

Abstract
Francisella tularensis is a facultative intracellular bacterium that infects many cell types including neutrophils. We demonstrated previously that F. tularensis inhibits NADPH oxidase assembly and activity and then escapes the phagosome to the cytosol, but effects on other aspects of neutrophil function are unknown. Neutrophils are short-lived cells that undergo constitutive apoptosis, and phagocytosis typically accelerates this process. We now demonstrate that F. tularensis significantly inhibited neutrophil apoptosis as indicated by morphological analysis as well as Annexin V and TUNEL staining. Thus, ~80% of infected neutrophils remained viable at 48 h as compared with ~50% of control cells, and ~40% of neutrophils that ingested opsonized zymosan. In keeping with this, processing and activation of procaspases-8, -9 and -3 were markedly diminished and delayed. F. tularensis also significantly impaired apoptosis triggered by Fas crosslinking. Of note, these effects were dose-dependent and could be conferred by either intracellular or extracellular live bacteria, but not by formalin-killed organisms or isolated LPS and capsule, and were not affected by disruption of wbtA2 or FTT1236/FTL0708, genes required for LPS O-antigen and capsule biosynthesis. In summary, we demonstrate for the first time that F. tularensis profoundly impairs constitutive neutrophil apoptosis via effects on the intrinsic and extrinsic pathways, and thereby define a new aspect of innate immune evasion by this organism. As defects in neutrophil turnover prevent resolution of inflammation, our findings also suggest a mechanism that may in part account for the neutrophil accumulation, granuloma formation and severe tissue damage that characterizes lethal pneumonic tularemia.
Keywords: human neutrophil, apoptosis, tularemia, inflammation, bacteria
Introduction
Polymorphonuclear leukocytes (PMNs, neutrophils) represent the largest leukocyte population in human blood and are rapidly mobilized to sites of infection (1). These cells phagocytose microbes and utilize a combination of reactive oxygen species (ROS) and cytotoxic granule components to generate a highly lethal intraphagosomal environment for killing of ingested microorganisms (2). In circulation, human PMNs have a relatively short lifespan (<24 h) and undergo rapid constitutive (spontaneous) apoptosis (1). However, upon recruitment to inflammatory foci PMN lifespan is modulated by cytokines, microbial components, and the local environment (1). In particular, phagocytosis of microbes and subsequent NADPH oxidase-dependent ROS production accelerate the apoptotic program, functioning to target spent neutrophils to tissue macrophages for removal from sites of infection (3–5). Timely apoptosis and clearance of PMNs are critical for resolution of the inflammatory response, minimizing tissue damage by down-regulating the phagocytic and proinflammatory capacity of neutrophils, preventing the release of cytotoxic PMN components into the extracellular compartment, and delivering dying cells to macrophages for disposal (6–8). Some pathogens manipulate PMN function, including apoptosis, to avoid killing and cause disease. For example, Anaplasma phagocytophilum, Chlamydia pneumoniae and Neisseria gonorrhoeae inhibit PMN apoptosis as a mechanism to protect their intracellular replicative niche (9–11). In contrast, Streptococcus pyogenes, Pseudomonas aeruginosa, Burkholderia cenocepacia, and Staphylococcus aureus markedly accelerate PMN apoptosis or redirect cell death towards necrosis to evade intracellular killing and eliminate neutrophils from sites of infection (3, 12–14).
Francisella tularensis is a facultative intracellular, Gram-negative bacterium and the causative agent of the zoonotic disease tularemia (15, 16). The clinical presentation and severity of tularemia depends upon the bacterial strain, dose, and route of infection (17). Human infection most commonly occurs following inoculation into the skin by infected arthropods (including ticks, mosquitoes, and deer flies) or through skin breaks when handling infected animal carcasses (15). However, a distinguishing feature of this organism is its extreme virulence when acquired via the respiratory route, whereby inhalation of as few as 10 CFU can cause severe pneumonic disease, sepsis, and death in humans (17). Consequently, F. tularensis was stockpiled by several countries for use as a biowarfare agent and is currently classified as a Category A Select Agent by the Centers for Disease Control and Prevention (15, 16).
The two subspecies of F. tularensis that account for nearly all cases of human tularemia differ in both geographic distribution and virulence. F. tularensis subspecies tularensis is found almost exclusively in North America and is highly virulent, whereas F. tularensis subspecies holarctica is distributed throughout the Northern Hemisphere and causes milder disease that is rarely fatal (16, 17). The attenuated live vaccine strain (LVS) of F. tularensis subspecies holarctica retains many of the pathogenic mechanisms of virulent F. tularensis strains during in vitro infections of eukaryotic cells and for this reason has been widely studied (15, 16, 18).
The ability of F. tularensis to cause rapid overwhelming disease or death at low inocula suggests that this organism has developed effective mechanisms to disrupt the innate immune response. Indeed, F. tularensis evades killing by macrophages, monocytes, dendritic cells and neutrophils, and resists the lytic effects of serum complement (16, 18–21). Specifically, we and others have shown that killing of virulent F. tularensis strains by human PMNs is inefficient in vitro (19, 22, 23), and that F. tularensis disrupts oxidant production and escapes the phagosome to the cytosol (19, 22). Of note, several studies have demonstrated F. tularensis-PMN interactions are important in the pathogenesis of tularemia. In both simian and murine models, infected neutrophils accumulate in the airways and alveoli, yet cannot eliminate the organism, and bacterial burden increases throughout the course of infection (24–27). Furthermore, blockade of PMN migration into the lung diminishes disease severity and enhances survival of mice infected with F. tularensis strain Schu S4, suggesting that PMN microbicidal mechanisms are not just ineffective, but are dysregulated and harmful to the host (28, 29). As toxic NADPH oxidase-derived ROS are key regulators of PMN apoptosis and this aspect of host defense is impaired by F. tularensis, we predicted that PMN lifespan may also be affected. We utilized a multifaceted approach to test this hypothesis, and our findings demonstrate that this pathogen not only failed to accelerate human neutrophil death, cell lifespan was instead profoundly prolonged via effects on the intrinsic and extrinsic apoptotic pathways.
Materials and Methods
Reagents
Cysteine heart agar was obtained from Becton, Dickinson and Company (Sparks, MD). Defibrinated sheep blood was from Remel (Lenexa, KS). Endotoxin-free Dulbecco’s PBS, HBSS, and HEPES buffer were from Mediatech Incorporated (Herndon, VA). Clinical grade dextran (m.w. 500,000 Da) was purchased from Pharmacosmos A/S (Holbaek, Denmark). Ficoll-Paque Plus was obtained from GE Healthcare Bio-sciences (Uppsala, Sweden). Endotoxin-free HEPES-buffered RPMI-1640 (with and without phenol red) was from Lonza (Walkersville, MD). Staurosporine was from Sigma-Aldrich (St. Louis, MO). PROTOCOL HEMA-3 staining kit was purchased from Fisher Scientific (Kalamazoo, MI). Annexin V-FITC conjugate was obtained from Invitrogen (Camarillo, CA). APO-BRDU™ kit was from BD Biosciences (San Jose, CA). The CytoTox-ONE™ Homogenous Membrane Integrity Assay and the Caspase-Glo® 3/7, -8, and -9 assay kits were purchased from Promega Corporation (Madison, WI). Mouse anti-Fas (human, activating) mAb (clone CH-11) was from Millipore Corporation (Temecula, CA). Rabbit anti-F. tularensis antiserum was from BD Diagnostics (Sparks, MD). Mouse anti-caspase-3 mAb (clone C33) and rabbit anti-active caspase-9 polyclonal Ab were from BioVision Research Products (Mountain View, CA). Mouse anti-caspase-8 mAb (clone IC12) was from Cell Signaling Technologies (Danvers, MA). Mouse anti-actin mAb (clone JLA20) was from Calbiochem (Darmstadt, Germany). Mouse mAb FB11, specific for F. tularensis LPS, was from QED Biosciences (San Diego, CA) and mouse mAb 11B7 to F. tularensis capsule (30) was a kind gift from Michael Apicella (University of Iowa, Iowa City, IA). Rhodamine-conjugated donkey-anti-rabbit F(ab′)2 was from Jackson ImmunoResearch Laboratories (West Grove, PA). Horseradish peroxidase-conjugated goat-anti-mouse IgG (H+L) was from Bio-Rad Laboratories (Hercules, CA). DAPI and Pierce SuperSignal West Pico Enhanced Chemiluminescence substrate were purchased from Thermo Scientific (Rockford, IL).
Neutrophil isolation
Heparinized venous blood was obtained from healthy adult volunteers in accordance with a protocol approved by the Institutional Review Board for Human Subjects at the University of Iowa. PMNs were isolated using dextran sedimentation followed by density gradient separation as described (31). Neutrophils were suspended in HBSS without divalent cations, counted, and diluted to 2×107/ml. Purity of the each preparation was assessed by HEMA-3 staining followed by microscopic analysis, and the suspensions were routinely 95–98% PMNs. In all cases, replicate experiments were performed using PMNs from different donors.
Bacterial strains and growth conditions
Fully virulent, wild-type F. tularensis subsp. tularensis (type A) strain Schu S4 and the attenuated F. tularensis subsp. holarctica live vaccine strain (LVS) (ATCC 29684) have been described (22). An LVS Himar transposon mutant lacking functional wbtA2 was the generous gift of Dara Frank (Medical College of Wisconsin, Milwaukee, WI) and has been described (32). FTL0708 was disrupted in LVS by group II intron retargeting using Sigma Targetron reagents as we described previously for disruption of the homologous gene FTT1236 in Schu S4 (33). All studies of the virulent type A strain were performed in a Biosafety Level 3 (BSL-3) facility with Select Agent approval and in accordance with all Centers for Disease Control and Prevention and National Institutes of Health regulatory and safety guidelines. Wild-type and mutant bacteria were inoculated onto cysteine heart agar supplemented with 9% defibrinated sheep blood (CHAB) and grown for 48 h at 37°C in 5% CO2. Bacteria were harvested from the plates and washed twice with HBSS (containing Ca2+ and Mg2+). Formalin-killed LVS (fkLVS) were prepared by incubating washed bacteria in 10% buffered formalin (Sigma-Aldrich) for 30 min at room temperature. Killed bacteria were subsequently washed two additional times with HBSS (containing Ca2+ and Mg2+) and sterility was confirmed by plating aliquots on CHAB.
