

Abstract
The tandem Friedel–Crafts conjugate addition/asymmetric protonation reaction between 2-substituted indoles and methyl 2-acetamidoacrylate is reported. The reaction is catalyzed by (R)-3,3′-dibromo-BINOL in the presence of stoichiometric SnCl4, and is the first example of a tandem conjugate addition/asymmetric protonation reaction using a BINOL•SnCl4 complex as the catalyst. A range of indoles furnished synthetic tryptophan derivatives in good yields and high levels of enantioselectivity, even on preparative scale. The convergent nature of this transformation should lend itself to the preparation of unnatural tryptophan derivatives for use in a broad array of synthetic and biological applications.
Introduction
Tryptophan and unnatural tryptophan derivatives are important building blocks for the total synthesis of natural products, as well as the development of new drugs,1 biological probes,2,3 and chiral small molecule catalysts.4 For example, functionalized tryptophan derivatives have served as key intermediates in the syntheses of the bioactive natural products indolactam V5 and stephacidin A.6 Alternatively, unnatural tryptophan derivatives have been employed as probes for studying protein conformational dynamics by way of Förster resonance energy transfer (FRET) experiments,2 as well as for elucidating cation-π binding interactions by linear free energy relationship studies.3 As a result, the development of new catalytic asymmetric methods to prepare enantioenriched unnatural tryptophans is an important area of chemical research.7
As part of our research program aimed at establishing new methods for the enantioselective synthesis of alkaloids, we are interested in developing convergent syntheses of tryptophans and cyclo-tryptophans (also known as pyrroloindolines) from simple indole starting materials. In 2010, we reported a new reaction for the preparation of enantioenriched pyrroloindolines (3) in which (R)-BINOL•SnCl4 catalyzes a formal (3 + 2) cycloaddition reaction between 1,3-disubstituted indoles (1) and benzyl 2-trifluoroacetamidoacrylate (2a) (Scheme 1, a).8 Good yields, moderate exo:endo diastereoselectivities, and high enantioselectivities were obtained for a variety of indole substrates (Table 1). The enantio- and diastereoselectivity of pyrroloindoline formation were found to be dependent on the identity of the acrylate; the highest ee values were observed using acrylate 2a. Use of the commercially available methyl 2-acetamidoacrylate (2c) provided higher drs but attenuated ees. N-Alkyl substitution on the indole substrates gave higher yields of the pyrroloindoline products relative to the N-protioindoles (Table 1, entry 1 versus entry 10).
Scheme 1.

Table 1.
Catalytic asymmetric synthesis of pyrroloindolines.
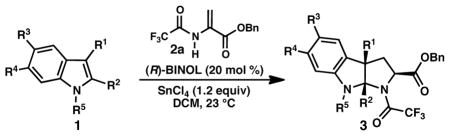 | ||
|---|---|---|
| entry | ee (%)a (exo/endo)b | |
| 1) |
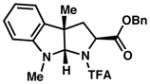 3a 86% yield dr = 4:1 |
94/91 |
| 2) |
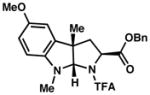 3b 93% yield dr = 3:1 |
93/92 |
| 3) |
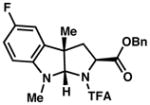 3c 61% yield dr = 3:1 |
93/90 |
| 4) |
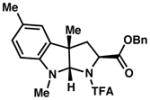 3d 84% yield dr = 5:1 |
94/91 |
| 5) |
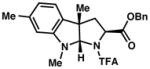 3e 91% yield dr = 4:1 |
94/90 |
| 6) |
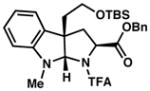 3f 54% yield dr = 6:1 |
92/90 |
| 7) |
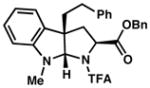 3g 80% yieldc dr = 4:1 |
92/90 |
| 8) |
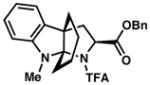 3h 65% yield dr = >18:1 |
86 |
| 9) |
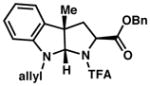 3i 90% yieldc dr = 3:1 |
93/90 |
| 10) |
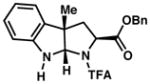 3j 18% yield dr = 8:1 |
95/93 |
Determined by chiral stationary phase SFC or HPLC.
Determined by 1H NMR analysis of mixture.
1.6 equiv SnCl4 were employed.
