

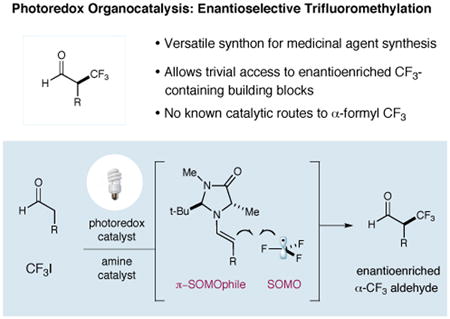
Organofluorine compounds possess unique physical properties that are exploited in a wide range of applications such as dyes, polymers, agrochemicals, and pharmaceuticals.1 In medicinal chemistry, for example, valuable physiological properties are often conferred on “drug-like” molecules via the incorporation of CF3 groups that enhance binding selectivity, elevate lipophilicity and/or improve metabolic stability.2 It is not surprising therefore, that broad research efforts have been focused on the enantioselective construction of trifluoromethyl-containing stereogenicity.3,4 While significant progress has been made in the arena of nucleophilic 1,2-trifluoromethylation of ketones,5 to date, the enantioselective α-alkylfluorination of carbonyl derived enolates (or enolate equivalents) remains an elusive goal.6,7 Herein we describe a conceptually new approach to the asymmetric α-trifluoromethylation of aldehydes via the successful merger of enamine8 and organometallic photoredox9,10 catalysis.
Design Plan
Recently, our laboratory introduced a new mode of organocatalytic activation, termed photoredox organocatalysis,9 whose mechanistic foundation relies on the propensity of electrophilic radicals (derived from the reduction of an alkyl halide by a photoredox catalyst (e.g. 1)) to combine with facially biased enamine intermediates (derived from aldehydes and chiral amine catalyst 2). Inspired by this strategy, we hypothesized that the enantioselective α-trifluoromethylation of aldehydes should also be possible by the marriage of two similar activation pathways. As detailed in Scheme 1, we anticipated that Ir(ppy)2(dtb-bpy)+ 1,11,12 previously employed as a photosynthesis mimic, should readily accept a photon from a variety of light sources within the visible spectrum (such as a household fluorescent bulb) to populate the *Ir(ppy)2(dtb-bpy)+ 7 excited state. Given its known tendency towards reductive quenching, we presumed that *Ir(ppy)2(dtb-bpy)+ 7 would readily accept a single electron from a sacrificial quantity of enamine (0.5 mol%) to form a strong reductant Ir(ppy)2(dtb-bpy) 8 (−1.51 V vs. SCE in CH3CN).13 At this stage we anticipated that this electron-rich iridium system 8 would participate in single electron transfer (SET) with trifluoromethyl iodide (−1.22 V vs. SCE in DMF)14 to render the electrophilic radical 3, while regenerating the photoredox catalyst 1. In concert with this radical formation pathway, we expected that an organocatalytic cycle would initiate by condensation of the imidazolidinone catalyst 2 with an aldehyde substrate to form the enamine 4. The merger of the two activation pathways would then occur in the key alkylation step via rapid addition of the trifluoromethyl radical 3 to the π -rich olefin 4 to form the α-amino radical 5. Given that radical 5 should have a low barrier to oxidation,15 a second electron transfer event with the *Ir(ppy)2(dtb-bpy)+ 7 excited state would close the photoredox cycle and deliver the iminium ion 6. Rapid hydrolysis of iminium 6 would then reconstitute the organocatalyst 2 while rendering the optically enriched α-CF3 aldehyde.
Scheme 1.
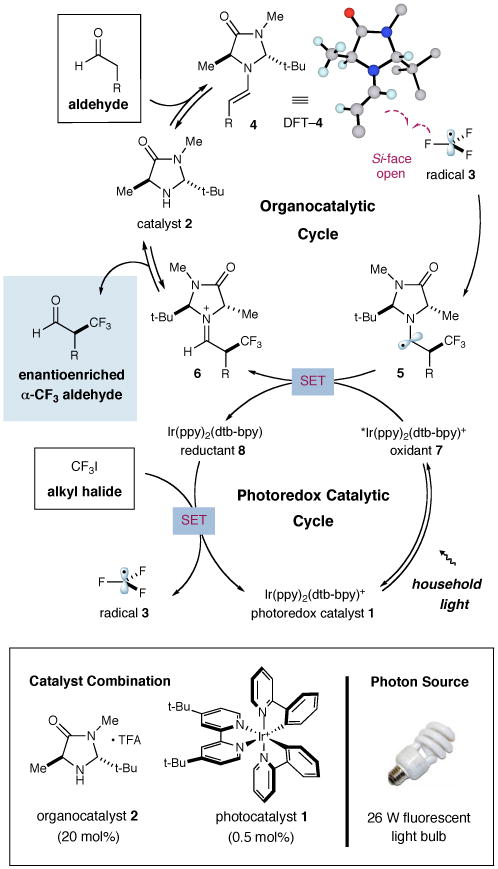
Proposed Merger of Catalytic Cycles for CF3-alkylation.