Opsonization and infection of neutrophils
Washed bacteria were quantified by measurement of absorbance at 600 nm. Unless otherwise indicated, wild-type F. tularensis (1×1010/ml) and yeast zymosan particles were opsonized in 50% pooled human serum for 30 min at 37°C, followed by two washes with HBSS prior to infection of PMNs. Conversely, the FTL0708 and wbtA2 mutants are exquisitely serum sensitive, and both mutant and wild-type bacteria were left unopsonized for studies of these strains. PMNs (5×106/ml) were diluted in Hepes-buffered RPMI-1640 (without serum) and mixed with zymosan at multiplicity of infection (MOI) 5:1 or with Francisella at MOIs ranging from 5:1–200:1 as indicated. One ml aliquots of each suspension were transferred into 5 ml polypropylene tubes and subsequently incubated at 37°C with 5% CO2 for 0–48 h.
Immunofluorescence microscopy
PMNs in polypropylene tubes were washed twice with cold PBS and then cytocentrifuged onto acid-washed coverslips. Cells were fixed with 10% formalin, permeablized in cold (1:1) methanol-acetone, and blocked in PBS supplemented 0.5 mg/ml NaN3 and 5 mg/ml BSA using our established methods (19, 34). Bacteria were detected using anti-F. tularensis antiserum and secondary antibodies conjugated to rhodamine, and DAPI was used to stain PMN nuclei. For studies of infection efficiency, 300 PMN per coverslip were scored for the presence of 0, 1–5, 6–10, 11–20 or > 20 bacteria per cell. In addition, differential staining was performed as we described previously to distinguish bound and internalized cell-associated LVS (34).
Quantitation of phagocytosis and intracellular growth by measurement of CFU
LVS phagocytosis and growth in PMN were quantified as we described (19) with minor modifications. In brief, PMNs (5×106/ml in RPMI) were infected with LVS at an MOI of 200:1. At the indicated time points, an aliquot of the suspension was removed and PMNs were lysed using 1% saponin. This sample was used for enumeration of total viable bacteria. The rest of the suspension was centrifuged at 300 × g to separate PMNs from extracellular bacteria, and the cell pellet was washed extensively with PBS to remove uningested LVS. After lysis in 1% saponin, cell pellets and supernatants were serially diluted in PBS and plated on CHAB. Viable intracellular and extracellular bacteria were determined by enumeration of CFU after 48–72 h at 37°C. To quantify intracellular growth specifically, PMNs were infected as described above. After 12 h, PMNs were pelleted and washed extensively with PBS. The medium was collected and depleted of bacteria by centrifugation and passage through a 0.2 μm filter, and its sterility was confirmed by plating on CHAB. Washed PMNs were resuspended in the sterile infection medium at 5 ×106 cells/ml and returned to 37°C. At the indicated time points, PMNs were washed with PBS, lysed in 1% saponin, diluted in PBS, and then plated on CHAB for enumeration of CFU.
Cytotoxicity assay
PMNs were left untreated or were mixed with F. tularensis (MOI 200:1) or zymosan (MOI 5:1) as described above. Aliquots (100 μl) containing 2.5×5105 PMNs were transferred into white-walled, clear-bottom 96-well plates and PMN viability was assessed using the CytoTox-ONE™ Homogenous Membrane Integrity Assay (Promega) according to the manufacturer’s instructions. This fluorometric assay measures activity of cytosolic lactate dehydrogenase (LDH) released from cells with damaged plasma membranes. LDH activity was assayed in triplicate wells using a FLUOstar OPTIMA Microplate fluorimeter (BMG LabTech Inc) with subtraction of the background signal obtained at time zero.
Morphological assessment of apoptosis
Aliquots of PMNs were cytocentrifuged onto coverslips at the indicated time points, fixed with 10% formalin for 15 min and then stained using the HEMA 3 kit (Fisher Scientific) according the manufacturers instructions. PMNs were examined by light microscopy and were scored as apoptotic when they exhibited nuclear condensation (4, 35). To quantify the percentage of apoptotic cells in the population, at least 300 cells per coverslip and condition were analyzed in each experiment.
Detection of externalized phosphatidylserine using Annexin V
Phosphatidylserine (PS) externalization was determined by flow cytometric analysis using an Annexin V-FITC conjugate (Invitrogen) according to the manufacturer’s instructions. Propidium iodide (PI) staining was included to distinguish early apoptotic cells from late apoptotic/necrotic PMNs. In brief, PMNs were costained with Annexin V-FITC and PI in buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) prior to analysis using a FACSCalibur flow cytometer (Becton Dickson). Twenty thousand events were collected for each sample and the data were analyzed using CellQuest (Becton Dickson) and FlowJo software (Tree Star, Inc.).
Fas-stimulated apoptosis
PMNs were treated with 500 ng/ml mouse anti-Fas IgM antibody for 6 h prior to analysis of nuclear morphology or staining with Annexin V and PI as described above. Where noted, PMNs were infected with LVS or Schu S4 (at MOI 200:1) for 1 h at 37°C prior to Fas crosslinking.
Assessment of DNA fragmentation
PMNs were stimulated with bacteria (MOI 200:1) or zymosan (MOI 5:1) or were left untreated and DNA fragmentation was determined using the Apo-BRDU™ apoptosis detection kit (BD Biosciences), a modified TUNEL assay. Samples were labeled according to the manufacturers instructions with minor modifications. In brief, PMNs were fixed with 4% paraformaldehyde for 60 min at 4°C, washed and stained for 90 min. Samples were analyzed using a FACSCalibur flow cytometer (Becton Dickson) and twenty thousand events were collected for each sample. Data were analyzed using CellQuest software.
Caspase activity assays
PMNs were left untreated, or treated with 1 μM staurosporine, 500 ng/ml anti-Fas mAb, zymosan (MOI 5:1), or F. tularensis (200:1) as described above. The activities of caspases 3, 8, and 9 were measured using Caspase-Glo 3/7, 8, and 9 Assay Kits (Promega) in accordance with the manufacturer’s instructions. In each case, caspase activity was assessed by quantifying the luminescence generated upon cleavage of a caspase-specific proluminogenic substrate. In brief, 100 μl aliquots of each sample containing 5×104 PMNs were transferred into a white-walled 96-well plate in triplicate. Equal volumes of the Caspase-Glo reagent containing either the tetrapeptide sequence DEVD recognized by caspase 3, LETD recognized by caspase 8, or LEHD recognized by caspase-9 were added to the wells, mixed, and incubated at room temperature for 45 min. Luminescence was detected using NovoStar (BSL-2 experiments) or LumiStar (BSL-3 experiments) luminometers (both from BMG LabTech Inc.). Caspase activity was expressed as mean luminescence intensity per 5×104 PMNs.
Caspase processing
PMNs (5×106) were left untreated or treated with 1 μM staurosporine, 500 ng/ml anti-Fas mAb, zymosan (MOI 5:1), or F. tularensis (MOI 200:1) as described above. At the indicated time points, PMNs were pelleted and then resuspended in ice-cold protease inhibitor cocktail (TBS containing aprotinin, leupeptin, PMSF, sodium orthovanadate, AEBSF, levamisole, bestatin, E-64, and pepstatin A) and incubated on ice for 10 min. PMNs were subsequently lysed using 1% NP-40 and clarified by centrifugation. Protein concentrations were determined using the BCA Protein Assay (Pierce). Twenty micrograms of each sample were separated using NuPAGE 4–12% Bis-Tris gradient gels and then transferred to polyvinylidene fluoride by electroblotting. Western blot analyses for processing of caspases 3, 8, and 9 were performed using mouse anti-caspase-3 mAb (1 μg/ml, BioVision), mouse anti-caspase-8 mAb (1:1000 dilution, Cell Signaling Technologies), and rabbit anti-active caspase-9 polyclonal Ab (1 μg/ml, BioVision). Mouse anti-actin mAb (1:10,000 dilution, Calbiochem) was used as the loading control. Bands were detected using horseradish peroxidase-conjugated secondary antibodies and Pierce Supersignal West Pico Enhanced chemiluminescence reagents.
CXCL8 ELISA
Where noted, tissue culture media from control or infected PMNs were collected, sterile filtered (0.2 μm), and assayed for the presence of CXCL8 by ELISA according to the manufacturer’s directions (R&D Systems, Minneapolis, MN).
Isolation of F. tularensis LPS and capsule
LVS was grown on CHAB for 48 h at 37°C in 5% CO2. LPS and capsule were isolated using a modification of a recently described method, but omitting the final steps in which LPS and capsule are separated from one another (30). Specifically, bacteria were scraped into PBS, pelleted, and resuspended in 2% SDS containing 10 mM EDTA and 60 mM Tris (pH 6.8). This suspension was brought to 50 μg/ml of proteinase K, heated to 65°C for 45 min, and then incubated at 37°C overnight. Samples were washed three times by precipitation in 0.3 M sodium acetate and 3 volumes of ethanol at −20°C. This was followed by nuclease treatment, hot phenol extraction, and then ethanol precipitation. To remove any remaining DNA and ethanol, this final pellet was subjected to two rounds of ultracentrifugation in 14 ml of water at 147,000 × g for 75 min at room temperature. The resulting clear pellet was raised in HPLC-grade water, lyophilized, and weighed. The final product lacked detectable protein as determined by colloidal gold staining and was devoid of DNA as indicated by lack of absorbance at 260 nm (not shown).
Immunoblot analysis of bacterial lysates and isolated LPS/capsule
Bacterial whole cell lysates (from 5×108 cells in 200 μl) were heated to 72°C for 10 min in SDS loading buffer. Fifteen microliters of each bacterial lysate or 5 μl of the LPS/capsule preparation (1.0 mg/ml) were separated on NuPAGE 4–12% Bis-Tris gradient gels and then transferred to polyvinylidene fluoride. Blocked membranes were probed with mAb11B7 (1:10,000) to detect capsule or mAb FB11 (1:10,000) to detect LPS. Bands were visualized using horseradish peroxidase-conjugated secondary antibodies and Pierce Supersignal West Pico Enhanced chemiluminescence reagents.
Statistical Analysis
Data from experiments containing a control group and one experimental group were analyzed using an unpaired two-sided Student t test. Data from studies containing multiple experimental groups were analyzed by one-way ANOVA followed by Tukey’s post-test. All analyses were performed using GraphPad Prism version 4.0 software. P values less than 0.05 were considered statistically significant.