Unexpectedly, our studies revealed that the initially formed exo- and endo-diastereomers of 3 were generated in opposite enantiomeric series. These findings led us to propose that pyrroloindoline formation proceeds by a stepwise mechanism, in which an initial conjugate addition of indole 1 to 2a is followed by a catalyst-controlled protonation to give 5 (Scheme 1, a). Subsequent cyclization of the amide onto the iminium ion provides the pyrroloindoline product (3). We hypothesized that the (R)-BINOL•SnCl4 complex (9a•SnCl4, see Figure 1) served as a chiral Lewis acid-assisted Brønsted acid (LBA)9,10 to effect an asymmetric protonation of the enolate intermediate. Whereas Yamamoto and coworkers initially developed (R)-BINOL•SnCl4 as an LBA to effect enantioselective protonation of silyl enolates, these complexes had never previously been used in tandem conjugate addition/asymmetric protonation reactions.
Figure 1.

(R)-BINOL derived catalysts.
Herein, we report our efforts to expand the scope of products accessible by (R)-BINOL•SnCl4-catalyzed conjugate addition/asymmetric protonation processes. These studies have resulted in the first direct, enantioselective synthesis of tryptophan derivatives by a tandem Friedel–Crafts conjugate addition/asymmetric protonation reaction (Scheme 1, b).11 The reactions require no pre-activation of the indole substrates, and provide convergent access to a range of substituted tryptophan derivatives in enantioen-riched form. Friedel–Crafts conjugate addition reactions in which a new stereogenic center is set solely at the α-position of the conjugate acceptor through an enantioselective protonation event are rare,12 and have only recently been reported with indole nucleophiles.13,14,15,16 Genet and Darses have reported the synthesis of α-amino acids by a Rh-catalyzed conjugate addition of aryl trifluoroborate salts to 2-amidoacrylates with in situ asymmetric protonation;14d,h however, there are no examples using indole-based nucleophiles to give tryptophan derivatives.
Results and Discussion
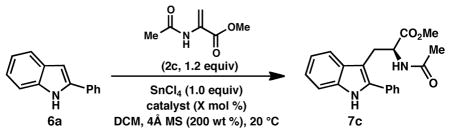
Our studies commenced with efforts to promote the Friedel–Crafts conjugate addition/asymmetric protonation reaction between 2-phenylindole (6a) and benzyl 2-trifluoroacetamidoacrylate (2a) under the reaction conditions previously optimized for the enantioselective formal (3 + 2) cycloaddition reaction. Somewhat surprisingly, the desired reaction was sluggish under these conditions: after 2 hours, trifluoroacetamido ester 7a was formed in low yield and poor enantiomeric excess (Table 2, entry 1). In an effort to improve the reactivity, a screen of additional 2-amidoacrylates was conducted. Gratifyingly, the use of commercially available methyl 2-acetamidoacrylate (2c) gave substantially improved results, providing acetamido ester 7c in 73% yield and 78% ee. As we observed in the enantioselective pyrroloindoline formation,8 SnCl4 promotes the reaction between 6a and 2c in the absence of (R)-BINOL (9a) (entry 4); however, 9a produces a substantial acceleration in the rate of the SnCl4-promoted reaction.17 No reaction is observed in the absence of SnCl4 (entry 5).
Table 2.
Optimization of reaction parameters.a
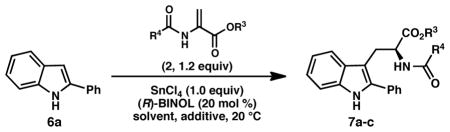 | ||||||
|---|---|---|---|---|---|---|
| entry | R3, R4 | pdt | solvent | additive | yield (%)b | ee (%)c |
| 1 | Bn, CF3 (2a) | 7a | DCM | -- | 12 | 35 |
| 2 | Me, CF3 (2b) | 7b | DCM | -- | 12 | 42 |
| 3 | Me, Me (2c) | 7c | DCM | -- | 73 | 78 |
| 4 | Me, Me (2c) | 7c | DCM | --d | 13 | -- |
| 5 | Me, Me (2c) | 7c | DCM | --e | 0 | -- |
| 6 | Me, Me (2c) | 7c | DCM | K2CO3 | 73 | 78 |
| 7 | Me, Me (2c) | 7c | DCM | 2,6-lutidine | 0 | -- |
| 8 | Me, Me (2c) | 7c | DCM | 4Å MS | 86 | 81 |
| 9 | Me, Me (2c) | 7c | DCE | 4Å MS | 87 | 79 |
| 10 | Me, Me (2c) | 7c | CHCl3 | 4Å MS | 80 | 72 |
Reactions conducted under inert atmosphere on 0.2 mmol scale for 2 h.
Isolated yield.
Determined by chiral stationary phase SFC.
No (R)-BINOL was employed.
No SnCl4was employed.
