

Abstract
Cardiac troponin T (cTnT), the tropomyosin binding subunit of the troponin complex, plays a pivotal regulatory role in the Ca2+-mediated interaction between actin thin filament and myosin thick filament. The post-translational modifications (PTMs) and alternative splicing of cTnT may represent important regulatory mechanisms of cardiac contractility. However, a complete characterization of PTMs and alternatively spliced isoforms in cTnT present in vivo is lacking. Top-down protein mass spectrometry (MS) analyzes whole proteins, thus providing a global view of all types of modifications, including PTMs and sequence variants, simultaneously in one spectrum without a priori knowledge. In this study, we applied an integrated immunoaffinity chromatography and top-down MS approach to comprehensively characterize PTMs and alternatively spliced isoforms of cTnT purified from healthy human and wild-type mouse heart tissue. High-resolution Fourier transform MS revealed that human cTnT (hcTnT) and mouse cTnT (mcTnT) have similar phosphorylation patterns, whereas higher molecular heterogeneity was observed for mcTnT than hcTnT. Further MS/MS fragmentation of monophosphorylated hcTnT and mcTnT by electron capture dissociation and collisionally activated dissociation unambiguously identified Ser1 as the conserved in vivo phosphorylation site. In contrast, we identified a single spliced isoform for hcTnT but three alternatively spliced isoforms for mcTnT. Moreover, we observed distinct proteolytic degradation products for hcTnT and mcTnT. This study also demonstrates the advantage of top-down MS/MS with complementary fragmentation techniques for the identification of modification sites in the highly acidic N-terminal region of cTnT.
Troponin (Tn), along with tropomyosin (Tm), is an important thin filament regulatory protein complex in striated muscle that controls the Ca2+-mediated interaction between actin thin filament and myosin thick filament.1,2 It consists of three distinct protein subunits: troponin C (TnC), the Ca2+-binding subunit that transduces Ca2+ signaling; troponin I (TnI), the inhibitory subunit that inhibits myosin ATPase activity; and troponin T (TnT), the tropomyosin binding subunit that anchors the Tn protein complex to the thin filament.1 During activation of contraction, at an increased cytosolic level Ca2+ binds to TnC and induces a conformational change in the Tn–Tm complex that releases the blocking of the actin–myosin interaction and thereby permits the formation of cross bridges, activation of myosin ATPase, and production of force.1-4
As the largest subunit of the Tn complex, TnT directly interacts with all key components in the thin filament regulatory systems and plays a pivotal role in the regulation of contractile function.3-5 TnT, a striated muscle-specific protein in vertebrates, is encoded by three homologous genes (TNNT2, TNNT1, and TNNT3) and expressed as three isoforms, cardiac (cTnT), slow skeletal (ssTnT), and fast skeletal (fsTnT) muscle TnT, respectively.3,5 Both cTnT and cTnI are released into the general circulation following necrotic death of heart muscle tissue and can be detected in human serum by an enzyme-linked immunosorbant assay (ELISA). Hence, cTnT and cTnI have become the biomarkers of choice for the detection of cardiac injury.6 Mutations of cTnT are known to be a common cause of familial hypertrophic cardiomyopathy7 and dilated cardiomyopathy.8
It has been increasingly recognized that alternatively spliced isoforms and post-translational modifications (PTMs) of cTnT may play important roles in the regulation of cardiac contractility.3-5,9-12 Three of the 17 exons in the cTnT gene are known to be alternatively spliced, which include exon 4 located in the N-terminal region, exon 13 in the central region, and exon 5 in the N-terminal region that is specifically expressed in mammalian cardiac muscle.3-5 Alternative RNA splicing generates a highly variable N-terminal region that is shown to modulate the function of TnT and cardiac contractility. 3,4,13,14 Phosphorylation, the major PTM studied for cTnT, has been shown to be regulated by a variety of Ser/Thr kinases, including protein kinase C (PKC) at Thr197, Ser201, Thr206, and Thr287,15 Rho-A-dependent protein kinase (ROCK-II) at Ser278 and Thr287,16 apoptosis signal-regulating kinase (ASK-1) at Thr197 and Ser201,17 and Raf-1 at Thr206.18 PKC-dependent phosphorylation of cTnT is believed to be functionally critical in the development of cardiac hypertrophy/failure syndrome.19 Early studies also showed evidence of phosphorylation of cTnT by a “specific” troponin T kinase and phosphorylase kinase at Ser1,5,20,21 but the significance of this site remains unclear.22 These previous studies were predominantly performed by in vitro kinase assays, and the heterogeneous phosphorylation sites reported among different mammalian species make the role of cTnT phosphorylation in cardiac regulation more elusive. Traditional analytical methods such as radioactive labeling and specific phosphoprotein antibody also precluded comprehensive characterization of all existing modifications in cTnT. Thus far, there has been a lack of direct and complete characterization of PTMs and alternative spliced isoforms present in vivo in human and mouse cTnT.
Mass spectrometry (MS) is able to provide universal information about protein modifications without a priori knowledge and therefore is the preferred method for analysis of protein modification.23,24 However, in the peptide-based “bottom-up” MS approach, proteins are digested prior to MS analysis so that the modification information at the whole protein level is often incomplete because of the limited protein coverage and loss of the correlation between the modifications on disparate portions of a protein.25 Recently, top-down protein MS26-35 has emerged as a powerful tool for the characterization of protein modifications present in vivo. It measures intact proteins directly, providing a global view of all of the existing modifications, including PTMs and sequence variants, simultaneously in one spectrum without a priori knowledge.36,37 Subsequently, a specific modified form can be isolated and fragmented in the mass spectrometer to map the modification sites by tandem mass spectrometry (MS/MS) techniques, including but not limited to collisionally activated dissociation (CAD)38 and electron capture dissociation (ECD).39 Importantly, ECD as a nonergodic fragmentation technique40 is especially suitable for the localization of labile PTMs because they are well-preserved during the fragmentation process.31-33,41 In previous studies, we have demonstrated the successful application of top-down MS with ECD/CAD fragmentation in the global characterization of PTMs in cTnI purified from human, pig, rat, and mouse heart tissue and identification of conserved basal PKA phosphorylation sites in cTnI.30,31,33,42 We have also characterized a single-amino acid polymorphism for rat cTnT at residue 192.42
In this work, we have applied immunoaffinity chromatography combined with top-down MS to comprehensively characterize the PTMs and alternatively spliced isoforms of cTnT directly purified from healthy human and wild-type mouse hearts. We have characterized PTMs, including acetylation, phosphorylation, and proteolytic degradation of human cTnT (hcTnT) and mouse cTnT (mcTnT). Our data revealed a conserved phosphorylation pattern between hcTnT and mcTnT and unambiguously identified Ser1 as the phosphorylation site. In contrast, the proteolytic degradation products of cTnT purified from human and mouse hearts are distinct. Moreover, we identified a single alternatively spliced isoform for hcTnT but three isoforms for mcTnT. To the best of our knowledge, this is the first complete and simultaneous characterization of cTnT modifications suggesting that phosphorylation, but not alternatively spliced isoform or proteolytic degradation, is conserved in human and mouse cTnT.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
All reagents were purchased from Sigma Chemical Co. (St. Louis, MO) unless noted otherwise. Complete protease and phosphatase inhibitor cocktail tablets were purchased from Roche Diagnostics Corp. (Indianapolis, IN). All solutions were prepared in Milli-Q water (Millipore Corp., Billerica, MA).
Heart Tissue Samples
Human heart tissue samples were surgically excised from the left ventricle of brain-dead healthy donors during heart transplant surgery (within 15 min after the heart stopped beating). These donor hearts were characterized with normal cardiac function but deemed unacceptable for heart transplantation for other reasons. The procedure for collection of human heart tissues was approved by the Institutional Review Board of the University of Wisconsin. Mouse heart samples were freshly collected from adult wild-type FVB and C57BL/6 mice with approval from the University of Wisconsin Animal Care and Use Committee as described previously.33 The heart samples were immediately snap-frozen and stored at −80 °C until protein extraction and purification.