As a critical design element, we anticipated high levels of enantiocontrol resulting from positioning of the 4π-electron system of activated enamine DFT-4 away from the bulky tert-butyl group, while also adopting an (E)-configuration to minimize non-bonding interactions.16 In this arrangement, the methyl group on the imidazolidinone scaffold effectively shields the Re face, leaving the Si face exposed towards electrophilic radical addition.
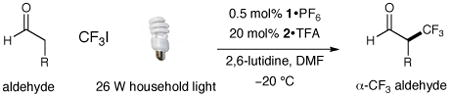
Our photoredox fluoroalkylation was first evaluated using octanal and trifluoromethyl iodide along with imidazolidinone catalysts 2 or 10, photoredox catalysts 1 or 9, and a 26 W fluorescent household lamp (Table 1). Initial experiments revealed that the proposed alkylation reaction was indeed possible using a combination of Ru(bpy)32+ (9) and the amine catalyst 10 (entry 1, 51% yield), albeit to render a racemic product. Importantly, exclusion of light from this protocol resulted in almost complete loss of catalyst efficiency (<5% yield), in accord with the photoredox mechanism outlined in Scheme 1. While the use of Ir(ppy)2(dtb-bpy)+ 1 allowed a significant increase in reaction yield (entry 3, 85%), it was not until sub–ambient temperatures were employed that enantioinduction was observed (entry 4, −20 °C, 52% ee). Moreover, implementation of the trans-tert-butyl-methyl imidazolidinone catalyst 2 (along with photocatalyst 1) provided almost perfect enantiocontrol in the trifluoromethylation step without detectable post-reaction racemization (entry 6, 79% yield, 99% ee).17 The superior levels of induction and efficiency exhibited by the combination of organocatalyst 2 with photoredox catalyst 1 in DMF at −20 °C prompted us to select these conditions for further exploration.18, 19
Table 1. Enantioselective α-Trifluoromethylation: Initial Studies.
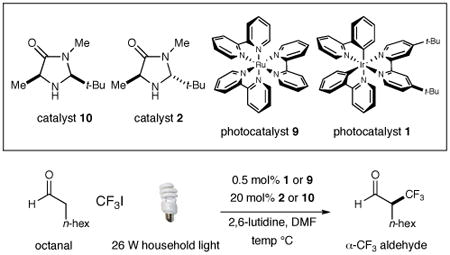 | ||||||
|---|---|---|---|---|---|---|
| entry | organocat. | photocat. | light | temp (°C) | % yield | % eea |
| 1 | 10 | 9 | yes | 23 | 51 | 0 |
| 2 | 10 | 9 | no | 23 | <5 | 0 |
| 3 | 10 | 1 | yes | 23 | 85 | 0 |
| 4 | 10 | 1 | yes | −20 | 92 | 52 |
| 5 | 2 | 9 | yes | −20 | 67 | 87 |
| 6 | 2 | 1 | yes | −20 | 79 | 99 |
Enantiomeric excess by chiral GC analysis of the corresponding alcohol.
We next performed a series of experiments to determine the scope of the aldehydic component in this asymmetric trifluoromethylation protocol. As revealed in Table 2, these mild redox conditions are compatible with a wide range of functional groups including ethers, esters, amines, carbamates, and aromatic rings (entries 2–4, 6, 8, 9, 61–86% yield, 93–98% ee). Moreover, significant variation in the steric demand of the aldehyde substituent can be accommodated without loss in enantiocontrol (entries 5–7, 10, and 11, ≥96% ee). Notably, this protocol enables the formation of a benzylic-CF3 α-formyl stereocenter without significant erosion in enantiopurity (entry 8, 61% yield, 93% ee).21 As highlighted in entries 10 and 11, exposure of enantiopure (R)-3-phenyl-butyraldehyde to catalyst (2S, 5R)-2 results in the diastereoselective production of the anti α,β-disubstituted isomer, while the use of (S)-3-phenyl-butyraldehyde with the same amine catalyst affords the corresponding syn adduct with high fidelity. These transformations clearly demonstrate the synthetic advantages of catalyst-enforced induction versus substrate-directed stereocontrol.