We suspected that the reaction of adventitious water and SnCl4 might generate HCl, which could erode the apparent ee of 7c by promoting a racemic background protonation reaction; thus, additives known to scavenge water or HCl were evaluated. Whereas insoluble inorganic bases such as K2CO3 showed no effect (entry 6), the use of soluble bases such as 2,6-lutidine completely inhibited the reaction (entry 7). On the other hand, the use of activated powdered 4Å molecular sieves increased both the yield and selectivity of the reaction, furnishing acetamido ester 7c in 86% yield and 81% ee, while also improving the reproducibility (entry 8).
At this stage, our efforts to further improve the enantioselectivity of this transformation turned to optimization of the catalyst structure. We were pleased to find that 3,3′-disubstitution with halides furnished improved selectivities and comparable yields (Table 3, entries 5 and 6). Interestingly, dimethoxycatalyst 9g provided acetamido ester 7c as a racemate in low yield (entry 7). It is proposed that the coordinating ability of the methoxy groups may permit alternative binding modes between SnCl4 and 9g, resulting in mixtures of less reactive and less selective catalyst systems. To probe whether the electronic or steric properties of the 3,3′-substituents were responsible for modulating the selectivities of catalysts 9a–g, several 6,6′-disubstituted BINOL derivatives were also evaluated. However, no linear dependence between the BINOL electronics and the ee of 7c was observed (Table 3, entries 1, 8–10). Of the catalysts evaluated, the commercially available (R)-3,3′-dibromo-BINOL catalyst 9f18 gave optimal results, delivering acetamido ester 7c in 76% yield and 93% ee (entry 6).19 Whereas 10 mol % 9f provided comparable selectivity for the formation of 7c (entry 13), use of 5 mol % 9f resulted in diminished ee (entry 14). The decreased selectivity likely results from competition by the achiral SnCl4-promoted background reaction at low catalyst loadings. Because 20 mol % catalyst imparted consistently higher enantioselectivities for more functionalized substrates (vide infra), this catalyst loading was utilized in subsequent experiments.
Table 3.
Catalyst optimization.a
 | ||||
|---|---|---|---|---|
| entry | catalyst | loading (mol %) | yield (%)b | ee (%)c |
| 1 | 9a | 20 | 86 | 81 |
| 2 | 9b | 20 | 17 | 37 |
| 3 | 9c | 20 | 83 | 87 |
| 4 | 9d | 20 | 76 | 84 |
| 5 | 9e | 20 | 85 | 90 |
| 6 | 9f | 20 | 76 | 93 |
| 7 | 9g | 20 | 7 | 1 |
| 8 | 9h | 20 | 86 | 54 |
| 9 | 9i | 20 | 88 | 78 |
| 10 | 9j | 20 | 82 | 78 |
| 11 | 9f | 40 | 76 | 93 |
| 12 | 9f | 15 | 77 | 93 |
| 13 | 9f | 10 | 75 | 92 |
| 14 | 9f | 5 | 72 | 88 |
Reactions conducted under inert atmosphere on 0.2 mmol scale for 2 h.
Isolated yield.
Determined by chiral stationary phase SFC.
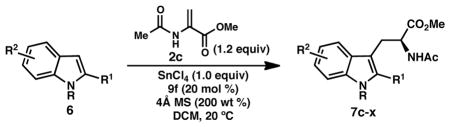
Having identified conditions to prepare acetamido ester 7c in high yield and enantiomeric excess, a survey of indole substrates was conducted to evaluate the scope of the reaction (Table 4). In contrast to our observations in the formal (3 + 2) cycloaddition,8 methylation or allylation of the indole nitrogen provides tryptophans 7d and 7e in slightly lower yields and ee (entries 2 and 3).20 Similarly, use of unsubstituted indole as the nucleophile provides N-α-acetyltryptophan methyl ester in 31% yield and 67% ee (not shown, see Supporting Information).21 Alternatively, substitution of the 2-phenylindole backbone at the 4, 5, 6, and 7-positions is well tolerated (entries 4–7). Whereas substrates bearing either electron-donating or electron-withdrawing substituents furnish products with high enantioselectivity, the more electron-poor indoles are less reactive and provide lower yields of the acetamido ester products even with increased loadings of SnCl4 (entries 9 and 10).
Table 4.
