

G protein-coupled receptors (GPCRs) are involved in the control of every aspect of our behavior and physiology. This is the largest class of receptors, with several hundred GPCRs identified thus far. Examples are receptors for hormones such as calcitonin and luteinizing hormone or neurotransmitters such as serotonin and dopamine. G protein-coupled receptors can be involved in pathological processes as well and are linked to numerous diseases, including cardiovascular and mental disorders, retinal degeneration, cancer, and AIDS. More than half of all drugs target GPCRs and either activate or inactivate them. Binding of specific ligands, such as hormones, neurotransmitters, chemokines, lipids, and glycoproteins, activates GPCRs by inducing or stabilizing a new conformation in the receptor (1, 2). Activated receptors (R*) can then activate heterotrimeric G proteins (composed of α.GDP, β, and γ subunits) on the inner surface of the cell membrane (3–5).
GPCRs have a common body plan with seven transmembrane helices. The intracellular loops that connect these helices form the G protein-binding domain reviewed by refs. 5–7. How do GPCRs activate G proteins and cause such specific responses in cells? What are the triggering changes in GPCRs on agonist binding? How do they fold, and what causes misfolding in so many genetic diseases? All of these unanswered questions in the field depend on detailed structural information. Recently, the first high-resolution structure of a GPCR, rhodopsin, the visual light receptor, was solved by the groups of Palczewski, Okada, Stenkamp, and Miyano (8). This structure reveals a wealth of information about how retinal is bound and how the rhodopsin ground state is stabilized. It also shows that critical residues for G protein activation (E134, R135) are buried and inaccessible to the rod photoreceptor G protein, transducin (Gt). However, this does not resolve the question of how an activated receptor activates a G protein, because the structure of the inactive receptor was solved, leaving open the question of the activation mechanism and the structure of the active receptor. Thus, new structural approaches are needed to address these questions. Four papers from Khorana's group provide several new approaches to these questions and important new information about the active conformation of rhodopsin and how it contacts the G protein (13–15, 35).
Rhodopsin signal transduction in rods and cones underlies our ability to see both in dim light (rod vision) and in color (cone vision). Different rhodopsins absorb light maximally at different light wavelengths, and on activation they activate rod or cone transducins. Transducins activate rod and cone cGMP phosphodiesterases, causing rapid light-activated cGMP breakdown, resultant closure of cGMP-sensitive channels, and photoreceptor cell hyperpolarization and inhibition of photoreceptor neurotransmitter release. The study of visual signal transduction has provided many firsts. The major breakthroughs in receptor structure, including the primary sequence (9, 10), the tertiary structure (8), and the conformational changes required for activation (11), all have first come from studies of rhodopsin and then have been verified to varying degrees in other G protein-coupled receptors. The visual G protein transducin was, in addition, the first heterotrimeric G protein whose structure was solved (12).
Khorana's group, in collaboration with Wayne Hubbell's laboratory, has pioneered the use of site-directed Cys mutagenesis to place reporters (13) and crosslinkers (14, 15) in specific sites for more detailed structural understanding of rhodopsin. Loewen et al. (13) use 19F-trifluoroethylthio groups to derivatize several sets of cysteines at particular sites in rhodopsin's first, second, and third cytoplasmic loops and α helix 8 (previously designated intracellular loop 4 before the crystal structure showed its helical nature). They then use 19F nuclear Overhauser effects between the fluorine labels to analyze distances between them. All of the 19F labels on single cysteines have distinct chemical shifts, but when pairs of cysteines are labeled, there are upfield or downfield shifts, suggesting proximity between residues 139 and 251 on rhodopsin's second and third loops and between residues 65 and 316 on the first loop and α helix 8. These measures of distances provide information on the structure of rhodopsin's cytoplasmic face in the native environment, which can complement crystallographic findings. In addition, they demonstrate a new method for examining tertiary contacts in proteins by using solution NMR, which is applicable to membrane proteins. This method will be particularly powerful in examining light-induced conformational changes in the cytoplasmic surface, which almost certainly underlie G protein activation. Extensive mutagenesis and biochemical experiments in rhodopsin as well as a variety of other G protein-coupled receptors suggest that receptor activation by ligand binding causes changes in the relative orientations of transmembrane helices 3 and 6 (11, 16–18, 31). These changes are then thought to affect the conformation of G protein-interacting intracellular loops of the receptor and thus uncover previously masked G protein-binding sites on the second, third, and fourth cytoplasmic loops (19–21). Fig. 1 shows a view of the receptor–G protein complex.
Figure 1.
