

Abstract
Transcription regulation by steroid hormones, vitamin derivatives, and metabolites is mediated by nuclear receptors (NRs), which play an important role in ligand-dependent gene expression and human health. NRs function as homodimers or heterodimers and are involved in a combinatorial, coordinated and sequentially orchestrated exchange between coregulators (corepressors, coactivators). The architecture of DNA-bound functional dimers positions the coregulators proteins. We previously demonstrated that retinoic acid (RAR-RXR) and vitamin D3 receptors (VDR-RXR) heterodimers recruit only one coactivator molecule asymmetrically without steric hindrance for the binding of a second cofactor. We now address the problem of homodimers for which the presence of two identical targets enhances the functional importance of the mode of binding. Using structural and biophysical methods and RAR as a model, we could dissect the molecular mechanism of coactivator recruitment to homodimers. Our study reveals an allosteric mechanism whereby binding of a coactivator promotes formation of nonsymmetrical RAR homodimers with a 2∶1 stoichiometry. Ligand conformation and the cofactor binding site of the unbound receptor are affected through the dimer interface. A similar control mechanism is observed with estrogen receptor (ER) thus validating the negative cooperativity model for an established functional homodimer. Correlation with published data on other NRs confirms the general character of this regulatory pathway.
Keywords: allostery, structure
The superfamily of nuclear receptors (NRs) comprises ligand-dependent transcription factors involved in the regulation of gene expression. They constitute key drug targets for human diseases such as cancer, osteoporosis, obesity, or type II diabetes (1–2). NRs share a common structural organization with a variable amino-terminal domain, a conserved DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) linked by a flexible hinge peptide. In addition to the ligand-binding pocket, the LBD comprises dimerization surfaces and the sites for co-regulator interactions. In the classic mode of action, in absence of ligand, some NRs are associated with corepressors (NCoRs) that harbor histone-deacetylase activity to maintain the chromatin in a transcriptionally silent state (3–4). Upon ligand binding, DNA-bound receptors recruit coactivators like the steroid receptor coactivator 1 (SRC-1), a member of p160 CoA family (5), to enhance target gene expression. The receptor interaction domain (RID) of the coactivators is responsible for the interaction with NRs and contains several copies of the short consensus interaction motif LXXLL (6).
The vast majority of NRs functions as dimers. RAR, like the vitamin D (VDR) and thyroid hormone (TR) receptors, heterodimerizes with rexinoid receptors (RXRs) (7). Their ability to form homodimers is also documented (8–10). RAR homodimers have been shown to be functional in yeast using a two-hybrid system, and their activity is further enhanced by the presence of SRC-2 coactivator (11). In vivo, the targeted ablation of all RXR isotypes in mouse Sertoli cells yields a phenotype distinct from that observed upon ablation of all RARs isotypes suggesting that in Sertoli cells an alternate RXR independent mechanism accounts for the RAR-mediated biological responses (12). Similarly, knockdown of RXRs by RNAi impair only a subset of T3 regulated genes (13).
We recently demonstrated that SRC-1 binds to RXR-RAR dimers via the RAR subunit (14). In absence of steric hindrance different affinities for RXR and RAR could account for the observed 2∶1 stoichiometry. However, the explanation cannot apply to homodimers although it has been shown that SRC-1 RID binds to ER dimers with a stoichiometry of one coactivator interacting domain bound per dimer (15–16). The present study tackles this problem using RAR as a model case and testing the observations on ERα, an established functional homodimer. The rationale behind the choice of RAR lies in the observation that with the same ligand its LBD could be crystallized as a monomer and a dimer, the latter in presence of a silicon analog of the ligand and a coactivator peptide (17–18). In contrast with other NRs we can thus have access to all different structural states, including RAR-RXR heterodimer, and correlate them to the binding of the coactivators and/or different ligands. In the present study we demonstrate that binding of the coactivator to one RAR subunit exerts an allosteric control over its own interaction with the second RAR protomer. The molecular mechanism involves modification of both the ligand-binding and dimerization functions of RAR. Extending our analysis to steroidal homodimers, estrogen receptor (ER) homodimers, we observe a similar molecular mechanism of control. The crystal structures of ERα LBD homodimers show an asymmetry consistent with the conclusions derived from RAR observations. This molecular mechanism of regulation opens a novel perspective for the design of more selective ligands.
Results
Crystal Structures of hRARβ LBD Homodimer with SRC-1 Coactivator Peptide.
We determined the crystal structures of the ternary complexes containing the RARβ LBD bound to a 25-mer peptide derived from NR-interaction motif 2 of the SRC-1 coactivator (SRC-1 NR2) and the agonists TTNPB or 9-cis RA (Table S1). Both complexes crystallizes into the P212121 space group and contain two LBDs in the asymmetric unit, each LBD being bound to one ligand and one SRC-1 NR2 peptide. The TTNPB and 9-cis RA complex structures were solved by molecular replacement and refined to 1.9 Å and 2.3 Å resolution, respectively. Packing analysis (Fig. 1) shows that crystals are built from dimeric entities similar to those observed in the crystal structures of ER (19), ERR (20), PPAR (21), or LXR (22–23). Both subunits adopt the active conformation, with the C-terminal activation helix H12 capping the ligand-binding pocket and the SRC-1 NR2 peptide bound to the surface formed by residues from helices H3, H4, and H12 (Fig. 2A). In contrast to symmetric homodimers, the overall RAR-RAR arrangement is asymmetric and reminiscent of that seen in RXR-RAR heterodimers (24–25) (Fig. 2 B–D). The dimerization interface involves residues from helices H7, H9, and H10 and loops L8-9 and L9-10 with a pseudo twofold axis relating each monomer. Compared to the RXRα-RARβ heterodimer (PDB ID code 1XDK) (25), more contacts are observed for the RAR-RAR interface at the C-terminal end of H10. Overall, the buried surface of each monomer within the homodimer (1,090 Å2) is larger than that of RAR in the heterodimer (930 Å2). A remarkable observation is the asymmetry of the dimer with different ligand conformation in each monomer. See below for a detailed description of the asymmetry in the RAR-RAR homodimer.