Table 2. Enantioselective α-Trifluoromethylation: Aldehyde Scope.
 | |||||
|---|---|---|---|---|---|
| entry | producta | yield,b eec | entry | producta | yield,b eec |
| 1 |
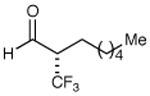
|
79% yield 99% ee | 7 |
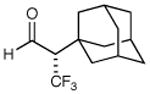
|
73% yieldd 90% ee |
| 2 |
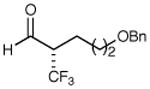
|
72% yield 95% ee | 8 |
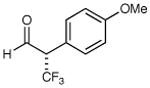
|
61% yield 93% ee |
| 3 |
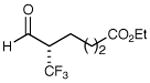
|
86% yield 97% ee | 9 |
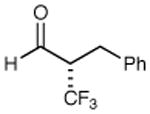
|
75% yield 97% ee |
| 4 |
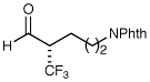
|
78% yield 98% ee | 10 |
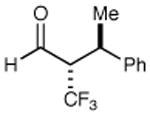
|
68% yield >20:1 dr 99% ee |
| 5 |
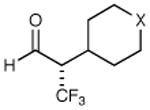
|
X = CH2 70% yield 99% ee | 11 |
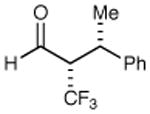
|
62% yield >20:1 dr 99% ee |
| 6 | X = NBoc 70% yield 98% ee | ||||
Stereochemistry assigned by chemical correlation or by analogy.
Isolated yields of the corresponding alcohol.
Enantiomeric excess determined by chiral SFC or HPLC analysis.
Using catalyst 11; ref 20.
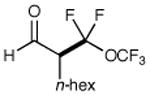
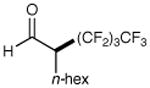
We have found that a broad range of perfluoroalkyl iodides and bromides also participate in this enantioselective alkylation reaction (Table 3). For example, n-perfluoroalkyl substrates of varying chain length undergo reductive radical formation and enamine addition without loss in enantiocontrol or reaction efficiency (entries 1–3 and 5–8, 66–88% yield, 96–99% ee). We have also found that aldehyde α-functionalization step can be performed with sterically demanding coupling partners such as perfluoro isopropyl iodides (entry 4, 72% yield, 98% ee). Moreover, benzylic, -ester, and α-ether difluoromethylene carbons are readily incorporated as part of this new enantioselective catalytic α-carbonyl alkylation.
Table 3. Enantioselective α-Perfluoroalkylation: Alkyl Iodide Scope.
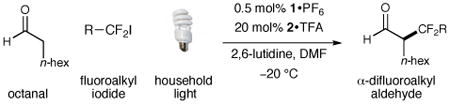 | ||||||
|---|---|---|---|---|---|---|
| entry | producta | yield,b eec | entry | producta | yield,b eec | |
| 1 |
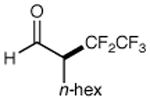
|
73% yield 96% ee | 5 |
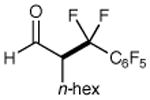
|
85% yield 98% ee | |
| 2 |
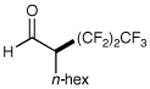
|
69% yield 99% ee | 6 |
|
71% yield 99% ee | |
| 3 |
|
67% yield 96% ee | 7 |
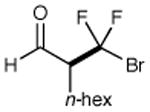
|
68% yieldd 99% ee | |
| 4 |
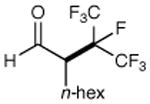
|
72% yield 98% ee | 8 |
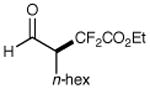
|
89% yieldd 99% ee | |
Stereochemistry assigned by chemical correlation or by analogy.
Isolated yields of the corresponding alcohol.
Enantiomeric excess determined by chiral HPLC analysis of corresponding 2-naphthoyl ester.
The perfluoroalkyl bromide was employed as starting material.
We fully expect that the α-trifluoromethyl aldehyde products generated in this study will be of value for the production of a variety of organofluorine synthons. As shown in Scheme 2, reduction or oxidation of the formyl group is possible to generate β-hydroxy and as α-trifluoromethyl acids (the latter we expect will be a key building block for the formation of heterocycles that incorporate CF3 at the benzylic position). Moreover, these aldehyde products can be employed in a reductive amination sequence without significant loss in enantioselectivity to produce β-trifluoromethyl amines. Last, and perhaps most important, aldehyde oxidation followed by a Curtius rearrangement allows enantioselective formation of α-trifluoromethyl amine containing stereocenters, a commonly employed amide isostere in medicinal chemistry.2 In this case, careful selection of base and reaction temperature is essential to maintain the enantiopurity obtained in the initial alkylation step.