Substrate scope of the tandem Friedel–Crafts conjugate addition/asymmetric protonation.a
 | |
|---|---|
| entry | |
|
|
| 1) | 76% yield, 93% ee |
|
|
| 2) | R = Me (7d) 63% yield, 85% ee |
| 3) | R = allyl (7e) 68% yield, 85% ee |
|
|
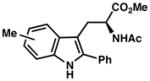
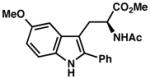
| 4) | 4-Me (7f): 88% yield, 96% ee |
| 5) | 5-Me (7g): 83% yield, 95% ee |
| 6) | 6-Me (7h): 80% yield, 89% ee |
| 7) | 7-Me (7i): 94% yield, 94% ee |
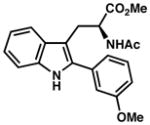
 7j |
|
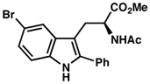
| 8) | 85% yield, 91% ee |
 7k |
|
| 9)b | 60% yield, 93% ee |
 7l |
|
| 10)b | 63% yield, 92% ee |
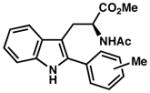
|
|
| 11) | 4-Me (7m): 86% yield, 94% ee |
| 12) | 2-Me (7n): 26% yield, 87% ee |
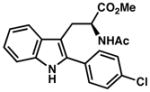 7o |
|
| 13) | 75% yield, 93% ee |
|
|
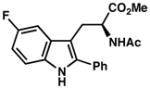
| 14) | 4-F (7p): 78% yield, 93% ee |
| 15) | 3-F (7q): 76% yield, 92% ee |
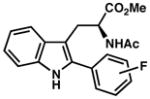
| 16) | 2-F (7r): 35% yield, 92% ee |
 7s |
|
| 17) | 88% yield, 92% ee |
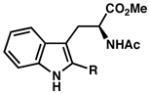
|
|
| 18) | R = Me (7t): 61% yield, 85% ee |
| 19) | R = n-Bu (7u): 72% yield, 91% ee |
| 20) | R = i-Pr (7v): 66% yield, 92% ee |
| 21) | R = t-Bu (7w): 29% yield, 84% ee |
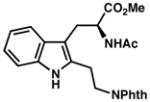 7x |
|
| 22) | 80% yield, 90% ee |
Reactions conducted under inert atmosphere on 0.1 or 0.2 mmol scale for 2 h. Isolated yields are reported. Enantiomeric excess was determined by chiral stationary phase SFC.
1.6 equiv SnCl4 were employed.
A range of substituents are tolerated at the 2-position of the indole, including both aryl and alkyl groups. 2-Arylindoles bearing substituents in either the m- or p-position of the arene are accommodated; on the other hand, o-substituted arenes are substantially less reactive (entries 12 and 16). For indoles containing 2-alkyl substituents, the ee improves in switching from a methyl group to the slightly larger n-butyl and i-propyl substituents (entries 18–20); however, both the yield and selectivity are diminished in the case of bulky t-butyl substitution (7w, entry 21). Notably, a phthalimide-protected amine functionality is also compatible, as demonstrated in 7x (entry 22). Attempts to further expand the scope of C2-substituents were unfruitful. For example, 2-iodoindole underwent decomposition under the reaction conditions, whereas 2-(trimethylsilyl)indole returned unreacted starting material.
Using 2-phenylindole (6a), this reaction has been conducted on a 5 mmol scale, providing acetamido ester 7c in 78% yield and 93% ee. Although our screening protocol was conducted in a glove box, this preparative scale reaction could be run using standard Schlenk techniques. We have also demonstrated that the acetamide and methyl ester groups can be hydrolyzed under orthogonal conditions. Heating 7c to 75 °C with HCl in aqueous methanol cleaves the acetamide group to deliver free amine 10 in 76% yield and 93% ee.22 Alternatively, exposure of 7c to aqueous LiOH in THF at 0 °C provides carboxylic acid 11 in 92% yield and 92% ee.23
Upon treatment with NBS and TFA, tryptophan 7c can be converted to bromo-dehydroindoline 12, which is formed as a 1:1 mixture of diastereomers. Interestingly, compound 12 does not undergo cyclization to the pyrroloindoline under the reaction conditions. On the other hand, exposure of N-methyl derivative 7d to NCS24 and TFA in acetonitrile provides the corresponding chloro-pyrroloindoline, as detected by HRMS. Subsequent silica gel-promoted hydrolysis then delivers the more stable hydroxy-pyrroloindoline 13 in 52% yield and 6:1 dr, favoring the endo diastereomer.
Mechanistically, it seems likely based on Yamamoto’s prior reports of catalytic asymmetric protonation of silyl enol ethers and silyl ketene acetals,9b,c that catalytically generated 9f•SnCl4 is serving as a chiral LBA to protonate an intermediate Sn-enolate. Whether 9f•SnCl4 is the species responsible for activating acrylate 2c toward conjugate addition by the indole is unclear. Although 9f•SnCl4 is proposed to serve as the catalyst, stoichiometric tin is required due to binding of the acetamido ester product to tin, resulting in product inhibition. Notably, no significant non-linear effects are observed when using scalemic BINOL to catalyze the reaction (Chart 1).25
Chart 1.