Fig. 1.

Crystal packings of RARβ LBD complexes. The asymmetric unit content is shown in green. (A) The dimeric RARβ-TTNPB-SRC-1 NR2 complex. (B) The RARβ-TTNPB monomer (PDB ID code 1XAP).
Fig. 2.

Crystal structure of RARβ LBD-TTNPB-SRC-1 NR2. Overall structures of RARβ homodimer (A) and RXR-RAR heterodimer (B) are similar. (A) The crystal structure of HsRARβ homodimer bound to TTNPB and SRC-1 NR2 peptide is shown in green (monomer 1, M1) and light green (monomer 2, M2). (B) Crystal structure of the heterodimer complex (PDB ID code 1XDK) with MmRXRα (cyan) and HsRARβ (green) bound to 9-cis RA and the coactivator peptide (purple). (C) Superposition of the RARβ-RARβ (pink) and RXRα-RARβ dimerization interface. Helices H9 and H10 are depicted as Cα traces. (D) Superposition of the RARβ-RARβ (pink) and RXRα-RXRα (PDB ID code 2ZY0) (blue) dimerization interface.
Although the overall intermolecular interaction pattern is conserved in the homodimer, some important differences with possible structural/functional implications can be observed. Indeed, the salt bridge between HsRARα Arg364 (H9) and MmRXRα Glu406 (H9) is absent in the homodimer (Fig. 2C). The position of Glu406 is occupied by Lys360. This difference is likely to account for a closer contact of RAR H9 with the second subunit in the heterodimer. Despite the occurrence of more H10-H10 contacts in the homodimer, the absence of this salt bridge can explain its lower stability relative to that of the heterodimer.
RARβ Forms Dimers in Solution in Presence of Ligand and Coactivator Peptide.
The different oligomerization states in the crystals of the RARβ-TTNPB in absence and in presence of the SRC-1 NR2 peptide raised the question of the oligomeric form of the complex in solution. We analyzed the solution properties of the RARβ LBD alone and complexed with ligand and NR2 peptide by several biophysical methods; namely, analytical ultracentrifugation in velocity mode, isothermal titration calorimetry (ITC), and size exclusion chromatography. In the latter, the unliganded RARβ LBD elutes as a monomer with an apparent molecular weight of 25–30 kDa. Addition of ligands (TTNPB or 9-cis RA) and SRC-1 NR2 resulted in the appearance of a second peak with an elution volume (Fig. 3A and Table S2) corresponding to an apparent molecular weight of 60 kDa, thus suggesting formation of RAR homodimers. Both the homogeneity and stability of the assayed samples were verified by native PAGE (Fig. S1A). Analysis of the sedimentation velocity profiles by analytical ultracentrifugation confirms that in absence of ligand, the RARβ LBD appears as a single species with a sedimentation coefficient and a molecular weight corresponding to that of a monomer (Fig. 3B). Two sedimentation species corresponding to a monomer and a dimer of RARβ LBD are observed upon addition of ligands (TTNPB or 9-cis RA) and 3 molar equivalents of SRC-1 NR2 (Fig. 3B). Under these conditions, the dimeric species represent approximately 1/3 of the protein content. Similar conclusions could be drawn for the other RAR, α and γ paralogues (Fig. S1 B and C).
Fig. 3.

Coactivator bound-RARβ LBD forms homodimers in solution. (A) Gel filtration profile of apo RARβ LBD and the TTNPB complexes with SRC-1 NR2 or SRC-1 RID showing that two oligomeric species are observed in presence of the peptide and only one species in presence of the RID. (Bottom) SDS gel stained by quantitative Sypro-Ruby of the RARβ-SRC-1 RID fraction that indicates a stoichiometry of 1 RID for 2 RARβ. (B) Sedimentation velocity analysis for RARβ LBD apo and its complexes with TTNPB and SRC-1 NR2 peptide by Lamm equation fits using the Sedfit program. The sedimentation distribution plots show one sedimentation species for the apo RARβ LBD and the TTNPB complex with a sedimentation coefficient 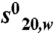 of 2.6 ± 0.1 Svedberg units. The calculated molecular mass value is 29 kDa and corresponds to the monomer. In presence of ligand (TTNPB or 9-cis RA) and SRC-1 NR2 peptide, two peaks are observed, one corresponding to the monomer (
of 2.6 ± 0.1 Svedberg units. The calculated molecular mass value is 29 kDa and corresponds to the monomer. In presence of ligand (TTNPB or 9-cis RA) and SRC-1 NR2 peptide, two peaks are observed, one corresponding to the monomer (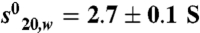 ; Mw = 31 kDa) and the second to the dimer (
; Mw = 31 kDa) and the second to the dimer (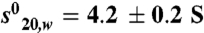 (Mw = 58 kDa). (C–D) Representative ITC titrations of SRC-1 NR2 peptide (C) and SRC-2 RID (D) into RARα LBD. RARα LBD binds SRC-1 NR2 with a stoichiometry of two peptides per homodimer and a dissociation constant Kd of 1.5 μM, whereas in the case of SRC-1 RID, RARα binds with a stoichiometry of 1 RID per homodimer and a Kd of 2.5 μM.
