

Abstract
The effects of mGlu1 and mGlu5 receptor activation on the depolarization-evoked release of [3H]D-aspartate ([3H]D-ASP) from mouse cortical synaptosomes were investigated. The mGlu1/5 receptor agonist 3,5-DHPG (0.1–100 μM) potentiated the K+(12 μM)-evoked [3H]D-ASP overflow. The potentiation occurred in a concentration-dependent manner showing a biphasic pattern. The agonist potentiated [3H]D-ASP exocytosis when applied at 0.3 μM; the efficacy of 3,5-DHPG then rapidly declined and reappeared at 30–100 μM. The fall of efficacy of agonist at intermediate concentration may be consistent with 3,5-DHPG-induced receptor desensitization. Facilitation of [3H]D-ASP exocytosis caused by 0.3 μM 3,5-DHPG was prevented by the selective mGlu5 receptor antagonist MPEP, but was insensitive to the selective mGlu1 receptor antagonist CPCCOEt. In contrast, CPCCOEt prevented the potentiation by 50 μM 3,5-DHPG, while MPEP had minimal effect. Unexpectedly, LY 367385 antagonized both the 3,5-DHPG-induced effects. A total of 0.3 μM 3,5-DHPG failed to facilitate the K+-evoked [3H]D-ASP overflow from mGlu5 receptor knockout (mGlu5−/−) cortical synaptosomes, but not from nerve terminals prepared from the cortex of animals lacking the mGlu1 receptors, the crv4/crv4 mice. On the contrary, 50 μM 3,5-DHPG failed to affect the [3H]D-ASP exocytosis from cortical synaptosomes obtained from crv4/crv4 and mGlu5−/−mice. Western blot analyses in subsynaptic fractions support the existence of both mGlu1 and mGlu5 autoreceptors located presynaptically, while immunocytochemistry revealed their presence at glutamatergic terminals. We propose that mGlu1 and mGlu5 autoreceptors exist on mouse glutamatergic cortical terminals; mGlu5 receptors may represent the “high affinity” binding sites for 3,5-DHPG, while mGlu1 autoreceptors represent the “low affinity” binding sites.
Keywords: Mouse cortical synaptosomes, [3H]D-aspartate release, mGlu1 autoreceptor, mGlu5 autoreceptor, crv4 Mice, mGlu5 Receptor knockout mice
1. Introduction
Presynaptic modulation of neurotransmitter release represents one of the fundamental mechanisms of regulation of central transmission; most of the central neurotransmitters, including glutamate, undergo this control, exerted either by auto or by heteroreceptors located presynaptically on nerve endings.
Of the known autoreceptor systems, the glutamatergic system appears the most complex (Raiteri, 2008 and references therein). Both ionotropic and metabotropic glutamate (mGlu) receptors belong to this system. However, although several lines of evidence support the existence and the role of glutamate ionotropic autoreceptors, such as NMDA (Luccini et al., 2007a and references therein), AMPA (Barnes et al., 1994) and kainate (Chittajallu et al., 1996; Pittaluga et al., 1997) receptors, mGlu autoreceptors appears predominant (Raiteri, 2008).
mGlu receptors belong to the family 3 of G protein coupled receptors (GPCRs, Bockaert and Pin, 1999) and possess a very large N-terminal domain containing the agonist binding sites. These receptors are structurally and pharmacologically heterogeneous and exist as eight subtypes (mGlu1–mGlu8 receptors) having discrete regional, cellular and subcellular localizations (Conn and Pin, 1997; Pin et al., 2003). Depending on the specific receptor involved, the main transduction mechanisms can be activation of the phosphatidylinositol (PI) pathway (group I mGlu receptors, that are mGlu1 and 5 receptors) or inhibition of adenylyl cyclase (AC) activity (group II mGlu receptors, namely mGlu2 and 3, and group III mGlu receptors, including mGlu4, 6, 7 and 8).
Because of the negative control on AC and K+ channels, group II and III mGlu autoreceptors function as negative feedback mechanisms that inhibit glutamate release (Corti et al., 2007; Raiteri, 2008 and references therein). In contrast, group I mGlu receptors favour Ca2+-dependent phosphorylative processes and facilitate glutamate release (Herrero et al., 1992; Raiteri, 2008 and references therein).
Group I mGlu receptors have been proposed to be located presynaptically at glutamatergic terminals (Raiteri, 2008 and references therein), but the relative contributions of mGlu1 and mGlu5 receptors to the facilitation of glutamate release remain matter of discussion.
To directly address this question we have pharmacologically characterized the receptor/s involved in the facilitation of the K+-evoked [3H]D-ASP release from mouse cortical glutamatergic terminals exerted by the group I selective agonist 3,5-DHPG. We also used transgenic mouse models lacking mGlu1 (crv4/crv4; Conti et al., 2006) or mGlu5 (mGlu5+/−) receptors. Our results are consistent with the existence of both mGlu1 and mGlu5 autoreceptors in cortex. Although activation of both these receptors increases evoked glutamate exocytosis, the two receptors do not compensate one for each other. The pharmacological profile as well as the results obtained with transgenic mice suggest that mGlu1 autoreceptors behave as “low affinity” binding site for 3,5-DHPG, while mGlu5 autoreceptors appears to be the high affinity binding sites.
2. Methods
2.1. Animals
Adult male mice (Swiss; 20–25 g) were used as control animals in all the experiments with the exception of the experiments carried out with cervelet-4 (crv4) mice and mGlu5−/− knockout mice. The crv4 mutation is a spontaneous recessive mutation occurred in the BALB/c/Pas inbred strain. It consists in an LTR intronic insertion which disrupts splicing of mGluR1 gene and causes absence of the protein (Conti et al., 2006). Crv4 homozygous mice present mainly with an ataxic phenotype. Affected (crv4/crv4) and control (+/+) mice are maintained on the same genetic background by inter-crossing +/crv4 mice at the animal facility of the National Institute of Cancer Research (Genoa, Italy). The genotype of the wild-type and crv4 mice was determined by PCR with use of tail genomic DNA and a pair of primers (5′-GAGTGTTCACTAGTTCACCCAAGA-3′ and 5′-TCAGGCAACAATAAGGCAAG-3′) which flank the insertion; the PCR products synthesized with these primers were 688 bp for the crv4 mutant and 498 bp for wild-type.
Heterozygous mGlu5 receptor knockout mice (129-Gprc1etmt1rod) were obtained from The Jackson Laboratories (Bar Harbor, ME, USA). Mice heterozygous for the targeted mutation were intercrossed to homozygosity at the “Istituto Neurologico Mediterraneo Neuromed” (Pozzilli, IS, Italy). Homozygous females and males were fertile, but poor breeders. Thus, all mice were generated by heterozygous breeding. Primers for the genotyping of knockout mice were from Jackson Laboratory. Transgenic mice were then sent to the animal facility of our laboratory (Department of Experimental Medicine, Pharmacology and Toxicology Section). Mice were kept under environmentally controlled conditions (ambient temperature 22 ± 1 °C, humidity 50%) on a 12-h light/dark cycle with food and water ad libitum. Mice were identified by PCR analysis on tail samples after birth.
The animals were killed by decapitation; the cortices rapidly removed and purified synaptosomes prepared within minutes. The experimental procedures were approved by the Department Ethical Committee, in accordance with the European legislation (European Communities Council directive of 24 November 1986, 86/609/ EEC). Experiments were performed following the Guidelines for Animal Care and Use of the National Institutes of Health.