Purification of Human and Mouse cTnT
Approximately 0.1–0.2 g of human heart tissue and four to six whole mouse hearts were used per preparation. Heart tissue was homogenized in wash buffer [500 mM NaH2PO4, 100 mM Na2HPO4, 100 mM MgCl2, 100 mM EGTA, 0.1 M NaCl, 1% Triton X-100, 5 mM dithiothreitol (DTT), protease and phosphatase inhibitor cocktail tablet (0.75 mg/mL), 1 mM PMSF, and 2 μg/mL leupeptin (pH 7.4)] using a Polytron electric homogenizer for 30 s on ice. The homogenate was centrifuged at 16000g (Eppendorf centrifuge 5415R, Hamburg, Germany) for 10 min at 4 °C; the supernatant was discarded, and the pellet was resuspended in 6 mL of protein extraction buffer [0.7 M LiCl, 25 mM Tris, 5 mM EGTA, 0.1 mM CaCl2, 5 mM DTT, 1 mM PMSF, 2 μg/mL leupeptin, and protease and phosphatase inhibitor cocktail (0.75 mg/mL) (pH 8.0)]. Protein extraction was performed with agitation on a nutating mixer (Fisher Scientific Inc., Pittsburgh, PA) at 4 °C for 45 min. The sample was then centrifuged at 16000g (Eppendorf centrifuge 5415R) for 5 min to collect the supernatant. The supernatant was further centrifuged at 55000 rpm (Beckman L-55 ultracentrifuge) (Beckman Coulter, Fullerton, CA) for 45 min to completely remove tissue debris before affinity chromatography purification. The supernatant was incubated with 0.25 mL of CNBr-activated Sepharose CL-4B conjugated with 1.25 mg of monoclonal cTnI antibody [anti-troponin I monoclonal antibody 14G5 and MF4 (Hytest)] for 35 min at 4 °C to ensure complete binding of the troponin complex to the antibody. After being washed with 2 mL of extraction buffer, the bound troponin complex was eluted with 100 mM glycine (pH 2.0) into four 0.4 mL fractions and neutralized immediately with 40 μL of 1 M MOPS (pH 9.0). Fractions were analyzed for enriched protein content on 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels stained with Coomassie blue. Analytical reproducibility was assessed with three replicates.
Top-Down Mass Spectrometry
Immunoaffinity-purified human and mouse cTn complexes were separated and desalted using an offline reverse phase protein microtrap (Michrom Biore-sources, Inc.), with a two-step reverse phase elution method, first with 1% acetic acid in a 50:50 methanol/water mixture and then 1% acetic acid in a 75:25 methanol/water mixture.30,33 Desalted samples were analyzed using a 7T linear ion trap/Fourier transform ion cyclotron resonance (FTICR) hybrid mass spectrometer (LTQ FT Ultra, Thermo Scientific Inc., Bremen, Germany) equipped with an automated chip-based nano ESI source (Triversa NanoMate) (Advion BioSciences, Ithaca, NY). The spray voltage was 1.2–1.6 kV versus the inlet of the mass spectrometer, resulting in a flow of 50–200 nL/min. Transmission of the ion into the linear trap and subsequently into the FTICR cell was automatically optimized for a maximal ion signal. The numbers of accumulated ions for the full scan linear trap (IT), FTICR cell (FT), MSn FTICR cell, and ECD were 3 × 104, 8 × 106, 8 × 106, and 8 × 106, respectively. The resolving power of the FTICR mass analyzer was typically set at 200,000 at m/z 400. For CAD and ECD fragmentation, individual charge states of protein molecular ions were first isolated and then dissociated using 15–25% normalized collision energy (CAD) or 2–3% electron energy (ECD) with a duration of 25–75 ms with no delay. Typically, 1000–3000 transients were averaged to ensure high-quality ECD spectra.
All FTICR spectra were processed with Xtract (FT programs 2.0.1.0.6.1.4, Xcalibur 2.0.5, Thermo Scientific Inc.) using a signal-to-noise ratio (S/N) threshold of 1.5 and a fit factor of 40% and validated manually. The resulting mass lists were further assigned using in-house developed “Ion Assignment” software (version 1.0) based on the protein sequence of hcTnT and mcTnT obtained from the UniProtKB/Swiss-Prot protein knowledgebase (http://www.uniprot.org/uniprot/) and the NCBI protein database (http://www.ncbi.nlm.nih.gov/). Allowance was made for possible PTMs such as the removal of the initial Met, acetylation of the new N-terminus, and variable phosphorylation sites (residues Ser, Thr, and Tyr), using tolerances of 10 and 20 ppm for precursor and fragment ions, respectively. The assigned ions were manually validated to ensure the quality of the assignments. For fragment ions containing possible phosphorylation sites, the masses of fragment ions were manually examined for 80 Da mass shifts to confirm or exclude the existence of phosphorylation. All reported masses are the most abundant masses.
RESULTS
SDS–PAGE and High-Resolution FTMS of Mouse and Human cTnT
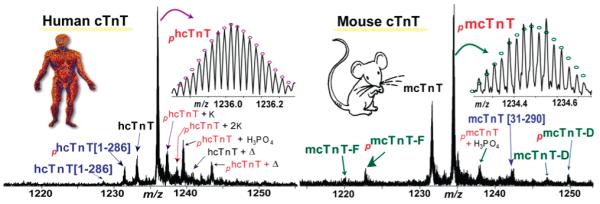
SDS–PAGE analysis of the immunoaffinity-purified proteins from human (Figure 1A) and mouse (Figure 1B) heart samples consistently revealed three major bands representing the three cTn protein subunits, cTnC, cTnI, and cTnT, in an approximately equal molar ratio. The comparatively weaker intensity of the cTnC band is probably due to the known relatively weaker Coomassie staining for cTnC.42 This indicated that the immunoaffinity method used in this study has high specificity for both human and mouse cTn samples. High-resolution and high-accuracy FTMS analysis provides a global view of all detectable protein forms present in human and mouse cTnT, respectively (Figure 2). The molecular masses of 34500.70 and 34580.59 Da from the hcTnT sample corresponded to unphosphorylated (hcTnT) and monophosphorylated (phcTnT) forms, respectively, of hcTnT sequence TNNT2_HUMAN isoform 6 (UniProtKB/Swiss-Prot entry P45379-6) (Figure 3) with the removal of the N-terminal Met and acetylation at the new N-terminus. N-Terminal acetylation was present in both protein forms, which has been observed extensively for eukaryotic proteins.43 There were also small amounts of potassium (+K) and noncovalent phosphoric acid adducts (+H3PO4) detected for hcTnT. A minor degradation product, cTnT[1–286], which resulted from the loss of the C-terminal Lys, was detected. We have observed only one isoform of hcTnT purified from healthy human hearts, in agreement with the previous report by Jin and co-workers.44 The other isoforms produced by alternative splicing as denoted in the UniProtKB/ Swiss-Prot protein database (http://www.uniprot.org/uniprot/P45379) (Figure S1 of the Supporting Information) were indiscernible here. Intriguingly, another peak with a mass corresponding to hcTnT isoform 6 plus 210 Da was observed in both monophosphorylated and unphosphorylated forms. However, the molecular masses do not match with any alternatively spliced isoforms or natural variants as reported in the UniProtKB/Swiss-Prot or NCBI database even with the consideration of the common N-terminal Met loss and acetylation. Therefore, it likely resulted from a less common PTM, e.g., myristoylation, which adds 210 Da to the protein. Because of the low abundance of this species and the limited amount of clinical tissue samples, we were unable to conduct MS/MS experiments to confirm such a possible assignment.
Figure 1.