(Mw = 58 kDa). (C–D) Representative ITC titrations of SRC-1 NR2 peptide (C) and SRC-2 RID (D) into RARα LBD. RARα LBD binds SRC-1 NR2 with a stoichiometry of two peptides per homodimer and a dissociation constant Kd of 1.5 μM, whereas in the case of SRC-1 RID, RARα binds with a stoichiometry of 1 RID per homodimer and a Kd of 2.5 μM.
A Single SRC-1 RID Domain Binds to RARβ LBD Homodimer.
The correlation between the binding of a short CoA peptide and the existence of a dimeric form of the receptor prompted us to evaluate the effect of interaction with the entire SRC-1 RID (616-779 aa) containing three LXXLL interaction motifs on the oligomerization state. Using size exclusion chromatography and native PAGE, we identified only one species with an apparent molecular weight corresponding to that of the ternary (RARβLBD)2 -TTNPB-SRC-1 RID complex (Fig. 3A and lanes 2 and 3 in Fig. S1A). While the SRC-1 RID is partially unfolded alone as seen by its gel-filtration profile (Table S2), the protein adopts a defined fold upon interaction with RARβ LBD as observed in the Kratky plot obtained by small angle X-ray scattering (SAXS) (Fig. S2). The stoichiometry of the complex suggested by Sypro–Ruby quantitative staining on the SDS/PAGE gel corresponds to one RARβ LBD homodimer bound to one molecule of coactivator domain (Fig. 3A). This result is supported by analytical ultracentrifugation in equilibrium mode (Fig. S3), a method that is independent of the shape of the molecules. The observed molecular mass (76,000 ± 3,000 Da) of the complex is in agreement with that of a 2∶1 complex (79,000 Da). From these observations, we conclude that the presence of coactivator peptide favors the formation of homodimers coexisting with the monomeric form of RAR in solution and that the crystallization conditions have selected the dimers.
Binding affinities were measured by ITC for the SRC-1 NR2 peptide and SRC-2 RID (Fig. 3 C–D and Table S3). The SRC-1 NR2 peptide binds to RARβ LBD with a stoichiometry of 2 (2 NR2 peptides per homodimer) and a mean Kd of 1.5 μM. In contrast only one SRC-2 RID binds to RAR dimer LBD with a mean Kd of 2.5 μM, suggesting a larger induced conformational change at the second binding site. These observations are in agreement with the crystallographic and biophysical data. A similar stoichiometry of one coactivator RID per dimer is observed for the heterodimer RXR-RAR.Interestingly the affinity of SRC-2 RID is higher for RXR-RAR than for the RAR homodimer.
Asymmetry of the RARβ Homodimer Complex and Suggested Mechanism.
In contrast to the twofold symmetry of the RXR LBD homodimers (26), the RARβ homodimers like the RXRα-RARβ heterodimer are asymmetric (Fig. 4A). A marked tilt of the dimer interface brings helix H7 of monomer (M1) closer to loop L8-9 of its partner (M2). Further asymmetric interactions involving residues of loop L8-9 are observed between the two monomers, as for example the salt-bridge interaction of Asp338 (loop L8-9) with Lys380 (H10). When the two monomers are superimposed, the Cα atoms of the two subunits M1 and M2 have an rmsd of 0.65 Å over 233 residues (Fig. 4B). Large differences are observed for loop L8-9 (rmsd of 0.94 Å), the C-terminal end of the proteins (rmsd of 1.13 Å on the Cα atoms of the last 24 residues from H11 to H12), and the SRC-1 NR2 peptide (rmsd of 1.4 Å). Similar values were obtained for the RARβ-9-cis RA-SRC-1 NR2 complex. This difference is not a consequence of the crystal packing because the coactivator peptide and the C-terminal part of M2 are not involved in crystal packing interactions. The structural superposition of the crystal structure of monomeric RARβ-TTNPB LBD (PDB ID code 1XAP) onto M1 and M2 subunits of the homodimer indicates that monomer M1 is much closer to the monomeric conformer (rmsd 0.46 Å versus 0.65 Å for M2). The comparison of RARβ M1 and M2 of the homodimer with the RARβ of the heterodimer (PDB ID code 1XDK) indicates that the monomer M1 is also the closest to the RARβ in the heterodimer. Furthermore, M1 is the monomer with the more tightly bound CoA peptide according to the observed contacts (Fig. 4B). Thus, M1 can be considered as the initial conformation and the CoA target and the conformational changes observed in M2 are most likely the result of an allosteric transfer through the dimer interface.
Fig. 4.