2.2. Preparation of synaptosomes
To isolate purified synaptosomes, the tissue was homogenized in 10 volumes of 0.32 M sucrose, buffered to pH 7.4 with TRIS (final concentration 0.01 M) using a glass Teflon tissue grinder (clearance 0.25 mm).The homogenate was centrifuged at 1000 g for 5 min, to remove nuclei and debris, and the supernatant was gently stratified on a discontinuous Percoll gradient (6%, 10% and 20% v/v in Tris-buffered sucrose) and centrifuged at 33,500 g for 5 min. The layer between 10% and 20% Percoll (synaptosomal fraction) was collected and washed by centrifugation. The synaptosomal pellets were resuspended in a physiological medium having the following composition (mM): NaCl, 125; KCl, 3; MgSO4, 1.2; CaCl2, 1.2; NaH2PO4,1; NaHCO3, 22; glucose, 10 (aeration with 95% O2 and 5% CO2); pH, 7.2–7.4.
2.3. Experiments of release from superfused synaptosomes
Synaptosomes were labelled with [3H]D-aspartate ([3H]D-ASP; final concentration 30 nM) at 37 °C, for 15 min, in a rotary water bath and in an atmosphere of 95% O2 and 5% CO2. After the labelling period, identical portions of the synaptosomal suspensions were layered on microporous filters at the bottom of parallel superfusion chambers (Ugo Basile, Comerio, Varese, Italy; Raiteri and Raiteri, 2000) thermostated at 37 °C and superfused at 0.5 ml/min with standard physiological solution aerated with 95% O2 and 5% CO2.
When studying the effect of 3,5-DHPG on [3H]D-ASP release evoked by high K+, synaptosomes were transiently (90 s) exposed at t = 39 min to 12 μM KCl containing medium (NaCl substituting for an equimolar concentration of KCl) in absence or in presence of the agonist. To evaluate whether presynaptic mGlu5 autoreceptors can desensitize and switch from facilitation to inhibition (Herrero et al., 1998; Rodriguez-Moreno et al., 1998; Sistiaga et al., 1998; Nicoletti et al., 1999), synaptosomes were pre-exposed to 3,5-DHPG from t = 30 min to 35 min of superfusion, then substituted until t = 39 min with standard physiological medium and then rechallenged with 3,5-DHPG concomitantly added with the depolarizing stimulus (12 or 30 μM K+). Antagonists were added 8 min before agonist. Starting from t = 36 min of superfusion, four consecutive 3 min fractions were collected. Fractions collected and superfused synaptosomes were counted for radioactivity.
2.4. Isolation of detergent-soluble fraction from synaptosomes
Purified synaptosomes were prepared as already described and collected by centrifugation for 15 min at 4 °C and 14,000 g. The pellet was diluted and lysed in 1 mL of ice-cold 20 μM Tris/HCl (pH 7.4, containing 10 μM NaCl and protease inhibitor cocktail). In order to isolate the detergent-soluble membrane fraction (DS-Syn; see Pedrazzi et al., 2006), after 15 min at 4 °C, 450 mg of synaptosome lysate (Tot-Syn) were centrifuged at 200,000 g,4 °C for 15 min and the pellet was solubilized in 800 μl of 20 μM Tris/HCL (pH 7.4, containing 140 μM NaCl, 0.2% Triton X-100 and protease inhibitor cocktail). After 15 min at 4 °C, samples were centrifuged at 200,000 g,4 °C for 15 min and the supernatant, the DS-Syn, collected. Proteins were quantified using Lowry Assay.
2.5. Immunoblotting
Proteins (25 μg/lane) were separated by SDS-7.5% PAGE, and then transferred onto PVDF membranes. Membranes were blocked in 20 μM sodium phosphate buffer (PBS, pH 7.4, containing 140 μM NaCl, 5% non fat dry milk, 0.1% Tween-20) and probed with one of the following primary antibodies (60 min at 20 °C): rabbit anti-mGlu1 receptor (1:500), rabbit anti-mGlu5 receptor (1:1000), mouse anti-syntaxin-1A (Stx-1A; 1:10000). After extensive washes, membranes were incubated for 1 h at 20 °C with the appropriate horseradish peroxidase-linked secondary antibody (1:4000), and immunoblots were visualized with an ECL (enhanced chemiluminescence) Plus Western blotting detection system.
2.6. Sub-synaptic fractionation of nerve terminals
Sub-synaptic fractionation was prepared as described in literature (Feligioni et al., 2006). Briefly, synaptosomes were prepared, as described above, from cortex of three mouse brains in order to obtain enough proteins for the preparation. The pellets from purified synaptosomes were resuspended in 300 μl of 0.32 M sucrose and 0.1 μM CaCl2. Protease inhibitors were used in all purification steps. Synaptosomes were then diluted 1:10 in ice-cold 0.1 μM CaCl2 and mixed with an equal volume of 2× solubilisation buffer (2% Triton X-100, 40 μM Tris, pH 6.0; 4 °C). Following a 30 min incubation at 4 °C, the insoluble material (synaptic junctions) was pelleted (40,000 g, 30 min, 4 °C) and the supernatant (non-synaptic synaptosomal protein; NSSP) was decanted and proteins precipitated with 6 volumes of acetone at −20 °C and centrifuged (18,000 g; 30 min; −15 °C). The synaptic junction pellet was resuspended in 10 volumes of 1× solubilisation buffer (1% Triton X-100, 20 μM Tris, pH 8.0; 4 °C) and incubated for 30 min at 4 °C and then centrifuged (40,000 g, 30 min, 4 °C). The pellet contained the insoluble postsynaptic density and the supernatant contained the presynaptic active zone. The protein in the supernatant (presynaptic fraction) was acetone precipitated and collected as above. Proteins obtained from this preparation contained about 1.70 μg/μl per fraction.
2.7. Sub-fractionation immunoblotting
A10 mg protein/lane where loaded on 10% SDS-PAGE gel and then transferred onto PVDF membranes. Non-specific binding sites were blocked over-night with Tris-buffered saline-Tween (t-TBS; 0.02 M Tris, 0.137 M NaCl, and 0.1% Tween 20) containing 5% non-fat dried milk and probed for protein of interest with the following primary antibodies: rabbit anti-mGlu1 receptor (1:500), rabbit anti-mGlu5 receptor (1:1,000), mouse anti-syntaxin-1A (Stx-1A; 1:10,000), mouse anti-Synaptophysin, (1:20,000), mouse anti-PSD95, (1:1,000). After washes, membranes were incubated for 1 h at room temperature with the appropriate horseradish peroxidase-linked secondary antibody (1:5000), and immunoblots were visualized with an ECL (enhanced chemiluminescence) Plus Western blotting detection system.
2.8. Immunocytochemical analysis in mouse cortical nerve terminals
For immunohistochemical analysis, cortical synaptosomes were obtained through discontinuous Percoll gradient and 80 mg of synaptosomal proteins were placed onto coverslips, previously coated with poly-L-ornithine, fixed with 2% paraformaldehyde for 15 min. and washed with PBS. The synaptosomes were permeabilized with 0.05% Triton X-100 PBS for 5 min. After extensive washes with 0.5% BSA PBS, synaptosomes were incubated with: rabbit anti-mGlu1 receptor (1:500); rabbit anti-mGlu5 receptor (1:500); mouse antisintaxin-1A (Stx-1A; 1:10,000) and guinea pig anti-vesicular glutamate transporters type 1 (VGLUT-1; 1:500) as indicated for 1 h at room temperature. The synaptosomes were then extensively washed with 0.5% BSA PBS and incubated for 1 h at room temperature with AlexaFluor-488 (green)-labelled donkey anti-mouse IgG antibodies, or AlexaFluor-647 (red) donkey anti-rabbit IgG antibodies or AlexaFluor-647 (red) donkey anti-mouse IgG antibodies or Donkey, FITC Conjugate (green), anti-guinea pig IgG antibodies (1:1,000 for all).
Fluorescence image acquisition was performed by a three-channel Leica TCS SP5 laser-scanning confocal microscope, equipped with 458, 476, 488, 514, 543 and 633 nm excitation lines. Images (512 × 512 × 8 bit) were taken through a planapochromatic oil immersion objective 100x/NA1.4. Light collection configuration was optimized according to the combination of chosen fluorochromes and sequential channel acquisition was performed to avoid cross-talk phenomena. Leica LCS software package was used for acquisition, storage and visualization. Each coverslip was analyzed by counting at least three different fields.