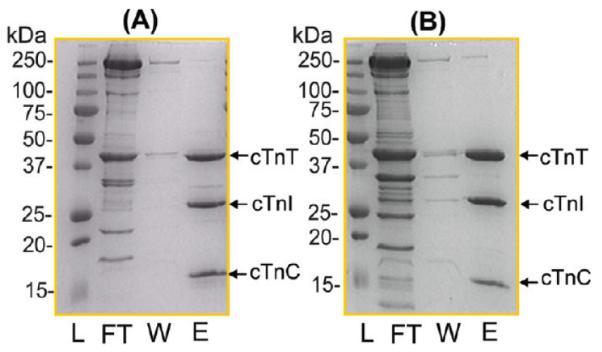
SDS–PAGE analysis of immunoaffinity-purified cTn complexes from (A) human and (B) mouse heart tissue samples. L, protein mass ladder; FT, sample flow through; W, wash; E, elution.
Figure 2.

High-resolution ESI/FTMS spectra of intact cTnT purified from (A) human and (B) mouse heart samples. hcTnT isoform 6 (P45379-6) and mcTnT isoform E (P50752-3) were denoted as hcTnT and mcTnT, respectively. mcTnT-D and mcTnT-F stand for isoform D (P50752-4) and isoform F (P50752-2) of mcTnT, respectively. The subscript p stands for phosphorylation (80 Da), +K for the potassium adduct (38 Da), +H3PO4 for the noncovalent adduct of phosphoric acid (98 Da), and Δ for 210 Da. Insets show high-accuracy measurements of un- and monophosphorylated hTnT (A) and mcTnT (B). Circles represent the theoretical isotopic abundance distributions of the isotopomer peaks corresponding to the assigned mass. Calcd denotes the calculated most abundant mass and exptl the experimental most abundant mass.
Figure 3.

Protein sequence homology alignment of hcTnT and mcTnT. The isoforms presented here are detected in healthy adult human and mouse hearts. Protein sequences were retrieved from the UniProtKB/Swiss-Prot database: isoform 6 of hcTnT (P45379-6_HUMAN) also known as TNT3, isoform E of mcTnT (P50752-3_MOUSE) also known as isoform A2 in the UniProtKB/Swiss-Prot database, isoform F of mcTnT (P50752-2_MOUSE) also known as isoform A3B in the UniProtKB/Swiss-Prot database, and isoform D of mcTnT (P50752-4_MOUSE) also known as isoform A1 in the UniProtKB/Swiss-Prot database. Exon numbers and boundaries are indicated. Asterisks indicate the conserved sequence regions.
In comparison, mcTnT samples purified from wild-type mouse heart showed significantly higher molecular heterogeneity (Figure 2B). Three mouse cTnT isoforms (Figure 3) have been detected, corresponding to isoform D (mcTnT-D, exptl, 34885.75 Da; calcd, 34885.62 Da), isoform E (mcTnT-E, exptl, 34457.54 Da; calcd, 34457.57 Da), and isoform F (mcTnT-F, exptl, 34129.41 Da; calcd, 34129.84 Da), with the removal of the N-terminal Met and acetylation at the new N-terminus. The predominant isoform was mcTnT-E (accession number P50752-3 in the UniProtKB/SwissProt database and NP_00112365 in the NCBI protein database), termed mcTnT hereafter. The molecular species at 34457.54 and 34537.49 Da (Figure 2B) corresponded to the unphosphorylated and monophosphorylated mcTnT, respectively, both with the removal of the N-terminal Met and addition of acetylation at the new N-terminus. The other two isoforms, mcTnT-D and -F (accession numbers P50752-4 and −2, respectively, in the UniProtKB/SwissProt database), are present in much lower abundance. These three observed mcTnT isoforms, along with three other larger mcTnT isoforms, were aligned to show the multiprotein sequence homology (Figure S2 of the Supporting Information). The larger isoforms, mcTnT-A, -B, and -C as reported in the NCBI protein database, all express fetal-specific exon 5 and thus were not detected in the adult mouse hearts. In addition to the alternatively spliced isoforms, we have also observed a proteolytic degradation product, mcTnT[P31–K290], consistent with our previous analysis of rat cTnT.42
Monophosphorylation was the predominant PTM in both human and wild-type mouse cTnT. For human cTnT, the relative percentages of unphosphorylated (hcTnT) and monophosphorylated (phcTnT) forms were 22.7 ± 2.5 and 77.3 ± 1.8%, respectively. For mouse cTnT isoform E (the major isoform), the relative percentages of unphosphorylated (mcTnT) and monophosphorylated (pmcTnT) forms were 46.3 ± 2.2 and 53.7 ± 1.6%, respectively. The two other minor isoforms (isoforms D and F) had monophosphorylation patterns similar to that of mcTnT isoform E. No bisphosphorylated form was detected for hcTnT or mcTnT (estimated to be <1% of the total cTnT population), indicating the absence of higher-order phosphorylation in human and mouse cTnT samples analyzed, or below the detection limit of the FTMS method used in this study (Figure 2). Subsequently, we have performed MS/MS to identify the phosphorylation sites in mcTnT and hcTnT (Figures 4-6) as well as the characterization of the alternative splicing isoforms and proteolytic degradation products (Figures 7 and 8).
Figure 4.

Representative MS/MS product ions of monophosphorylated mouse cTnT for the identification of phosphorylation sites in wild-type mouse heart samples. (A–F) ECD product ions. No neutral loss of phosphate group is observed in ECD spectra. The triangle shows the expected position for the loss of H3PO4 ( −98 Da) peak that is not observed here. (G–L) CAD product ions. The neutral loss of the phosphate moiety is detected in phosphorylated b ions. −98 indicates the loss of H3PO4. Note that the masses of b ions (e.g., b6) equal those for the loss of HPO3 ( −80 Da) from their phosphorylated counterparts (e.g., pb6). The dashed arrow indicates the theoretical position for the labeled ions that is not observed here. The subscript p stands for product ions carrying phosphorylations. Circles show the theoretical isotopic abundance distribution of the isotopomer peaks corresponding to the assigned mass.
Figure 6.

MS/MS product ion maps for localization of cTnT phosphorylation sites. (A) Fragmentation map of monophosphorylated cTnT (pmcTnT) in wild-type mouse heart samples from two ECD and two CAD data. (B) Fragmentation map of monophosphorylated cTnT (phcTnT) in healthy human heart samples summarized from one ECD and two CAD data. Phosphorylation sites are highlighted in circles. The same phosphorylation site (Ser1) was found for both pmcTnT and phcTnT.
Figure 7.

MS/MS characterization of the alternatively spliced isoform of mcTnT. (A) High-accuracy mass measurement of monophosphorylated mTnT isoform F (pmcTnT-F). (B–E) Representative fragment ions from CAD of pmcTnT-F. −98 Da indicates the neutral loss of H3PO4. (F) Fragmentation map of one single CAD spectrum of pmcTnT-F that suggests that the N-terminus is both acetylated (−Ac) and phosphorylated (circled).
Figure 8.

MS/MS characterization of the proteolytic degradation product of mcTnT. (A) High-accuracy mass measurement of mTnT-[P31–K290]. (B–E) Representative fragment ions from CAD of mTnT[P31–K290]. (F) Fragmentation map of one single CAD spectrum of mTnT[P31–K290] that suggests the N-terminus is not acetylated or phosphorylated.
Localization of the Phosphorylation Site in Mouse cTnT
To determine the phosphorylation site in mouse cTnT, a single charge state of precursor ions of monophosphorylated mouse cTnT (mcTnT-E) was isolated and fragmented by both ECD and CAD (Figure 4). One representative ECD spectrum generated 112 validated high-quality MS/MS peaks (Figure S3A of the Supporting Information), including 22 c ions and 90 z• ions. Much more extensive protein backbone cleavage at the C-terminus was observed compared to what occurred at the N-terminus. This is likely due to the highly acidic N-terminus (28 Glu and 3 Asp residues in the first 60 residues at the N-terminus) in the primary sequence of mouse cTnT. These negatively charged residues have a low electron affinity that substantially reduced the fragmentation efficiency of ECD. Figure 4A–F shows representative c and z• ions observed for monophosphorylated mcTnT. All c ions (c27–c289) were detected only in the monophosphorylated form with no unphosphorylated counterpart (Figure 6A). The smallest c ion observed was monophosphorylated c27 (pc27), suggesting the phosphorylation was located in the first 27 residues at the N-terminus (Figure 4A). Given the fact that only two possible residues for phosphorylation were present in this N-terminal region (Ser1 and Tyr10), Ser1 was likely to be the phosphorylation site in this monophosphorylated mouse cTnT, because the occurrence of Tyr phosphorylation is much less frequent than that of Ser/Thr phosphorylation (~90% for Ser, ~10% for Thr, and ~<0.05% Tyr).45 Because ECD is a nonergotic method that is known to keep labile modifications such as phosphorylation, the absence of unphosphorylated c ions (indicated by the dashed arrows in Figure 4A–F) was also able to provide evidence of nearly exclusive phosphorylation occupancy at Ser1. In agreement, all z• ions were detected in their unphosphorylated forms, with the largest z• ions being z•280, suggesting that the first 280 amino acids counting from the C-terminus were not phosphorylated, which confirmed that the phosphorylation site is located in the first 10 amino acid residues of the N-terminal region (note that the full-length mcTnT has a total of 290 amino acids after the removal of the N-terminal Met).