Asymmetry in the RARβ homodimer. (A) View along the pseudo-twofold axis of the homodimer LBD of hRARβ. Red arrows highlight the dissymmetrical distances between RARβ’s helix H7 and loop L8-9. The closest residues (Cα atoms) are at 5.7 Å and 8.9 Å, respectively. (B) Superimposition of monomer M1 (green) on monomer M2 (light green) showing the large differences at the C-terminal end with a tilt of H11 and a shift of H12 (rmsd of 1.1 Å on Cα) and a larger shift of the CoA peptide (rmsd of 1.4 Å). (C) Stereoview of the class II specific salt bridge in the superimposed monomers. Amino acids of M1 monomer are in green; those of the M2 that exhibit a different conformation are represented in orange. One of the conserved salt bridges (Asp267—Arg385) is disrupted in M2. (D) Close-up view of the two conformations adopted by the TTNPB ligand in each monomer of the homodimeric hRARβ LBD superimposition was done on the protein Cα atoms. The TTNPB molecules of M1 and M2 are shown in red and dark blue, respectively.
We then examined some key residues that define the signature motif of heterodimers and compared their position to that identified in the RXR-RAR complex (7). The intramolecular salt bridge between Glu325 of H8 and Arg367 of loop L9-10 is similar in both monomers (Fig. 4C). In contrast, the residues forming the second specific intramolecular salt bridge (Asp267 of H5 and Arg339 of loop L8-9) have different orientations in M1 and M2. In M1, Asp267 interacts with Arg385, whereas in M2 Asp267 interacts with Arg339. A similar asymmetry of the monomers was observed in RARβ complexes with both TTNPB and 9-cis RA.
The mutation RARα Glu393Gln (Fig. S4) has been shown to induce weaker transcriptional activity and to be important in the allosteric communication between ligand and the dimerization interface (27). In the context of our work, we characterized the ability of this mutant to form homodimers and to recruit coactivator. The analysis of the sedimentation velocity profiles by analytical ultracentrifugation of RARα Glu393Gln in absence of ligand and in complex with TTNPB and SRC-1 NR2 reveals that this mutant still allows the formation of dimer induced by the peptide (Fig. S4B). Binding affinities were measured using ITC for the SRC-1 NR2 peptide and SRC-2 RID (Table S3). The SRC-1 NR2 peptide binds to RARα Glu393Gln LBD with a lower affinity than the wild type (Kd of 7 μM instead of 1.5 μM). Analyzing the data with a sequential binding, the binding affinities for the first and second peptides are 3 and 16 μM, respectively. The data show that the mutation affects mainly the binding of the second peptide as a consequence of the role in allosteric communication of this residue.
Ligand Binding and Coactivator Recruitment.
The asymmetry is also observed in the ligand-binding pocket with different conformations of the aromatic part of the TTNPB molecules in each monomer (Fig. 4D). In the M1 monomer, the TTNPB ligand exhibits the same conformation as in the RARβ monomer and forms similar interactions with residues of the ligand-binding pocket. Differences between the two monomers are observed for the positions of the 5,6,7,8-tetrahydronaphthalene skeleton (Fig. 4D). Some contacts are affected, such as fewer weak interactions involving TTNPB in M1 [e.g., contact with Ile410 (H12) of 4.3 Å instead of 3.7 Å in M2 and with Leu414 (H12) of 4.1 Å instead of 3.8 Å in M2] (Fig. S5). The lower resolution of the structure and the more symmetrical shape of 9-cis RA do not allow the detection of a similar conformational change with this ligand. Both SRC-1 NR2 peptides form an amphipathic α-helix that interacts with the hydrophobic cleft that is generated by the C-terminal part of H3, the loop L3-4, H4, and H12. These interactions are similar to those described for other NRs. In particular, in both monomers, Lys244 from H3 and Glu412 from H12 form a charge clamp by forming hydrogen bonds with the main-chain peptide bond of the LXXLL motif. The hydrophobic interactions formed by the leucine residues of the peptide are similar in the two monomers, but marked differences are observed in the N-terminal flanking residues of the LHRLL motif that has different orientations in the two monomers. An interaction between His687 at position −3 of the motif with Lys262 (2.8 Å) is present in M1 and absent in M2. An rmsd of 1.4 Å is observed for the coactivator peptide in the superimposed M1-M2 (Fig. 4B). The interatomic distances are longer in the M2-CoA contacts such as for example Val240 with Leu694 (4.0 Å in M2 instead of 3.8 Å in M1), Met413 with Leu690 (4.0 Å in M2 instead of 3.8 Å in M1) thereby explaining the weaker binding.
ERα Homodimers Are Asymmetric in Presence of SRC-1 NR2 Peptide.
To investigate the possibility that the observations could apply to other NRs known to function as homodimers, we analyzed the crystal structure of the ternary complex formed by ERα LBD bound to SRC-1 NR2 peptide and the synthetic estrogen ligand, RU100132. The complex crystallizes into the P21 space group and contains one homodimer in the asymmetric unit, each LBD being bound to one ligand and one SRC-1 NR2 peptide. The structure was solved by molecular replacement and refined to 2.3-Å resolution (Table S1). Both subunits adopt the active agonist conformation and the SRC-1 NR2 peptide bound to the surface formed by residues from helices H3, H4, and H12 (Fig. S6A). The dimerization interface involves residues from helices H7, H9 and H10 and loops L8-9 and L9-10 as previously described (19, 28) with a tilt of the dimer interface (Fig. S6B). A detailed analysis reveals an asymmetry within the dimer. Superimposition of the two monomers M1 and M2 leads to rmsd of only 0.52 Å over the Cα atoms of 243 residues but large variation are observed for loop L1-3, and more importantly significant differences affect the C-terminal end of the proteins (last 24 residues from H11 to H12).