Nonlinear effects study.
Given the proposed mechanistic similarities between the reactions to give pyrroloindoline 3 and tryptophan 7, we were interested in whether the conditions optimized for tryptophan formation would catalyze the formal (3 + 2) cycloaddition reaction with improved selectivity. Treatment of 1a and methyl 2-acetamidoacrylate (2c) with 1.0 equiv SnCl4 and 20 mol % catalyst 9f in the presence of 4Å MS furnished pyrroloindoline 3k in 58% yield as an 8:1 mixture of exo and endo diastereomers in 87 and 85% ee, respectively (Table 5, entry 2). Although these conditions provide pyrroloindoline 3k with improved dr and ee relative to the originally reported conditions (Table 5, entry 1), they are less enantioselective then when benzyl 2-trifluoromethylacetamidoacrylate (2a) is employed (entry 3). Finally, use of acrylate 2a with catalyst 9f and 4Å MS provided pyrroloindoline 3a with high dr and exceptional ee; unfortunately 3a was isolated in unacceptably low yield. The lower reactivity of acrylate 2a is consistent with the reactivity trend observed for tryptophan formation (see Table 2, entry 1). Taken together, these data highlight that an appropriate matching of the acrylate and catalyst is required to obtain 3 with both high yields and high enantioselectivity.
Table 5.
Comparison of conditions for pyrroloindoline formation.
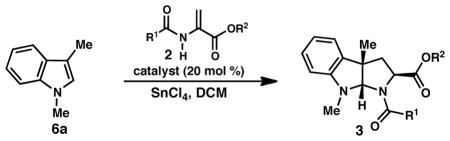 | ||||||
|---|---|---|---|---|---|---|
| entry | conditions | R1, R2 | pdt | yielda (%) | drb | ee (%)c |
| 1d | 9a | Me, Me (2c) | 3k | 70 | 5:1 | 65/80 |
| 2e | 9f, 4Å MS | Me, Me (2c) | 3k | 58 | 8:1 | 87/85 |
| 3d | 9a | CF3, Bn (2a) | 3a | 86 | 4:1 | 94/91 |
| 4e | 9f, 4Å MS | CF3, Bn (2a) | 3a | 39 | 7:1 | 98/95 |
Isolated yield.
Determined by 1H NMR analysis of mixture.
Determined by chiral stationary phase SFC or HPLC.
Reaction run with 1.0 equiv acrylate, 1.2 equiv SnCl4.
Reaction run with 1.2 equiv acrylate, 1.0 equiv SnCl4.
Conclusions
In conclusion, 9f•SnCl4 catalyzes a tandem Friedel–Crafts conjugate addition/asymmetric protonation reaction between 2-substituted indoles (6) and methyl 2-acetamidoacrylate (2c). A range of indoles furnished synthetic tryptophan derivatives 3 in good yields and high levels of enantioselectivity, even on preparative scale. We have shown that such tryptophan derivatives can be orthogonally deprotected, or converted to more functionalized derivatives. This is the first example of a chiral diol•SnCl4-catalyzed Friedel–Crafts conjugate addition reaction in which the new stereogenic centers are set solely at the α-position of the conjugate acceptor by an asymmetric protonation. The convergent nature of this transformation should lend itself to the preparation of unnatural tryptophan derivatives for use in a broad array of synthetic and biological applications. Further mechanistic studies and the development of related asymmetric protonation reactions are the subject of continued research in our laboratory.