2.9. Calculations
The amount of radioactivity released into each superfusate fraction was expressed as % of the total synaptosomal tritium content at the start of the fraction collected (fractional efflux). The K+-induced tritium overflow was expressed as % induced overflow and was estimated by subtracting the neurotransmitter content into the first and the fourth fractions collected (basal release) from that in the second and in the third fractions collected during and after the depolarization pulse (evoked release). Analysis of variance was performed by ANOVA followed by Newman Keuls multiple-comparison test; direct comparisons were performed by applying Student’s t-test. Data were considered significant for p < 0.05 at least. Appropriate controls with antagonists were always run in parallel. Western blots were quantified by densitometric analysis using NIH Image J software.
The quantitative estimation of colocalized proteins in immunocytochemical studies has been performed calculating the co-localization coefficients (Manders et al., 1993) from the red- and green-channel scatterplot. Co-localization coefficients express the fraction of colocalizing molecular species in each component of a dual-colour image and are based on the Pearson’s correlation coefficient, a standard procedure for matching one image with another in pattern recognition (Gonzalez and Wintz, 1987). If two molecular species are colocalized, the overlay of their spatial distributions has a correlation value higher than what would be expected by chance alone. Costes et al. (2004) developed an automated procedure to evaluate correlation between the green and red channels with a significance level >95%. The same procedure automatically determines an intensity threshold for each colour channel based on a linear least-square fit of the green and red intensities in the image’s 2D correlation cytofluorogram. Costes’ approach has been accomplished by macro routines integrated as plug-ins (WCIF Co-localization Plugins, Wright Cell Imaging Facility, Toronto Western Research Institute, Canada) in the ImageJ 1.39 software (Wayne Rasband, NIH, USA).
2.10. Drugs
1-[7,8 3H]D-aspartate (specific activity 16.3 Ci/mmol) was from Amersham Radiochemical Center (Buckinghamshire, UK). (RS)-3,5-Dihydroxyphenylglycine (3,5-DHPG) was from Ascent Scientific (Weston Super-Mare, UK); 2-methyl-6-(phenylethynyl) pyridine hydrochloride (MPEP), 7-(hydroxyimino)cyclo propa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt) and (S)-(+)-a-Amino-4-carboxy-2-methylbenzene acetic acid (LY 367385) were obtained from Tocris Bioscience (Bristol, UK). Anti-mGlu5 receptor polyclonal rabbit immunoaffinity purified IgG, anti-mGlu1 receptor polyclonal rabbit immunoaffinity IgG, and mouse anti-PSD95 were from Upstate Biotechnology (Lake Placid, NY, USA). Anti-syntaxin-1A monoclonal mouse IgG was from Synaptic System, Germany; mouse anti-Syn-aptophysin, was from Calbiochem, San Diego, CA, USA; Guinea Pig anti-VGLUT-1 and Donkey FITC Conjugated anti-Guinea Pig antibodies were purchased from Chemicon (CA, USA). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies and the ECL Plus Western blotting detection system were purchased from GE Healthcare. Protease Inhibitor Cocktail was from Sigma–Aldrich. AlexaFluor-647 or AlexaFluor-488 antibodies are from Molecular Probes (Alfagene) (Oregon, USA).
3. Results
3.1. 3,5-DHPG potentiates biphasically the release of [3H]D-ASP evoked by KCl
We investigated the effects of 3,5-DHPG, a broad spectrum mGlu1/5 receptor agonist, on the release of [3H]D-ASP evoked by 12 μM K+ from mouse cortical nerve endings. It is known that, when mouse cortical synaptosomes are exposed in superfusion to mild K+ depolarization, a Ca2+-dependent, exocytotic-like release of prelabelled [3H]D-ASP occurs. Indeed, recent work showed that the 12 μM K+-induced release of [3H]D-ASP represents a reliable measure of endogenous glutamate from this synaptosomal preparation (Raiteri et al., 2007).
Fig. 1 shows that 3,5-DHPG (0.1–100 μM) facilitated the 12 μM KCl-evoked release of [3H]D-ASP in a concentration-dependent manner; a significant facilitation of [3H]D-ASP exocytosis was observed when the agonist was added at concentrations around 0.3 and above 10 μM 3,5-DHPG.
Fig. 1.
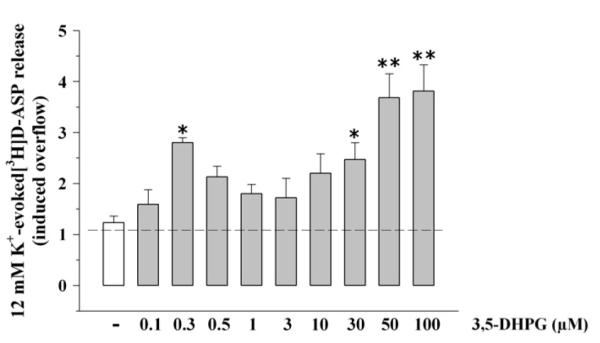
Concentration–effect relationship of 3,5-DHPG on the 12 μM K+-evoked release of [3H]D-aspartate ([3H]D-ASP) from mouse cortical synaptosomes. Mouse cortical synaptosomes were exposed to 12 μM KCl in absence (white bar) or in presence (grey bar) of 3,5-DHPG (concentration as indicated). Results are expressed as induced overflow. Data are mean ± SEM of 3–12 experiments run in triplicate (three determinations for each experimental conditions). *p < 0.05 vs control; **p < 0.01 vs control.
3.2. Effects of selective mGlu1 and mGlu5 receptor antagonists on the 12 μM K+-induced [3H]D-ASP overflow
The pharmacological characterization of the mGlu receptors involved in the facilitation by 3,5-DHPG of [3H]D-ASP exocytosis was then carried out using the selective mGlu1 receptor non-competitive antagonist CPCCOEt, the competitive antagonist LY 367385 and the selective non-competitive mGlu5 receptor antagonist MPEP. Preliminary experiments investigated the effects of these antagonists, added alone, on the K+-evoked release of [3H]D-ASP.
LY 367385 and CPCCOEt (0.5–10 μM) failed to affect the K+-evoked release of [3H]D-ASP, whereas MPEP (0.3–3 μM) decreased it significantly (Table 1). Notably, the inhibition by MPEP of glutamate exocytosis appears well in line with the results by Wang and Sihra (2004) who suggested the existence mGlu5 autoreceptors constitutively activated in cortical glutamatergic terminals. The inhibition by MPEP of the K+-evoked release of [3H]D-ASP appeared maximal when the mGlu5 receptor antagonist was applied at 1 μM and it was unaffected by the contemporary addition of 5 μM CPCCOEt (Table 1).
Table 1.
Effects of group I mGlu receptor antagonists on the K+-evoked release of [3H]D-ASP from mouse cortical synaptosomes
| 12 mMK+-evoked release of [3H]D-ASP |
||
|---|---|---|
| (% Induced overflow) | (% of Changes vs control) | |
| Control | 1.61± 0.13 | |
| 0.01 μM LY 367385 | 1.68± 0.09 | +4.3 |
| 0.01 μM LY 367385 | 1.62± 0.07 | +0.6 |
| 0.01 μM LY 367385 | 1.67± 0.11 | +3.7 |
| 0.5 μM CPCCOEt | 1.72± 0.11 | +6.8 |
| 5 μM CPCCOEt | 1.64± 0.08 | +1.8 |
| 10 μM CPCCOEt | 1.74± 0.26 | +0.8 |
| 0.3 μM MPEP | 1.26± 0.04 | −21.8 |
| 1 μM MPEP | 1.11± 0.11a | −31.9 |
| 3 μM MPEP | 1.52± 0.13 | −5.6 |
| 5 μM CPCCOEt+1 μM MPEP | 1.13± 0.11a | −29.9 |
Results are expressed as % induced overflow or as % of changes respect respective control. Data are means± SEM of at least three experiments run in triplicate.
p< 0.05 at least vs control.