The asymmetry is also observed in the ligand-binding pocket with differences in the position of the ligands and in their contacts within the LBP of each monomer (Fig. S7). The interactions mediated by the SRC-1 NR2 peptides are similar to those described (17). In particular, in both monomers, Lys362 from H3 and Glu542 from H12 form a charge clamp by forming hydrogen bonds with the main-chain peptide bond of the LXXLL motif. The hydrophobic interactions formed by the Leucine residues of the peptide are similar in the two monomers, but an overall shift of 1.7 Å for the backbone M2 bound peptide suggests different binding strengths (Fig. 5).
Fig. 5.

Asymmetry of the ERα LBD-SRC-1 complex. Superimposition of monomer M1 (orange) on monomer M2 (light orange) showing the differences of the CoA peptides (violet and pink) and of their interactions with each monomer. Small but significant differences are observed in the interactomic distances that are longer in the M2-CoA contacts such as for example Ile358 with Leu693 (3.6 Å in M2 instead of 3.3 Å in M1), Val376 with His691 (4.0 Å in M2 instead of 3.5 Å in M1).
One Molecule of SRC-1 RID Binds to ER Dimer.
Previous studies (15–16) have shown that SRC-1 RID bind to ER dimers with a stoichiometry of one coactivator interacting domain bound to the ER dimer. A twofold stabilizing effect in the affinity was also reported in case of SRC-1 RID compared to SRC-1 NR2 peptide binding to ERα (29). We performed ITC experiments on ERα complexes (Table S3). The SRC-2 RID binds to liganded ERα LBD dimers with a stoichiometry of 1 coactivator per homodimer and a Kd of 0.65 μM. Analytical ultracentrifugation experiments were also performed on ERβ dimer in complex with SRC-1 RID. Analysis of the sedimentation velocity profiles confirms the stoichiometry of one SRC-1 RID bound to ERβ homodimer. The ERβ LBD-estradiol-SRC-2 RID (623–722) appears as a single species with a sedimentation coefficient and a molecular weight of 84500 Da in agreement with a 2∶1 stoichiometry (Fig. S8). In case of an ER mutant (ERα Glu523Gln) in H11 at the dimer interface that is involved in allosteric communication between ligand and the dimerization interface and has been shown to affect transcription activity (27), the binding affinity for the SRC-2 RID is slightly lower than in the case of the non mutated ERα (Table S3).
Solution Structure of RARΔAB-DNA Homodimers.
To extend our observation of the asymmetry of SRC-1 bound RAR LBD homodimer to the full-length receptor, we looked at the ability of RARαΔAB to form homodimers in presence of DNA response elements and SRC-1. By fluorescence anisotropy we measured the affinity of RARαΔAB for a DR2 response element fluorescently labeled in absence and presence of the 25-mer SRC-1 NR2 peptide (Fig. S9). SRC-1 NR2 peptide stabilizes the DNA binding of RAR homodimer (Kd = 25 ± 2 nM). These results are consistent with the observed enhanced DNA binding of TR homodimers in presence of SRC-1 (13). SAXS was used to obtain structural information on the dimers bound to SRC RID. Monodisperse concentrated solutions of RARαΔAB in complex with DNA and SRC RID were analyzed (Fig. S10). The structural parameters including the radius of gyration (Rg) and the maximum particle dimension (Dmax) computed from the experimental scattering patterns are given in Table S4. Two natural DRs were used, DR5 from the mRARβ2 and DR2 from the Hoxa10 promoters. The large differences between the Rg and Dmax values suggest an elongated shape for the RARαΔAB-DNA homodimer complexes similar to those of the heterodimer (Fig. S10C). We have previously shown that the structure of RXR-RAR-DR5 complex shows an asymmetric conformation with distinct DBD and LBD modules with the LBDs positioned at the 5′ end of target DNA (14). The scattering pattern of the complex between RARαΔAB-DNA and SRC-2 RID is very similar to those obtained for SRC-2 RID complex to RXRαΔAB-RARαΔAB-DNA using a RXR binding cleft mutant that is unable to bind CoA (14). The SAXS data are consistent with a structural model in which SRC RID is asymmetrically bound to NR dimers in contrast to a tethered binding of the coregulators to the dimers as suggested by the PPARγ-SRC-1 crystal structure (21). This asymmetry was also described by Margeat et al. (15) for ERα homodimer bound to SRC-1 RID.
Discussion
A combination of structural and thermodynamic data provides the structural basis for an allosteric mechanism controlling the binding of coactivators. A molecular pathway can be inferred from the observed deviations from symmetry. The binding of an SRC-1 peptide to the RAR LBD promotes its dimerization through residues in the N-terminal region of the LXXLL in contact with loop L8-9. An asymmetry is then propagated to the other monomer through H10, which in turn induces a conformational change at the C-terminal end (H11-H12) of the second monomer destabilizing the binding of the second peptide (Fig. 6). The ligand conformation of the second unit is also affected, opening new perspectives for drug design. H11 has previously been identified as a ligand sensor acting on the activation helix with a conformational change from a beta-strand-to-alpha-helix transition upon corepressor-coactivator exchange (30). The choice of RAR as a model system to dissect the molecular mechanism of allostery was essential because in absence of RXR and cofactors the LBD is a monomer in solution. It allowed us to unambiguously detect the origin of the conformational changes. Whether RAR homodimers may play a role in the regulation of specific target genes acting as additional modulators of physiological response to retinoic acid is not fully admitted, but some results support the possibility (11–12, 31). The formation of such homodimers is contingent upon either the nature of the coactivator interacting with RAR or the promoter context of target genes (11, 13). Other NR LBDs that are known to function as heterodimers with RXR such as PPAR (21), LXRα (22) or LXRβ (23) have also been crystallized as homodimers, with the same interface as RAR, in presence of coactivator peptides. For PPARγ-SRC-1 (PDB ID code 2PRG), while an rmsd of 0.65 Å on the overall Cα structure is observed between the two monomers, the C-terminal region (H11-H12) shows a significant difference with an rmsd value of 1.18 Å. In the LXRβ structure, different conformations of the ligand were also observed. A recent study of RXR complex to 9-cis retinoic acid and SRC-1 NR2 revealed a difference in the conformation of the 9-cis RA in the coactivator bound RXR (32).