Results
Francisella tularensis LVS prolongs human neutrophil lifespan
Neutrophils are intrinsically short-lived phagocytes that undergo constitutive apoptosis (1, 4, 5). This inherent death program is accelerated markedly by ROS production that typically accompanies phagocytosis (3–5) and does not occur in PMNs from persons with chronic granulomatous disease (CGD) (1, 36). As neither virulent strains of F. tularensis nor LVS trigger NADPH oxidase activation in PMNs (19, 22), we hypothesized that F. tularensis may alter PMN lifespan.
To begin to address this question, purified human neutrophils were incubated in suspension with preopsonized F. tularensis LVS in serum-free RPMI-1640 media. This established approach for studies of PMN apoptosis (37) is preferred as it avoids confounding effects of growth factors and other serum components on cell viability (38–40). Microscopic analysis demonstrated that uptake of LVS under these conditions was inefficient at low doses, and an MOI of 200:1 was used to ensure that the majority of PMNs were infected. Representative confocal images are shown in Figure 1A with quantitation of infection efficiency in Figure 1B. Specifically, we show that ~40% of PMNs were infected with 1–5 LVS each by 3–6 hours post-infection (hpi). Between 12 and 48 hpi we observed a marked increase in both the percentage of infected cells and the bacterial load per cell. At 24 hpi 80% of PMNs were infected, but the number of bacteria per cell was heterogeneous as indicated by the mixture of low, moderate and heavily-infected cells. By 36 hpi at least 50% of PMNs were heavily infected (>20 LVS/cell), and by 48 hpi the vast majority of PMNs contained large numbers of bacteria. Quantitation of total, PMN-associated and extracellular CFU (Fig. 1C) revealed that LVS was viable in serum-free RPMI but did not replicate in this media, whereas the number of PMN-associated bacteria increased 19-fold over the time course examined, and differential staining demonstrated that >97% of PMN-associated LVS were intracellular (not shown). These data indicate that LVS accumulated in PMNs over 48 h, but do not distinguish intracellular growth from continued phagocytosis of extracellular organisms. To quantify intracellular replication directly, we infected PMNs with LVS for 12 h and then removed extracellular bacteria by extensive washing of the cells and sterile filtration of the media. Samples were returned to 37°C, and viable intracellular LVS were quantified by plating PMN lysates for enumeration of CFU at 12, 24, 36 and 48 hpi. By this assay, intracellular LVS replicated eightfold between 12 and 36 hpi, with no further increase at 48 hpi (Fig. 1D).
FIGURE 1.

F. tularensis LVS infects human neutrophils in serum-free media. A, Representative confocal images of PMNs incubated with LVS at a MOI 200:1 for 6, 12, 24, 36 or 48 h at 37°C. Bacteria are shown in red and PMNs were detected by DIC optics and DAPI-staining of nuclear DNA (blue). Arrows indicate infected cells. B, Infection efficiency was quantified by confocal microscopy. Data indicate the percentage of PMNs containing 0, 1–5, 6–10, 11–20, or >20 bacteria/cell at each time point and are the mean ± SEM (n=3). C, Total, extracellular (media), and cell-associated (PMN pellet) LVS were quantified by CFU measurement. Data are the mean ± SEM (n=3). D, Intracellular growth. PMNs were infected with LVS at MOI 200:1 for 12 h, washed extensively to remove extracellular bacteria, and returned to 37°C in sterile media. At the indicated time points, PMNs were lysed with saponin and viable intracellular bacteria were quantified by enumeration of CFU. Data are the mean ± SEM (n=3).
Next, we quantified the viability of control and LVS-infected PMNs using the LDH cytotoxicity assay to detect loss of plasma membrane integrity and cell death. As a positive control neutrophils were stimulated with serum-opsonized zymosan (OpZ), phagocytosis of which accelerates PMN apoptotic death (10). Unstimulated neutrophils undergo constitutive apoptosis following 12–24 h of in vitro incubation (4, 5, 35). Consistent with this, we show that untreated PMNs began losing membrane integrity ~12 h after isolation, and the fraction of dead cells increased progressively over the time course examined (Fig. 2). As expected, death of PMNs that engulfed OpZ was accelerated significantly relative to untreated controls. Conversely, the vast majority of neutrophils incubated with LVS remained viable, and LDH release was significantly diminished at 48 h compared with both control and OpZ-treated cells. These data demonstrate that LVS not only failed to induce PMN death, but also prolonged cell viability.
FIGURE 2.
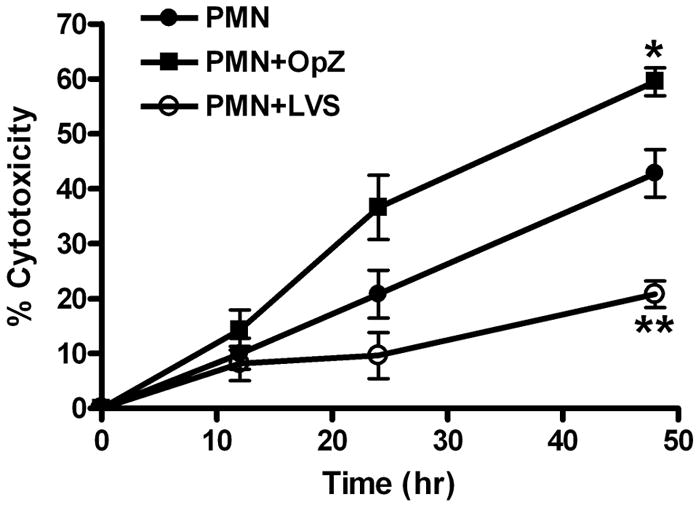
F. tularensis LVS prolongs neutrophil viability. PMNs were left untreated or were mixed with opsonized zymosan (OpZ; MOI 5:1) or LVS (MOI 200:1) at 37°C. Loss of neutrophil plasma membrane integrity was quantified at 0, 12, 24, and 48 h by measuring release of cytosolic lactate dehydrogenase into the culture medium. Data are the mean ± SEM from three or more independent experiments. * P <0.05 and ** P <0.01 vs. PMNs alone.
Infection with Francisella tularensis LVS delays PMN apoptosis
As the LDH release assay does not discriminate between different forms of cell death, and LDH activity can be affected by media composition (41), we utilized several experimental approaches to assess directly the extent to which LVS modulated the kinetics of PMN apoptosis. Neutrophils undergo distinctive morphological changes during apoptosis, including cell shrinkage, cytoplasmic vacuolation, and nuclear condensation that are amenable to single cell analysis and can be quantified by light microscopy (4, 35). Using this approach, we demonstrated that LVS significantly delayed the progression of neutrophils toward an apoptotic morphology (Fig. 3A). By 12 h, the percentage of apoptotic cells in the control and LVS-treated populations diverged, with control PMNs exhibiting condensed nuclei with significantly faster kinetics than the infected cells at later time points (Figure 3B). Thus, 90–100% of control neutrophils appeared apoptotic by 36 h as compared with only ~40% of LVS-treated PMNs. These data confirm and extend the data shown in Figure 2, and strongly suggest that LVS modulates neutrophil apoptosis to extend cell lifespan.
FIGURE 3.

F. tularensis LVS delays the progression of human neutrophils to an apoptotic morphology. Untreated PMNs or cells infected with LVS (MOI 200:1) for the indicated times were fixed and stained using HEMA 3 reagents, and nuclear morphology was analyzed by light microscopy. A, Representative images of untreated and LVS-infected PMNs. Arrows at 24 and 36 h indicate PMNs with condensed, apoptotic nuclei. Arrowheads indicate intracellular LVS that also stain with HEMA 3. B, Pooled data indicate the percentage of cells with apoptotic nuclei determined by microscopic evaluation of >300 cells per time point and are the mean ± SEM (n=4). ** P <0.01 and *** P <0.001.
Morphological changes associated with apoptosis are a late manifestation of this programmed cell death pathway. On the other hand, redistribution of PS from the inner leaflet of the plasma membrane to the cell surface occurs early in apoptosis (42). Using FITC-tagged Annexin V, which binds with high specificity to externalized PS, we used flow cytometry to identify and quantify the number of apoptotic cells in the PMN population. In addition, cells were costained with the membrane-impermeable dye PI. This double-staining approach allows three subgroups of cells to be distinguished: viable, nonapoptotic cells are Annexin V-negative/PI-negative (lower left quadrant); early apoptotic cells with intact plasma membranes are Annexin V-positive/PI-negative (lower right quadrant); and late apoptotic cells with compromised plasma membranes are Annexin V-positive/PI-positive (upper right quadrant). Our data demonstrate that at 12, 24 and 48 h, the control and LVS-infected neutrophil populations diverged significantly. At 24 and 48 hpi, markedly fewer LVS-treated PMNs were Annexin V-positive compared with uninfected controls. Representative FACS plots are shown in Figure 4A, and pooled data from 12 independent experiments are shown in Figure 4B. PI staining of the infected neutrophil population was also reduced at 24 h and 48 hpi (Fig. 4A), confirming that LVS maintained PMN plasma membrane integrity as indicated by the LDH cytotoxicity data shown above. Of note, these effects of LVS were dose- dependent, with a profound decline in PS externalization achieved at an MOI of 5:1, and maximum inhibition achieved at an MOI of ~100:1 (Fig. 4C).
FIGURE 4.

F. tularensis LVS reduces Annexin V staining of neutrophils in a dose-dependent manner. Uninfected cells or PMNs infected with LVS were double-stained with Annexin V-FITC and propidium iodide (PI) and then analyzed by flow cytometry. A, Representative dot plots of control PMNs and cells infected with LVS (MOI 200:1) at 6, 12, 24 and 48 h. The percentage of cells in each quadrant is indicated. B, Pooled flow cytometry data indicate the fraction of Annexin-V-positive PMNs at the indicated time points. Data are the mean ± SEM (n=12). *** P <0.001 for control vs. LVS-infected PMNs. Where not visible, error bars are smaller than symbols. C, Neutrophils were infected with increasing doses of LVS for 24 h and the Annexin V-positive cells were quantified by flow cytometry. Data are the mean ± SEM (n=3).