3.3. Effects of selective mGlu1 and mGlu5 receptor antagonists on the 3,5-DHPG potentiation of the K+-evoked [3H]D-ASP overflow
MPEP (0.1–1 μM) prevented the 3,5-DHPG (0.3 μM) potentiation of 12 μM K+-induced [3H]D-ASP overflow, which was totally abrogated when the antagonist was applied at 1 μM (Fig. 2A). On the contrary, CPCCOEt (5–10 μM) was ineffective (Fig. 2A). The antagonism brought about by 1 μM MPEP was unmodified by the contemporary addition of 5 μM CPCCOEt (Fig. 2A). Selective blockade of mGlu1 receptors by CPCCOEt counteracted the 3,5-DHPG (50 μM) potentiation of 12 μM K+-induced [3H]D-ASP overflow, which was totally prevented at the concentration of 5 μM (Fig. 2B). MPEP (0.1–1 μM) slightly reduced the effect of 50 μM 3,5-DHPG (Fig. 2B). The antagonism by 5 μM CPCCOEt was not modified by the contemporary addition of 1 μM MPEP (Fig. 2B). As expected, the competitive mGlu1 antagonist LY 367385 inhibited in a concentration-dependent fashion the 3,5-DHPG (50 μM) potentiation of 12 μM K+-evoked [3H]D-ASP release, being maximally effective when added up to 0.1 μM (Fig. 3B). Surprisingly, the facilitation of glutamate exocytosis caused by 0.3 μM 3,5-DHPG was significantly affected by 0.1–1 μM LY 367385 (Fig. 3A).
Fig. 2.

Effects of CPCCOEt and MPEP on the release of [3H]D-aspartate evoked by 12 μM K+ in presence of 3,5-DHPG. Mouse cortical synaptosomes were exposed to 12 μM KCl in absence (white bar) or in presence (grey bar) of 3,5-DHPG (0.3 μM, panel A; 50 μM, panel B). CPCCOEt (rising right grey bar), MPEP (rising left grey bar) or both agonists (cross-hatched grey bar) were added starting from t = 30 min of superfusion till the end of the experiment. Results are expressed as induced overflow. Data are mean ± SEM of 4–10 experiments run in triplicate. *p < 0.05 at least vs 12 μM K+; #p < 0.05 at least vs 12 μM K+ in the presence of 0.3 μM 3,5-DHPG; §p < 0.05 at least vs 12 μM K+ in the presence of 50 μM 3,5-DHPG.
Fig. 3.
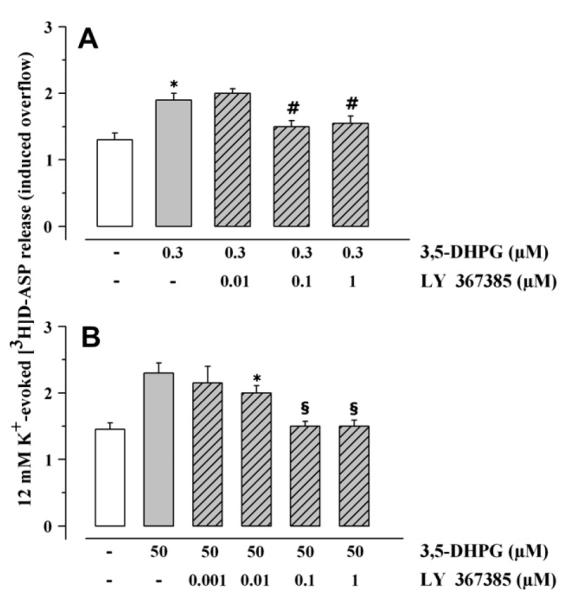
Effects of LY 367385 on the release of [3H]D-aspartate evoked by 12 μM K+ in presence of 3,5-DHPG. Mouse cortical synaptosomes were exposed to 12 μM KCl in absence (white bar) or in presence (grey bar) of 3,5-DHPG (0.3 μM, panel A; 50 μM, panel B). LY 367385 (rising right grey bar) was added starting from t = 30 min of superfusion till the end of the experiment. Results are expressed as induced overflow. Data are means ± SEM of seven experiments run in triplicate. *p < 0.05 at least vs 12 μM K+; #p < 0.05 at least vs 12 μM K+ in the presence of 0.3 μM 3,5-DHPG; §p < 0.05 at least vs 12 μM K+ in the presence of 50 μM 3,5-DHPG.
3.4. Deletion of mGlu1 or mGlu5 receptors prevents selective activation of mGlu1 or mGlu5 autoreceptors
To further confirm the existence of mGlu1 and mGlu5 autoreceptors controlling [3H]D-ASP exocytosis from mouse cortical glutamatergic terminals, we used mouse models lacking mGlu1 (crv4/crv4 mice, Conti et al., 2006) or mGlu5 (mGlu5−/− mice) receptors.
Preliminary experiments were carried out to evaluate whether receptor deletion had caused adaptative changes in the efficiency of release. The absence of mGlu5 receptors did not affect the spontaneous release of [3H]D-ASP (control 0.81 ± 0.15%; mGlu5−/− = 0.73 ± 0.04%; n = 5; n.s.; data expressed as % of the total synaptosomal content in the first fraction collected), nor it affected the 12 μM K+-evoked release of [3H]D-ASP (Fig. 4A). The lack of mGlu1 receptors did not modify the spontaneous release of [3H]D-ASP from cortical synaptosomes (control = 1.24 ± 0.11%; crv4/crv4 = 1.23 ± 0.08%; n = 6; n.s.; data expressed as % of the total synaptosomal content in the first fraction collected), but it significantly affected the amount of tritium released by depolarizing stimulus. The 12 μM K+-induced [3H]D-ASP overflow from crv4/crv4 cortical synaptosomes was significantly greater (36.05 ± 4.3%, p < 0.05 at least) than that released from control synaptosomes (Fig. 4B).
Fig. 4.
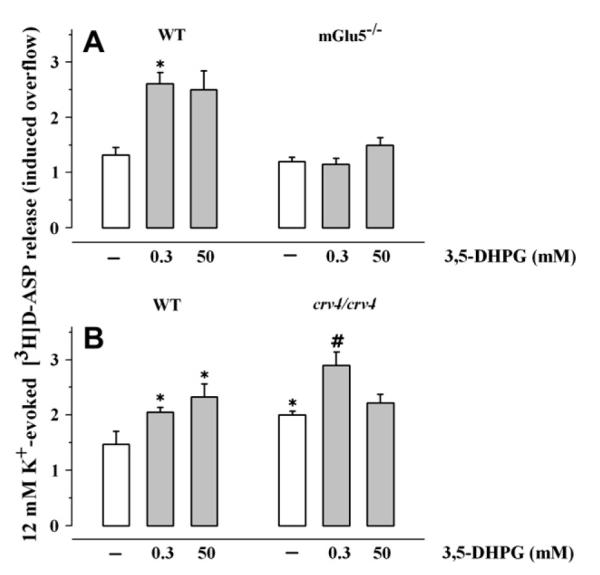
Effects of 3,5-DHPG on the release of [3H]D-aspartate evoked by 12 μM K+ from synaptosomes isolated from the cortex of mouse lacking mGlu1 or mGlu5 receptors. Synaptosomes were prepared from the cortex of transgenic mice lacking mGlu5 receptors (mGlu5−/− mouse, panel A) or mGlu1 receptors (crv4/crv4 mouse, panel B) and from relative controls. Synaptosomes were exposed to 12 μM KCl in absence (white bar) or in presence (grey bar) of 3,5-DHPG (concentration as indicated). Results are expressed as induced overflow. Data are mean ± SEM of 12 experiments run in triplicate. *p < 0.05 at least vs respective control. WT, wild type.