Fig. 6.

Proposed mechanism for the allosteric control by coactivator binding to nuclear hormone receptors. (A) Binding of the NR interacting domain RID to NR monomer M1 triggers the formation of dimers with an induced conformational change of the C-terminal end of monomer M2 and its ligand. Consequently, the binding of the CoA on M2 is affected. (B) Molecular level representation of our proposed mechanism of allosteric regulation.
In the complex with a larger domain of SRC-1, the asymmetry is even more pronounced with a single domain bound asymmetrically on one side of RAR or ER homodimer, even in presence of a large excess of CoA domain. Our results suggest that the N-terminal flanking residues of the LXXLL motif that join the dimer interface are the most effective in the control mechanism. An allosteric communication involving the N-terminal end of PGC-1α peptide and ERRα homodimer via the loop 8–9 has been characterized (20). The functional implications of asymmetry were illustrated in the case of the RXR-RAR heterodimer bound to the response element of RARβ2 gene promoter (14). The architecture of DNA-bound functional dimers positions the coregulators proteins. The asymmetric structure orients unambiguously the bound coactivator toward its target. In the case of homodimers the selection of the primary target (M1 of Fig. 6) determines the position of the coactivator and the final architecture. DNA and/or additional transcription factors may play a role in this preselection process. DNA may also play a role in the allosteric control of coactivator binding and dimerization as suggested for TR where the binding of the SRC-1 RID is influenced by the nature of the DNA response elements (10, 33). An allosteric communication between SRC-1 and DNA was reported for RXR-TR (34) and RXR-VDR (35). The influence of natural RARβ response elements was also shown to trigger the formation of RARα homodimers (8).
In the present study we show that the mechanism controling coactivator binding can be extended to others NRs homodimers such as ERs. In addition, the crystal structure of ERRα bound to a PGC-1α fragment exhibits a similar asymmetry in the coactivator peptide binding mode and confirms the general character of the model (20). The structural explanation of the negative cooperativity is that the conformational changes in the second monomer affect the binding surface and thus the affinity for the same CoA domain. It is also possible that the allosterically induced structural differences would favor the recruitment of other coactivators. The close structural similarity between RAR homodimers and RXR-RAR heterodimers further suggests that this allosteric mechanism could also apply to heterodimers. The binding of coactivators to RAR would thus regulate their binding to RXR.
Methods
Ligands and Peptides.
9-cis RA, TTNPB and estradiol were purchased from BioMol. All ligands were dissolved in ethanol. The SRC-1 NR2 (676-CPSSHSSLTERHKILHRLLQEGSPS-700) and (686-RHKILHRLLQEGSPS-700) peptides were synthesized by Pascal Eberling (IGBMC peptide synthesis common facility).
Expression and Purification.
The HsRARα LBD ( 176–421 aa), HsRARβ LBD (169–414 aa) and HsRARγ LBD (178–423 aa), HsRARαΔAB (82–462), HsSRC-1 RID (616–779), HsSRC-2 RID (622–828 and 632–772) HsERα LBD (305–552), HsERα C530A LBD (305–552), and HsERβ LBD (255–509) were expressed and purified as described in refs. 14, 36, and 37. Ligands were added in a twofold excess to saturate the receptors. For the RAR LBD-CoA, ER LBD-CoA and RARΔAB-DNA-CoA complexes, the coactivator peptides were added in threefold molar excess or two equivalent of purified RID. The complexes were characterized by Native-PAGE and biochemical methods. Sypro-Ruby dye (Bio-Rad) was used to quantify the amount of proteins in the complex.
Resolution of the Crystal Structures.
Crystallization of RARβ complexes were obtained as described (36). Both RAR structures were refined using the known hRARβ LBD-TTNPB structure (PDB ID code 1XAP) as a starting model. The resolution was 2.3 Å and 1.9 Å for the 9-cis RA and TTNPB complexes. ERα structures were refined to 2.3 using published ERα LBD structure as starting model (PDB ID code 3ERD). Model building and refinement using restrained refinement and individual B-factor refinements and were done as described (17).
Analytical Ultracentrifugation.
Sedimentation velocity and sedimentation equilibrium experiments were performed at a temperature of 4 °C using a Beckman Optima XL-A analytical ultracentrifuge with absorbance monitoring in a buffer containing 20 mM Tris (pH 8.0), 200 mM NaCl. Protein concentrations were in the range of 0.8 to 2 mg/mL. For sedimentation equilibrium experiments, samples were spun at 15,000 and 20,000 rpm, and systems were first allowed to equilibrate for 12 h before absorbance profiles were compared at different times to ensure that system had reached equilibrium. Using nonlinear least-squares analysis with Sedphat program, these datasets were fitted using single component model and several equilibrium models. For sedimentation velocity, samples were spun at 50,000 rpm. Consecutive scans were automatically recorded at regular intervals and analyzed with the software Sedfit which directly models boundary profiles in terms of a continuous distribution of discrete and noninteracting species.