Scheme 2.
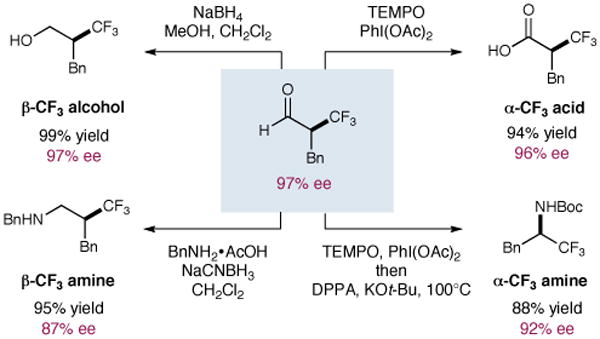
Access to Enantioenriched Organofluorine Synthons.
In summary, the first enantioselective, organocatalytic α-trifluoromethylation and α-perfluoroalkylation of aldehydes have been accomplished using a readily available iridium photocatalyst and a commercial imidazolidinone catalyst. Full details of this survey will be forthcoming.
Supplementary Material
Acknowledgments
Financial support was provided by the NIHGMS (R01 GM078201-01-01) and kind gifts from Merck and Amgen. M.E.S. acknowledges the Natural Sciences and Research Council (NSERC) of Canada for postdoctoral funding. We thank Dr. David Nicewicz for preliminary experimental contributions.
Footnotes
Supporting Information Available. Experimental procedures, and spectral data are provided (PDF).
References
- 1.(a) Chambers RD. Fluorine in Organic Chemistry. Wiley; New York: 1973. [Google Scholar]; (b) Banks RE, Smart BE, Tatlow JC. Organofluorine Chemistry: Principles & Applications. Plenum Press; New York: 1994. [Google Scholar]
- 2.(a) Filler R, Kobayashi Y, Yagupolskii LM. Organofluorine Compounds in Medicinal Chemistry & Biomedical Applications. Elsevier; New York: 1993. [Google Scholar]; (b) Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (c) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (d) Hagmann WK. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 3.For a review of strategies for trifluoromethylation, see: Ma JA, Cahard D. J Fluorine Chem. 2007;128:975–996.
- 4.For reviews of asymmetric trifluoromethylations, see: Ma JA, Cahard D. Chem Rev. 2008;108:PR1–PR43. doi: 10.1021/cr800221v.Shibata N, Mizuta S, Kawai H. Tetrahedron: Asymmetry. 2008;19:2633–2644.Billard T, Langlois BR. Eur J Org Chem. 2007:891–897.
- 5.For seminal contributions towards the nucleophilic trifluoromethylation of carbonyls, see: Ruppert I, Schlich K, Volbach W. Tetrahedron Lett. 1984;25:2195–2198.Prakash GKS, Krishnamurti R, Olah GA. J Am Chem Soc. 1989;111:393–395.Prakash GKS, Yudin AK. Chem Rev. 1997;97:757–786. doi: 10.1021/cr9408991.Singh RP, Shreeve JM. Tetrahedron. 2000;56:7613–7632.Ait-Mohand S, Takechi N, Médebielle M, Dolbier WR., Jr Org Lett. 2001;3:4271–4273. doi: 10.1021/ol016933x.
- 6.For seminal contributions towards the electrophilic or radical trifluoromethylation of enamines, enolates, and other carbonyl derivatives, see: Cantacuzène D, Dorme R. Tetrahedron Lett. 1975;16:2031–2034.Miura K, Taniguchi M, Nozaki K, Oshima K, Utimoto K. Tetrahedron Lett. 1990;31:6391–6394.Umemoto T, Ishihara S. J Am Chem Soc. 1993;115:2156–2164.Ma JA, Cahard D. J Org Chem. 2003;68:8726–8729. doi: 10.1021/jo034881e.Itoh Y, Mikami K. Org Lett. 2005;7:4883–4885. doi: 10.1021/ol0517574.Kieltsch I, Eisenberger P, Togni A. Angew Chem Int Ed. 2007;46:754–757. doi: 10.1002/anie.200603497.Sato K, Yuki T, Yamaguchi R, Hamano T, Tarui A, Omote M, Kumadaki I, Ando A. J Org Chem. 2009;74:3815–3819. doi: 10.1021/jo9004348.