We then evaluated to what extent the lack of mGlu5 receptor expression could affect the facilitation by 0.3 or by 50 μM 3,5-DHPG of the 12 μM K+-induced [3H]D-ASP overflow. Interestingly, the 12 μM K+-induced release of glutamate from mGlu5−/− cortical synaptosomes was unmodified when 0.3 or 50 μM 3,5-DHPG was added concomitantly to the depolarizing stimulus (Fig. 4A). On the contrary, the facilitation caused by 50 μM, but not that caused by 0.3 μM, 3,5-DHPG was absent in cortical synaptosomes isolated from crv4/crv4 animals (Fig. 4B).
3.5. Mouse cortical synaptosomal membranes contain mGlu1 and mGlu5 receptor proteins
The functional results obtained with mGlu1 and mGlu5 receptor antagonists and with mouse models are compatible with the existence of mGlu1 and mGlu5 autoreceptors on glutamatergic terminals. To substantiate this view, we firstly investigated whether the detergent soluble membrane fraction (DS-syn) isolated from cortical synaptosomes was endowed with mGlu1 and mGlu5 receptor proteins. We focussed on this synaptosomal preparation, since DS-Syn contains all the synaptosomal membrane proteins, including those from intraterminal vesicles and organelles, with the exception of the soluble proteins. To this aim, mouse cortical synaptosomal lysates (Tot-Syn) and the corresponding DS-Syn were probed with anti mGlu1 and anti mGlu5 receptor antibodies. The results obtained reported in Fig. 5A are consistent with the existence of both receptors in mouse cortical synaptosomal membranes. Unfortunately, however, such an approach does not account for the exact location of receptor proteins. In order to assess if mGlu1 and mGlu5 receptor proteins are indeed located presynaptically, as suggested by the functional results, we applied a fractionation procedure that permit to purify the presynaptic active zone (Phillips et al., 2001; Pinheiro et al., 2003; Feligioni et al., 2006). Experiments were carried out to validate the separation of the presynaptic active zone and of the postsynaptic density from other presynaptic proteins not located in the active zone, here termed non-synaptic synaptosomal proteins (NSSP). PSD95 was found to be predominant in the postsynaptic fraction, syntaxin-1A (Stx-1A) in the presynaptic component (although traces are also present in the NSSP) while Synaptophysin in the NSSP fraction (Fig. 5B). The mGlu5 receptor protein was expressed both pre- and postsynaptically and to a lower extent extrasynaptically, while the mGlu1 receptor protein was found to be preferentially located pre- and postsynaptically (Fig. 5B).
Fig. 5.

mGlu1 and mGlu5 receptor protein expressions in mouse cortical synaptosomes. (A) Western blotting of mGlu1 and mGlu5 receptor proteins in the synaptosomal lysate (Tot-Syn) and in its detergent soluble fraction (Ds-Syn). Protein weights are expressed in kDa. (B) Immunoblot of synapse fractionation showing mGlu1 receptor and mGlu5 receptor relative distribution in synaptosomal compartments in the NSSP fraction (NSSP), in the postsynaptic fraction (Post) and in the presynaptic fraction (Pre). Anti-PSD95, anti-synaptophysin and anti-Stx-1A were used as markers for ultrasynaptic fraction to determinate the purity of the preparation that is more than 90%. Protein weights are expressed in kDa. The blots are representative of four to six analyses.
3.6. Identification of mGlu1 and mGlu5 receptors in mouse cortical glutamatergic terminals
Considering that the above fractionation procedure does not permit discrimination between families of nerve terminals, it was important to ascertain whether mGlu1 and mGlu5 receptor immunoreactivities could be detected in glutamatergic nerve endings. Glutamatergic synaptosomes were identified by using antibodies against the vesicular glutamate transporter type 1 (VGLUT-1) and against Stx-1A and monitoring the co-localization of VGLUT-1 and Stx-1A.
The mouse cortical synaptosomal preparations efficiently stained for VGLUT-1 (Fig. 6A, a, green) and Stx-1A (Fig. 6A, b, red) and merging these images revealed that a great percentage of Stx-1A-positive particles was also positive for VGLUT-1 protein (Fig. 6A, c). Analysis of six different image couples indicated that about 77 ± 1% of the Stx-1A-positive particles were also VGLUT-1-positive. The mouse cortical synaptosomal preparations efficiently stained for mGlu5 receptor (Fig. 6A, e and h, red), and significant percentages of the Stx-1A-positive terminal particles (Fig. 6A, d, green) and of the VGLUT-1-positive particles (Fig. 6A, g, green) were positive for mGlu5 receptor (Fig. 6B). Similarly, the mouse cortical synaptosomal preparations stained efficiently for mGlu1 receptor (Fig. 6, panel A, k and n, red), and significant percentages of the Stx-1A-positive terminal particles (Fig. 6A, j, green) and of the VGLUT-1-positive particles (Fig. 6A, m, green) were also positive for mGlu1 receptor (Fig. 6B).
Fig. 6.

mGlu1 and mGlu5 receptors are present on mouse cortical glutamatergic terminals. (A) Representative double-labelling images of anti mGlu5 (panels d–i) and of mGlu1 (panels j–o) with anti A-1 (marker of all nerve terminals) and with anti VGLUT-1 (specific marker of glutamatergic nerve terminals). (B) Percent of co-localization of mGlu1 and mGlu5 receptor immunoreactivities with VGLUT-1 or Stx-1A staining. Results are expressed as co-localization of Stx-1A and VGLUT-1 with mGlu1 and mGlu5 receptor proteins, respectively. Data are the mean ± SEM of 6–8 different image couples.
3.7. Effects of 3,5-DHPG pre-exposure on the 3,5-DHPG potentiation of the K+-evoked [3H]D-ASP overflow
Group I mGlu receptors were reported to undergo desensitization (Catania et al., 1991; Herrero et al., 1994) shortly after a brief pre-exposure to agonist. Since this event was also observed in cortical synaptosomes isolated from mGlu1−/− animals (Sistiaga et al., 1998), it had been proposed that mGlu5 receptors were those preferentially involved in the agonist-induced modifications. To investigate whether this phenomenon also occurred in our experimental conditions, control or crv4/crv4 cortical synaptosomes were exposed to 3,5-DHPG (0.3 μM), then washed to remove the agonist and finally re-challenged with 3,5-DHPG (0.3 μM) in the presence of a depolarizing stimulus (12 μM). 3,5-DHPG (0.3 μM), added contemporary to the depolarizing stimulus, potentiated the 12 μM K+-evoked release of [3H]D-ASP from cortical synaptosomes isolated from control and crv4/crv4 mice (see results in Figs. 1–4). Accordingly to results in literature, the positive effect of 3,5-DHPG was abolished when control or crv4/crv4 synaptosomes were pre-exposed to the agonist (Table 2).
Table 2.
Effects of pre-exposure to 3,5-DHPG on the 3,5-DHPG-induced facilitation of K+-evoked [3H]D-aspartate release from control and crv4/crv4 mice
| Control mice |
crv4/crv4 mice |
|||
|---|---|---|---|---|
| % Induced overflow |
% of Changes vs control |
% Induced overflow |
% of Changes vs control |
|
| 12 mMK+ | 1.80±0.15 | 2.52 ±0.09a | ||
| 12 mMK+/0.3 μM 3,5-DHPG | 3.22 ±0.28a | +78.9a | 3.93 ±0.22c | + 55.9a |
| pre-exposure to 0.3 μM 3,5-DHPG12 mMK+/0.3 μM 3,5-DHPG |
1.45±0.25b | −19.5 | 2.84 ±0.18d | +12.7 |
| 30 mMK+ | 10.06 ±1.01 | 14.53 ±1.65e | ||
| 30 mMK+/3 μM 3,5-DHPG | 10.85 ±1.54 | +7.85 | 14.57 ±2.01 | +0.2 |
| Pre-exposure to 3 μM 3,5-DHPG30 mMK+/3 μM 3,5-DHPG |
8.04 ±0.95 | −20.1 | 13.03 ±1.89 | −10.4 |
Results are expressed as % induced overflow or as % of changes respect respective control. Data are means ± SEM of at least three experiments run in triplicate.
p<0.05 at least vs 12 mM K+-induced overflow from control synaptosomes.
p<0.05 at least vs (12 mMK+/0.3 μM 3,5-DHPG)-induced overflow fromcontrol synaptosomes.
p<0.05 at least vs 12 mM K+-induced overflow from crv4/crv4 synaptosomes.
p<0.05 at least vs (12 mM K+/0.3 μM 3,5-DHPG)-induced overflow from crv4/crv4 synaptosomes.
p<0.05 at least vs 30 mMK+-induced overflow from control synaptosomes.