We also performed fragmentation of monophosphorylated mouse cTnT by CAD, a complementary MS/MS technique, to provide additional information especially near the highly acidic N-terminus where few c fragmentation ions from ECD were detected. Through fine-tuned CAD conditions, we were able to generate consistent backbone cleavages at the N-terminus (Figure S3B of the Supporting Information). CAD fragmentation produced continuous cleavages in the N-terminal region of pmcTnT, generating a continuous monophosphorylated b ion ladder, including pb5–pb13 (Figure 6A). The neutral loss of phosphoric acid (H3PO4, −98 Da) is evident in CAD spectra (Figure 4G–J and Figure S3B of the Supporting Information). This is consistent with the energetic dissociation nature of CAD that may potentially knock off labile phosphorylation because of the high-energy collision with inert gas.46 Figure 4G–L shows representative b and y ions from CAD fragmentation. These monophosphorylated b6 and b7 ions (Figure 4G,H) provide strong evidence supporting the possibility that phosphorylation occurred at the first six amino acids, suggesting that Ser1 is the phosphorylation site in the monophosphorylated mcTnT. The monophosphorylated b ions were also accompanied by a small amount of corresponding b ions at −80 Da. The mass of the unphosphorylated b ion is the same as that of the monophosphorylated b ion after the loss of metaphosphoric acid (HPO3, −80 Da). These CAD fragment ions all resulted from cleanly isolated monophosphorylated mcTnT precursor ions (like “gas phase” purification).33 Therefore, these ions at −80 Da can be either the loss of HPO3( −80 Da) from its monophosphorylated counterpart or unphosphorylated b ions resulting from a minor amount of positional isomers with phosphorylation occupancy at another hypothetical site beyond the N-terminus. However, our ECD data did not support such a possibility of another phosphorylation site beyond the N-terminus. Moreover, the loss of HPO3 was detected as the predominating neutral loss in the CAD spectrum of human cTnI.31
Overall, two ECD spectra of pmcTnT generate 58 c ions and 120 z• ions with 30 complementary pairs covering the entire sequence. On the other hand, two CAD data produced 23 b ions and 48 y ions with 3 complementary pairs (Figure 6A). These combined ECD and CAD data unambiguously identified Ser1 as the phosphorylation site in the monophosphorylated cTnT from the wild-type mouse.
Localization of the Phosphorylation Site in hcTnT
We performed both ECD and CAD fragmentation to determine the phosphorylation site in the monophosphorylated hcTnT. There is a high degree of sequence homology (~88.5%) between the hcTnT sequence and mcTnT isoform E (the major isoform) (Figure 3). Notably across species, the C-terminal and central region of cTnT is well-conserved whereas the N-terminal region is hypervariable. Nevertheless, hcTnT also has an acidic N-terminal region similar to that of mcTnT. Hence, ECD fragmentation was not able to generate efficient cleavage at the N-terminus of hcTnT because of the highly negatively charged sequence. In contrast, extensive cleavages were observed at the C-terminus (Figures 5 and 6B). Representative ECD product ions are shown in Figure 5A–F. All the c ions were detected consistently only in the monophosphorylated form, and the smallest monophosphorylated c ion was the pc69 ion (Figure 6B). This indicates the nearly exclusive phosphorylation among the first 69 N-terminal amino acid residues. Importantly, z• ions were present only in their unphosphorylated forms, and no phosphorylated z• ions were detected. The largest z• ion observed is unphosphorylated z•286 containing the entire sequence except the first amino acid (the full-length sequence has 287 amino acid residues), which provided evidence that the phosphorylation site was located in the first amino acid residue at the N-terminus, Ser1. Overall, a single ECD spectrum of phcTnT generated 35 c ions and 95 z• ions with 21 complementary pairs covering the entire sequence.
Figure 5.

Representative MS/MS product ions for phosphorylation site mapping in hcTnT purified from normal human heart samples. (A–F) ECD product ions of pcTnT. No neutral loss of a phosphate group is observed in ECD spectra. The triangle shows the expected position for the loss of H3PO4 ( −98 Da) peak that is not detected here. (G–L) CAD product ions of monophosphorylated hcTnT (pcTnT). The neutral loss of the phosphate moiety is detected in phosphorylated b ions. −98 indicates the loss of H3PO4. The masses of b ions (e.g., b6) equal those for the loss of HPO3 ( −80 Da) from their phosphorylated counterparts (e.g., pb6). The dashed arrow indicates the theoretical position for the labeled ions that is not observed here. The subscript p stands for product ions with phosphorylation. Circles show the theoretical isotopic abundance distribution of the isotopomer peaks corresponding to the assigned mass.
Moreover, CAD fragmentation of phosphorylated hcTnT generated extensive protein backbone cleavages at both N- and C-termini (Figure S5 of the Supporting Information). A continuous sequence ladder was generated in the N-terminal sequence, including monophosphorylated pb4–pb15, which confirms that Ser1 is the phosphorylation site in the monophosphorylated hcTnT (Figures 5G–L and 6B). Similar to the mcTnT data, the neutral loss of H3PO4 ( −98 Da) and HPO3 ( −80 Da) was evident in CAD spectra (Figure 5 and Figure S5 of the Supporting Information). All the y ions were detected only in their unphosphorylated forms (their monophosphorylated counterparts were not detectable) (Figure 6B). The fact that the largest y ion, y284 (including the first 284 amino acids counting from the C-terminus) (Figure S5 of the Supporting Information), was observed only in its unphosphorylated form confirmed the phosphorylation was on the first three amino acids (Ser-Asp-Ile), with Ser1 as the only possible phosphorylation site. Here two CAD spectra produced 32 b ions and 70 y ions with 14 complementary pairs (Figure 6B). Taken together, these two complementary fragmentation methods of CAD and ECD consistently support the suggestion that Ser1 is the exclusive phosphorylation site in hcTnT.
Characterization of Alternatively Spliced Isoforms and Proteolytic Degradation in mcTnT
We have obtained a high-accuracy mass measurement of monophosphorylated isoform F of mcTnT (pmcTnT-F, exptl, 34209.55 Da; calcd, 34209.70 Da, 4.4 ppm) as shown in Figure 7A. Subsequently, a single charge state of pmcTnT-F (M28+) was isolated and subjected to CAD. High-quality CAD data were obtained despite the relatively low abundance of precursor ions with representative b/y fragment ions shown in Figure 7B–D. The loss of the phosphorylation group was evident in b ions (Figure 7B,C), which is consistent with that observed in the CAD spectra of the most abundant isoform E of mcTnT. The neutral loss of phosphate as HPO3 or H3PO4 is commonly observed in CAD of phosphoprotein/peptides.46,47 From a single CAD spectrum, we have detected a total of 14 b ions and 39 y ions with 3 complementary pairs covering the entire sequence (Figure 7F). The observed fragment ions are in good agreement with mcTnT-F (UniProtKB/SwissProt entry P50752-2) with acetylation and phosphorylation at the N-terminus.
