

Abstract
Restriction factors, such as the retroviral complementary DNA deaminase APOBEC3G, are cellular proteins that dominantly block virus replication1-3. The AIDS virus, human immunodeficiency virus type 1 (HIV-1), produces the accessory factor Vif, which counteracts the host’s antiviral defence by hijacking a ubiquitin ligase complex, containing CUL5, ELOC, ELOB and a RING-box protein, and targeting APOBEC3G for degradation4-10. Here we reveal, using an affinity tag/purification mass spectrometry approach, that Vif additionally recruits the transcription cofactor CBF-β to this ubiquitin ligase complex. CBF-β, which normally functions in concert with RUNX DNA binding proteins, allows the reconstitution of a recombinant six-protein assembly that elicits specific polyubiquitination activity with APOBEC3G, but not the related deaminase APOBEC3A. Using RNA knockdown and genetic complementation studies, we also demonstrate that CBF-β is required for Vif-mediated degradation of APOBEC3G and therefore for preserving HIV-1 infectivity. Finally, simian immunodeficiency virus (SIV) Vif also binds to and requires CBF-β to degrade rhesus macaque APOBEC3G, indicating functional conservation. Methods of disrupting the CBF-β–Vif interaction might enable HIV-1 restriction and provide a supplement to current antiviral therapies that primarily target viral proteins.
Mammals have evolved cellular proteins termed restriction factors that function to prevent the spread of mobile genetic elements including retroviruses1-3. As a counter-defence, most retroviruses, including the human pathogen HIV-1, have developed mechanisms to prevent restriction, often through subversion of the host’s ubiquitin–proteasome system. In eukaryotic cells, 8.6-kDa ubiquitin moieties are added to a target protein by sequential action of one of two ubiquitin-activating enzymes (E1), which transfer ubiquitin to a pool of dozens of ubiquitin-conjugating enzymes (E2) that, in turn, collaborate with hundreds of ubiquitin ligases (E3) to catalyse transfer to specific substrates11. If more than four ubiquitins are joined together through K48 linkages, the target protein is usually degraded by the 26S proteasome12. At least three HIV-1 proteins, Vif, Vpu and Vpr, hijack cullin-RING E3 ligases consisting of CUL5, CUL1 and CUL4A to promote ubiquitination and degradation of APOBEC3 family members (for example, APOBEC3G, A3G), BST2/tetherin and an unknown, putative restriction factor, respectively2. Understanding the composition of cullin-RING E3 ligase complexes and the underlying cellular signalling components may provide therapeutic routes for treating a variety of human diseases, including infection by HIV-1.
HIV-1 Vif is recruited to CUL5 by virtue of its SOCS box, which contains an elongin C binding helix (the BC-box), a conserved HCCH Zn binding motif and a short Cullin Box4-6. Although a structure of the BC-box peptide in complex with the heterodimer of Elongin B and C (ELOBC) has been reported13, the architecture of the full-length Vif in complex with host factors has remained elusive, in part because Vif complexes have poor solubility and activity. We therefore reasoned that Vif may bind an additional host factor and that such a factor may render it more tractable in vitro.
We took an unbiased proteomic approach to identify host factors that bind all 18 HIV processed and polyproteins using an affinity tag/purification mass spectrometry (AP–MS) approach14,15. To this end, 2×Strep and 3×Flag was fused to the carboxy (C) terminus of these factors, including Vif. The tagged Vif construct was both transiently transfected into HEK293 cells and used to make a stable, tetracycline-inducible Vif–Strep–Flag Jurkat T cell line (Fig. 1a). Epitope-tagged Vif was purified from both cell types using antibodies specific to either Strep or Flag and aliquots of the co-purifying proteins were subjected to SDS–polyacrylamide gel electrophoresis (SDS–PAGE) (Fig. 1b). Materials from each step were analysed by mass spectrometry14.
Figure 1. AP–MS experiments identify CBF-β as a Vif-dependent component of the Vif–CUL5 ubiquitin ligase complex.