Evidences have been also provided showing that 3,5-DHPG is unable to affect the release of glutamate caused by a depolarizing stimulus stronger than 12 μM K+ (i.e. 30 μM K+, Herrero et al., 1998), but that pre-exposure to the agonist unveil a switch from facilitation to inhibition of group I mGlu receptors (Herrero et al., 1998; Rodriguez-Moreno et al., 1998), a molecular event thought to be involved in the dual role of group I antagonists in neurotoxicity and neuroprotection (Nicoletti et al., 1999). Also in this case the effect was retained in synaptosomes isolated from mGlu1−/− mice, suggesting the prominent role of mGlu5 receptors (Sistiaga et al., 1998). To investigate whether this phenomenon could be observed also in our experimental conditions, synaptosomes were exposed to 3 μM 3,5-DHPG and then re-challenged with 30 μM K+/3 μM 3,5-DHPG. Results were compared to those obtained from synaptosomes that were not pre-exposed to the agonist. The release of [3H]D-ASP evoked by 30 μM K+ from wild type or crv4/crv4 cortical synaptosomes was unaffected by 3 μM 3,5-DHPG contemporary added to the depolarizing stimulus (Table 2). A slight inhibition of the 30 μM K+/3 μM 3,5-DHPG-evoked release was observed shortly after a preliminary exposure of wild type, but not of crv4/crv4, cortical synaptosomes to the agonist (Table 2). Differences in animal models as well as in the experimental approaches applied could, perhaps, account for the discrepancies between our and previous results (Herrero et al., 1998; Rodriguez-Moreno et al., 1998).
Notably, the amount of glutamate release elicited by high K+ from cortical crv4/crv4 glutamatergic terminals was altered when compared to control synaptosomes (Fig. 4B, Table 2). Further investigations are needed to clarify this aspect.
4. Discussion
The major findings of this work are: (i) mouse cortical glutamatergic terminals are endowed with both mGlu1 and mGlu5 autoreceptors, whose activation favours the release of glutamate induced by a mild depolarizing stimulus (i.e. 12 μM K+); (ii) mGlu5 autoreceptors may represent the “high affinity” binding sites, while mGlu1 autoreceptors may represent the “low affinity” binding sites for glutamate. Data obtained with Western blot after subsynaptic fractionation of synaptosomal membranes and with immunocytochemistry support the functional results and sustain the location of both mGlu1 and mGlu5 on glutamatergic nerve endings of the cerebral cortex.
The present demonstration of the existence of both presynaptic mGlu1 and mGlu5 autoreceptors is of interest; in fact there has been much controversy on whether group I mGlu autoreceptors are mGlu1 or mGlu5. mGlu1 receptor subtypes were proposed to have a presynaptic location by Moroni et al. (1998), based on “in vivo” microdialysis studies in rat parietal cortex. Sistiaga et al. (1998), however, argued against this view and proposed the existence of mGlu5 autoreceptors on mouse cortical nerve endings. The existence of mGlu1 autoreceptors was then reconsidered by Reid et al. (1999), who showed that the 3,5-DHPG-evoked increase of [3H]glutamate release from rat cortical synaptosomes was prevented by the preferential mGlu1 receptor antagonist AIDA. More recently, however, Fazal et al. (2003) provided evidence favouring the existence of mGlu5, but not of mGlu1, autoreceptors on cortical glutamatergic terminals. Thus, our results, consistent with the existence of both mGlu1 and mGlu5 presynaptic autoreceptors, deserve particular attention.
Functional experiments were performed by monitoring release from superfused monolayers of purified synaptosomes, a technique particularly appropriate to study release-regulating presynaptic receptors (Raiteri and Raiteri, 2000). Using this experimental approach we found that the addition of the mGlu1/mGlu5 receptor agonist 3,5-DHPG at 0.3 μM enhanced glutamate release evoked by mild K+ depolarization; the effect of 3,5-DHPG was much weaker and not significant between 0.3 and 10 μM, but it reappeared when the agonist was added at concentrations higher than 10 μM.
These dual effects seemed predictive of the existence of “high affinity” and “low affinity” binding sites for 3,5-DHPG. In line with this idea, the pharmacological profile of the receptors involved obtained by using non-competitive antagonists revealed that the two effects were due to activation of different receptors. In particular, the response caused by low 3,5-DHPG concentrations was totally antagonized by the selective mGlu5 receptor antagonist MPEP, suggesting that the “high affinity” site may correspond to the mGlu5 autoreceptor. On the other hand, CPCCOEt blocked the potentiating effect caused by high 3,5-DHPG concentrations, indicating that the “low affinity” site could coincide with the mGlu1 autoreceptor. Consistent with the results obtained with CPCCOEt, LY 367385, a selective competitive antagonist at mGlu1 receptor subtype, totally antagonized the potentiation by 50 μM 3,5-DHPG of glutamate exocytosis, but, unexpectedly, it also affected significantly the facilitation caused by 0.3 μM 3,5-DHPG. These results are in a way surprising enough, in particular when considering that the effect caused by 0.3 μM 3,5-DHPG was CPCCOEt-insensitive, but MPEP-sensitive. Since the binding of LY 367385 to mGlu5 receptors appears unlikely, an alternative explanation might consider that, in cortical glutamatergic terminals, mGlu1 homodimers may colocalize with mGlu5 homodimers, influencing their functions. It is known that mGlu homodimers can adopt a symmetric open conformation in the absence of the agonist that turns to an asymmetric open-closed conformation in presence of glutamate. Both conformations have been observed in plasmamembranes, also in the absence of the agonist, suggesting that the equilibrium between the two forms exist (De Blasi et al., 2001). Assuming that mGlu1 and mGlu5 homodimers coexist on glutamatergic cortical terminals, we speculate that LY 367385 can force mGlu1 homodimers to adopt the open conformation and that this modification to the dimer structure could result in changes to homodimer–homodimer interactions. Future studies will be useful to clarify this hypothesis.
In recent years, mouse models lacking expression of a given receptor have been used to corroborate functional results from wild type animals. In order to substantiate the existence of “high affinity” mGlu5 and of “low affinity” mGlu1 autoreceptors and to shed light on their reciprocal role as presynaptic modulators of glutamate exocytosis, we took advantage of animals lacking mGlu1 (crv4/crv4) or mGlu5 (mGlu5−/−) receptors, although, in general, receptor deletion were shown to induce compensative adaptations to central neurotransmission. In particular, as group I mGlu receptor is concerned, changes to mGlu1 receptor-mediated functions were reported to be caused by mGlu5 receptor deletion (Volk et al., 2006), while mGlu5 receptor expression and functions appeared unaltered in mGlu1 knockout animals (Herrero et al., 1998; Sistiaga et al., 1998).