Accurate mass measurement was also performed on a molecular ion with a mass of 31032.39 Da, which matched with a proteolytic fragment of mcTnT, mcTnT[P31–K290] (calcd, 31032.79 Da) (Figure 8A). The subsequent CAD of the precursor ions resulted in high-quality b/y fragment ions (Figure 8B–E) with high mass accuracy (1–5 ppm). Because of the low abundance of the precursor ions, 11 b ions and 23 y ions were observed from one single CAD spectrum (Figure 8F). Nonetheless, we have observed 5 b/y complementary pairs (e.g., b143/y117, b97/y163) covering the full sequence. The fragment ions are consistent with the sequence of mcTnT[P31–K290] with no modification at the N-terminus.
DISCUSSION
Physiological Significance of cTnT Alternative Splicing
Alternative RNA splicing is recognized as an important feature of cTnT.3,4 The mammalian cTnT gene contains three alternatively spliced exons (exons 4, 5, and 13) in addition to the 14 constitutively expressed exons.3,5 Exon 5 in the N-terminal variable region is expressed only in the embryonic isoforms of mammalian cTnTs, whereas exons 4 and 13 of cTnT gene are alternatively spliced in a manner independent of the developmental stage.3,4 Although there are a total of 10 isoforms produced by alternative splicing as annotated in the UniProtKB/SwissProt protein database (http://www.uniprot.org/uniprot/P45379) (Figure S1 of the Supporting Information), eight isoforms contain the exon 5-expressed sequence and are thus embryonic isoforms, and only two isoforms (isoform 6 with accession number P45379-6 and isoform 7 with accession number of P45379-7) are adult isoforms. Reexpression of embryonic forms of hcTnT in adult heart has been associated with the development of heart failure and reduced heart efficiency.4,13 Here we have observed only isoform 6 of hcTnT (also known as TNT3) in healthy human hearts, which agrees with the previous report by Jin and co-workers using Western blot analysis.44 Exon 4 is not expressed in hcTnT isoform 7 (also known as TNT4), resulting in a lower molecular mass (<499 Da) compared to that of hcTnT isoform 6 (TNT3) because of the difference of five amino acids (Ala-Ala-Vla-Glu-Glu). HcTnT isoform 7 (TNT4) has been detected only in the hypertrophic and failing hearts. The coexpression of exon 4-excluded hcTnT isoform 7 (TNT4) with isoform 6 (TNT3) in human failing hearts suggests a pathological correlation with cTnT alternative splicing.44,48,49 Therefore, the fact that we observed only a single adult isoform 6, TNT3, in our top-down FTMS data is expected because our human sample is obtained from healthy adult human heart.
In contrast, we have detected one major and two minor alternatively spliced isoforms of mcTnT purified from wild-type healthy adult mice (Figure 2) that matched with the three adult mouse cTnT isoforms as reported in the UniProtKB/SwissProt database (http://www.uniprot.org/uniprot/P50752) based on a previous cDNA expression report by Jin et al.50 In addition to the three adult forms, only one embryo-specific isoform is annotated in the UniProtKB/SwissProt database, whereas at least three embryo-specific isoforms of mcTnT are reported in the NCBI protein database (http://www.ncbi.nlm.nih.gov/protein/?term=mouse+cTnT) (Figure S2 of the Supporting Information). These cTnT isoforms are produced from alternative splicing of exons 4, 5, and 13. Interestingly, exon 4 is not expressed in the major mcTnT isoform E detected here. None of the three mcTnT isoforms observed (Figure 2B) contain the embryo-specific exon 5 coding sequence, which is consistent with the expectation for the adult mouse hearts studied here. The alternative splicing of the N-terminal hypervariable region does not eliminate the core function of cTnT.4 A recent transgenic mouse study by Jin and co-workers indicates the coexistence of embryonic cTnT, and exon 7 reduces myocardial efficiency.13 Note that the three isoforms (isoforms D–F) observed here are the natural variants for mcTnT from the healthy adult mouse hearts. Nevertheless, the functional and physiological significance of these three mcTnT isoforms for the mouse cardiac function remains to be further elucidated.
To address the concern about whether the affinity method may fail to detect some cTnT splicing isoforms because of the lack of recognition of these isoforms by the anti-cTnT antibody employed, we have used anti-cTnI monoclonal antibodies instead of anti-cTnT antibodies. The cTnT was copurified with cTnI in an equal molar ratio because of its strong interaction with cTnI in the formation of the cTn complex.1,33,42 Moreover, we have used two types of anti-cTnI monoclonal antibodies with distinct epitopes at cTnI amino acid residues 1–23 and 190–197, which yielded comparable results (data not shown). Therefore, we reason that the antibodies employed in the immunoaffinity purification here should have a minimal influence on the selection of cTnT splicing isoforms.
Conserved Phosphorylation Site in hcTnT and mcTnT
Our top-down MS/MS data showed that both hcTnT and mcTnT are predominantly monophosphorylated and unequivocally identified the phosphorylation site as Ser1. Our previous study of rat cTnT also showed that it is predominantly monophosphorylated but was unable to localize the site of phosphorylation with any precision even though the data argued strongly against phosphorylation at the C-terminus and were consistent with Ser1 phosphorylation.42 These results are in accord with the well-known structure of cTnT in which the N-terminal region of cTnT adopts an extended conformation because of its highly charged residues and therefore makes the Ser1 residue easily accessible to kinase activities. Early studies of cTnT also determined that Ser1 was the predominant phosphorylation site in rabbit fsTnT and bovine cTnT and suggested that troponin T kinase20 and phosphorylase b kinase21 were involved in phosphorylation via a combination of in vitro 32P labeling kinase assay, enzymatic digestion, and Edman sequencing.5,20,21,51,52 Phosphorylase b kinase can phosphorylate Ser1 in the presence or absence of Ca2+ and also phosphorylate other sites, including Ser149/150 and Ser156/157.5,21 However, these early studies have not been pursued further so the significance of the N-terminal Ser1 phosphorylation remains elusive.22
The N-terminal region of TnT is hypervariable in length and amino acid sequence among vertebrate species in contrast to the highly conserved central and C-terminal region.3,4 Phylogenetic data suggested that the N-terminal region has evolved as an addition to the conserved core structure of TnT from nearly absent in some fish fsTnT to more than 70 amino acids in avian and mammalian cTnT.3 The N-terminal variable region does not bind other thin filament components.3,4 However, results from Jin and co-workers suggested that the structure of the N-terminal variable region modulates the overall conformation of the protein with a potential functional significance.3 Earlier results of Tobacman et al. suggested that the variable region influences the overall response of the thin filament to Ca2+, supporting the hypothesis that the regulation of this region by mRNA splicing may modulate muscle function.53 A more recent study by the same group showed that the N-terminal tail of cTnT is an important element in establishing the diastolic state and cTnT[1–153] alone can induce a blocked state of the myofilaments in the complete absence of cTnI.54 Removal of the N-terminal region from cTnT, fsTnT, and ssTnT was shown to decrease the maximal force and increase the binding affinity for Tm compared with that of intact TnT.14,55,56 Although the core structure of TnT confers the basal function in maintaining muscle contractility, the evolutionary addition of the hypervariable N-terminal domain was shown to regulate the interaction of TnT within the thin filament systems through charge variation-based long-range conformational effects, thereby modulating the conformation and function of TnT core to fine-tune muscle contractility.3,4,14