- 7.For examples of moderately stereoselective methods, see Kitazume T, Ishikawa N. J Am Chem Soc. 1985;107:5186–5191.Umemoto T, Adachi K. J Org Chem. 1994;59:5692–5699.Itoh Y, Mikami K. Tetrahedron. 2006;62:7199–7203.
- 8.(a) Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]; (b) MacMillan DWC. Nature. 2008;455:304–308. doi: 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]
- 9.Nicewicz DA, MacMillan DWC. Science. 2008;322:77–80. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For recent examples of photoredox catalysis applications in organic synthesis, see: Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f.Koike T, Akita M. Chem Lett. 2009;38:166–167.Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582.Müller C, Bauer A, Bach T. Angew Chem, Int Ed. 2009;48 doi: 10.1002/anie.200901603.Du J, Yoon TP. J Am Chem Soc. 2009;131 doi: 10.1021/ja903732v.
- 11.Although Ir(ppy)2(dtb-bpy)PF6 1 was optimal for this transformation, commercially available Ru(bpy)3Cl2 could also be used to afford the desired products in slightly diminished yields and enantioselectivities (Table 1, entry 5). 1 is available in a single step from commercially available [Ir(ppy)2Cl]2 (Sigma-Aldrich, CAS-No 92220-65-0). See ref 12.
- 12.For synthesis and photophysical properties of Ir(ppy)2(dtb-bpy)PF6 1, see: Slinker JD, Gorodetsky AA, Lowry MS, Wang J, Parker S, Rohl R, Bernhard S, Malliaras GG. J Am Chem Soc. 2004;126:2763–2767. doi: 10.1021/ja0345221.For a review on the photo-properties of iridium complexes, see: Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top Curr Chem. 2007;281:143–203.
- 13.Fluorescence quenching experiments using preformed enamine 4 (R = Bn) confirmed quenching of *Ir(ppy)2(dtb-bpy)PF6, Stern-Volmer constant = 185, see Supporting Information for details.
- 14.(a) Andrieux CP, Gelis L, Médebielle M, Pinson J, Saveant JM. J Am Chem Soc. 1990;112:3509–3520. [Google Scholar]; (b) Bonesi SM, Erra-Balsells R. J Chem Soc, Perkin Trans 2. 2000:1583–1595. [Google Scholar]
- 15.Wayner DDM, Dannenberg JJ, Griller D. Chem Phys Lett. 1986;131:189. [Google Scholar]
- 16.Performed using B3LYP/6-311+G(2d,p)// B3LYP/6-31G(d). See ref 9.
- 17.We have recently shown the disruption of a post-reaction racemization pathway via design of catalyst 2 (versus catalyst 10), see: Amatore M, Beeson TD, Brown SP, MacMillan DWC. Angew Chem Int Ed. 2009;48:5121–5124. doi: 10.1002/anie.200901855.
- 18.The transformation does not proceed in the absence of the amine catalyst. The addition of 2,6-lutidine is necessary to remove hydroiodic acid that is formed during the reaction. Rapid racemization of the α-CF3 formyl adduct was observed at room temperature but not at −20 °C.
- 19.Lower reaction efficiency appears to lead to lower enantiopurity as the product is exposed to unconsumed base (i.e. Table 1, entry 5, catalyst 9).
- 20.A hitherto unreported amine catalyst 11, was employed in Entry 7 (Table 2), see Supporting Information.
- 21.Efficient construction of a benzylic CF3 bond on the unsubstituted parent compound, phenylacetaldehyde is also attainable (cf. 89% 19F NMR yield), however facile post-reaction racemization is observed.
- 22.General Procedure for the Enantioselective α–Trifluoromethylation of Aldehydes. To a dry test tube was added organocatalyst 2 (0.20 equiv), photocatalyst 1 (0.005 equiv) and DMF (0.3M). The resultant yellow solution was degassed by alternating vacuum evacuation/argon backfill (×3) at −78 °C before addition of CF3I (∼8 equiv), followed by aldehyde (0.76 mmol, 1.0 equiv) and 2,6-lutidine (1.1 equiv). The reaction vessel was placed near a 26 W compact fluorescent light bulb in a −20 °C bath for 7.5–8 hours. Upon reaction completion, the α– trifluoromethyl aldehydes were typically reduced in situ with NaBH4 (10 equiv) in CH2Cl2 and MeOH to afford β-trifluoromethyl alcohols in high yield and enantioselectivity.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.