The results obtained when studying the effects of 3,5-DHPG on glutamate exocytosis from cortical synaptosomes isolated from crv4/crv4 mice sustained, in our opinion, the existence of a “high affinity” mGlu5 and a “low affinity” mGlu1 autoreceptors. Actually, in this synaptosomal preparation, 0.3 μM 3,5-DHPG facilitated glutamate exocytosis, while the potentiation caused by 50 μM 3,5-DHPG was undetectable. The role of mGlu5 receptors as the “high affinity” binding site for 3,5-DHPG was also supported by the finding that the facilitation by 0.3 μM 3,5-DHPG of glutamate exocytosis was abrogated in mGlu5−/− cortical synaptosomes. Differently from expectation, however, 50 μM 3,5-DHPG failed to potentiate the K+-induced release in mGlu5−/− cortical synaptosomes. Although unexpected, this finding could be consistent with the above-mentioned alterations in mGlu1 receptor functions observed in mGlu5−/− animals (Volk et al., 2006) and might further support the hypothesis that mGlu1 and mGlu5 receptors can coexist and reciprocally interact on glutamate nerve terminals.
The pattern of the concentration–response curve of 3,5-DHPG deserves a comment, particularly regarding the fall of efficacy at the intermediate concentrations (0.5–10 μM). Our explanation may be found taking into account that mGlu5 receptors can undergo rapid agonist-induced desensitization (results in Table 2, but see also Catania et al., 1991; Herrero et al., 1994, 1998; Rodriguez-Moreno et al., 1998; Fazal et al., 2003); therefore the fall of efficacy of 3,5-DHPG at intermediate concentrations might represent the resultant of an agonist-induced mGlu5 autoreceptor desensitization that precedes the 3,5-DHPG-induced activation of mGlu1 autoreceptors.
Contradictory results are present in the literature on the presynaptic location of mGlu1 and mGlu5 receptors investigated by immunocytochemistry (Fothui et al., 1993; Shigemoto et al., 1993; Romano et al., 1995; Lujan et al., 1996; Marino et al., 2001; Hubert et al., 2001; Muly et al., 2003). When we looked at the protein subsynaptic distribution, we found that mGlu1 and mGlu5 receptor proteins were present in the presynaptic zone and largely expressed in the postsynaptic density. Furthermore, restricting the analysis to the glutamatergic nerve terminals, immunocytochemistry revealed that mGlu1 and mGlu5 receptor proteins were present in a significant percentage of VGLUT-1 positive glutamatergic nerve terminals. By combining the data obtained in functional studies with those obtained in Western Blot experiments after subsynaptic fractionation and by immunocytochemistry, it seems reasonable to conclude that presynaptic mGlu1 and mGlu5 autoreceptors exist on mouse cortical glutamatergic nerve terminals.
The apparent affinity of 3,5-DHPG for mGlu5 autoreceptors is three orders of magnitude greater than that for mGlu1 autoreceptors. Assuming that this also occurs when glutamate is the autoreceptor agonist, one may speculate that the two receptors, although able to mediate glutamate release, play different roles. In fact, mGlu5 autoreceptors would be expected to be activated by physiological concentrations of glutamate, while ”pathological” concentrations of the endogenous ligand would be needed to activate mGlu1 receptors. We do not know whether mGlu1 and mGlu5 autoreceptors colocalize on glutamatergic terminals or if there exist a mixed populations of nerve terminals bearing either mGlu1 or mGlu5 autoreceptors, although co-existence would seem more compatible with our findings as well as with the differential implication of the receptors in synaptic plasticity and excitotoxic events. Notably, mGlu1 and mGlu5 heteroreceptors were already shown to be co-localized at the presynaptic terminals, in human and rodent cortical and hippocampal noradrenergic nerve endings (Luccini et al., 2007b). Differently from what observed on noradrenergic terminals (Luccini et al., 2007b), however, mGlu1 and mGlu5 autoreceptors do not compensate one for each other, but exert distinct modulations of glutamate release.
Notably, there is abundant evidence that group I mGlu receptors and NMDA receptors reciprocally interact in neurons (Luccini et al., 2007b and references therein). NMDA receptors also exist on glutamatergic terminals, where they mediate different modes of glutamate release (exocytosis or carrier-mediated release) depending on the glutamate concentrations acting at the receptor complex (Luccini et al., 2007a). If NMDA autoreceptors and group I mGlu autoreceptors colocalize on glutamate nerve terminals, one could hypothesize that glutamate exocytosis evoked by low glutamate concentrations acting at release-enhancing NMDA autoreceptors could facilitate mGlu5 autoreceptor activation. On the other side, mGlu1 receptors should be preferentially activated by the “pathological”, carrier-mediated release of glutamate induced by high glutamate concentrations acting at NMDA presynaptic autoreceptors. Further studies, however, are needed to substantiate this hypothesis.
Acknowledgements
We wish to thank Maura Agate for careful editorial assistance. This work was supported by grants from Istituto Superiore di Sanità (V Programma Nazionale di Ricerca sull’AIDS: Progetto “Patologia, Clinica e Terapia dell’AIDS), from Italian “Ministero dell’Istruzione, dell’Università e della Ricerca Scientifica”(MIUR) and from IRCCS Istituto Neurologico Mediterraneo NEUROMED (Pozzilli, IS, Italy).
References
- Barnes JM, Dev KK, Henley JM. Cyclothiazide unmasks AMPA-evoked stimulation of [3H]-L-glutamate release from rat hippocampal synaptosomes. British Journal of Pharmacology. 1994;113:339–341. doi: 10.1111/j.1476-5381.1994.tb16902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockaert J, Pin J-P. Molecular tinkering of G-protein-coupled receptors: an evolutionary success. EMBO Journal. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania MV, Aronica E, Sortino MA, Canonico PL, Nicoletti F. De-sensitization of metabotropic glutamate receptors in neuronal cultures. Journal of Neurochemistry. 1991;56:1329–1335. doi: 10.1111/j.1471-4159.1991.tb11429.x. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Vignes M, Dev KK, Barnes JM, Collingridge GL, Henley JM. Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature. 1996;379:78–81. doi: 10.1038/379078a0. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annual Review of Pharmacology and Toxicology. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Conti V, Aghaie A, Cilli M, Martin N, Caridi G, Musante L, Candiano G, Castagna M, Fairen A, Ravazzolo R, Guenet JL, Puliti A. crv4, a mouse model for human ataxia associated with kyphoscoliosis caused by an mRNA splicing mutation of the metabotropic glutamate receptor 1 (Grm1) International Journal of Molecular Medicine. 2006;18:593–600. [PubMed] [Google Scholar]
- Corti C, Battaglia G, Molinaro G, Riozzi B, Pittaluga A, Corsi M, Mugnaini M, Nicoletti F, Bruno V. The use of knock-out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. Journal of Neuroscience. 2007;27:8297–8308. doi: 10.1523/JNEUROSCI.1889-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophysical Journal. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Blasi A, Conn PJ, Pin JP, Nicoletti F. Molecular determinants of metabotropic glutamate receptor signaling. Trends in Pharmacological Sciences. 2001;22:114–120. doi: 10.1016/s0165-6147(00)01635-7. [DOI] [PubMed] [Google Scholar]
- Fazal A, Parker F, Palmer AM, Croucher MJ. Characterisation of the actions of group I metabotropic glutamate receptor subtype selective ligands on excitatory amino acid release and sodium-dependent re-uptake in rat cerebrocortical mini slices. Journal of Neurochemistry. 2003;86:1346–1358. doi: 10.1046/j.1471-4159.2003.01932.x. [DOI] [PubMed] [Google Scholar]