Isothermal Titration Calorimetry (ITC).
ITC measurements were performed at 30 °C on a MicroCal ITC200 (MicroCal). Purified proteins and peptides were dialyzed extensively against the same buffer used in the ITC experiments. The buffer contained 20 mM Tris pH 8.0, 200 mM sodium chloride and 1 mM TCEP. In a typical experiment 2 μl aliquots of SRC-1 NR2 peptide (676-CPSSHSSLTERHKILHRLLQEGSPS-700) at 1.3 mM or SRC-2 RID at 350–700 μM were injected into a 20–50 μM RARα LBD-TTNPB or ERα LBD-estradiol solution (200 μl sample cell). The delay between injections was 120 to 180 s to permit the signal to return to baseline before the next injection. ITC titration curves were analyzed using the software Origin 7.0 (OriginLab). Standard free energies of binding and entropic contributions were obtained, respectively, as ΔG = -RT ln(Ka) and TΔS = ΔH - ΔG, from the Ka and ΔH values derived from ITC curve fitting.
SAXS Experiments and Data Processing.
The synchrotron radiation X-ray scattering data were collected at the storage ring DORIS III of the Deutsches Elektronen Synchrotron (DESY). All studied complexes were measured for at least three protein concentrations ranging from 2 to 5 mg/mL. The data were collected and processed using standard procedures as described (14).
Supplementary Material
Acknowledgments.
We thank Proskelia–Aventis, C. Birck for help in analytical ultracentrifugation, E. Ennifar for the help in ITC, N. Ramalanjoana for fluorescence experiments, and P. Antony and I. Davidson for critical reading of the manuscript. We are also grateful to D. I. Svergun and the technical staff of Deutsches Elektronen Synchrotron (EMBL, Hamburg) for technical support on the beam line. We thank the staff of the beamline at the European Synchrotron Radiation Facility for the experimental assistance during data collection. We thank P. Eberling for the peptide synthesis. This work was funded by Centre National de Recherche Scientifique, Institut National de Santé et de Recherche Médicale, Université de Strasbourg, by Agence Nationale de la Recherche (ANR-09-BLAN-0282 -NUREDIM) and the EU I3 grant for the access to European Molecular Biology Laboratory beamline. J.O. was supported by a long-term EMBO fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 3610 (volume 109, number 10).
Data deposition: The atomic coordinates, structure factors, and crystallography have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4DM6, 4DM8, and 4DMA).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118192109/-/DCSupplemental.
References
- 1.Mark M, Ghyselinck NB, Chambon P. Function of retinoic acid receptors during embryonic development. Nucl Recept Signal. 2009;7:e002. doi: 10.1621/nrs.07002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sonoda J, Pei L, Evans RM. Nuclear receptors: Decoding metabolic disease. FEBS Lett. 2008;582:2–9. doi: 10.1016/j.febslet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagy L, et al. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev. 1999;13:3209–3216. doi: 10.1101/gad.13.24.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 5.Oñate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 6.Ding XF, et al. Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): Multiple motifs with different binding specificities. Mol Endocrinol. 1998;12:302–313. doi: 10.1210/mend.12.2.0065. [DOI] [PubMed] [Google Scholar]
- 7.Brelivet Y, Kammerer S, Rochel N, Poch O, Moras D. Signature of the oligomeric behaviour of nuclear receptors at the sequence and structural level. EMBO Rep. 2004;5:423–429. doi: 10.1038/sj.embor.7400119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venepally P, Reddy LG, Sani BP. Analysis of homo- and heterodimerization of retinoid receptors in solution. Arch Biochem Biophys. 1997;343:234–242. doi: 10.1006/abbi.1997.0158. [DOI] [PubMed] [Google Scholar]
- 9.Takeshita A, Ozawa Y, Chin WW. Nuclear receptor coactivators facilitate vitamin D receptor homodimer action on direct repeat hormone response elements. Endocrinology. 2000;141:1281–1284. doi: 10.1210/endo.141.3.7441. [DOI] [PubMed] [Google Scholar]
- 10.Velasco LF, et al. Thyroid hormone response element organization dictates the composition of active receptor. J Biol Chem. 2007;282:12458–12466. doi: 10.1074/jbc.M610700200. [DOI] [PubMed] [Google Scholar]
- 11.Walfish PG, et al. Yeast hormone response element assays detect and characterize GRIP1 coactivator-dependent activation of transcription by thyroid and retinoid nuclear receptors. Proc Natl Acad Sci USA. 1997;94:3697–3702. doi: 10.1073/pnas.94.8.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vernet N, et al. Prepubertal testis development relies on retinoic acid but not rexinoid receptors in Sertoli cells. EMBO J. 2006;25:5816–5825. doi: 10.1038/sj.emboj.7601447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diallo EM, Wilhelm KG, Jr, Thompson DL, Koenig RJ. Variable RXR requirements for thyroid hormone responsiveness of endogenous genes. Mol Cell Endocrinol. 2007;264:149–156. doi: 10.1016/j.mce.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rochel N, et al. Common architecture of nuclear receptor heterodimers on DNA direct repeat elements with different spacing. Nat Struct Mol Biol. 2011;18:564–570. doi: 10.1038/nsmb.2054. [DOI] [PubMed] [Google Scholar]