Experimental Section
General Information
Unless otherwise stated, reactions were performed under a nitrogen atmosphere using freshly dried solvents. Methylene chloride, ether, tetrahydrofuran, and dioxane were dried by passing through activated alumina columns. Dichloroethane and chloroform were distilled over calcium hydride. Powdered 4Å molecular sieves were flame-dried under vacuum immediately prior to use. Potassium carbonate was dried for 12 h at 130 °C under vacuum and 2,6-lutidine was distilled over AlCl3. All other commercially obtained reagents were used as received unless specifically indicated. (R)–BINOL (9a), 2-phenylindole (6a) and 2-methylindole (6r) were purchased from Alfa Aesar, N-methyl-2-phenylindole (6b) was obtained from Sigma-Aldrich, and 1 M SnCl4 in CH2Cl2 was purchased from Acros Organics. (R)-3,3′-diphenyl-BINOL (9b),26 (R)-3,3′-dimethyl-BINOL (9c),27 (R)-3,3′-dichloro-BINOL (9e),28 (R)-3,3′-dibromo-BINOL (9f),18 (R)-3,3′-dimethoxy-BINOL (9g),18 (R)-6,6′-dimethyl-BINOL (9i)29 and (R)-6,6′-dibromo-BINOL (9j)30 were prepared according to literature procedures. All reactions were monitored by thin-layer chromatography using EMD/Merck silica gel 60 F254 pre-coated plates (0.25 mm). Silica gel column chromatography was performed either as described by Still et al.31 using silica gel (particle size 0.032–0.063) purchased from Silicycle or using pre-packaged RediSep®Rf columns on a CombiSilica gel Rf system (Teledyne ISCO Inc.). 1H and 13C NMR spectra were recorded on a Varian Inova 500 (at 500 MHz and 125 MHz respectively) or a Varian Inova 600 (at 600 MHz and 150 MHz respectively), and are reported relative to internal chloroform (1H, δ = 7.26, 13C, δ = 77.0). Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Multiplicity and qualifier abbreviations are as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. IR spectra were recorded on a Perkin Elmer Paragon 1000 spectrometer and are reported in frequency of absorption (cm−1). Analytical SFC was performed with a Mettler SFC supercritical CO2 analytical chromatography system with Chiralcel AD-H, OD-H, AS-H, and OB-H columns (4.6 mm × 25 cm). HRMS were acquired using either an Agilent 6200 Series TOF with an Agilent G1978A Multimode source in electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI) or mixed (MM) ionization mode, or obtained from the Caltech Mass Spectral Facility.
General Procedure for the Synthesis of Tryptophan Derivatives
An oven-dried vial was charged with the indole (1.00 equiv), methyl 2-acetamidoacrylate (2c, 1.20 equiv), (R)-3,3′-dibromo-BINOL (9f, 0.20 equiv) and pumped into a glove box. To the vial was added flame-dried powdered 4Å molecular sieves (200 wt % relative to indole). The vial was charged with CH2Cl2 to an indole concentration of 0.12 M, and SnCl4 (1.00 equiv as a 1 M solution in CH2Cl2) was added. The reaction was stirred at 20 °C for 2 hours, after which time it was removed from the glove box and quenched by dilution with 1 M HCl (5 mL) and CH3CN (1 mL). The aqueous layer was extracted with EtOAc (2 × 5 mL) and the (5 combined organic layers were washed with saturated aqueous NaHCO3 mL), dried (Na2SO4), filtered, and concentrated. The crude residue was purified by silica gel chromatography.
Preparative Scale Procedure for the Synthesis of Tryptophan 7c
To a flame-dried flask under nitrogen containing freshly activated powdered 4Å molecular sieves (200 wt %) was added 2-phenylindole (1a, 1.00 g, 5.20 mmol, 1.00 equiv), methyl 2-acetamidoacrylate (2c, 890 mg, 6.20 mmol, 1.20 equiv), and (R)-3,3′-dibromo-BINOL (9f, 457 mg, 1.00 mmol, 0.20 equiv). The flask was charged with 40 mL DCM and SnCl4 (1 M in DCM, 5.20 mL, 5.20 mmol, 1.00 equiv) was added. The reaction was stirred at room temperature for 2 hours, then quenched by addition of 1 M HCl (50 mL). The aqueous layer was extracted with EtOAc (2 × 50 mL) and the combined organic layers were washed with saturated aqueous NaHCO3 (50 mL), dried (Na2SO4), filtered and concentrated. The crude residue was purified by silica gel chromatography (40:60 to 100:0 EtOAc:hexanes) to yield 1.33 g (77% yield) of 7c as a pale yellow foam. The enantiomeric excess was determined to be 93% by chiral SFC analysis (Chiracel AD-H, 2.5 mL/min, 30% IPA in CO2, λ = 254 nm): tR(major) = 5.7 min tR(minor) = 6.9 min.
Supplementary Material
Scheme 2.

Functionalization of tryptophan 7c and 7d.
Acknowledgments
We thank Prof. Brian Stoltz, Dr. Scott Virgil, and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment, and Dr. David VanderVelde for assistance with NMR structure determination. Fellowship support was provided by the NSF (M. E. K., Graduate Research Fellowship under Grant No. DGE-1144469) and the ACS Division of Organic Chemistry (L. M. R., sponsored by Genentech). Nadine Currie is acknowledged for assistance in the preparation of several indole substrates. Financial support from the California Institute of Technology, the NIH (NIGMS RGM097582A), and the donors of the ACS Petroleum Research Foundation are gratefully acknowledged.
References
- 1.For example, tadalafil: Daugan A, Grondin P, Ruault CC, Le Monnier de Gouville A-C, Coste H, Linget JM, Kirilovsky J, Hyafil FO, Labaudiniere R. J Med Chem. 2003;46:4533. doi: 10.1021/jm0300577.