a, Flow-chart of the proteomic analysis performed during the study. b, Affinity-tagged versions of Vif, Vpu and Vpr were purified using 3×Flag from HEK293 and Jurkat cells, subjected to SDS–PAGE and stained with silver. Visible bands corresponding to interactions that are known for each accessory factor are labelled. Note Vif and CBF-β run at a similar place on the gel. Tagged versions of Vpr and Vpu were used as specificity controls. c, A network representation of Vif–host protein–protein interactions from both HEK293 (blue) and Jurkat T cells (red) after subjecting the data derived from the AP–MS analysis to the MiST scoring system15. The intensity of the node colours corresponds to the quantitative MiST score. Blue edges represent interactions derived during this work; black edges are previously described interactions between host factors; dashed edges correspond to previously described Vif–host interactions present in the database VirusMint. d, The double purification approach, which allows for the identification of stable, stoichiometric protein complexes. e, Double purifications were performed in triplicate using 3×Flag-tagged CUL5, A3G or CBF-β with 2×Strep-tagged Vif in HEK293 cells. Proteins that were identified in all three double purifications, after trypsin digestion and analysis by mass spectrometry, are represented. The coverage corresponds to the percentage of protein identified by tryptic peptides. f, Immunoblots showing that Vif recruits CBF-β to theCUL5/ELOBC/RBX2 ubiquitin ligase complex. HA-tagged ELOB or CUL5 were immunoprecipitated in the absence or presence of increasing amounts of Vif, and endogenous CBF-β was monitored by immunoblot. g, HIV and SIV Vif co-immunoprecipitate CBF-β and ELOC. GFP and HIV Nef were analysed in parallel as specificity controls.
Using a new scoring system for data derived from AP–MS studies, termed Mass Spectrometry Interaction Statistics (MiST)15, we identified 24 Vif–human protein–protein interactions with seven of them found in both cell types (Fig. 1c). Seventeen of these were verified independently by co-immunoprecipitation (Supplementary Fig. 1). Among these were the components of the E3 ubiquitin ligase complex, CUL5, ELOB and ELOC, known to interact with Vif and trigger A3G degradation4-6,8-10. Although the RING-box protein RBX1 was originally reported as part of this complex4, only RBX2 was above the MiST score threshold used15 consistent with recent work showing that it binds CUL5 (refs 16, 17). We did not find endogenous A3G, probably because of its poor expression in HEK293 and Jurkat cell lines exacerbated by further depletion through Vif-mediated degradation. We did find Vif associating with two proteins that function in autophagy, AMRA1 and SQSTM, as well as with the transcriptional co-repressor complex NCOR1/HDAC3/GPS2/TBL1R (the last only in T cells) (Fig. 1c). Also, in both cell types, Vif was found to interact with the transcription cofactor CBF-β, which is known to heterodimerize with the RUNX family of transcription factors18.
To determine if any of the newly defined Vif interactors belong to the Vif–CUL5 complex, we performed double affinity purifications using cells co-transfected with Vif–2×Strep and either A3G– or CUL5–3×Flag (Fig. 1d, e). After purification first with Strep-Tactin and second with anti-Flag beads, mass spectrometry analysis of the final elution revealed the presence of CUL5, ELOB, ELOC, RBX2 and invariably CBF-β, strongly suggesting that this last protein may be a new component of the Vif E3 ubiquitin ligase complex (other factors from single Vif purifications depicted in Fig. 1c were not present). To confirm this interaction and the composition of the complex, we performed an additional double affinity purification experiment using Vif–2×Strep and CBF-β–3×Flag. This strategy also yielded CUL5, ELOB, ELOC and RBX2, in addition to the epitope-tagged bait proteins (Fig. 1d, e).
To determine if the association of CBF-β with the CUL5 ligase complex was dependent on Vif, we immunoprecipitated CUL5-haemagglutinin (HA) or ELOB-HA in the presence or absence of Vif in HEK293 cells and blotted for endogenous CBF-β. Only in the presence of Vif did CBF-β co-immunoprecipitate with tagged CUL5 or ELOB, indicating that recruitment of CBF-β to the CUL5 ligase is dependent on Vif (Fig. 1f). SIV Vif also associated with CBF-β by immunoprecipitation, suggesting the interaction is conserved (Fig. 1g).
We next asked if the Vif–CUL5 ligase could be reconstituted with CBF-β using recombinant proteins purified from Escherichia coli. Initial purification attempts without CBF-β yielded aggregated and inactive complexes, assayed by size-exclusion chromatography and autoubiquitination activity, suggesting that CBF-β may be required for complex formation (data not shown). Therefore, full-length Vif, ELOB, ELOC and CBF-β were co-expressed, purified to homogeneity and found to form a stable, monodisperse complex with recombinant CUL5/RBX2, as shown by size-exclusion chromatography and SDS–PAGE analysis (Fig. 2a, b). Pull-down experiments performed with purified, His-tagged APOBEC3 enzymes immobilized on cobalt-chelating resin showed that the four protein complex containing Vif, CBF-β and ELOBC binds A3G, but not the related Vif-resistant deaminase, A3A (Fig. 2c). These observations suggested that Vif, CBF-β and ELOBC form a substrate adaptor for CUL5/RBX2 that enables specific interaction with susceptible A3 proteins.