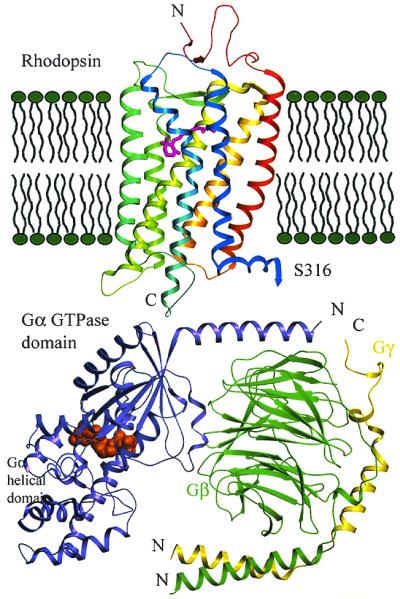
Orientation of rhodopsin, transducin, and the membrane. Refined rhodopsin structure is from ref. 36, and Gt is from ref. 23. Models are based on the crystal structures and are to scale. The carboxyl-terminal residues after S316 are not shown. The orientation of Gt with respect to rhodopsin and the membrane is based on the charge and hydrophobicity of the surface, the known rhodopsin-binding sites on Gt, and the sites of lipidation of Gα and Gβγ (23).
The activated receptor causes heterotrimeric G protein activation (Fig. 1) by causing GDP release from its binding site on the Gα subunit. GTP binds to Gα, and Gα-GTP has reduced affinity for Gβγ and receptor; both Gα-GTP and Gβγ are then free to activate downstream effectors. G protein activation leads to activation of various second messenger systems and intracellular responses, leading to physiological responses of tissues and organisms. In the inactive heterotrimeric state, GDP is bound to the Gα subunit. The three-dimensional structures of the heterotrimeric G proteins Gt and Gi (22, 23) show the overall shape of the GDP-bound heterotrimer and the residues on the surface that can interact with other proteins. The amino terminal region of the α subunit and the carboxyl-terminal region of the γ subunit, which are both sites of lipid modification, are relatively close together, suggesting a site of membrane attachment (Fig. 1).
How does light-activated rhodopsin interact with transducin? Cai et al. (15) and Itoh et al. (14) used particular cysteines on rhodopsin's cytoplasmic face to crosslink transducin by using two different heterobifunctional crosslinking reagents, one photoactivatable and one chemically preactivated. They could crosslink transducin from a variety of different sites on the second and third intracellular loops with each of these reagents, consistent with current expectations that these loops are involved in G protein binding. In the case where the crosslinkers derivatized Cys-240 on the third intracellular loop, the sites on transducin that had been crosslinked were analyzed by using mass spectroscopy to identify the insertion site. Interestingly, the two crosslinkers yielded different insertion points on transducin. The photoactivatable reagent inserted in two sites at the carboxy terminus of the α subunit, at residues 342–345 at the extreme C terminus (350 is the last residue in Gαt) and within the α4-β6 loop at residues 310–313. Both of these sites had been know receptor-binding sites (refs. 24–27; reviewed in refs. 3 and 4), and the findings are important independent evidence of these contacts by using a different methodology. The chemically preactivated crosslinker, which typically inserts into the uncharged ɛ-amino groups of lysines, derivatized residues 19–28 at the amino terminal helix of αt. These data suggest that amino and carboxyl-terminal receptor-binding regions on Gα are very close together in the receptor-bound Gt. In the ground state of Gt (1GOT), the closest approach is between the side chains of Leu-32, Val-30, and Ile-339 (<4 Å). This pocket of hydrophobic interactions couples the amino and carboxyl-terminal regions of Gα. Another point of great interest is that with crosslinkers on Cys-240 of the third intracellular loop, not much crosslinking was seen to Gβγ. Again, there is good evidence in the literature that receptors do directly contact the Gβγ subunit (28–30), and recent studies implicate a different part of the receptor's cytoplasmic surface: α helix 8, previously called intracellular loop 4 (20, 21).
As seen in Fig. 2, the receptor-facing surface of Gt is large with respect to the cytoplasmic surface of rhodopsin. A real puzzle in thinking about receptor–G protein contact is that in the ground states of both rhodopsin and Gt (Fig. 2), the known points of contact are not all achievable, and therefore it is likely that large conformational changes occur in both proteins to produce the active complex. For example, the carboxyl-terminal region is shown to contact the third intracellular loop (14, 15), in agreement with Kostenis and Wess (31), but the carboxy terminus of Gα also contacts α helix 8 of rhodopsin (20, 21), which is close by (Fig. 2). However, the carboxyl terminus of Gγ also interacts with α helix 8 of rhodopsin (20, 21). Asn-343 on α and Glu-66 on γ are removed from each other by 42 Å in the crystal structure of heterotrimeric Gt! An alternative possibility is that the functional unit activating a G protein is a receptor dimer. There is convincing evidence that a number of GPCRs do homo- and heterodimerize (32). It is clear, therefore, that there is a compelling need for detailed structural studies of the sort pioneered by Khorana's group (13), as well as the studies using site-directed spin labeling done in collaboration with Hubbell's group (11, 16) to understand the nature of the activated receptor–G protein complex. Similar site-directed spin-labeling work on the conformation of Gt when it is in the complex is also needed. Of course, the crystal structure of the complex is eagerly awaited!