Phosphorylation of cTnT is also known to be regulated by other kinases, including the PKC,9,12,15 ROCK-II,16 ASK-1,17 and Raf-1,18 mainly on the basis of in vitro phosphorylation experiments. PKC phosphorylation sites of cTnT at Thr197, Ser201, Thr206, and Thr287 in the C-terminal region were identified as being functionally important.9,12,15 Moreover, a recent study by Sumandea et al. on isometric tension development and actomyosin Mg-ATPase activity identified Thr206 as a functionally critical site for cTnT because this site alone is sufficient to reduce the maximal tension.19 Recently, Thr206 of cTnT was also identified as a substrate for a novel cTnT kinase, Raf-1.18 Vahebi et al. demonstrated that ROCK-II phosphorylates TnT at Ser278 and Thr287 and depresses the maximal myofilament tension, ATPase activity, and Ca2+ sensitivity likely by a mechanism dependent on phosphorylation of cTnT.16 ASK-1 is strongly expressed in cardiac muscle and has been shown to specifically phosphorylate cTnT at the ASK1 phosphorylation consensus sequence (T194/S198) by a mutagenesis study.17 cTnT phosphorylation by ASK1 has been shown to inhibit shortening and the Ca2+ transient in adult cardiomyocytes and, thus, plays an important role in the regulation of cardiac contractile function and cytokine/ROS-induced pathogenesis of cardiomyopathy and heart failure.17 Unlike cTnI, cTnT was known to be a poor substrate for protein kinase A (PKA).5 In contrast to these in vitro and ex vivo findings, our top-down MS data unambiguously identified Ser1 as the nearly exclusive phosphorylation site under basal in vivo conditions in both healthy human and mouse hearts. Because every analytical method has its detection limit, there is always the possibility that other phosphorylation sites are present but at extremely low occupancy (i.e., <1%), which may be below the detection limit.33 In addition, PKC phosphorylation may be transient and relevant on a beat-to-beat basis responding to stress.57 More importantly, it is reasonable to believe that other phosphorylation sites regulated by PKC, ROCK-II, and Raf-1 might result from maladaptive signaling in the cardiomyocytes so that they are present in only diseased hearts. Likewise, we have observed conserved phosphorylation at only Ser22/23 (PKA sites) in cTnI purified from healthy adult hearts of human, swine, and rodents.30,31,33,42 PKC phosphorylation of Ser43/45 in cTnI is believed to be maladaptive, which is associated with contractile dysfunction and may contribute to the progression of failure.11,58
Distinct Proteolytic Pattern in hcTnT and mcTnT
We have observed minor proteolytic products for both hcTnT and mcTnT, albeit with distinct patterns. For human cTnT, we have observed a minor proteolytic fragment, hcTnT[1–286], with the loss of the last C-terminal amino acid residue (Lys287) from the full-length sequence. The observation of phosphorylation of this proteolytic fragment confirmed the intactness of its N-terminal sequence and the presence of Ser1. For mouse cTnT, we have consistently detected a single proteolytic product that matched the sequence of mcTnT[P31–K290]. The lack of acetylation at the N-terminus of this truncated mcTnT form confirms that it indeed resulted from proteolysis of full-length mcTnT rather than via transcriptional or translational mechanisms in which the N-terminus is intact and prone to acetylation. This is consistent with our previous study of rat cTnT in which a proteolytic degradation product of cTnT[P30–K288] was observed42 (note that mcTnT has one extra amino acid in the N-terminal region compared with rat cTnT). The proteolytic loss of the N-terminal amino acid residues is conceivable because the N-terminal domain of cTnT is highly acidic, which is predicted to be disordered and unstable.42 Previously, Jin and co-workers reported that a truncated cTnT with the removal of 71 residues from the N-terminus was produced during myocardial ischemia reperfusion, suggesting the thin filament adapted to a sustained cardiac function under stress conditions.59,60 We have not observed such a proteolytic fragment here, which is conceivable because the cTnT analyzed in this study was purified from healthy heart tissues. Nevertheless, the apparent distinct patterns detected for hcTnT and mcTnT may reflect a species difference between rodents and humans. Similarly, disparity between species has also been reported for proteolytic degradation of cTnI, for which a degradation product of cTnI was found in rat myocardium but not in large animal or in human myocardium, suggesting that the cTnI degradation might be species-dependent.61
Complementary MS/MS Techniques for Comprehensive Characterization of Acidic Proteins
cTnT is a fairly acidic molecule, especially at the N-terminus. hcTnT contains 31 Glu and 4 Asp residues in the first 60 residues in the N-terminal region, compared with mcTnT, which contains 28 Glu and 3 Asp residues. As mentioned above, the ECD fragmentation efficiency is hindered because of the low electron affinity of the negatively charged residues. Here in ECD spectra of mcTnT and hcTnT (Figure 6), we have observed very limited fragment ions in the N-terminal region, whereas extensive fragment ions were detected at the relatively basic C-terminus. Thus, to achieve a comprehensive characterization of the whole protein sequence, we have used two complementary MS/MS techniques, ECD and CAD. The extensive fragmentation by CAD in the N-terminal region is observed, which can be rationalized by the highly acidic residue composition of the N-terminal sequence. Although these acidic residues (Glu) are poor electron acceptors because of their negative charges, these sites are preferential cleavage sites in CAD because of their high proton affinity, and therefore, continuous backbone cleavages were generated along the sequence of these acidic residues.62-64 On the other hand, ECD produced many more fragment ions than CAD because of the nonergodic and nonselective nature of ECD, which essentially can cleave any backbone bonds except Pro bonds.27,39 Moreover, although CAD can effectively localize stable modification sites such as acetylation, proteolytic degradation products, and alternatively spliced isoforms, it has difficulty in the accurate determination of phosphorylation occupancy because of the facile neutral loss of phosphate as H3PO4 ( −98 Da) and/or HPO3( −80 Da), which is a favored fragmentation event in CAD of phosphoproteins and peptides.46,47 In particular, the loss of 80 Da from a phosphorylated fragment ion is indistinguishable from the original unphosphorylated fragment ion without neutral loss, which makes it difficult to assess the existence of potential positional isomers and determine the phosphorylation occupancy. For example, unphosphorylated b6 fragment ions (Figures 4 and 5) can result from either the loss of HPO3 ( −80 Da) from its phosphorylation b6 fragment ions or a positional isomer with a hypothetical phosphorylation site beyond the first six amino acids (for a detailed discussion of the positional isomers, see our review paper37). In contrast, no neutral loss of the phosphate moiety was observed in ECD data, which makes it especially valuable to determine the phosphorylation occupancy and accurately quantify the phosphorylated positional isomers.31 Here the lack of detection of unphosphorylated c ions in ECD data confirmed the exclusive phosphorylation at the N-terminus. Therefore, top-down MS/MS with combined ECD and CAD data provides a comprehensive characterization of this acidic N-terminus of cTnT, including acetylation, proteolysis, phosphorylation, and the alternatively spliced isoforms. This demonstrated the power of these two complementary fragmentation techniques in top-down MS for the identification of PTM sites and alternatively spliced isoforms in acidic proteins.
Supplementary Material
ACKNOWLEDGMENT
We thank Drs. Holly Norman, Takushi Kohmoto, and Richard Moss for providing the human heart tissue samples and Dr. Jing Zhang and the late Dr. Jeffery Walker for the mouse heart tissue samples. We are grateful to Dr. Jian-Ping Jin for the insightful discussions and Drs. Raquel Sancho Solis and Beth Altschafl for critical reading of the manuscript.
Funding Sources
Financial support was kindly provided by the Wisconsin Partnership Fund for a Healthy Future and an American Heart Association Scientist Development Grant (to Y.G.).