Figure 2. CBF-β is a stoichiometric component of the Vif E3 ubiquitin ligase.
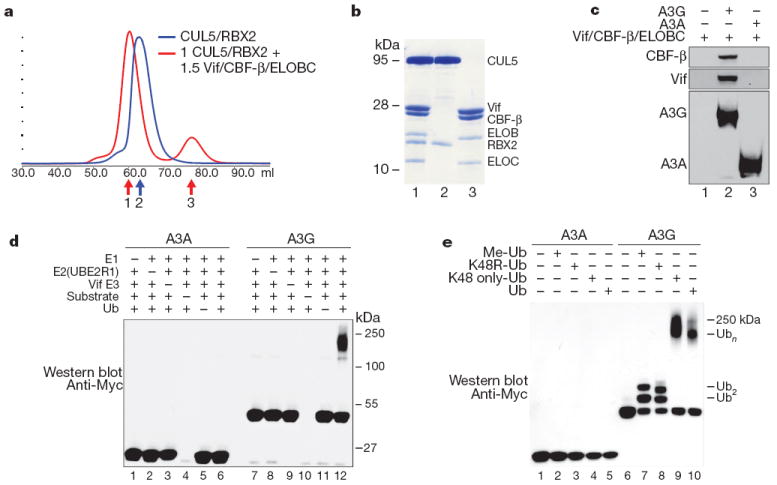
a, Size exclusion chromatography of recombinant purifiedCUL5/RBX2 (blue) overlaid with CUL5/RBX2 mixed with 1.5 equivalents of purified Vif substrate adaptor containing Vif, ELOBC and CBF-β (red). b, Coomassie-stained SDS–PAGE of fractions labelled 1–3 in a indicating the Vif substrate adaptor and a six-protein assembly (CRL5–Vif–CBF-β) co-purify as stable monodisperse species. c, A3G, but not A3A, directly binds the tetrameric Vif substrate adaptor in pull-down experiments in vitro. d, CRL5–Vif–CBF-β is an E3 ligase that promotes polyubiquitination of A3G, but not A3A (detected using an anti-c-Myc antibody to the C-terminal tag on the deaminases). Ub, ubiquitin. e, CRL5–Vif–CBF-β and UBE2R1 catalyse formation of K48-linked chains on A3G. Immunoblots showing substrate in ubiquitination reactions containing UBE2R1 as E2, no ubiquitin, Me-ubiquitin, K48R-ubiquitin, K48-only ubiquitin or wild-type ubiquitin. Reactions with Me-ubiquitin indicate at least two distinct sites are modified on A3G; K48R recapitulates the pattern observed with Me-ubiquitin, whereas both wild type and K48R-only ubiquitin result in extensive polyubiquitin chains.
To test the activity of the reconstituted six protein complex, CUL5/RBX2/ELOB/ELOC/Vif/CBF-β (CRL5–Vif–CBF-β), we assayed substrate and Vif ubiquitination activities using two distinct and well characterized ubiquitin conjugating enzymes, UBE2R1 (hCDC34a) and UBCH5b, which are capable of forming specific K48 and heterogenous ubiquitin chain linkages, respectively19,20. With UBE2R1, CRL5–Vif–CBF-β catalysed formation of high-molecular mass K48 chains on A3G, but not A3A (Fig. 2d, e), mirroring the chain linkage and substrate specificity observed in cells4,6,21-23. As with most ubiquitin ligase assemblies, the CRL5–Vif–CBF-β complex also possessed autoubiquitination activity that was only marginally affected by substrate A3s (Supplementary Fig. 2). These experiments were done with NEDD8-modified CUL5, because NEDD8ylation is required for CUL5 to degrade A3G in vivo4 (Supplementary Fig. 3). Similarly, with UBCH5b, CRL5–Vif–CBF-β was able to promote the specific polyubiquitination of A3G and elicit Vif autoubiquitination activity (Supplementary Fig. 4). We conclude that the reconstituted Vif E3 ligase is specific for A3G, supports K48 chain formation and can function with at least two ubiquitin conjugating enzymes in vitro. It is conceivable that these two ubiquitin-conjugating enzymes work together in cells to promote multi-monoubiquitination of A3G followed by specific chain elongation, as described for other RING E3s24,25, but additional work will be necessary to rule out other E2s in vivo.