During the late stages of apoptosis caspase-activated DNases cleave nuclear DNA. The TUNEL assay labels with a fluorescent marker the exposed 3-hydroxy termini of cleaved, fragmented DNA, enabling apoptotic cells to be identified and quantified by flow cytometry (42). Using this approach, we now demonstrate that relative to untreated control PMNs, DNA fragmentation was significantly delayed by LVS at all time points examined between 12–48 hpi (Figs. 5A–5B). Approximately 60% of the untreated PMNs were TUNEL positive at 24 h, whereas only 30% of LVS-infected PMNs exhibited fragmented DNA. As an additional control, we confirmed that OpZ accelerated PMN apoptosis, as indicated by increased TUNEL labeling at 24 h (untreated vs. OpZ-stimulated PMNs: 60% and 77% TUNEL-positive, respectively) (Fig 5A). Collectively, these data indicate that F. tularensis LVS markedly impairs PMN apoptosis as indicated by morphological analysis as well as Annexin V and TUNEL staining.
FIGURE 5.

F. tularensis LVS inhibits neutrophil DNA fragmentation. PMNs were left untreated or incubated with opsonized zymosan (OpZ, MOI 5:1) or LVS (MOI 200:1). Nuclear DNA fragmentation was detected using a modified TUNEL assay and flow cytometry. A, Representative histograms obtained for control, OpZ or LVS treated PMNs after 24 h at 37°C. B, Pooled data indicate the percentage of TUNEL-positive cells and are the mean ± SEM (n≥3). *P <0.05 and ** P <0.01 for control vs. LVS-infected PMNs.
Francisella tularensis LVS inhibits caspase-3 processing and activity in PMNs
Caspase-3 is the primary effector caspase in human neutrophils and is responsible for the proteolytic cleavage of cellular targets that ultimately lead to apoptotic cell death (43, 44). Caspase-3 is synthesized as an inactive proenzyme that is processed into large (17 kDa) and small (12 kDa) subunits which associate to form a mature active enzyme (45). Using Western blotting and densitometric analysis, we followed the time course of procaspase-3 processing in untreated and LVS-infected PMNs, and used OpZ- and staurosporine-treated treated cells as positive controls (Fig 6A). In untreated PMNs undergoing constitutive apoptosis, trace processing of procaspase-3 to its mature form was detected at 6 h. This increased markedly by 12 h, and progressed to near complete depletion of procaspase-3 by 48 h. In PMNs treated with staurosporine, a potent proapoptotic stimulus (46), procaspase-3 processing was rapid, with substantial amounts of the mature enzyme present by 6 h, and near complete depletion of procaspase-3 by 12h. Uptake of OpZ also accelerated procaspase-3 processing relative to untreated PMNs as indicated by increased mature caspase-3 at 6 h, and more rapid depletion of procaspase-3 at later time points, consistent with the ability of this particulate stimulus to accelerate PMN apoptotic death. In stark contrast, procaspase-3 processing was markedly diminished and delayed by LVS relative to untreated control PMNs as well as cells exposed to control stimuli, and did not go to completion by 48 h.
FIGURE 6.

Procaspase-3 processing and caspase-3 activity are impaired by LVS. PMNs were left untreated or were incubated with staurosporine (stauro., 1 μM), opsonized zymosan (OpZ, MOI 5:1), or LVS (MOI 200:1). A, At the indicated time points, procaspase-3 processing was detected by Western blotting of cell lysates. The caspase-3 mAb recognizes both procaspase-3 (37 kDa) and the large subunit of active caspase-3 (17 kDa). Actin was used as a loading control. Data shown are from one experiment representative of four. Lower panel shows the percentage of mature caspase-3 in each sample determined by densitometric analysis of each immunoblot normalized to the actin loading control. #, 48 h time point not determined for staurosporine-treated PMNs. B, Caspase-3 activity was assessed using a caspase-3-specific proluminogenic substrate. Data indicate relative luminescence units (RLU) and are the mean ± SEM of triplicate samples from one representative experiment (n≥3). ** P <0.01 and *** P <0.001 for LVS-infected vs. control PMN.
Of note, processing of procaspases to their mature forms is essential but not sufficient for activity, as enzyme function is modulated further by direct association with cytoplasmic regulatory factors (41). We therefore used a luminescence assay to measure caspase-3 activity (Fig 6B). In agreement with the Western blotting data, caspase-3 activity in untreated neutrophils increased steadily beginning 12 h after isolation. This pattern was conserved using PMNs from different donors, and was accelerated by both staurosporine and OpZ (compare Fig. 6B and Supplemental Fig. 1A). Specifically, staurosporine-triggered caspase-3 activity routinely peaked at 6 or 12 h and then declined, whereas OpZ-stimulated activity peaked at 12 or 18 h. In all cases, caspase-3 activity in PMNs undergoing constitutive apoptosis increased progressively 12–24 h post-isolation, and analyses of later time points suggest that caspase-3 activity in these cells peaked at about 30 h and began to decline by 36 h (Supplemental Figs. 1B and 1C). On the other hand, capsase-3 activity was consistently and profoundly inhibited by LVS at all time points examined from 12 to 30 hpi (the longest time point examined) (Fig. 6B and Supplemental Figs. 1A and 1C). Marked inhibition was also observed using LVS at MOI 50:1 (Supplemental Figs. 1A and 1B). Taken together, our data demonstrate that the ability of LVS to delay PMN apoptosis is due, at least in part, to defects in processing and activation of executioner capsase-3.
Francisella tularensis LVS modulates both the intrinsic and extrinsic apoptotic pathways in PMNs
The initiator caspases-8 and -9 are upstream activators of caspase-3. Caspase-8 is the initiator caspase of the extrinsic apoptotic pathway and is activated upon stimulation of surface death receptors (47). The intrinsic apoptotic pathway requires caspase-9, which is activated following release of cytochrome c from permeabilized mitochondria and assembly of the apoptosome (47). We therefore determined if LVS impaired caspase-3 processing and activity via effects on either initiator caspase.
The extrinsic pathway was initiated using antibodies to crosslink and activate Fas at the PMN surface. Similar to caspase-3, activation of caspase-8 can be followed by Western blotting, as procaspase-8 (55/57 kDa) is cleaved into intermediate p43/p41 forms and subsequently cleaved further to generate a p18 subunit. Consistent with previous reports (48), robust processing of procaspase-8 was observed within 6 h in PMNs treated with anti-Fas IgM (Fig. 7A). As expected, procaspase-8 processing was slower in PMNs undergoing spontaneous apoptosis, with a marked increase in mature caspase-8 18 h after cell isolation. In contrast, only trace amounts of mature caspase-8 were detected in LVS-infected PMNs at 18–24 hpi (Fig. 7A). Consistent with the blotting data, caspsase-8 activity peaked 12–18 h after Fas crosslinking, increased progressively over 24 h in PMNs undergoing spontaneous apoptosis, and was markedly lower in cells infected with LVS (Fig. 7B).
FIGURE 7.

LVS inhibits processing and activation of the initiator caspases -8 and -9. Untreated PMNs or cells treated with staurosporine (1 μM), anti-Fas IgM (500 ng/ml), or LVS (MOI 200:1) were incubated at 37°C. A, Immunoblots of cell lysates obtained at the indicated time points show full length procaspase-8 (57 kDa), cleavage intermediates p43/p41, and mature caspase-8 (p18). Actin immunoblots demonstrate equal loading. Data shown are from one experiment representative of two. B, Caspase-8 activity was assessed using a caspase-8-specific proluminogenic substrate. Data indicate relative luminescence units (RLU) and are the mean ± SEM of triplicate samples from a representative experiment (n≥3). ** P <0.01 and *** P <0.001 for LVS-infected vs. control PMNs. C, Mature caspase-9 was detected in PMN lysates using an antibody specific for the 37 kDa processed enzyme. Actin served as a loading control. Data shown are representative of two independent experiments. D, Caspase-9 activity was measured using a caspase-9-specific proluminogenic substrate. Data indicate relative luminescence unit (RLU) and are the mean ± SEM of triplicate samples from one representative experiment (n≥3). *P <0.05, ** P <0.01 and *** P <0.001 for LVS-treated vs. control PMNs as indicated.
The intrinsic (mitochondrial) apoptotic pathway plays a critical role in the constitutive turnover of human neutrophils and is also activated by exposure to staurosporine (1, 47, 49–51). Using immunoblotting and luminescence assays, we examined the time course of caspase-9 activation in either control, staurosporine-treated, or LVS-infected PMNs (Figs. 7C and 7D). We observed a time-dependent increase in caspase-9 processing and activity in control neutrophils between 12 and 24 h after isolation that was accelerated by staurosporine. Thus, caspase-9 activity in control PMNs at 24 h was similar to cells treated with staurosporine for only 6 h. Infection with LVS had the opposite effect, as accumulation of mature caspase-9 was delayed relative to both control and staurosporine-treated cells. Moreover, caspase-9 activity remained very low in LVS-infected neutrophils and was significantly reduced compared to control cells at all time points examined over 3–24 h. Altogether, these data demonstrate that relative to control PMNs, activation of both caspases-8 and -9 was significantly impaired by LVS, and as such suggest that this organism may act at multiple points in the apoptotic cascade to curtail activation of executioner caspase-3 and extend cell lifespan.
F. tularensis LVS inhibits Fas-mediated PMN apoptosis
The data shown above suggest that F. tularensis can inhibit the extrinsic pathway. To address this in more detail, we tested the ability of LVS to affect apoptosis triggered by Fas crosslinking. PMNs were left untreated or were infected with LVS for 1 h prior to the addition of Fas-activating antibodies, and apoptosis was quantified using Annexin V-staining and flow cytometry. Consistent with the data shown in Figures 7A–B, 71% of PMNs externalized PS within 6 h of Fas crosslinking, as compared with only 10% of control neutrophils or cells exposed to LVS alone (Fig. 8). At the same time, prior exposure to LVS significantly inhibited Fas-stimulated PS externalization, as the percentage of Annexin V-positive cells was reduced by 36% (P<0.05). These data strongly suggest that LVS inhibits or overrides the Fas-mediated apoptotic pathway in neutrophils.
FIGURE 8.
