

Abstract
Cancer cells require large amounts of micronutrients, particularly iron, for their rapid growth and frequent divisions. Cellular iron uptake is regulated by the transferrin receptor and the hemochromatosis protein (HFE) system. Two frequent mutations in the HFE gene, H63D and C282Y, are associated with hemochromatosis type I, an inherited iron overload disease and, possibly, with cancer. In this study, we evaluated the frequency of the H63D and C282Y mutations in a cohort of 677 consecutive cases of woman with gynecological pathologies. Cases included 80 women with tumor-free pathologies normal ovary (NOV), 124 with benign ovarian tumors (BOV), 96 with epithelial ovarian cancer (EOC) tumors of low malignant potential (LPM), 264 with invasive tumors of the ovary (TOV) and 113 with endometrial cancer. We found that the C282Y allele frequency in EOC patients was higher than that in the control NOV group (5.8% vs. 1.3%, p < 0.001) and was associated with an increased risk of ovarian cancer (OR = 4.88; 95% CI 1.15–20.61; p = 0.018). The effect of the two HFE mutations on patient survival was also analyzed. Kaplan-Meier analyses did not find any significant association between the H63D allele and patient survival. However, EOC patients with at least one C282Y allele had a decreased overall survival compared to those with no C282Y allele (p = 0.001). These results indicate that the C282Y mutation may increase the risk of developing ovarian cancer and may be further associated with poor outcomes.
Keywords: ovarian cancer, HFE mutations, hemochromatosis, iron, prognosis
Iron is an essential trace element that can be carcinogenic through a variety of mechanisms including acting as an essential nutrient for proliferating tumor cells,1 catalyzing the formation of mutagenic hydroxyl radicals,2 and by suppressing the host immune response.3,4 In the past decades, several genes were identified as being central to the maintenance of iron homeostasis.5 One such gene is HFE, a major histocompatibility class I-like (MHC-I-like) molecule, that, when mutated, may cause hereditary hemochromatosis (HH).6 HH is a common genetic disorder in Caucasian populations that is characterized by high levels of iron absorption from diet, which results in the presence of high circulating iron levels and iron accumulation in the body.7 As with other MHC-I molecules, HFE needs to associate with β2-microglobulin (β2m) for its appropriate expression at the cell surface.8 The two most common mutations in HFE are C282Y, a guanine-to-adenine transition leading to a cysteine-to-tyrosine change and H63D, a cysteine-to-guanine transition causing a histidine-to-aspartic acid change.6
HFE associates with the major protein responsible for cellular iron uptake, namely the transferrin receptor (TfR),9 also called CD71. The association of HFE with TfR at the cell surface lowers TfR affinity for the circulating iron-transporter transferrin, thereby limiting iron uptake and thus directly implicating HFE in the modulation of cellular iron levels.9–12 The C282Y mutation prevents the functional interaction between the HFE and TfR proteins thereby increasing iron uptake. While the interaction between HFE and TfR is not affected by H63D, this mutation fails to decrease the affinity of the TfR protein for transferrin, and could similarly result in increased cellular iron intake.9
Tumor cells express high levels of TfR and internalize iron from transferrin at a tremendous rate.1 In vitro, overexpression of HFE in cancer cell lines leads to a decrease in transferrin-mediated iron uptake and the consequent development of an iron-deficient phenotype.11,13–16 By inference, a failure to appropriately express HFE at the cell surface, as it happens with the C282Y mutated HFE protein, may result in an enhanced ability to capture iron and may hence be advantageous for tumor cell proliferation.
