Louneva N, Huaman G, Fertala J, Jimenez SA. Inhibition of systemic sclerosis dermal fibroblast type I collagen production and gene expression by simvastatin. Arthritis Rheum. 2006;54:1298–308. doi: 10.1002/art.21723. [DOI] [PubMed] [Google Scholar]
- 33.Allanore Y, Borderie D, Perianin A, Lemarechal H, Ekindjian OG, Kahan A. Nifedipine protects against overproduction of superoxide anion by monocytes from patients with systemic sclerosis. Arthritis Res Ther. 2005;7:R93–100. doi: 10.1186/ar1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Firuzi O, Fuksa L, Spadaro C, Bousova I, Riccieri V, Spadaro A, et al. Oxidative stress parameters in different systemic rheumatic diseases. J Pharm Pharmacol. 2006;58:951–7. doi: 10.1211/jpp.58.7.0010. [DOI] [PubMed] [Google Scholar]
- 35.Allanore Y, Borderie D, Lemarechal H, Ekindjian OG, Kahan A. Acute and sustained effects of dihydropyridine-type calcium channel antagonists on oxidative stress in systemic sclerosis. Am J Med. 2004;116:595–600. doi: 10.1016/j.amjmed.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 36.Akhmetshina A, Venalis P, Dees C, Busch N, Zwerina J, Schett G, et al. Treatment with imatinib prevents fibrosis in different preclinical models of systemic sclerosis and induces regression of established fibrosis. Arthritis Rheum. 2009;60:219–24. doi: 10.1002/art.24186. [DOI] [PubMed] [Google Scholar]
- 37.Samuel G, Bujor A, Nakerakanti S, Hant F, Trojanowska M. Autocrine transforming growth factor beta signaling regulates extracellular signal-regulated kinase 1/2 phosphorylation via modulation of protein phosphatase 2A expression in scleroderma fibroblasts. Fibrogenesis Tissue Repair. 2010;3:25. doi: 10.1186/1755-1536-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haj FG, Markova B, Klaman LD, Bohmer FD, Neel BG. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-1B. J Biol Chem. 2003;278:739–44. doi: 10.1074/jbc.M210194200. [DOI] [PubMed] [Google Scholar]
- 39.Byon JC, Kusari AB, Kusari J. Protein-tyrosine phosphatase-1B acts as a negative regulator of insulin signal transduction. Mol Cell Biochem. 1998;182:101–8. [PubMed] [Google Scholar]
- 40.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–95. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 41.Flint AJ, Tiganis T, Barford D, Tonks NK. Development of "substrate-trapping" mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–5. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aoki N, Matsuda T. A cytosolic protein-tyrosine phosphatase PTP1B specifically dephosphorylates and deactivates prolactin-activated STAT5a and STAT5b. J Biol Chem. 2000;275:39718–39726. doi: 10.1074/jbc.M005615200. [DOI] [PubMed] [Google Scholar]
- 43.Chiarugi P, Cirri P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem Sci. 2003;28:509–14. doi: 10.1016/S0968-0004(03)00174-9. [DOI] [PubMed] [Google Scholar]
- 44.Lou YW, Chen YY, Hsu SF, Chen RK, Lee CL, Khoo KH, et al. Redox regulation of the protein tyrosine phosphatase PTP1B in cancer cells. FEBS J. 2008;275:69–88. doi: 10.1111/j.1742-4658.2007.06173.x. [DOI] [PubMed] [Google Scholar]
- 45.Sfrent-Cornateanu R, Mihai C, Stoian I, Lixandru D, Bara C, Moldoveanu E. Antioxidant defense capacity in scleroderma patients. Clin Chem Lab Med. 2008;46:836–41. doi: 10.1515/CCLM.2008.132. [DOI] [PubMed] [Google Scholar]
- 46.Ammendola R, Ruocchio MR, Chirico G, Russo L, De Felice C, Esposito F, et al. Inhibition of NADH/NADPH oxidase affects signal transduction by growth factor receptors in normal fibroblasts. Arch Biochem Biophys. 2002;397:253–257. doi: 10.1006/abbi.2001.2641. [DOI] [PubMed] [Google Scholar]
- 47.Kappert K, Sparwel J, Sandin A, Seiler A, Siebolts U, Leppanen O, et al. Antioxidants relieve phosphatase inhibition and reduce PDGF signaling in cultured VSMCs and in restenosis. Arterioscler Thromb Vasc Biol. 2006;26:2644–51. doi: 10.1161/01.ATV.0000246777.30819.85. [DOI] [PubMed] [Google Scholar]
- 48.Kovalenko M, Denner K, Sandstrom J, Persson C, Gross S, Jandt E, et al. Site-selective dephosphorylation of the platelet-derived growth factor beta-receptor by the receptor-like protein-tyrosine phosphatase DEP-1. J Biol Chem. 2000;275:16219–26. doi: 10.1074/jbc.275.21.16219. [DOI] [PubMed] [Google Scholar]