- 2.Royer CA. Chem Rev. 2006;106:1769. doi: 10.1021/cr0404390.For a specific example, see: Lepthien S, Hoesl MG, Merkel L, Budisa N. Proc Natl Acad Sci, USA. 2008;105:16095. doi: 10.1073/pnas.0802804105.
- 3.Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA. Proc Natl Acad Sci, USA. 1998;95:12088. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Austin JF, Kim SG, Sinz CJ, Xiao WJ, MacMillan DWC. Proc Nat Acad Sci. 2004;101:5482–7. doi: 10.1073/pnas.0308177101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ishihara K, Fushimi N, Akakura M. Acc Chem Res. 2007;40:1049. doi: 10.1021/ar700083a. [DOI] [PubMed] [Google Scholar]
- 5.Xu Z, Zhang F, Zhang L, Jia Y. Org Biomol Chem. 2011;9:2512. doi: 10.1039/c0ob01115k. [DOI] [PubMed] [Google Scholar]
- 6.Artman GD, Grubbs AW, Williams RM. J Am Chem Soc. 2007;129:6336. doi: 10.1021/ja070259i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Existing catalytic asymmetric methods: Zheng BH, Ding CH, Hou XL, Dai LX. Org Lett. 2010;12:1688. doi: 10.1021/ol100161n.Sui Y, Liu L, Zhao J-L, Wang D, Chen Y-J. Tetrahedron. 2007;63:5173.Castle SL, Srikanth GSC. Org Lett. 2003;5:3611. doi: 10.1021/ol035236x.Drury WJ, Ferraris D, Cox C, Young B, Lectka T. J Am Chem Soc. 1998;120:11006. doi: 10.1021/ja016838j.Asymmetric hydrogenation: Townsend JM, Blount JF, Sun RC, Zawoiski S, Valentine D., Jr J Org Chem. 1980;45:2995.
- 8.Repka LM, Ni J, Reisman SE. J Am Chem Soc. 2010;132:14418. doi: 10.1021/ja107328g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Ishihara K, Nakashima D, Hiraiwa Y, Yamamoto H. J Am Chem Soc. 2003;125:24. doi: 10.1021/ja021000x. [DOI] [PubMed] [Google Scholar]; (b) Nakamura S, Kaneeda M, Ishihara K, Yamamoto H. J Am Chem Soc. 2000;122:8120. [Google Scholar]; (c) Ishihara K, Nakamura S, Kaneeda M, Yamamoto H. J Am Chem Soc. 1996;118:12854. [Google Scholar]; (d) Ishihara K, Kaneeda M, Yamamoto H. J Am Chem Soc. 1994;116:11179. [Google Scholar]
- 10.For a review of combined acid catalysis, see: Yamamoto H, Futatsugi K. Angew Chem Int Ed. 2005;44:1924. doi: 10.1002/anie.200460394.
- 11.The use of achiral Lewis acids to give racemic tryptophan derivatives has been reported: Gentilucci L, Cerisoli L, Marco RD, Tolomelli A. Tetrahedron Lett. 2010;51:2576.Angelini E, Balsamini C, Bartoccini F, Lucarini S, Piersanti G. J Org Chem. 2008;73:5654. doi: 10.1021/jo800881u.Blaser G, Sanderson JM, Batsanov AS, Howard JAK. Tetrahedron Lett. 2008;49:2795.
- 12.(a) Sibi MP, Coulomb J, Stanley LM. Angew Chem Int Ed. 2008;47:9913. doi: 10.1002/anie.200804221. [DOI] [PubMed] [Google Scholar]; (b) Fu N, Zhang L, Li J, Luo S, Cheng JP. Angew Chem, Int Ed. 2011;50:11451. doi: 10.1002/anie.201105477. [DOI] [PubMed] [Google Scholar]
- 13.After the submission of this manuscript, the use of indole nucleophiles for tandem Friedel-Crafts conjugate addition/asymmetric protonation was reported. See reference 12b.
- 14.Non-Friedel–Crafts conjugate addition/protonation reactions: Jousseaume T, Wurz NE, Glorius F. Angew Chem Int Ed. 2011;50:1410. doi: 10.1002/anie.201006548.Poisson T, Yamashita Y, Kobayashi S. J Am Chem Soc. 2010;132:7890. doi: 10.1021/ja102555a.Morita M, Drouin L, Motoki R, Kimura Y, Fujimori I, Kanai M, Shibasaki M. J Am Chem Soc. 2009;131:3858. doi: 10.1021/ja9005018.Navarre L, Martinez RM, Genet JP, Darses S. J Am Chem Soc. 2008;130:6159. doi: 10.1021/ja710691p.Leow D, Lin S, Chittimalla SK, Fu X, Tan CH. Angew Chem Int Ed. 2008;47:5641. doi: 10.1002/anie.200801378.Wang B, Wu F, Wang Y, Liu X, Deng L. J Am Chem Soc. 2007;129:768. doi: 10.1021/ja0670409.Sibi MP, Tatamidani H, Patil K. Org Lett. 2005;7:2571. doi: 10.1021/ol050630b.Navarre L, Darses S, Genet JP. Angew Chem Int Ed. 2004;43:719. doi: 10.1002/anie.200352518.Moss RJ, Wadsworth KJ, Chapman CJ, Frost CG. Chem Commun. 2004:1984. doi: 10.1039/b406905f.Hamashima Y, Somei H, Shimura Y, Tamura T, Sodeoka M. Org Lett. 2004;6:1861. doi: 10.1021/ol0493711.