- Feligioni M, Holman D, Haglerod C, Davanger S, Henley JM. Ultrastructural localisation and differential agonist-induced regulation of AMPA and kainate receptors present at the presynaptic active zone and postsynaptic density. Journal of Neurochemistry. 2006;99:549–560. doi: 10.1111/j.1471-4159.2006.04087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fothui M, Sharp AH, Glatt CE, Hwang PM, Vonkrosigk M, Snyder SH, Dawson TM. Differential localization of phosphoinositide-linked metabotropic glutamate receptor (mGluR1) and the inositol 1,4,5-trisphosphate receptor in rat brain. Journal of Neuroscience. 1993;13:2001–2012. doi: 10.1523/JNEUROSCI.13-05-02001.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez RC, Wintz P. Digital Image Processing. second ed Addison Wesley Publication Company; Mass, USA: 1987. [Google Scholar]
- Herrero I, Miras-Portugal MT, Sanchez-Prieto J. Positive feedback of glutamate exocytosis by metabotropic presynaptic receptor stimulation. Nature. 1992;360:163–166. doi: 10.1038/360163a0. [DOI] [PubMed] [Google Scholar]
- Herrero I, Miras-Portugal MT, Sanchez-Prieto J. Rapid desensitization of the presynaptic metabotropic receptor that facilitates glutamate exocytosis. European Journal of Neuroscience. 1994;6:163–166. doi: 10.1111/j.1460-9568.1994.tb00253.x. [DOI] [PubMed] [Google Scholar]
- Herrero I, Miras-Portugal MT, Sanchez-Prieto J. Functional switch from facilitation to inhibition in the control of glutamate release by metabotropic glutamate receptors. Journal of Biological Chemistry. 1998;273:1951–1958. doi: 10.1074/jbc.273.4.1951. [DOI] [PubMed] [Google Scholar]
- Hubert GW, Paquet M, Smith Y. Differential subcellular localization of mGluR1a and mGluR5 in the rat and monkey substantia nigra. Journal of Neuroscience. 2001;21:1838–1847. doi: 10.1523/JNEUROSCI.21-06-01838.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luccini E, Musante V, Neri E, Raiteri M, Pittaluga A. N-methyl-D-aspartate autoreceptors respond to low and high agonist concentrations by facilitating, respectively, exocytosis and carrier-mediated release of glutamate in rat hippocampus. Journal of Neuroscience Research. 2007a;85:3657–3665. doi: 10.1002/jnr.21446. [DOI] [PubMed] [Google Scholar]
- Luccini E, Musante V, Neri E, Brambilla Bas M, Severi P, Raiteri M, Pittaluga A. Functional interactions between presynaptic NMDA receptors and metabotropic glutamate receptors co-expressed on rat and human noradrenergic terminals. British Journal of Pharmacology. 2007b;151:1087–1094. doi: 10.1038/sj.bjp.0707280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perysinaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. European Journal of Neuroscience. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA. Measurement of colocalization of objects in dual-colour confocal images. Journal of Microscopy. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Wittmann M, Bradley Risso, Hubert GW, Smith Y, Conn PJ. Activation of group I metabotropic glutamate receptors produces a direct excitation and disinhibition of GABAergic projection neurons in the substantia nigra pars reticulata. Journal of Neuroscience. 2001;21:7001–7012. doi: 10.1523/JNEUROSCI.21-18-07001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni F, Cozzi A, Lombardi G, Sourtcheva S, Leonardi P, Carfì M, Pellicciari R. Presynaptic mGlu1 type receptors potentiate transmitter output in the rat cortex. European Journal of Pharmacology. 1998;347:189–195. doi: 10.1016/s0014-2999(98)00124-1. [DOI] [PubMed] [Google Scholar]
- Muly EC, Maddox M, Smith Y. Distribution of mGluR1a and mGluR5 immunolabelling in primate prefrontal cortex. Journal of Comparative Neurology. 2003;467:521–535. doi: 10.1002/cne.10937. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Bruno V, Catania MV, Battaglia G, Copani A, Barbagallo G, Cena V, Sanchez-Prieto J, Spano PF, Pizzi M. Group-I metabotropic glutamate receptors: hypothesis to explain their dual role in neurotoxicity and neuroprotection. Neuropharmacology. 1999;38:1477–1484. doi: 10.1016/s0028-3908(99)00102-1. [DOI] [PubMed] [Google Scholar]
- Pedrazzi M, Raiteri L, Bonanno G, Patrone M, Ledda S, Passalacqua M, Milanese M, Melloni E, Raiteri M, Pontremoli S, Sparatore B. Stimulation of excitatory amino acid release from adult mouse brain glia subcellular particles by high mobility group box 1 protein. Journal of Neurochemistry. 2006;99:827–838. doi: 10.1111/j.1471-4159.2006.04120.x. [DOI] [PubMed] [Google Scholar]
- Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon RE, Gawinowicz MA, Zhao Y, Colman DR. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- Pin J-P, Galvez T, Prézeau L. Evolution, structure, and activation mechanism of family 3/C G protein-coupled receptors. Pharmacology and Therapeutics. 2003;98:325–354. doi: 10.1016/s0163-7258(03)00038-x. [DOI] [PubMed] [Google Scholar]
- Pinheiro PS, Rodrigues RJ, Silva AP, Cunha RA, Oliveira CR, Malva JO. Solubilization and immunological identification of presynaptic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors in the rat hippocampus. Neuroscience Letters. 2003;336:97–100. doi: 10.1016/s0304-3940(02)01217-x. [DOI] [PubMed] [Google Scholar]
- Pittaluga A, Bonfanti A, Raiteri M. Differential desensitization of ionotropic non-NMDA receptors having distinct neuronal location and function. Naunyn Schmiedeberg’s Archives of Pharmacology. 1997;356:29–38. doi: 10.1007/pl00005025. [DOI] [PubMed] [Google Scholar]
- Raiteri M. Presynaptic metabotropic glutamate and GABAB receptors. Handbook of Experimental Pharmacology. 2008;184:373–407. doi: 10.1007/978-3-540-74805-2_12. [DOI] [PubMed] [Google Scholar]
- Raiteri L, Raiteri M. Synaptosomes still viable after 25 years of superfusion. Neurochemical Research. 2000;25:1265–1274. doi: 10.1023/a:1007648229795. [DOI] [PubMed] [Google Scholar]
- Raiteri L, Zappettini S, Milanese M, Fedele E, Raiteri M, Bonanno G. Mechanisms of glutamate release elicited in rat cerebrocortical nerve endings by ‘pathologically’ elevated extraterminal K+ concentrations. Journal of Neurochemistry. 2007;103:952–961. doi: 10.1111/j.1471-4159.2007.04784.x. [DOI] [PubMed] [Google Scholar]
- Reid ME, Toms NJ, Bedingfield JS, Roberts PJ. Group I mGlu receptors potentiate synaptosomal [3H]glutamate release independently of exogenously applied arachidonic acid. Neuropharmacology. 1999;38:477–485. doi: 10.1016/s0028-3908(98)00217-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Sistiaga A, Lerma J, Sanchez-Prieto J. Switch from facilitation to inhibition of excitatory synaptic transmission by group I mGluR desensitization. Neuron. 1998;21:1477–1486. doi: 10.1016/s0896-6273(00)80665-0. [DOI] [PubMed] [Google Scholar]
- Romano C, Sesma MA, McDonald CT, O’Malley K, Van den Pol AN, Olney JW. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. Journal of Comparative Neurology. 1995;335:455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Nomura S, Ohishi H, Nakanishi S, Mizuno N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neuroscience Letters. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- Sistiaga A, Herrero I, Conquet F, Sanchez-Prieto J. The metabotropic glutamate receptor 1 is not involved in the facilitation of glutamate release in cerebrocortical nerve terminals. Neuropharmacology. 1998;37:1485–1492. doi: 10.1016/s0028-3908(98)00129-4. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Daly CA, Huber KM. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. Journal of Neurophysiology. 2006;95:2427–2438. doi: 10.1152/jn.00383.2005. [DOI] [PubMed] [Google Scholar]
- Wang S-J, Sihra TS. Noncompetitive metabotropic glutamate5 receptor antagonist (E)-2-methyl-6-styryl-pyridine (SIB1893) depresses glutamate release through inhibition of voltage-dependent Ca2+ entry in rat cerebrocortical nerve terminals (synaptosomes) Journal of Pharmacology and Experimental Therapeutics. 2004;309:951–958. doi: 10.1124/jpet.103.064881. [DOI] [PubMed] [Google Scholar]