To determine if CBF-β is required for Vif folding and/or stability in living cells, we transfected a constant amount of Vif into HEK293T cells expressing either a scrambled short hairpin (sh)RNA or a CBF-β-specific shRNA. The levels of steady-state Vif were threefold lower in CBF-β-depleted cells than in the scrambled control cells (Fig. 3a). Proteasome inhibitor MG132 reversed this effect, suggesting that Vif degradation is accelerated without CBF-β. Analogous data were obtained when Vif was expressed from a proviral plasmid in CBF-β-depleted cells and complemented with a CBF-β expression plasmid (Supplementary Fig. 5).
Figure 3. CBF-β and Vif collaborate to degrade APOBEC3G and enable HIV-1 infectivity.
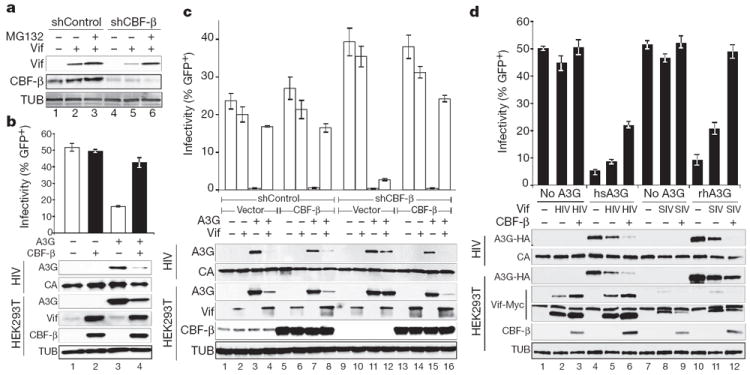
a, CBF-β-depleted HEK293T cells have lower steady-state Vif levels, which recover upon treatment with 2.5 μM MG132. b, Infectivity of replication-competent, Vif-proficient HIV-1 in the presence and absence of CBF-β and A3G (n = 3; mean, s.d.). Immunoblots are shown for the indicated proteins in virus-producing cells and viral particles. c, Infectivity of HIV-GFP produced using HEK293T-shCBF-β or HEK293T-shControl clones transfected with the single-cycle virus cocktail, A3G, Vif and CBF-β as indicated (n = 3; mean, s.d.). The corresponding immunoblots are shown below. d, Infectivity of a Vif-deficient HIV-1 molecular clone produced in the presence or absence of human or rhesus A3G-HA, HIV or SIV Vif–Myc, and CBF-β as indicated (n = 3; mean, s.d.). Immunoblots are shown for the indicated proteins in virus-producing cells and viral particles with two exposures of the anti-Myc (Vif) blot shown to clarify the SIV Vif signal (the longer exposure also shows endogenous c-Myc).
Based on these observations, we predicted that CBF-β knockdown should result in less functional Vif and less infectious HIV-1 particles when produced in the presence of A3G. To test this prediction, shRNA was used to deplete CBF-β stably in HEK293T cells, and a knockdown clone was used to produce replication competent Vif-proficient HIV-1 in the presence or absence of A3G and CBF-β expressed from plasmids (Fig. 3b). In CBF-β depleted cells, steady-state Vif levels were very low despite equivalent levels of virus production as indicated by capsid. Moreover, Vif levels increased when CBF-β was replenished by complementation, and this correlated with decreases in cellular and viral A3G levels and corresponding increases in viral infectivity. In the absence of A3G, no difference in infectivity was observed regardless of cellular CBF-β or Vif levels. Titration experiments showed that CBF-β complementation is dose-responsive (Supplementary Fig. 6). Analogous results were obtained with a multi-vector HIV/green fluorescent protein (GFP) system (Fig. 3c and Supplementary Fig. 7). The Vif/CBF-β interaction was confirmed in virus-producing cells by co-immunoprecipitation experiments (Supplementary Fig. 8). Furthermore, SIVmac239 Vif requires CBF-β to degrade rhesus macaque A3G and promote viral infectivity (Fig. 3d). Interestingly, in contrast to HIV Vif, lower steady-state levels of SIV Vif were observed in the presence of CBF-β, which may be functionally significant or may be a consequence of the heterologous assay system (that is, expressing SIV/rhesus proteins in human cells). Nevertheless, these results demonstrate the essential and conserved nature of CBF-β for Vif function in promoting A3G degradation and efficient virus replication.