ABBREVIATIONS
- cTnT
cardiac troponin T
- hcTnT
human cTnT
- mcTnT
mouse cTnT
- PTMs
post-translational modifications
- MS
mass spectrometry
- Tn
troponin
- TnT
troponin T
- Tm
tropomyosin
- TnC
troponin C
- TnI
troponin I
- fsTnT
fast skeletal troponin T
- ssTnT
slow skeletal troponin T
- ELISA
enzyme-linked immunosorbant assay
- PKC
protein kinase C
- ROCK-II
Rho-A-dependent protein kinase
- ASK-1
apoptosis signal-regulating kinase
- PKA
protein kinase A
- MS/MS
tandem mass spectrometry
- CAD
collisionally activated dissociation
- ECD
electron capture dissociation
- FTICR
Fourier transform ion cyclotron resonance
- DTT
dithiothreitol
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- RP-HPLC
reverse phase high-performance liquid chromatography
- S/N
signal-to-noise ratio
Footnotes
ASSOCIATED CONTENT
Supporting Information. Amino acid sequence alignment of multiple hcTnT isoforms (Figure S1), sequence alignment of multiple mcTnT isoforms (Figure S2), representative MS/MS spectra of phosphorylated mcTnT for identification of the phosphorylation site in cTnT purified from wild-type mouse heart samples (Figure S3), representative ECD spectra of phcTnT for the identification of the phosphorylation site in cTnT purified from normal human heart samples (Figure S4), and representative CAD spectra of phcTnT for the identification of the phosphorylation site in cTnT purified from normal human heart samples (Figure S5). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- (2).Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- (3).Jin JP, Zhang ZL, Bautista JA. Isoform diversity, regulation, and functional adaptation of troponin and calponin. Crit. Rev. Eukaryotic Gene Expression. 2008;18:93–124. doi: 10.1615/critreveukargeneexpr.v18.i2.10. [DOI] [PubMed] [Google Scholar]
- (4).Wei B, Jin JP. Troponin T isoforms and posttranscriptional modifications: Evolution, regulation and function. Arch. Biochem. Biophys. 2011;505:144–154. doi: 10.1016/j.abb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Perry SV. Troponin T: Genetics, properties and function. J. Muscle Res. Cell Motil. 1998;19:575–602. doi: 10.1023/a:1005397501968. [DOI] [PubMed] [Google Scholar]
- (6).Babuin L, Jaffe AS. Troponin: The biomarker of choice for the detection of cardiac injury. Can. Med. Assoc. 2005;173:1191–1202. doi: 10.1503/cmaj.050141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: Linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Failure Rev. 2005;10:237–248. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- (8).Chang AN, Potter JD. Sarcomeric protein mutations in dilated cardiomyopathy. Heart Failure Rev. 2005;10:225–235. doi: 10.1007/s10741-005-5252-6. [DOI] [PubMed] [Google Scholar]
- (9).Noland TA, Kuo JF. Protein kinase C phosphorylation of cardiac troponin-I and troponin-T inhibits Ca2+-stimulated MgATPase activity in reconstituted actomyosin and isolated myofibrils, and decreases actin-myosin interactions. J. Mol. Cell. Cardiol. 1993;25:53–65. doi: 10.1006/jmcc.1993.1007. [DOI] [PubMed] [Google Scholar]
- (10).Katoh N, Wise BC, Kuo JF. Phosphorylation of cardiac troponin inhibitory subunit (troponin-I) and tropomyosin-binding subunit (troponin-T) by cardiac phospholipid-sensitive Ca2+-dependent protein-kinase. Biochem. J. 1983;209:189–195. doi: 10.1042/bj2090189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Solaro RJ, Rosevear P, Kobayashi T. The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochem. Biophys. Res. Commun. 2008;369:82–87. doi: 10.1016/j.bbrc.2007.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jideama NM, Noland TA, Raynor RL, Blobe GC, Fabbro D, Kazanietz MG, Blumberg PM, Hannun YA, Kuo JF. Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J. Biol. Chem. 1996;271:23277–23283. doi: 10.1074/jbc.271.38.23277. [DOI] [PubMed] [Google Scholar]
- (13).Feng HZ, Jin JP. Coexistence of cardiac troponin T variants reduces heart efficiency. Am. J. Physiol. 2010;299:H97–H105. doi: 10.1152/ajpheart.01105.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Biesiadecki BJ, Chong SM, Nosek TM, Jin JP. Troponin T core structure and the regulatory NH2-terminal variable region. Biochemistry. 2007;46:1368–1379. doi: 10.1021/bi061949m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sumandea MP, deTombe PP, Solaro RJ. PKC dependent regulation of myofilament function through cardiac troponin T phosphorylation. Circulation. 2003;108:592. [Google Scholar]
- (16).Vahebi S, Kobayashi T, Warren CM, de Tombe PP, Solaro RJ. Functional effects of Rho-kinase-dependent phosphorylation of specific sites on cardiac troponin. Circ. Res. 2005;96:740–747. doi: 10.1161/01.RES.0000162457.56568.7d. [DOI] [PubMed] [Google Scholar]
- (17).He X, Liu Y, Sharma V, Dirksen RT, Waugh R, Sheu S-S, Min W. ASK1 associates with troponin T and induces troponin T phosphorylation and contractile dysfunction in cardiomyocytes. Am. J. Pathol. 2003;163:243–251. doi: 10.1016/S0002-9440(10)63647-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pfleiderer P, Sumandea M, Rybin V, Wang C, Steinberg S. Raf-1: A novel cardiac troponin T kinase. J. Muscle Res. Cell Motil. 2009;30:67–72. doi: 10.1007/s10974-009-9176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac Troponin T. J. Biol. Chem. 2003;278:35135–35144. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- (20).Gusev NB, Dobrovolskii AB, Severin SE. Isolation and some properties of troponin-T kinase from rabbit skeletal-muscle. Biochem. J. 1980;189:219–226. doi: 10.1042/bj1890219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Moir AJG, Cole HA, Perry SV. Phosphorylation sites of troponin-T from white skeletal-muscle and effects of interaction with troponin-C on their phosphorylation by phosphorylase kinase. Biochem. J. 1977;161:371–382. doi: 10.1042/bj1610371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Solaro R, Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. J. Biol. Chem. 2011;286:9935–9940. doi: 10.1074/jbc.R110.197731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mann M, Jensen ON. Proteomic analysis of posttranslational modifications. Nat. Biotechnol. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- (24).McLafferty FW, Fridriksson EK, Horn DM, Lewis MA, Zubarev RA. Biochemistry-Biomolecule mass spectrometry. Science. 1999;284:1289–1290. doi: 10.1126/science.284.5418.1289. [DOI] [PubMed] [Google Scholar]
- (25).Chait BT. Mass Spectrometry: Bottom-up or Top-Down? Science. 2006;314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- (26).Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 1999;121:806–812. [Google Scholar]
- (27).Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park J-H, Begley TP, McLafferty FW. Top down characterization of larger proteins (45 kDa) by electron capture dissociation mass spectrometry. J. Am. Chem. Soc. 2002;124:672–678. doi: 10.1021/ja011335z. [DOI] [PubMed] [Google Scholar]
- (28).Han XM, Jin M, Breuker K, McLafferty FW. Extending top-down mass spectrometry to proteins with masses greater than 200 kDas. Science. 2006;314:109–112. doi: 10.1126/science.1128868. [DOI] [PubMed] [Google Scholar]
- (29).Kelleher NL. Top-down proteomics. Anal. Chem. 2004;76:196A–203A. [PubMed] [Google Scholar]
- (30).Zhang J, Dong X, Hacker TA, Ge Y. Deciphering modifications in swine cardiac troponin I by top-down high-resolution tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2010;21:940–948. doi: 10.1016/j.jasms.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zabrouskov V, Ge Y, Schwartz J, Walker JW. Unraveling molecular complexity of phosphorylated human cardiac troponin I by top-down electron capture dissociation/electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics. 2008;7:1838–1849. doi: 10.1074/mcp.M700524-MCP200. [DOI] [PubMed] [Google Scholar]