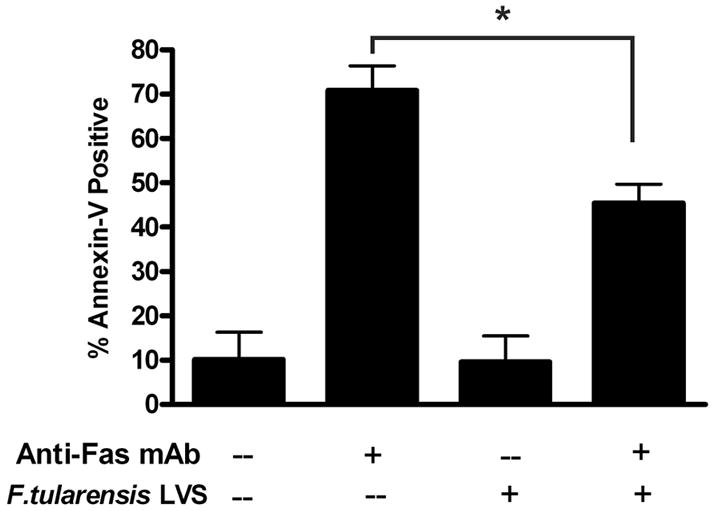
F. tularensis LVS inhibits Fas-induced apoptosis. Neutrophils were preincubated with LVS (MOI 200:1) for 1 h, and then treated with 500 ng/ml anti-Fas IgM for an additional 6 h. Apoptosis was measured using Annexin V staining and flow cytometry. Data indicate the percentage of Annexin V-positive cells and are the mean ± SEM (n=3).* P <0.05.
Live and killed bacteria differentially affect PMN apoptosis and CXCL8 secretion
For certain pathogens, such as A. phagocytophilum, killed bacteria retain the ability to prolong PMN lifespan (9), but whether this is also true for F. tularensis is unknown. For this study, LVS was killed by exposure to 10% formalin, washed, opsonized, and then incubated with PMNs. We now show, as judged by LDH release, that fkLVS had a distinct phenotype as these organisms lost the ability to significantly prolong neutrophil viability compared with untreated control cells (Fig. 9A), yet also failed to accelerate or induce PMN death, as do most particles including OpZ (Figs. 2 and 5A). In marked contrast to live F. tularensis, fkLVS also failed to alter the constitutive rate of PS externalization detected by Annexin V-staining (Fig. 9B), indicating an inability of killed bacteria to delay the onset of apoptosis. At the same time, the ability of fkLVS to affect caspase-3 activity was diminished but not ablated. Thus, caspase-3 activity increased more slowly in fkLVS-infected cells than in untreated controls, yet more rapidly than in cells infected with live bacteria (Fig. 9C and Supplemental Fig. 1B). Control experiments indicated that live and fkLVS infected PMNs to a similar extent (data not shown).
FIGURE 9.

Formalin-killed LVS does not delay neutrophil apoptosis. PMNs were left untreated or were infected with live or formalin-killed LVS (fkLVS) at MOI 200:1. A, PMN lysis was quantified by measuring LDH activity in cell-free supernatants. Data indicate percent cytotoxicity and are the mean ± SEM (n=3). B, PMN apoptosis was quantified using Annexin V staining and flow cytometry. Data are the mean ± SEM (n=3). C, Caspase-3 activity was assessed using a specific luminescence assay. RLU data are the mean ± SEM of triplicate samples from one experiment representative of two. For panels A–C, statistically significant differences between control and LVS-infected PMNs are * P <0.05 and ** P <0.01. D, CXCL8 in media of control, LVS- or fkLVS-infected PMNs was quantified by ELISA. Data shown are from one experiment representative of three.
Live C. pneumoniae stimulates secretion of the antiapoptotic cytokine CXCL8 as a means to prolong PMN lifespan (11). We show here that fkLVS stimulated CXCL8 secretion that was detected as early as12 hpi, and increased sharply 24–48 hpi, whereas live bacteria did not (Fig. 9D). Taken together, our data reinforce the notion that live and killed LVS have distinct effects on PMNs (19), and indicate that although fkLVS stimulated secretion of CXCL8 and had some capacity to impair caspase-3 activity, this was not sufficient to delay spontaneous PMN apoptosis.
Virulent F. tularensis subsp. tularensis Schu S4 inhibits caspase-3 activation and prolongs PMN viability
To determine if the ability to delay PMN apoptosis was conserved in human pathogenic strains of F. tularensis, we infected PMNs with virulent F. tularensis subsp tularensis strain Schu S4. Parallel samples were left untreated, infected with LVS, or treated with staurosporine. Similar to LVS, Schu S4 prolonged PMN viability as judged by LDH release (Fig. 10A). As expected, staurosporine accelerated PMN apoptosis relative to untreated controls as judged by analysis of nuclear morphology (Fig. 10B) and quantitation of caspase-3 activity (Fig. 10C) over 30 h at 37°C. In marked contrast, both these apoptotic parameters were profoundly diminished and delayed by Schu S4 and LVS. In particular, the fraction of PMNs exhibiting condensed nuclei was reduced more than fivefold at 30 h (***P<0.001) (Fig. 10B), and caspase-3 activity was markedly inhibited (***P<0.001) by both F. tularensis strains at 12, 18, 24 and 30 hpi (Fig. 10C). Reproducibility of Schu S4-mediated capsase-3 inhibition was confirmed using PMNs from another donor (Supplemental Fig. 1C). Finally, we show that similar to LVS, Schu S4 also inhibited apoptosis triggered by Fas crosslinking (Fig. 10D).
FIGURE 10.

Virulent F. tularensis stain Schu S4 impairs spontaneous and Fas-triggered apoptosis. Neutrophils were left untreated, infected with Schu S4 or LVS (at MOI 200:1), or treated with staurosporine (1 μM) as indicated. A, PMN death was quantified by measurement of LDH release. Data shown are from one experiment representative of three. B, Data indicate the percentage of cells with apoptotic nuclei determined by microscopic evaluation of >300 cells per time point and are the mean ± SEM (n=2). *** P <0.001 vs. untreated control. C, Caspase-3 activity was assessed as described above. RLU data are the mean ± SEM of triplicate samples from a representative experiment. *** P <0.001 vs. untreated controls. D, Like LVS, Schu S4 inhibits apoptosis triggered by Fas crosslinking. PMNs were preincubated with Schu S4 or LVS (MOI 200:1) for 1 h, and then treated with 500 ng/ml anti-Fas IgM for an additional 6 h. Apoptosis was assessed by analysis of nuclear morphology after HEMA-3 staining. Data indicate the percentage of cells with condensed nuclei and are the mean ± SEM (n=2). ** P <0.01 and ***P<0.001 as indicated.
Delayed apoptosis does not require direct contact between neutrophils and LVS
We show here that infection of human PMNs by F. tularensis in serum-free media was inefficient during the first several hours of infection (Figs. 1A–1B). Nevertheless, exposure of neutrophils to LVS or Schu S4 for only 1 h prior to Fas crosslinking was sufficient to inhibit neutrophil apoptosis initiated via the extrinsic pathway (Figs. 8 and 10D). Taken together, these data suggest that extracellular bacteria may play a role in modulating PMN apoptosis in our infection model. To test this hypothesis, we utilized Transwells equipped with 0.4 μm pore membranes to prevent contact between LVS and PMNs. For each experiment, neutrophils were added to the lower chamber of the Transwell, and LVS was added either to the upper chamber (to prevent direct contact with PMNs) or to the lower chamber (which allowed direct contact and resembled our typical experimental conditions), and PMNs incubated in the absence of LVS were used as controls. We also confirmed by measurement of CFU that LVS did not cross the Transwell membrane (data not shown). Consistent with data shown above, conditions that allowed direct contact between LVS and PMNs markedly delayed neutrophil apoptosis as judged by Annexin V-staining (**P<0.01) (Fig. 11A). However, LVS on the opposite side of the Transwell filter also significantly delayed PMN apoptosis relative to untreated controls (*P<0.05) (Fig. 11A). Thus, these results extend our findings to show that direct contact enhances but is not essential for the ability of LVS to delay PMN apoptosis.
FIGURE 11.

Delayed apoptosis does not require bacterial binding to PMNs, capsular polysaccharides, or LPS O-antigen. A, Delayed apoptosis does not require LVS binding. PMNs were added to the lower chamber of a Transwell containing a 0.4 μm filter support. Where noted, LVS was added to the lower (same) or upper (opposite) chamber of the Transwell at MOI 200:1. Apoptosis was quantified after 24 h at 37°C. Data indicate the percentage of Annexin V-positive PMNs and are the mean ± SEM (n=3). * P <0.05 and ** P <0.01 vs. PMNs alone. B–D, Major surface polysaccharides of LVS are dispensable for delayed PMN apoptosis. B, Lysates prepared from wild-type LVS or isogenic FTL0708 and wbtA2 mutants were analyzed for the presence of capsule and LPS O-antigen by immunoblotting with mAbs 11B7 and FB11, respectively. An LPS and capsule-enriched preparation from wild-type LVS was used as positive control. C, Apoptosis of control PMNs, cells infected with LVS (MOI 200:1), or cells treated with the indicated amounts of isolated LPS and capsule was assayed after 24 h at 37°C using Annexin V staining and flow cytometry. Data are the mean ± SEM (n=4). ** P <0.01 for LVS-infected vs. control PMNs. D, PMNs were left untreated or were infected with wild-type LVS, FTL0708, or wbtA2 (at MOI 200:1). 24 h later, the percentage of Annexin V-positive cells was determined. Data indicate the mean ± SEM (n=4). * P <0.05 and ** P <0.01 vs. PMNs alone.
F. tularensis LPS and capsular polysaccharides are neither sufficient nor required for delayed apoptosis
The data in Figure 11A suggest that one or more factors released or secreted by live F. tularensis may delay PMN death. Major surface components of this organism include LPS and capsule (30), and most Gram-negative bacteria can shed LPS and other surface components via release of outer membrane vesicles or by other mechanisms. We isolated a fraction containing both LPS and capsule from LVS using established procedures (30, 33) (and see Methods), and confirmed sample composition by Western blotting (Fig. 11B). This material was then tested directly for its ability to modulate PMN lifespan. Data shown in Figure 11C demonstrate that in marked contrast to the effects of whole F. tularensis, Annexin V-staining was not affected by our capsule and LPS-enriched samples at any of the concentrations tested. Thus, under these conditions, isolated LPS and capsule were not sufficient to delay PMN apoptosis.