- 15.Margeat E, et al. The human estrogen receptor alpha dimer binds a single SRC-1 coactivator molecule with an affinity dictated by agonist structure. J Mol Biol. 2001;306:433–442. doi: 10.1006/jmbi.2000.4418. [DOI] [PubMed] [Google Scholar]
- 16.Tamrazi A, Carlson KE, Daniels JR, Hurth KM, Katzenellenbogen JA. Estrogen receptor dimerization: Ligand binding regulates dimer affinity and dimer dissociation rate. Mol Endocrinol. 2002;16:2706–2719. doi: 10.1210/me.2002-0250. [DOI] [PubMed] [Google Scholar]
- 17.Germain P, et al. Rational design of RAR-selective ligands revealed by RARbeta crystal stucture. EMBO Rep. 2004;5:877–882. doi: 10.1038/sj.embor.7400235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Büttner MW, et al. Silicon analogues of the retinoid agonists TTNPB and 3-methyl-TTNPB, disila-TTNPB and disila-3-methyl-TTNPB: Chemistry and biology. Chembiochem. 2007;8:1688–1699. doi: 10.1002/cbic.200700182. [DOI] [PubMed] [Google Scholar]
- 19.Shiau AK, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 20.Greschik H, et al. Communication between the ERRalpha homodimer interface and the PGC-1alpha binding surface via the helix 8–9 loop. J Biol Chem. 2008;283:20220–20230. doi: 10.1074/jbc.M801920200. [DOI] [PubMed] [Google Scholar]
- 21.Nolte RT, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 22.Fradera X, et al. X-ray structures of the LX Ralpha LBD in its homodimeric form and implications for heterodimer signaling. J Mol Biol. 2010;399:120–132. doi: 10.1016/j.jmb.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Williams S, et al. X-ray crystal structure of the liver X receptor beta ligand binding domain: Regulation by a histidine-tryptophan switch. J Biol Chem. 2003;278:27138–27143. doi: 10.1074/jbc.M302260200. [DOI] [PubMed] [Google Scholar]
- 24.Bourguet W, et al. Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol Cell. 2000;5:289–298. doi: 10.1016/s1097-2765(00)80424-4. [DOI] [PubMed] [Google Scholar]
- 25.Pogenberg V, et al. Characterization of the interaction between retinoic acid receptor/retinoid X receptor (RAR/RXR) heterodimers and transcriptional coactivators through structural and fluorescence anisotropy studies. J Biol Chem. 2004;280:1625–1633. doi: 10.1074/jbc.M409302200. [DOI] [PubMed] [Google Scholar]
- 26.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature. 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 27.Nettles KW, et al. Allosteric control of ligand selectivity between estrogen receptors alpha and beta: Implications for other nuclear receptors. Mol Cell. 2004;13:317–327. doi: 10.1016/s1097-2765(04)00054-1. [DOI] [PubMed] [Google Scholar]
- 28.Brzozowski AM, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 29.Cheskis BJ, et al. Hierarchical affinities and a bipartite interaction model for estrogen receptor isoforms and full-length steroid receptor coactivator (SRC/p160) family members. J Biol Chem. 2003;278:13271–13277. doi: 10.1074/jbc.M211031200. [DOI] [PubMed] [Google Scholar]
- 30.le Maire A, et al. A unique secondary-structure switch controls constitutive gene repression by retinoic acid receptor. Nat Struct Mol Biol. 2010;17:801–807. doi: 10.1038/nsmb.1855. [DOI] [PubMed] [Google Scholar]
- 31.Puttagunta R, et al. RA-RAR-β counteracts myelin-dependent inhibition of neurite outgrowth via Lingo-1 repression. J Cell Biol. 2011;193:1147–1156. doi: 10.1083/jcb.201102066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia G, et al. Structure, energetics, and dynamics of binding coactivator peptide to the human retinoid X receptor α ligand binding domain complex with 9-cis-retinoic acid. Biochemistry. 2011;50:93–105. doi: 10.1021/bi101288y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeshita A, et al. Thyroid hormone response elements differentially modulate the interactions of thyroid hormone receptors with two receptor binding domains in the steroid receptor coactivator-1. J Biol Chem. 1998;273:21554–21562. doi: 10.1074/jbc.273.34.21554. [DOI] [PubMed] [Google Scholar]
- 34.Putcha BD, Fernandez EJ. Direct interdomain interactions can mediate allosterism in the thyroid receptor. J Biol Chem. 2009;284:22517–22524. doi: 10.1074/jbc.M109.026682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, et al. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat Struct Mol Biol. 2011;18:556–563. doi: 10.1038/nsmb.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kammerer S, et al. RARbeta ligand-binding domain bound to an SRC-1 co-activator peptide: Purification, crystallization and preliminary X-ray diffraction analysis. Acta Crystallogr D Biol Crystallogr. 2004;60:2048–2050. doi: 10.1107/S0907444904021201. [DOI] [PubMed] [Google Scholar]
- 37.Cura V, Gangloff M, Eiler S, Moras D, Ruff M. Cleaved thioredoxin fusion protein enables the crystallization of poorly soluble ERalpha in complex with synthetic ligands. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;64:54–57. doi: 10.1107/S1744309107066444. [DOI] [PMC free article] [PubMed] [Google Scholar]