In women, excess iron may contribute to endogenous oxidative stress that is produced in target tissues by estrogen metabolites.2 In fact, there is increasing evidence in support of an association between inflammation and oxidative DNA damage in the estrogen-dependent target organs of cancers, such as breast, ovary and uterus.17 At these sites, in addition to its independent role as a proxidant, high levels of free iron may accentuate the effects of other carcinogenic agents, such as estradiol, ethanol and ionizing radiation.18
Studies on the association of HFE mutations and breast cancer risk have led to contradictory results, with some studies finding a positive association for both the C282Y19–21 and H63D22,23 mutations, while others reporting no association with either mutation.24 In contrast, there is a surprising lack of information whether HFE mutations may be associated with other estrogen-dependent cancers, such as ovarian cancers. Furthermore, no studies have yet evaluated the potential impact of these mutations on disease progression. The main objective of the present study was thus to determine the frequency of the C282Y and the H63D HFE mutations in epithelial ovarian cancer (EOC) and to evaluate their impact on patient survival.
Material and Methods
Patients and blood samples
Blood samples from consecutive patients, who underwent surgery within the Division of Gynecologic Oncology at the Centre Hospitalier de l’Université de Montréal (CHUM) from 1996 to 2004, were collected and immediately processed for DNA extraction. Histopathology, tumor grade and disease stage, as defined by the International Federation of Gynecology and Obstetrics, were determined by an independent pathologist who reviewed and graded the tumor samples.25 Clinical data were extracted from the “Système d’Archivage des Données en Oncologie” (SARDO), which includes entries on tumor grade and stage. Ovarian cancer patients were characterized as either benign (BOV) or low malignancy potential (LMP) and invasive (TOV), which comprise the EOC tumors (mean age of 55.1, 50.6 and 59.8 years, respectively) (Table 1). A control group of patients with nontumor gynecologic pathologies (NOV) (mean age of 49.5 years), who had undergone surgery, had the same diagnostic test as the cases, and were found tumor-free, was also included in the study. Women in this group were diagnosed with diverse types of non-neoplasic pathologies such as: cysts, endometriosis, ectopic pregnancies, fallopian tube pathologies, polyps, fibromas and benign mesonephromas. In addition, 113 patients were diagnosed with endometrial cancer and were included in the study (mean age of 60.9 years). We excluded from the study patients that had cancers that were not primary to the ovary or endometrius (mostly lung and colon metastasic cancers), and patients with germ cell and stromal cell tumors.
Table 1.
Study population
| Variables | Total | NOV | Endometrial cancer | Ovarian tumors
|
||
|---|---|---|---|---|---|---|
| EOC
|
TOV | |||||
| BOV | LMP | |||||
|
|
|
|
|
|||
| n = 677 | n = 80 | n = 113 | n = 124 | n = 96 | n = 264 | |
| Age at diagnosis | ||||||
|
| ||||||
| Median | 54.5 | 49.0 | 61.0 | 53.0 | 49.5 | 60.0 |
|
| ||||||
| Average | 55.2 | 49.5 | 60.9 | 55.1 | 50.6 | 59.8 |
|
| ||||||
| St. dev. | 12.1 | 8.9 | 11.6 | 13.0 | 15.2 | 11.8 |
|
| ||||||
| Range | 14–91 | 26–78 | 31–83 | 27–91 | 14–80 | 14–89 |
|
| ||||||
| Histopathological subtypes | ||||||
|
| ||||||
| Serous (%) | 290 (59.9) | – | – | 65 (52.4) | 46 (47.9) | 179 (67.8) |
|
| ||||||
| Mucinous (%) | 96 (19.8) | – | – | 40 (32.3) | 48 (50.0) | 8 (3.0) |
|
| ||||||
| Endometrioid (%) | 36 (7.4) | – | – | (0.0) | 1 (1.0) | 35 (13.3) |
|
| ||||||
| Other (%) | 46 (9.5) | – | – | 3 (2.4) | 1 (1.0) | 42 (15.9) |
|
| ||||||
| Unclassified (%) | 209 (3.3) | 80 (100) | 113 (100) | 16 (12.9) | – | – |
|
| ||||||
| Grade | ||||||
|
| ||||||
| Grade 0 (%) | 99 (14.6) | – | – | 3 (2.4) | 95 (99.0) | 1 (0.4) |
|
| ||||||
| Grade 1 (%) | 76 (11.2) | – | 45 (39.8) | 1 (0.8) | 1 (1.0) | 29 (11.0) |