Next, we used mutants with defects in LPS O-antigen and capsule biosynthesis to define better the role of surface sugars in modulation of PMN lifespan by LVS. The ΔwbtA2 mutant (32) was the generous gift of Dara Frank (Medical College of Wisconsin) and is devoid of capsule and O-antigen as judged by Western blotting of bacterial lysates (Fig. 11B), confirming published data (30, 32). FTL0708 was disrupted by group II intron retargeting as described in the Materials and Methods and is the LVS homologue of Schu S4 FTT1236 (33). Disruption of FTL0708 also prevented O-antigen synthesis. However, lysates prepared from this mutant retained weak reactivity with the anti-capsule mAb 11B7, whereas ΔwbtA2 lysates did not (Fig. 11B).
As both mutants are serum-sensitive [(32) and our unpublished data], studies of these strains utilized unopsonized wild-type and mutant bacteria. We now demonstrate that both mutant strains retained the ability to delay PMN apoptosis to a similar extent as wild-type LVS, as indicated by Annexin V-staining performed 24 hpi (Fig. 11D). These results are consistent with the data shown in Figure 11C, and further suggest that LPS O-antigen and capsular polysaccharides are dispensable for the ability of LVS to delay PMN apoptosis.
Discussion
Innate immune defense against invading bacteria relies heavily upon the aggressive response of neutrophils at sites of infection. These phagocytes are equipped with potent antimicrobial systems that collaborate to create a highly lethal intraphagosomal environment. However, effective neutrophil function at infection sites extends beyond containment and killing of bacterial invaders. Neutrophils also play a critical role in orchestrating the resolution phase of inflammation, undergoing controlled cellular demolition through apoptosis to down-regulate their proinflammatory capacity and target spent cells to macrophages for disposal (4, 6, 7). Therefore, neutrophil apoptosis at sites of infection is a characteristic feature of an effective immune response and is essential for resolution of the inflammation following bacterial infection (52).
In the present study, we examined the extent to which the facultative intracellular pathogen F. tularensis modulates human neutrophil apoptosis. We utilized a serum-free assay system developed by Henson and colleagues to avoid confounding effects of growth factors and other serum components on PMN lifespan (37), and followed established guidelines to quantify the rate and extent of cell death and apoptosis (41). In our hands, untreated control PMNs began to exhibit signs of apoptosis at 12 h, and 60–70% of cells were apoptotic by 24 h, confirming published data (4, 5, 35, 39). As reported previously, we show that procaspases-8, -9 and -3 were processed and activated during constitutive PMN apoptosis (47, 48, 53), and confirmed the ability of OpZ, staurosporine and anti-Fas IgM to significantly accelerate the onset of apoptosis relative to untreated controls (10, 46, 54).
With regard to neutrophils and F. tularensis, we utilized the LDH release assay to quantify cell death as indicated by loss of plasma membrane integrity, and show for the first time that F. tularensis significantly prolongs human neutrophil lifespan. Thereafter, we employed multiple complementary biochemical assays to demonstrate definitively that the constitutive apoptosis program of human neutrophils was impaired for at least 48 h. Specifically, our data indicate that relative to control cells, F. tularensis profoundly diminished the fraction of PMN exhibiting morphological signs of apoptosis such as nuclear condensation. Consistent with this, DNA fragmentation detected by TUNEL staining was also significantly impaired, as was the rate of PS externalization detected using Annexin V-FITC. Of particular note, the rate and extent of executioner caspase-3 processing and activation were markedly inhibited. Similar defects in processing and activation of upstream initiator caspases-8 and -9 indicate that F. tularensis affected both the extrinsic and intrinsic apoptotic pathways. Finally, we demonstrate that in our assay system uptake of F. tularensis was inefficient, yet PMNs accumulated large numbers of bacteria over the 48 h time course examined. Additional experiments revealed that neutrophil bacterial load increased 19-fold overall, and direct measurement of intracellular growth revealed eightfold replication of LVS between 12 and 36 hpi. In contrast, bacteria were viable but did not replicate in the tissue culture medium, consistent with the absence of cysteine, a critical nutrient for Francisella (55), in serum-free RPMI-1640. These data are of interest as F. tularensis undergoes robust replication in macrophages, dendritic and epithelial cells (16, 18), but to our knowledge, its ability to replicate in neutrophils has not been demonstrated previously. Overall, our findings suggest that both intracellular replication and continued uptake of extracellular LVS contributed to PMN bacterial load in our system. Similar data were obtained for Schu S4 (not shown). Inasmuch as loss of functional capacity, including the ability to engulf particles by phagocytosis, is a hallmark of apoptotic neutrophils (1), these data are consistent with the ability of F. tularensis to extend PMN lifespan.
In addition to its effects on constitutive neutrophil turnover, we show that F. tularensis can also significantly inhibit cell death triggered by anti-Fas IgM, indicating an ability to override the effects of a specific proapoptotic stimulus. This finding is of interest as neutrophils differ from other leukocytes in their ability to synthesize both Fas ligand and Fas (54), and autocrine stimulation of the extrinsic pathway via these molecules may contribute to PMN turnover at sites of infection. Thus, it is attractive to predict that inhibition of this pathway contributes to the ability of F. tularensis to prolong PMN viability.
The ability of NADPH oxidase-derived ROS to accelerate PMN apoptotic death is established (1, 36, 56), and oxidants may also be sufficient to counteract certain antiapoptotic signals. For instance, organisms such as live N. gonorrhoeae and heat-killed E. coli can delay PMN apoptosis at very low MOI (0.01 – 1:1), but this effect is rapidly negated and reversed by ROS at higher bacterial loads (5, 10, 57). In contrast, our published data demonstrate that F. tularensis prevents oxidant production via effects on NADPH oxidase assembly and activity (19, 22), and we show here that this bacterium inhibits PMN apoptosis in a dose-dependent manner (Fig. 4C) and under conditions where intracellular bacterial burdens were high (Fig. 3A). Thus, disruption of the oxidative burst likely accounts in large part for the fact that F. tularensis does not induce PMN apoptosis. However, CGD neutrophils exhibit no apparent defects in constitutive apoptosis despite their profound inability to accelerate the apoptotic program upon stimulation (1, 36). Therefore, defects in oxidant production likely cannot account for the ability of F. tularensis to inhibit basal PMN turnover.
Although the majority of neutrophils were infected with F. tularensis by 12 h under the conditions used throughout this study (Figs. 1A and 1B), and cells containing large numbers of bacteria did not exhibit signs of apoptosis at 24 or 36 hpi (Fig. 3A), we found to our surprise that inhibition of apoptosis could be uncoupled from phagocytosis and that bacterial uptake was not essential for this process. This was suggested first by the ability of F. tularensis to impair apoptosis at markedly lower MOI (EC50 of 5:1, Fig. 4C), even though only a subset of cells were infected. Furthermore, using Transwell supports to prevent direct contact between neutrophils and bacteria we observed that PMN lifespan was significantly prolonged, albeit with somewhat diminished efficiency (Fig. 11A). We favor a model in which intracellular and extracellular bacteria collaborate to extend PMN viability, but whether intracellular and extracellular F. tularensis have independent, sequential or synergistic effects on neutrophil apoptosis remains to be determined.
How F. tularensis extends PMN lifespan is unknown, but the ability of the organism to act at a distance suggested a role for factors released into the extracellular milieu. LPS is shed by many bacteria, and C. pneumoniae is thought to extend PMN longevity via the ability of its LPS to stimulate secretion of the antiapoptotic cytokine CXCL8 (11). In contrast, we show here that samples containing Francisella LPS and capsule had no apparent effect on PMN viability and turnover, and that live bacteria did not trigger release of CXCL8. These results are in keeping with the relatively inert nature of F. tularensis LPS (16), and strongly suggest that Francisella and Chlamydia inhibit apoptosis by different mechanisms. At the same time, it has long been known that LPS O-antigen and capsular polysaccharides act in concert to render F. tularensis resistant to the lytic effects of serum complement (16, 21, 33). Our studies of isolated capsule as well as mutant strains with defects in capsule and O-antigen synthesis revealed that these surface sugars are neither necessary nor sufficient for delayed PMN apoptosis. These data are noteworthy as we recently reported that a Schu S4 mutant lacking functional FTT1236, the homologue of FTL0708, triggers rapid lysis and death of human macrophages (33). Thus, our findings support a large body of data which indicates that pathogens manipulate apoptosis and other mechanisms of death in a cell type-specific manner (58).
Perhaps the best characterized pathogen known to delay neutrophil apoptosis is A. phagocytophilum. This obligate intracellular bacterium uses a multifaceted approach to extend the lifespan of its replicative niche for up to 90 h. Similar to F. tularensis, A. phagocytophilum acts in a dose-dependent manner to disrupt production of ROS by the NADPH oxidase, inhibit caspase-3, -8 and -9 processing and activity, and counteract the effects of Fas crosslinking (9), and this is achieved via effects on multiple intracellular signaling pathways as well as PMN gene expression (9). Unlike F. tularensis, killed A. phagocytophilum also significantly inhibits apoptosis (9, 59), and it has been proposed that surface molecules of live and killed bacteria initiate signaling that impairs apoptosis early in infection, whereas PMN viability is prolonged further by mechanisms specific for live organisms (59). Consistent with this, recent data indicate that Ats-1, delivered into PMNs by live A. phagocytophilum, acts directly on mitochondria to preserve their integrity and prevent release of proapoptotic factors into the cytosol (9). The extent to which F. tularensis modulates neutrophil signaling, gene expression, or mitochondrial integrity as a means to prolong cell lifespan is currently under investigation.