Figure 2.

grasp (http://trantor.bioc.columbia.edu/grasp) views of the interacting surfaces between rhodopsin's cytoplasmic face and Gt's rhodopsin-interacting surface. Imaginary folding on the dotted line will dock the two molecules in the “best guess” complementary surface. The cytoplasmic face of rhodopsin is relatively small and has a distinct orientation, because rhodopsin is a transmembrane protein. Coordinates from the refined rhodopsin structure [Protein Data Bank (PDB) no. 1HZX (37)] and Gt [PDB entry 1GOT (23)] were used in grasp to examine complementary surfaces of the two molecules. The extreme carboxyl-terminal residues of rhodopsin after S316 are not involved in G protein binding and occlude the intracellular loops. These residues were removed from the grasp view for clarity. This is the ground state of rhodopsin, and critical activating residues such as the ERY sequence are buried in the structure. The loops making up the cytoplasmic face are somewhat disordered in the crystal structure. Still, there is an overall complementarity of shape between the sites of interaction that might already be used to guide mutagenesis on Gα. Notice the overall charge complementarity and the more explicit charge complementarity between residues K341, K248, K141, and R147 on rhodopsin and D311 and E212 on Gα at the bottom of both molecules. Also notice that the deep pockets made up of the interhelical space in rhodopsin (where the flexible carboxyl-terminal residues from Gα could bind?) and the βγ cleft (where CIII residues could fit?) may be complementary.
Rhodopsin also serves as a model receptor for folding diseases of GPCRs (33), because a large number of mutations throughout rhodopsin's primary sequence have been found to cause retinitis pigmentosa, a progressive retinal degenerative disease (34) resulting from misfolding and improper targeting of rhodopsin. In their fourth paper in the series, Hwa et al. (35) again used mass spectroscopic analysis to identify the cysteines involved in disulfide formation. Conserved cysteines at the extracellular border of the molecule in the large family of GPCRs take part in disulfide formation and participate in the folding process. In wild-type rhodopsin, the disulfide is between Cys-110 and Cys-187. Several rhodopsin mutants that are known to fold improperly were revealed to form a disulfide bond between Cys-185 and Cys-187. Formation of the improper disulfide would disallow folding into the proper conformation, providing a molecular explanation for the misfolding of rhodopsin in mutant products of disease alleles. Such a mechanism may be relevant in other diseases caused by misfolding and improper surface delivery of regulatory molecules (36).
These papers show how useful site-directed cysteine mutagenesis of rhodopsin is proving for the investigation of the structure of normal rhodopsin and the molecular basis of folding abnormalities in disease-causing mutations of rhodopsin. The understanding of how receptors fold and how they interact with and activate G proteins and other regulatory proteins has enormous implications for physiology, pathology, and drug design.
Footnotes
References
- 1.Scheer A, Cotecchia S. J Recept Signal Transduction Res. 1997;17:57–73. doi: 10.3109/10799899709036594. [DOI] [PubMed] [Google Scholar]
- 2.Gether U. Endocr Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 3.Bourne H R. Curr Opin Cell Biol. 1997;9:134–142. doi: 10.1016/s0955-0674(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 4.Hamm H E. J Biol Chem. 1998;273:669–672. doi: 10.1074/jbc.273.2.669. [DOI] [PubMed] [Google Scholar]
- 5.Wess J. FASEB J. 1997;11:346–354. [PubMed] [Google Scholar]
- 6.Schoneberg T, Schultz G, Gudermann T. Mol Cell Endocrinol. 1999;151:181–193. doi: 10.1016/s0303-7207(99)00017-9. [DOI] [PubMed] [Google Scholar]
- 7.Strange P. Biochem Pharmacol. 1999;7:1081–1088. doi: 10.1016/s0006-2952(99)00144-6. [DOI] [PubMed] [Google Scholar]