|
| ||||||
| Grade 2 (%) | 82 (12.1) | – | 22 (19.5) | – | – | 60 (22.7) |
|
| ||||||
| Grade 3 (%) | 192 (28.4) | – | 21 (18.6) | – | – | 171 (64.8) |
|
| ||||||
| Missing (%) | 228 (33.7) | 80 (100) | 25 (22.1) | 120 (96.8) | – | 3 (1.1) |
|
| ||||||
| Mortality (n = 85) | ||||||
|
| ||||||
| Grade 0 (%) | – | – | – | – | – | 1 (1.2) |
|
| ||||||
| Grade 1 (%) | – | – | – | – | – | 2 (2.4) |
|
| ||||||
| Grade 2 (%) | – | – | – | – | – | 18 (21.2) |
|
| ||||||
| Grade 3 (%) | – | – | – | – | – | 61 (71.8) |
|
| ||||||
| Missing (%) | – | – | 2 (2.4) | – | – | 1 (1.2) |
|
| ||||||
| Mortality (n = 85) | ||||||
|
| ||||||
| Serous (%) | – | – | – | – | – | 66 (77.6) |
|
| ||||||
| Mucinous (%) | – | – | – | – | – | 0 (0.0) |
|
| ||||||
| Endometrioid (%) | – | – | – | – | – | 6 (7.1) |
|
| ||||||
| Other (%) | – | – | 2 (2.4) | – | – | 11 (12.9) |
NOV: normal ovary; BOV: benign tumor of the ovary; LMP: low malignant potential; TOV: invasive tumor of the ovary; EOC: epithelial ovarian cancer.
The characteristics of the tumors and patient outcomes for the included 677 patients are summarized in Table 1. The mean age of the patients was 55.2 ± 12.1 years, and the mean follow-up time was 32.7 ± 34.9 months. TOV patients were significantly older than NOV patients (p < 0.001, ANOVA). Endometrial cancer patients were of similar age as the TOV patients (p = 0.937, ANOVA) and served as a second control population for the development of EOC. For survival analysis, inclusion criteria were: epithelial ovarian cancer, clinical follow-up of at least 18 months or until death and malignant disease (TOV grades 1 to 3). Survival was defined as survival time from surgery to death due to the disease.
This project was approved by institutional Ethics Committee of the Centre de recherche du CHUM. Informed consent was obtained from all patients.
Assessment of HFE genotype
Hemochromatosis genotype was determined by amplification and melting curve analysis performed in a LightCycler™ instrument (Roche Diagnostics Gmbh, Mannheim, Germany). Reaction conditions, design of primers and fluorescent labeled hybridization probes have been described previously.26 The two common HFE mutations were analyzed in separate PCR reactions instead of a multiplex PCR. For participants carrying HFE mutations additional genotyping was performed for confirmation.
Statistical analyses
All statistical analyses were performed using the SPSS software, version 11.0 (SPSS, Chicago, IL) and statistical significance was set at p < 0.05. Nonparametric Mann-U tests were used to evaluate differences in allele and genotypic distributions. Risk assessments were estimated using odds ratio and Chi-Square. For survival and progression analyses, we used Kaplan-Meier curves coupled with the log rank test. Multivariate analysis was performed using a forward stepwise hazard Cox regression model. To avoid an over-fitting situation, only four categorical variables were included in the multivariate Cox regression model.
Results
Allelic frequency and HFE genotype in patients with gynecological pathologies
A total of 677 consecutive patients were genotyped for the H63D and C282Y mutations in the HFE gene. As it can be seen in Table 2, these patients were diagnosed with benign tumors (BOV, n= 124) as well as with epithelial ovarian cancer (EOC, n = 360), with this last group including 96 patients with low malignancy potential (LMP) and 264 patients with invasive tumor of the ovary (TOV). Another subgroup of 113 patients were diagnosed with endometrial cancer. The control group (NOV), was described in detail in the material and method section, and included 80 cases. In our study, 37% of the participants (n = 251) carried at least one HFE mutation. When our cohort was considered jointly, the Y282 allele frequency was 4.4% and that of the D63 allele was 16.4%. As a reference, the allele frequency in Québec neonates has been reported as being 4.3% for the Y282 and 18% for the D63 allele.27
Table 2.