Neutrophils are key regulators of the inflammatory response, and granuloma formation, prolonged PMN viability and tissue necrosis are all hallmarks of an aberrant and defective inflammatory response (52, 56, 60). Studies of primates, rabbits and mice with tularemia indicate that PMN accumulation, pyogranuloma formation and tissue necrosis are also prominent histological features of tissues infected with F. tularensis (24–26). Moreover, blockade of PMN migration into the lung diminishes tissue damage and favors survival of mice infected with this organism (28). As PMN apoptosis is essential to limit tissue injury, particularly in the lung (61), and we demonstrate here that F. tularensis profoundly inhibits this process, our findings support a model in which neutrophils play a prominent role in dysregulation of the inflammatory response during tularemia. The effects of delayed PMN apoptosis can be exacerbated by defects in corpse removal by macrophages, which increases the probability that dying neutrophils will progress to secondary necrosis with spilling of toxic cell contents and alarmins that amplify inflammation and tissue damage (6–8, 60, 62). During tularemia, efferocytosis is directly undermined by intramacrophage bacteria (63), and may be compromised further by local macrophage depletion (64). Moreover, end-stage tularemia is characterized by overwhelming sepsis (63, 65, 66), which is itself associated with decreased neutrophil apoptosis (61). Koedel et al. demonstrated a benefit of inducing neutrophil apoptosis during pneumococcal meningitis, where tissue damage is associated with increased longevity of neutrophils recruited to the brain (67). Therefore, one strategy proposed to manage infectious diseases where neutrophils play a role in disease pathology is to combine antibiotic therapy with drugs that induce PMN apoptosis (6). Whether a similar approach would be of therapeutic value in tularemia is as yet unclear.
In summary, the results of this study extend previous work to demonstrate for the first time that F. tularensis acts at multiple points to disrupt constitutive apoptosis and prolong human neutrophil lifespan. This is achieved by a mechanism that can be uncoupled from phagocytosis, is independent of major surface carbohydrates of the organism, and cannot be recapitulated by isolated LPS. Moreover, as PMN apoptosis is essential for resolution of the inflammatory response, our data define a new mechanism of innate immune evasion by F. tularensis and suggest a model to account, at least in part, for the in vivo histopathology that is characteristic of this disease. Although the bacterial components involved in these phenomena remain obscure, the ability of extracellular F. tularensis to act at a distance to modulate PMN lifespan at low MOI may contribute to pathogenicity by altering neutrophil function as soon as these cells enter infected tissues, a notion consistent with recent data (68). Collectively, the results of this study substantially advance our understanding of tularemia pathogenesis.
Supplementary Material
Acknowledgments
We thank Dr. Dara Frank (Medical College of Wisconsin) for sharing the LVS ΔwbtA2 mutant, and Dr. Michael Apicella (University of Iowa) for antibodies to F. tularensis capsule.
Abbreviations
- BSL
Biosafety Level
- CGD
chronic granulomatous disease
- CHAB
cysteine heart agar supplemented with sheep blood
- fk
formalin-killed
- hpi
hours post-infection
- LDH
lactate dehydrogenase
- LVS
live vaccine strain
- MOI
multiplicity of infection
- OpZ
opsonized zymosan
- PMN
polymorphonuclear leukocyte
- PI
propidium iodide
- PS
phosphatidylserine
- ROS
reactive oxygen species
Footnotes
This work was supported in part by funds from the NIH to L.-A.HA (R01AI073835 and P01AI044642) and J.T.S. (T32AI007511 predoctoral fellowship).
Conflicts of Interest: None of the authors has a conflict of interest.
Author Contributions
JTS designed and performed experiments, interpreted data and co-wrote the manuscript. JHB designed and performed experiments, interpreted data and edited the manuscript. JK, DCF and JMM designed and performed experiments and interpreted data. L-AHA conceived of the study, obtained financial support, designed experiments, interpreted data and co-wrote the manuscript.
References
- 1.Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res. 2009;43:25–61. doi: 10.1007/s12026-008-8049-6. [DOI] [PubMed] [Google Scholar]
- 2.Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi SD, Braughton KR, Whitney AR, Voyich JM, Schwan TG, Musser JM, DeLeo FR. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci, USA. 2003;100:10948–10953. doi: 10.1073/pnas.1833375100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson RW, Redmond HP, Wang JH, Condron C, Bouchier-Hayes D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol. 1996;156:3986–3992. [PubMed] [Google Scholar]
- 6.Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun. 2010;2:216–227. doi: 10.1159/000284367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi SD, Voyich JM, Braughton KR, DeLeo FR. Down-regulation of proinflammatory capacity during apoptosis in human polymorphonuclear leukocytes. J Immunol. 2003;170:3357–3368. doi: 10.4049/jimmunol.170.6.3357. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi SD, Voyich JM, Somerville GA, Braughton KR, Malech HL, Musser JM, DeLeo FR. An apoptosis-differentiation program in human polymorphonuclear leukocytes facilitates resolution of inflammation. J Leukoc Biol. 2003;73:315–322. doi: 10.1189/jlb.1002481. [DOI] [PubMed] [Google Scholar]
- 9.Rikihisa Y. Mechanisms of obligatory intracellular infection with Anaplasma phagocytophilum. Clin Microbiol Rev. 2011;24:469–489. doi: 10.1128/CMR.00064-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simons MP, Nauseef WM, Griffith TS, Apicella MA. Neisseria gonorrhoeae delays the onset of apoptosis in polymorphonuclear leukocytes. Cell Microbiol. 2006;8:1780–1790. doi: 10.1111/j.1462-5822.2006.00748.x. [DOI] [PubMed] [Google Scholar]
- 11.van Zandbergen G, Gieffers J, Kothe H, Rupp J, Bollinger A, Aga E, Klinger M, Brade H, Dalhoff K, Maass M, Solbach W, Laskay T. Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J Immunol. 2004;172:1768–1776. doi: 10.4049/jimmunol.172.3.1768. [DOI] [PubMed] [Google Scholar]
- 12.Bylund J, Campsall PA, Ma RC, Conway BA, Speert DP. Burkholderia cenocepacia induces neutrophil necrosis in chronic granulomatous disease. J Immunol. 2005;174:3562–3569. doi: 10.4049/jimmunol.174.6.3562. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi SD, Braughton KR, Palazzolo-Ballance AM, Kennedy AD, Sampaio E, Kristosturyan E, Whitney AR, Sturdevant DE, Dorward DW, Holland SM, Kreiswirth BN, Musser JM, DeLeo FR. Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J Innate Immun. 2010;2:560–575. doi: 10.1159/000317134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Usher LR, Lawson RA, Geary I, Taylor CJ, Bingle CD, Taylor GW, Whyte MK. Induction of neutrophil apoptosis by the Pseudomonas aeruginosa exotoxin pyocyanin: a potential mechanism of persistent infection. J Immunol. 2002;168:1861–1868. doi: 10.4049/jimmunol.168.4.1861. [DOI] [PubMed] [Google Scholar]
- 15.Oyston PC, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 16.McLendon MK, Apicella MA, Allen LAH. Francisella tularensis: taxonomy, genetics, and Immunopathogenesis of a potential agent of biowarfare. Annu Rev Microbiol. 2006;60:167–185. doi: 10.1146/annurev.micro.60.080805.142126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellis J, Oyston PC, Green M, Titball RW. Tularemia. Clin Microbiol Rev. 2002;15:631–646. doi: 10.1128/CMR.15.4.631-646.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chong A, Celli J. The Francisella intracellular life cycle: toward molecular mechanisms of intracellular survival and proliferation. Front Microbiol. 2010;1:138–149. doi: 10.3389/fmicb.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCaffrey RL, Allen LAH. Pivotal Advance: Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J Leukoc Biol. 2006;80:1224–1230. doi: 10.1189/jlb.0406287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandstrom G, Lofgren S, Tarnvik A. A capsule-deficient mutant of Francisella tularensis LVS exhibits enhanced sensitivity to killing by serum but diminished sensitivity to killing by polymorphonuclear leukocytes. Infect Immun. 1988;56:1194–1202. doi: 10.1128/iai.56.5.1194-1202.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clay CD, Soni S, Gunn JS, Schlesinger LS. Evasion of complement-mediated lysis and complement C3 deposition are regulated by Francisella tularensis lipopolysaccharide O-antigen. J Immunol. 2008;181:5568–5578. doi: 10.4049/jimmunol.181.8.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCaffrey RL, Schwartz JT, Lindemann SR, Moreland JG, Buchan BW, Jones BD, Allen LAH. Multiple mechanisms of NADPH oxidase inhibition by type A and type B Francisella tularensis. J Leukoc Biol. 2010;88:791–805. doi: 10.1189/jlb.1209811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lofgren S, Tarnvik A, Bloom GD, Sjoberg W. Phagocytosis and killing of Francisella tularensis by human polymorphonuclear leukocytes. Infect Immun. 1983;39:715–720. doi: 10.1128/iai.39.2.715-720.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall WC, Kovatch RM, Schricker RL. Tularaemic pneumonia: pathogenesis of the aerosol-induced disease in monkeys. J Pathol. 1973;110:193–201. doi: 10.1002/path.1711100302. [DOI] [PubMed] [Google Scholar]
- 25.Schricker RL, Eigelsbach HT, Mitten JQ, Hall WC. Pathogenesis of tularemia in monkeys aerogenically exposed to Francisella tularensis 425. Infect Immun. 1972;5:734–744. doi: 10.1128/iai.5.5.734-744.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tulis JJ, Eigelsbach HT, Kerpsack RW. Host-parasite relationship in monkeys administered live tularemia vaccine. Am J Pathol. 1970;58:329–336. [PMC free article] [PubMed] [Google Scholar]
- 27.Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, Kawula TH. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun. 2008;76:5843–5852. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malik M, Bakshi CS, McCabe K, Catlett SV, Shah A, Singh R, Jackson PL, Gaggar A, Metzger DW, Melendez JA, Blalock JE, Sellati TJ. Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J Immunol. 2007;178:1013–1020. doi: 10.4049/jimmunol.178.2.1013. [DOI] [PubMed] [Google Scholar]