- 8.Palczewski K, Kumasaka T, Hori T, Behnke C A, Motoshima H. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 9.Ovchinnikov Y A, Abdulaev N G, Feigina M Y, Artamonov I D, Zolotarev A S. Bioorg Khim. 1982;8:1011–1014. [Google Scholar]
- 10.Hargrave P A, Mcdowell J H, Curtis D R, Wang J K, Juszczak E. Biophys Struct. 1983;9:235–244. doi: 10.1007/BF00535659. [DOI] [PubMed] [Google Scholar]
- 11.Farrens D L, Altenbach C, Yang K, Hubbell W L, Khorana H G. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 12.Noel J P, Hamm H E, Sigler P B. Nature (London) 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 13.Loewen M C, Klein-Seetharaman J, Getmanova E V, Reeves P J, Schwalbe H, Khorana H G. Proc Natl Acad Sci USA. 2001;98:4888–4892. doi: 10.1073/pnas.051633098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itoh Y, Cai K, Khorana H G. Proc Natl Acad Sci USA. 2001;98:4883–4887. doi: 10.1073/pnas.051632998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai K, Itoh Y, Khorana H G. Proc Natl Acad Sci USA. 2001;98:4877–4882. doi: 10.1073/pnas.051632898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altenbach C, Yang K, Farrens D L, Farahbakhsh Z T, Khorana H G. Biochemistry. 1996;35:12470–12478. doi: 10.1021/bi960849l. [DOI] [PubMed] [Google Scholar]
- 17.Chen S, Lin F, Xu M, Hwa J, Graham R M. EMBO J. 2000;19:4265–4271. doi: 10.1093/emboj/19.16.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jordan B A, Devi L A. Nature (London) 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franke R R, Konig B, Sakmar T P, Khorana H G, Hofmann K P. Science. 1990;250:123–125. doi: 10.1126/science.2218504. [DOI] [PubMed] [Google Scholar]
- 20.Ernst O, Meyer C, Marin E, Henklein P, Fu W. J Biol Chem. 2000;275:1937–1943. doi: 10.1074/jbc.275.3.1937. [DOI] [PubMed] [Google Scholar]
- 21.Marin E, Krishna A, Zvyaga T, Isele J, Siebert F. J Biol Chem. 2000;275:1930–1936. doi: 10.1074/jbc.275.3.1930. [DOI] [PubMed] [Google Scholar]
- 22.Wall M A, Coleman D E, Lee E, Iniguez-Lluhi J A, Posner B A. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 23.Lambright D G, Sondek J, Bohm A, Skiba N P, Hamm H E. Nature (London) 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 24.Kostenis E, Conklin B R, Wess J. Biochemistry. 1997;36:1487–1495. doi: 10.1021/bi962554d. [DOI] [PubMed] [Google Scholar]
- 25.Onrust R, Herzmark P, Chi P, Garcia P D, Lichtarge O. Science. 1997;275:381–384. doi: 10.1126/science.275.5298.381. [DOI] [PubMed] [Google Scholar]
- 26.Bae H, Anderson K, Flood L A, Skiba N P, Hamm H E, Graber S G. J Biol Chem. 1997;272:32071–32077. doi: 10.1074/jbc.272.51.32071. [DOI] [PubMed] [Google Scholar]
- 27.Bae H, Cabrera-Vera T M, Depree K M, Graber S G, Hamm H E. J Biol Chem. 1999;274:14963–14971. doi: 10.1074/jbc.274.21.14963. [DOI] [PubMed] [Google Scholar]
- 28.Kisselev O, Ermolaeva M V, Gautam N. J Biol Chem. 1994;269:21399–213402. [PubMed] [Google Scholar]
- 29.Taylor J M, Jacob-Mosier G G, Lawton R G, Vandort M, Neubig R R. J Biol Chem. 1996;271:3336–3339. doi: 10.1074/jbc.271.7.3336. [DOI] [PubMed] [Google Scholar]
- 30.Yasuda H, Lindorfer M A, Woodfork K A, Fletcher J E, Garrison J C. J Biol Chem. 1996;271:18588–18495. doi: 10.1074/jbc.271.31.18588. [DOI] [PubMed] [Google Scholar]
- 31.Kostenis E, C, Wess J. Biochem J. 1997;36:1487–1495. doi: 10.1021/bi962554d. [DOI] [PubMed] [Google Scholar]
- 32.Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M. Nature (London) 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 33.Spiegel A M. Annu Rev Physiol. 1996;58:143–170. doi: 10.1146/annurev.ph.58.030196.001043. [DOI] [PubMed] [Google Scholar]
- 34.Berson E L. Proc Natl Acad Sci USA. 1996;93:4526–4528. doi: 10.1073/pnas.93.10.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwa J,, Klein-Seetharaman J, Khorana H G. Proc Natl Acad Sci USA. 2001;98:4872–4876. doi: 10.1073/pnas.061632798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuznetsov G, Nigam S K. N Engl J Med. 1998;339:1688–1695. doi: 10.1056/NEJM199812033392307. [DOI] [PubMed] [Google Scholar]
- 37.Teller, D. T., Behnke, C., Palczewsk, K. & Stenkamp, R. (2001) Curr. Top., in press. [DOI] [PMC free article] [PubMed]