- 15.A radical conjugate addition of alkyl halides to 2-amidoacrylates followed by hydrogen atom transfer to prepare α-amino acids with moderate enantioselectivity has been reported. See: Sibi MP, Asano Y, Sausker JB. Angew Chem Int Ed. 2001;40:1293. doi: 10.1002/1521-3773(20010401)40:7<1293::aid-anie1293>3.0.co;2-y.
- 16.Seminal examples of asymmetric Friedel–Crafts conjugate additions of unactivated indoles to set stereogenic centers in the β-position: Boersma AJ, Feringa BL, Roelfes G. Angew Chem Int Ed. 2009;48:3346. doi: 10.1002/anie.200900371.Rueping M, Nachtsheim BJ, Moreth SA, Bolte M. Angew Chem Int Ed. 2008;47:593. doi: 10.1002/anie.200703668.Evans DA, Fandrick KR, Song HJ. J Am Chem Soc. 2005;127:8942. doi: 10.1021/ja052433d.Palomo C, Oiarbide M, Kardak BG, Garcia JM, Linden A. J Am Chem Soc. 2005;127:4154. doi: 10.1021/ja0423217.Evans DA, Scheidt KA, Fandrick KR, Lam HW, Wu J. J Am Chem Soc. 2003;125:10780. doi: 10.1021/ja036985c.Austin JF, MacMillan DWC. J Am Chem Soc. 2002;124:1172. doi: 10.1021/ja017255c.Zhou J, Tang Y. J Am Chem Soc. 2002;124:9030. doi: 10.1021/ja026936k.Jensen KB, Thorhauge J, Hazell RG, Jørgensen KA. Angew Chem Int Ed. 2001;40:160. doi: 10.1002/1521-3773(20010105)40:1<160::AID-ANIE160>3.0.CO;2-S.
- 17.See Supporting Information.
- 18.9f is easily prepared on multi-gram scale in 3 steps from (R)-BINOL. See: Ooi T, Kameda M, Maruoka K. J Am Chem Soc. 2003;125:5139. doi: 10.1021/ja021244h.
- 19.The absolute stereochemistry of 7c was assigned by comparison of the optical rotation to previously reported literature data: Ruiz-Rodríguez J, Albericio F, Lavilla R. Chem Eur J. 2010;16:1124. doi: 10.1002/chem.200902676.
- 20.Alternatively, the corresponding N-substituted tryptophan products are accessible from corresponding the N-protio products.
- 21.The major byproduct of this reaction was determined to be an indole dimer, formed in 30% yield. See Supporting Information.
- 22.Endo Y, Shudo K, Itai A, Hasegawa M, Sakai S-I. Tetrahedron. 1986;42:5905. [Google Scholar]
- 23.Morieux P, Stables JP, Kohn H. Bioorg Med Chem. 2008;16:8968. doi: 10.1016/j.bmc.2008.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Use of NBS instead of NCS resulted in bromination of the indole backbone.
- 25.Unfortunately, deuterium labelling experiments aimed at elucidating the asymmetric protonation process were rendered inconclusive by rapid proton-deuterium exchange at N1 and C3 of deuterated indole substrates under standard reaction conditions.
- 26.Zhang X. WO 2002040491. PCT Int Appl. 2002
- 27.Wu TR, Shen L, Chong JM. Org Lett. 2004;6:2701. doi: 10.1021/ol0490882. [DOI] [PubMed] [Google Scholar]
- 28.Ito K, Takahashi M, Hoshino T, Nishiki M, Ohba Y. Lett Org Chem. 2006;3:735. [Google Scholar]
- 29.Verga D, Percivalle C, Doria F, Porta A, Freccero M. J Org Chem. 2011;76:2319. doi: 10.1021/jo1025892. [DOI] [PubMed] [Google Scholar]
- 30.Rueping M, Sugiono E, Steck A, Thiessmann T. Adv Synth Catal. 2010;352:281. [Google Scholar]
- 31.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.