- (32).Ge Y, Rybakova IN, Xu QG, Moss RL. Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc. Natl. Acad. Sci. U.S.A. 2009;106:12658–12663. doi: 10.1073/pnas.0813369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ayaz-Guner S, Zhang J, Li L, Walker JW, Ge Y. In vivo phosphorylation site mapping in mouse cardiac troponin I by high resolution top-down electron capture dissociation mass spectrometry: Ser22/23 are the only sites basally phosphorylated. Biochemistry. 2009;48:8161–8170. doi: 10.1021/bi900739f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jebanathirajah JA, Pittman JL, Thomson BA, Budnik BA, Kaur P, Rape M, Kirschner M, Costello CE, O'Connor PB. Characterization of a new qQq-FTICR mass spectrometer for post-translational modification analysis and top-down tandem mass spectrometry of whole proteins. J. Am. Soc. Mass Spectrom. 2005;16:1985–1999. doi: 10.1016/j.jasms.2005.08.008. [DOI] [PubMed] [Google Scholar]
- (35).Ryan CM, Souda P, Bassilian S, Ujwal R, Zhang J, Abramson J, Ping PP, Durazo A, Bowie JU, Hasan SS, Baniulis D, Cramer WA, Faull KF, Whitelegge JP. Posttranslational modifications of integral membrane proteins resolved by top-down Fourier transform mass spectrometry with collisionally activated dissociation. Mol. Cell. Proteomics. 2010;9:791–803. doi: 10.1074/mcp.M900516-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nat. Methods. 2007;4:817–821. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhang H, Ge Y. Comprehensive analysis of protein modifications by top-down mass spectrometry Circ. Cardiovasc. Res. 2011 doi: 10.1161/CIRCGENETICS.110.957829. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Senko MW, Speir JP, McLafferty FW. Collisional activation of large multiply-charged ions using Fourier-transform mass-spectrometry. Anal. Chem. 1994;66:2801–2808. doi: 10.1021/ac00090a003. [DOI] [PubMed] [Google Scholar]
- (39).Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- (40).Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 1998;120:3265–3266. [Google Scholar]
- (41).Cooper HJ, Hakansson K, Marshall AG. The role of electron capture dissociation in biomolecular analysis. Mass Spectrom. Rev. 2005;24:201–222. doi: 10.1002/mas.20014. [DOI] [PubMed] [Google Scholar]
- (42).Solis RS, Ge Y, Walker JW. Single amino acid sequence polymorphisms in rat cardiac troponin revealed by top-down tandem mass spectrometry. J. Muscle Res. Cell Motil. 2008;29:203–212. doi: 10.1007/s10974-009-9168-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Polevoda B, Sherman F. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 2003;325:595–622. doi: 10.1016/s0022-2836(02)01269-x. [DOI] [PubMed] [Google Scholar]
- (44).Huang QQ, Feng HZ, Liu J, Du J, Stull LB, Moravec CS, Huang X, Jin JP. Co-expression of skeletal and cardiac troponin T decreases mouse cardiac function. Am. J. Physiol. 2008;294:C213–C222. doi: 10.1152/ajpcell.00146.2007. [DOI] [PubMed] [Google Scholar]
- (45).Mann M, Ong SE, Gronborg M, Steen H, Jensen ON, Pandey A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002;20:261–268. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- (46).McLachlin DT, Chait BT. Analysis of phosphorylated proteins and peptides by mass spectrometry. Curr. Opin. Chem. Biol. 2001;5:591–602. doi: 10.1016/s1367-5931(00)00250-7. [DOI] [PubMed] [Google Scholar]
- (47).Nelson CA, Szczech JR, Dooley CJ, Xu QG, Lawrence MJ, Zhu HY, Jin S, Ge Y. Effective enrichment and mass spectrometry analysis of phosphopeptides using mesoporous metal oxide nanomaterials. Anal. Chem. 2010;82:7193–7201. doi: 10.1021/ac100877a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Anderson PAW, Greig A, Mark TM, Malouf NN, Oakeley AE, Ungerleider RM, Allen PD, Kay BK. Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circ. Res. 1995;76:681–686. doi: 10.1161/01.res.76.4.681. [DOI] [PubMed] [Google Scholar]
- (49).MesnardRouiller L, Mercadier JJ, ButlerBrowne G, Heimburger M, Logeart D, Allen PD, Samson F. Troponin T mRNA and protein isoforms in the human left ventricle: Pattern of expression in failing and control hearts. J. Mol. Cell. Cardiol. 1997;29:3043–3055. doi: 10.1006/jmcc.1997.0519. [DOI] [PubMed] [Google Scholar]
- (50).Jin JP, Wang J, Zhang JY. Expression of cDNAs encoding mouse cardiac troponin T isoforms: Characterization of a large sample of independent clones. Gene. 1996;168:217–221. doi: 10.1016/0378-1119(95)00803-9. [DOI] [PubMed] [Google Scholar]
- (51).Risnik VV, Dobrovolskii AB, Gusev NB, Severin SE. Phosphorylase-kinase phosphorylation of skeletal-muscle troponin-T. Biochem. J. 1980;191:851–854. doi: 10.1042/bj1910851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Risnik VV, Gusev NB. Some properties of the nucleotide-binding site of troponin-T kinase-casein kinase type-II from skeletal-muscle. Biochim. Biophys. Acta. 1984;790:108–116. doi: 10.1016/0167-4838(84)90213-9. [DOI] [PubMed] [Google Scholar]
- (53).Tobacman LS, Lee R. Isolation and functional comparison of bovine cardiac troponin-T isoforms. J. Biol. Chem. 1987;262:4059–4064. [PubMed] [Google Scholar]
- (54).Tobacman LS, Nihli M, Butters C, Heller M, Hatch V, Craig R, Lehman W, Homsher E. The troponin tail domain promotes a conformational state of the thin filament that suppresses myosin activity. J. Biol. Chem. 2002;277:27636–27642. doi: 10.1074/jbc.M201768200. [DOI] [PubMed] [Google Scholar]
- (55).Pan BS, Gordon AM, Potter JD. Deletion of the first 45 NH2-terminal residues of rabbit skeletal troponin T strengthens binding of troponin to immobilized tropomyosin. J. Biol. Chem. 1991;266:12432–12438. [PubMed] [Google Scholar]
- (56).Chandra M, Montgomery DE, Kim JJ, Solaro RJ. The N-terminal region of troponin T is essential for the maximal activation of rat cardiac myofilaments. J. Mol. Cell. Cardiol. 1999;31:867–880. doi: 10.1006/jmcc.1999.0928. [DOI] [PubMed] [Google Scholar]
- (57).Solaro RJ, van der Velden J. Why does troponin I have so many phosphorylation sites? Fact and fancy. J. Mol. Cell. Cardiol. 2010;48:810–816. doi: 10.1016/j.yjmcc.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Scruggs SB, Walker LA, Lyu T, Geenen DL, Solaro RJ, Buttrick PM, Goldspink PH. Partial replacement of cardiac troponin I with a non-phosphorylatable mutant at serines 43/45 attenuates the contractile dysfunction associated with PKCε phosphorylation. J. Mol. Cell. Cardiol. 2006;40:465–473. doi: 10.1016/j.yjmcc.2005.12.009. [DOI] [PubMed] [Google Scholar]
- (59).Feng HZ, Biesiadecki BJ, Yu ZB, Hossain MM, Jin JP. Restricted N-terminal truncation of cardiac troponin T: A novel mechanism for functional adaptation to energetic crisis. J. Physiol. 2008;586:3537–3550. doi: 10.1113/jphysiol.2008.153577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zhang ZL, Biesiadecki BJ, Jin JP. Selective deletion of the NH2-terminal variable region of cardiac troponin T inischemia reperfusion by myofibril-associated μ-calpain cleavage. Biochemistry. 2006;45:11681–11694. doi: 10.1021/bi060273s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Canty JM, Lee TC. Troponin I proteolysis and myocardial stunning: Now you see it-Now you don't. J. Mol. Cell. Cardiol. 2002;34:375–377. doi: 10.1006/jmcc.2002.1531. [DOI] [PubMed] [Google Scholar]
- (62).Yu W, Vath JE, Huberty MC, Martin SA. Identification of the facile gas-phase cleavage of the Asp-Pro and Asp-Xxx peptide bonds in matrix-assisted laser desorption time-of-flight mass spectrometry. Anal. Chem. 1993;65:3015–3023. doi: 10.1021/ac00069a014. [DOI] [PubMed] [Google Scholar]
- (63).Tsaprailis G, Somogyi A, Nikolaev EN, Wysocki VH. Refining the model for selective cleavage at acidic residues in arginine-containing protonated peptides. Int. J. Mass Spectrom. 2000;195:467–479. [Google Scholar]
- (64).Gu C, Tsaprailis G, Breci L, Wysocki VH. Selective gas-phase cleavage at the peptide bond C-terminal to aspartic acid in fixed-charge derivatives of Asp-containing peptides. Anal. Chem. 2000;72:5804–5813. doi: 10.1021/ac000555c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.