- 29.KuoLee R, Harris G, Conlan JW, Chen W. Role of neutrophils and NADPH phagocyte oxidase in host defense against respiratory infection with virulent Francisella tularensis in mice. Microbes Infect. 2011;13:447–456. doi: 10.1016/j.micinf.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 30.Apicella MA, Post DM, Fowler AC, Jones BD, Rasmussen JA, Hunt JR, Imagawa S, Choudhury B, Inzana TJ, Maier TM, Frank DW, Zahrt TC, Chaloner K, Jennings MP, McLendon MK, Gibson BW. Identification, characterization and immunogenicity of an O-antigen capsular polysaccharide of Francisella tularensis. PLoS One. 2010;5:e11060. doi: 10.1371/journal.pone.0011060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nauseef WM. Isolation of human neutrophils from venous blood. Methods Mol Biol. 2007;412:15–20. doi: 10.1007/978-1-59745-467-4_2. [DOI] [PubMed] [Google Scholar]
- 32.Maier TM, Casey MS, Becker RH, Dorsey CW, Glass EM, Maltsev N, Zahrt TC, Frank DW. Identification of Francisella tularensis Himar1-based transposon mutants defective for replication in macrophages. Infect Immun. 2007;75:5376–5389. doi: 10.1128/IAI.00238-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindemann SR, Peng K, Long ME, Hunt JR, Apicella MA, Monack DM, Allen LAH, Jones BD. Francisella tularensis Schu S4 O-antigen and capsule biosynthesis gene mutants induce early cell death in human macrophages. Infect Immun. 2011;79:581–594. doi: 10.1128/IAI.00863-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schulert GS, Allen LAH. Differential infection of mononuclear phagocytes by Francisella tularensis: role of the macrophage mannose receptor. J Leukoc Biol. 2006;80:563–571. doi: 10.1189/jlb.0306219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Payne CM, Glasser L, Tischler ME, Wyckoff D, Cromey D, Fiederlein R, Bohnert O. Programmed cell death of the normal human neutrophil: an in vitro model of senescence. Microsc Res Tech. 1994;28:327–344. doi: 10.1002/jemt.1070280408. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi SD, Deleo FR. An apoptosis differentiation programme in human polymorphonuclear leucocytes. Biochem Soc Trans. 2004;32:474–476. doi: 10.1042/BST0320474. [DOI] [PubMed] [Google Scholar]
- 37.Gardai SJ, Whitlock BB, Xiao YQ, Bratton DB, Henson PM. Oxidants inhibit ERK/MAPK and prevent its ability to delay neutrophil apoptosis downstream of mitochondrial changes and at the level of XIAP. J Biol Chem. 2004;279:44695–44703. doi: 10.1074/jbc.M405313200. [DOI] [PubMed] [Google Scholar]
- 38.Brach MA, deVos S, Gruss HJ, Herrmann F. Prolongation of survival of human polymorphonuclear neutrophils by granulocyte-macrophage colony-stimulating factor is caused by inhibition of programmed cell death. Blood. 1992;80:2920–2924. [PubMed] [Google Scholar]
- 39.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80:2012–2020. [PubMed] [Google Scholar]
- 40.Perianayagam MC, V, Balakrishnan S, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils via an extracellular signal-regulated kinase and Bad-mediated signalling pathway. Eur J Clin Invest. 2004;34:50–56. doi: 10.1111/j.1365-2362.2004.01273.x. [DOI] [PubMed] [Google Scholar]
- 41.Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, Cidlowski JA, Ciechanover A, Cohen GM, De Laurenzi V, De Maria R, Deshmukh M, Dynlacht BD, El-Deiry WS, Flavell RA, Fulda S, Garrido C, Golstein P, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Jaattela M, Kepp O, Kimchi A, Klionsky DJ, Knight RA, Kornbluth S, Kumar S, Levine B, Lipton SA, Lugli E, Madeo F, Malomi W, Marine JC, Martin SJ, Medema JP, Mehlen P, Melino G, Moll UM, Morselli E, Nagata S, Nicholson DW, Nicotera P, Nunez G, Oren M, Penninger J, Pervaiz S, Peter ME, Piacentini M, Prehn JH, Puthalakath H, Rabinovich GA, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Scorrano L, Simon HU, Steller H, Tschopp J, Tsujimoto Y, Vandenabeele P, Vitale I, Vousden KH, Youle RJ, Yuan J, Zhivotovsky B, Kroemer G. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–1107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voyich JM, DeLeo FR. Host-pathogen interactions: leukocyte phagocytosis and associated sequelae. Methods Cell Sci. 2002;24:79–90. doi: 10.1023/a:1024154200702. [DOI] [PubMed] [Google Scholar]
- 43.Daigle I, Simon HU. Critical role for caspases 3 and 8 in neutrophil but not eosinophil apoptosis. Int Arch Allergy Immunol. 2001;126:147–156. doi: 10.1159/000049506. [DOI] [PubMed] [Google Scholar]
- 44.Pongracz J, Webb P, Wang K, Deacon E, Lunn OJ, Lord JM. Spontaneous neutrophil apoptosis involves caspase 3-mediated activation of protein kinase C-delta. J Biol Chem. 1999;274:37329–37334. doi: 10.1074/jbc.274.52.37329. [DOI] [PubMed] [Google Scholar]
- 45.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 46.Paunel-Gorgulu A, Zornig M, Logters T, Altrichter J, Rabenhorst U, Cinatl J, Windolf J, Scholz M. Mcl-1-mediated impairment of the intrinsic apoptosis pathway in circulating neutrophils from critically ill patients can be overcome by Fas stimulation. J Immunol. 2009;183:6198–6206. doi: 10.4049/jimmunol.0901264. [DOI] [PubMed] [Google Scholar]
- 47.Cabrini M, Nahmod K, Geffner J. New insights into the mechanisms controlling neutrophil survival. Curr Opin Hematol. 2010;17:31–35. doi: 10.1097/MOH.0b013e3283333b29. [DOI] [PubMed] [Google Scholar]
- 48.Conus S, Perozzo R, Reinheckel T, Peters C, Scapozza L, Yousefi S, Simon HU. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med. 2008;205:685–698. doi: 10.1084/jem.20072152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 2004;11:143–153. doi: 10.1038/sj.cdd.4401320. [DOI] [PubMed] [Google Scholar]
- 50.van Raam BJ, Verhoeven AJ, Kuijpers TW. Mitochondria in neutrophil apoptosis. Int J Hematol. 2006;84:199–204. doi: 10.1532/IJH97.06131. [DOI] [PubMed] [Google Scholar]
- 51.Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, Edwards SW. The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol. 2003;170:1964–1972. doi: 10.4049/jimmunol.170.4.1964. [DOI] [PubMed] [Google Scholar]
- 52.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 53.van Raam BJ, Drewniak A, Groenewold V, van den Berg TK, Kuijpers TW. Granulocyte colony-stimulating factor delays neutrophil apoptosis by inhibition of calpains upstream of caspase-3. Blood. 2008;112:2046–2054. doi: 10.1182/blood-2008-04-149575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liles WC, Kiener PA, Ledbetter JA, Aruffo A, Klebanoff SJ. Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med. 1996;184:429–440. doi: 10.1084/jem.184.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larsson P, Oyston PC, Chain P, Chu MC, Duffield M, Fuxelius HH, Garcia E, Halltorp G, Johansson D, Isherwood KE, Karp PD, Larsson E, Liu Y, Michell S, Prior J, Prior R, Malfatti S, Sjostedt A, Svensson K, Thompson N, Vergez L, Wagg JK, Wren BW, Lindler LE, Andersson SG, Forsman M, Titball RW. The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nat Genet. 2005;37:153–159. doi: 10.1038/ng1499. [DOI] [PubMed] [Google Scholar]
- 56.Kobayashi SD, Voyich JM, Braughton KR, Whitney AR, Nauseef WM, Malech HL, DeLeo FR. Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol. 2004;172:636–643. doi: 10.4049/jimmunol.172.1.636. [DOI] [PubMed] [Google Scholar]
- 57.Matsuda T, Saito H, Inoue T, Fukatsu K, Lin MT, Han I, Furukawa S, Ikeda S, Muto T. Ratio of bacteria to polymorphonuclear neutrophils (PMNs) determines PMN fate. Shock. 1999;12:365–372. [PubMed] [Google Scholar]
- 58.Lamkanfi M, V, Dixit M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 59.Borjesson DL, Kobayashi SD, Whitney AR, Voyich JM, Argue CM, Deleo FR. Insights into pathogen immune evasion mechanisms: Anaplasma phagocytophilum fails to induce an apoptosis differentiation program in human neutrophils. J Immunol. 2005;174:6364–6372. doi: 10.4049/jimmunol.174.10.6364. [DOI] [PubMed] [Google Scholar]
- 60.Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Resp Crit Care Med. 1999;160:S5–11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- 61.Fialkow L, Fochesatto Filho L, Bozzetti MC, Milani AR, Rodrigues Filho EM, Ladniuk RM, Pierozan P, de Moura RM, Prolla JC, Vachon E, Downey GP. Neutrophil apoptosis: a marker of disease severity in sepsis and sepsis-induced acute respiratory distress syndrome. Crit Care. 2006;10:R155. doi: 10.1186/cc5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silva MT. Bacteria-induced phagocyte secondary necrosis as a pathogenicity mechanism. J Leukoc Biol. 2010;88:885–896. doi: 10.1189/jlb.0410205. [DOI] [PubMed] [Google Scholar]
- 63.Mares CA, Sharma J, Li Q, Rangel EL, Morris EG, Enriquez MI, Teale JM. Defect in efferocytosis leads to alternative activation of macrophages in Francisella infections. Immunol Cell Biol. 2011;89:167–172. doi: 10.1038/icb.2010.81. [DOI] [PubMed] [Google Scholar]
- 64.Wickstrum JR, Bokhari SM, Fischer JL, Pinson DM, Yeh HW, Horvat RT, Parmely MJ. Francisella tularensis induces extensive caspase-3 activation and apoptotic cell death in the tissues of infected mice. Infect Immun. 2009;77:4827–4836. doi: 10.1128/IAI.00246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eisen RJ, Yockey B, Young J, Reese SM, Piesman J, Schriefer ME, Beard CB, Petersen JM. Short report: time course of hematogenous dissemination of Francisella tularensis A1, A2, and Type B in laboratory mice. Am J Trop Med Hyg. 2009;80:259–262. [PubMed] [Google Scholar]
- 66.Sharma J, Mares CA, Li Q, Morris EG, Teale JM. Features of sepsis caused by pulmonary infection with Francisella tularensis Type A strain. Microb Pathog. 2011;51:39–47. doi: 10.1016/j.micpath.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koedel U, Frankenberg T, Kirschnek S, Obermaier B, Hacker H, Paul R, Hacker G. Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog. 2009;5:e1000461. doi: 10.1371/journal.ppat.1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moreland JG, Hook JS, Bailey G, Ulland T, Nauseef WM. Francisella tularensis directly interacts with the endothelium and recruits neutrophils with a blunted inflammatory phenotype. Am J Physiol Lung Cell Mol Physiol. 2009;296:L1076–1084. doi: 10.1152/ajplung.90332.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.