Our proteomic, biochemical and genetic studies combine to suggest a model in which HIV-1 Vif hijacks the cellular transcription factor CBF-β to facilitate Vif folding and/or stability as well as nucleation of the rest of the E3 ubiquitin ligase complex (Fig. 4). CBF-β is required for A3G substrate binding and, ultimately, for polyubiquitination and degradation, thereby enabling the production of infectious viral particles. Because genetic studies have shown that Vif is also capable of degrading APOBEC3F and several other human APOBEC3 proteins2,3,23 most of which are expressed in primary CD41 T lymphocytes26,27, it is quite likely that CBF-β is required for counteracting multiple endogenous APOBEC3s and thus for rendering T lymphocytes permissive for HIV-1 replication. We anticipate that the development of antiviral therapies that antagonize the CBF-β–Vif interaction will be more powerful than those that specifically target the A3G–Vif interaction, because they have the potential to unleash the simultaneous restriction potential of multiple APOBEC3s analogous to current combinatorial therapies.
Figure 4. Model for Vif–CBF-β E3 ligase formation and APOBEC3G polyubiquitination and degradation.
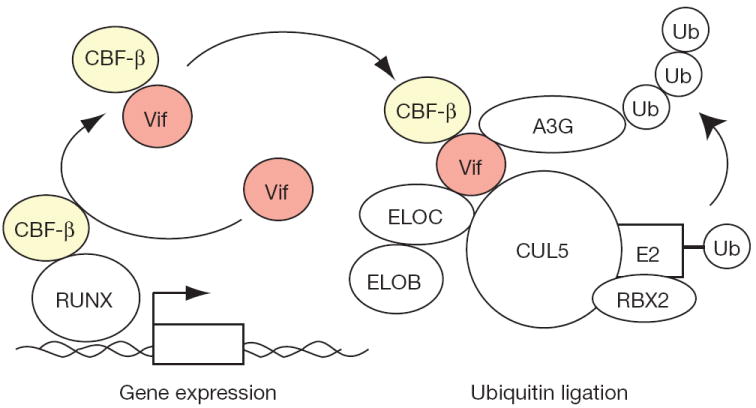
Vif is depicted hijacking cellular CBF-β to the E3 ubiquitin ligase complex required for A3G polyubiquitination and degradation. Vif may recruit newly translated CBF-β (not shown) and/or hijack existing CBF-β from RUNX transcription complexes.
METHODS SUMMARY
Affinity tagging, purification14 and ubiquitination assays28 were performed as described. Recombinant A3A and A3G were purified as Myc-His tagged proteins from HEK293 cells. HIV-1 infectivity studies used an HIV-1IIIB proviral DNA construct (with or without Vif) or an HIV-GFP reporter plasmid set. Control (RHS4346) and CBF-β (RHS4430-99161432) shRNA constructs were obtained from Open Biosystems. A CBF-β complementary DNA matching NM_001755.2 was cloned from the CEM T cell line by RT–PCR. Immunoblots used antibodies to A3G (National Institutes of Health (NIH) ARRRP 10201 courtesy of J. Lingappa), CBF-β (Santa Cruz Biotechnology), HA (HA.11; Covance), TUB (tubulin; Covance), c-Myc (Sigma), Vif (NIH ARRRP 2221 courtesy of D. Gabuzda) and p24/capsid (NIH ARRRP 3537 courtesy of B. Chesebro and K. Wehrly). Details are provided in the Supplementary Methods.
Supplementary Material
Acknowledgments
We thank members of the Krogan, Gross and Harris laboratories for comments, J. R. Johnson for mass spectrometry, B. Leonard for sharing unpublished data, M. Shales for help with figures, and B. Chesebro, D.Gabuzda, J.Lingappa, M.Malim, K. Wehrly, X. F. Yu, A. Bullock, B. Schulman and the AIDS Research and Reference Reagent Program for reagents. This research was funded by grants from QB3 at University of California, San Francisco, and the National Institutes of Health (P50 GM082250, P01 AI090935 and P50 GM081879 to N.J.K.; U54 RR022220 to A.S.; R01 AI064046 and P01 GM091743 to R.S.H.; P50 GM082250 to J.D.G. and C.S.C.; P41RR001614 and P50GM081879 to A.B.). N.J.K. is a Searle Scholar and a Keck Young Investigator.
Footnotes
Author Contributions K.S., R.S.L. and E.K. made equal secondary contributions to this work. S.J. and K.F.S. generated the Vif protein–protein interaction map (Fig. 1); S.J. performed the co-immunoprecipitation confirmation assays (Supplementary Fig. 1); P.C. developed and implemented the MiST scoring system (Fig. 1c); S.J. and E.K. performed double purification analyses (Fig. 1c, d); C.M. and J.K. performed immunoprecipitation analyses (Fig. 1f, g); D.Y.K., L.Y. and D.S. reconstituted the Vif E3 ligase from recombinant components and performed ubiquitination assays (Fig. 2 and Supplementary Figs 2–4); M.L. expressed and purified A3 proteins and did in vitro pulldowns (Fig. 2); K.S., J.F.H. and R.S.L. performed CBF-β knockdown, complementation and virus infectivity experiments (Fig. 3 and Supplementary Figs 5–7); and B.D.A. and J.F.H. did the CBF-β–Vif co-immunoprecipitation experiments (Supplementary Fig. 8). A.B., A.S., C.S.C., R.S.H., J.D.G. and N.J.K. supervised the research; S.J., D.Y.K., J.F.H., K.S., R.S.H., J.D.G. and N.J.K. wrote and revised the manuscript.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The authors declare no competing financial interests.
References
- 1.Goff SP. Retrovirus restriction factors. Mol Cell. 2004;16:849–859. doi: 10.1016/j.molcel.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Malim MH, Emerman M. HIV-1 accessory proteins—ensuring viral survival in a hostile environment. Cell Host Microbe. 2008;3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Albin JS, Harris RS. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev Mol Med. 2010;12:e4. doi: 10.1017/S1462399409001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu X, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 5.Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004;18:2861–2866. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mehle A, et al. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- 7.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 8.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nature Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 9.Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 10.Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 11.Passmore LA, Barford D. Getting into position: the catalytic mechanisms of protein ubiquitylation. Biochem J. 2004;379:513–525. doi: 10.1042/BJ20040198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanley BJ, et al. Structural insight into the human immunodeficiency virus Vif SOCS box and its role in human E3 ubiquitin ligase assembly. J Virol. 2008;82:8656–8663. doi: 10.1128/JVI.00767-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jäger S, et al. Purification and characterization of HIV-human protein complexes. Methods. 2011;53:13–19. doi: 10.1016/j.ymeth.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jäger S, et al. Global landscape of HIV–human protein complexes. Nature. doi: 10.1038/nature10719. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamura T, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004;18:3055–3065. doi: 10.1101/gad.1252404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang DT, et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell. 2009;33:483–495. doi: 10.1016/j.molcel.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong WF, Kohu K, Chiba T, Sato T, Satake M. Interplay of transcription factors in T-cell differentiation and function: the role of Runx. Immunology. 2011;132:157–164. doi: 10.1111/j.1365-2567.2010.03381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petroski MD, Deshaies RJ. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. 2005;123:1107–1120. doi: 10.1016/j.cell.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 20.Kim HT, et al. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem. 2007;282:17375–17386. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- 21.DeHart JL, Bosque A, Harris RS, Planelles V. Human immunodeficiency virus type 1 Vif induces cell cycle delay via recruitment of the same E3 ubiquitin ligase complex that targets APOBEC3 proteins for degradation. J Virol. 2008;82:9265–9272. doi: 10.1128/JVI.00377-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nature Struct Mol Biol. 2010;17:222–229. doi: 10.1038/nsmb.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hultquist JF, et al. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol. 2011;85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodrigo-Brenni MC, Morgan DO. Sequential E2s drive polyubiquitin chain assembly on APC targets. Cell. 2007;130:127–139. doi: 10.1016/j.cell.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 25.Wu K, Kovacev J, Pan ZQ. Priming and extending: a UbcH5/Cdc34 E2 handoff mechanism for polyubiquitination on a SCF substrate. Mol Cell. 2010;37:784–796. doi: 10.1016/j.molcel.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koning FA, et al. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. 2009;83:9474–9485. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Refsland EW, et al. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38:4274–4284. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32:21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.