Distribution of hemochromatosis (HFE) gene mutations among tumor patients and non-tumor controls (NOV)
| Genotype | NOV n = 80 |
Endometrial cancer n = 113 |
BOV n = 124 |
LMP n = 96 |
TOV n = 264 |
EOC (LMP+TOV) n = 360 |
|---|---|---|---|---|---|---|
| WT/WT (%) | 78 (97.5) | 108 (95.6) | 113 (91.1) | 84 (87.5) | 236 (89.4) | 320 (88.9) |
| WT/Y (%) | 2 (2.5) | 3 (2.7) | 10 (8.1) | 9 (9.4) | 23 (8.7) | 32 (8.9) |
| Y/Y (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (0.8) | 2 (0.6) |
| NOV vs. E | E vs. TOV | BOV vs. NOV | LMP vs. NOV | TOV vs. NOV | EOC vs. NOV | |
| p = 0.9311 | p = 0.0211 | p = 0.0971 | p = 0.0541 | p = 0.041 | p = 0.0381 | |
| E vs. EOC | BOV vs. TOV | |||||
| p = 0.0191 | p = 0.6321 | |||||
| WT/WT (%) | 60 (75.0) | 71 (62.8) | 90 (72.6) | 67 (69.8) | 187 (70.8) | 254 (70.6) |
| WT/D (%) | 17 (21.3) | 36 (31.9) | 28 (22.6) | 25 (26.0) | 67 (25.4) | 92 (25.6) |
| D/D (%) | 3 (3.8) | 4 (3.5) | 5 (4.0) | 1 (1.0) | 7 (2.7) | 8 (2.2) |
| NOV vs. E | E vs. TOV | BOV vs. NOV | LMP vs. NOV | TOV vs. NOV | EOC vs. NOV | |
| p = 0.1241 | p = 0.1411 | p = 0.7721 | p = 0.7481 | p = 0.6071 | p = 0.6301 | |
| E vs. EOC | BOV vs. TOV | |||||
| p = 0.1081 | p = 0.8251 | |||||
| D/Y (%) | 0 (0.0) | 2 (1.8) | 1 (0.8) | 3 (3.1) | 3 (1.1) | 6 (1.7) |
| Alleles | n = 160 | n = 226 | n = 248 | n = 192 | n = 528 | n = 720 |
| Y (%) | 2 (1.3) | 5 (2.2) | 11 (4.4) | 12 (6.3) | 30 (5.7) | 42 (5.8) |
| NOV vs. E | E vs. TOV | BOV vs. NOV | LMP vs. NOV | TOV vs. NOV | EOC vs. NOV | |
| p = 0.3201 | p = 0.0061 | p = 0.0101 | p = 0.0011 | p = 0.0011 | p = 0.0011 | |
| E vs. EOC | BOV vs. TOV | |||||
| p = 0.0031 | p = 0.4371 | |||||
| D (%) | 23 (14.4) | 46 (20.4) | 39 (15.7) | 30 (15.6) | 84 (15.9) | 114 (15.8) |
| NOV vs. E | E vs. TOV | BOV vs. NOV | LMP vs. NOV | TOV vs. NOV | EOC vs. NOV | |
| p = 0.0171 | p = 0.0301 | p = 0.5941 | p = 0.3691 | p = 0.3601 | p = 0.3361 | |
| E vs. EOC | BOV vs. TOV | |||||
| p = 0.0241 | p = 0.7111 |
Nonparametric Mann-U test. NOV: normal ovary; BOV: benign tumor of the ovary; LMP: low malignant potential; TOV: invasive tumor of the ovary; EOC: epithelial ovarian cancer
We then analyzed whether the Y282 and D63 allele frequencies varied among patients with distinct gynecological pathologies. As shown in Table 2, most of the Y282 alleles (88.3%, n = 53/60) were found in patients diagnosed with BOV, LMP or TOV pathologies. When compared to the NOV group (allelic frequency of 1.3%), the Y282 allele was statistically more present in BOV patients (4.4%, p = 0.010, Mann-U) as well as in the ovarian cancer LMP (6.3%, p = 0.001) and TOV (5.7%, p = 0.001) groups. Similarly, when endometrial (2.2%) and ovarian cancer patients were compared, a significantly higher frequency of the Y282 allele was found in the ovarian cancer patient group (p = 0.003) and, within EOC, in the TOV group (p = 0.006). Regarding the frequency of genotypes, individuals heterozygous for the C282Y mutation (WT/Y) were more present in the BOV group (8.1%) compared to the NOV group (2.5%), although this difference was not statistically significant (p = 0.097; Table 2). However, compared to the NOV group (2.5%), patients with the WT/Y genotype were more present in the EOC group (8.9%, p = 0.038) and in the malignant TOV subgroup (8.7%, p = 0.040). Moreover, the WT/Y genotype was also more present in the EOC and TOV groups than in patients with endometrial cancer (2.7%, p = 0.019 and p = 0.021, respectively). Our cohort also contained two C282Y homozygous women (Y/Y), which were exclusively TOV patients.
In contrast to the Y282 allele, we did not observe any significant difference in the D63 allelic frequency between the NOV and the EOC population (14.4% vs. 15.8%, p = 0.336) or between NOV and BOV patients (15.7%, p = 0.594; Table 2). An increase in the frequency of the D63 allele was found in endometrial cancer patients (20.4%) compared to NOV (p = 0.017), EOC (p = 0.024) and invasive TOV patients (15.9%, p = 0.030). However, the number of individuals heterozygous (WT/D) or homozygous (D/D) for the H63D mutation was very similar between the groups with different tumor types (endometrial cancer, BOV, LMP or TOV; p > 0.05; Table 2).
We also identified nine compound heterozygous patients carrying both mutations (D/Y compound genotype) (Table 2). The number of these patients was evenly distributed amongst the endometrial cancer, BOV, LMP and TOV groups, and absent in NOV. However, no significant associations were observed, possibly due to the lower occurrences observed.
The odds ratios (ORs) for the H63D and C282Y mutations were also calculated to estimate the ovarian cancer risk (Table 3). The C282Y carriers have significantly higher risk of EOC than noncarriers (OR = 4.88; 95% CI 1.15 – 20.61; p = 0.018). A higher risk was similarly observed when the analysis was restricted to patients with LMP (OR = 5.57; 95% CI 1.21–25.69; p = 0.015) and TOV tumors (OR = 4.63; 95% CI 1.08–19.87; p = 0.024). In contrast, the H63D mutation was not associated with an increased risk of developing ovarian cancer.
Table 3.
Odds ratio (OR) of the C282Y and H63D HFE mutations among epithelial ovarian cancer and nontumor controls (NOV)
| Number of carriers1/total number of subjects (%) | OR | 95% CI | p value2 | ||
|---|---|---|---|---|---|
| Lower | Upper | ||||
| C282Y mutation | |||||
| NOV | 2/80 (2.5) | 1.00 | – | ||
| Endometrial cancer | 5/113 (4.4) | 1.81 | 0.34 | 9.55 | 0.481 |
| BOV | 11/124 (8.9) | 3.80 | 0.82 | 17.60 | 0.069 |
| LMP | 12/96 (12.5) | 5.57 | 1.21 | 25.69 | 0.015 |
| TOV | 28/264 (10.6) | 4.63 | 1.08 | 19.87 | 0.024 |
| EOC (LMP + TOV) | 40/360 (11.1) | 4.88 | 1.15 | 20.61 | 0.018 |
| H63D mutation | |||||
| NOV | 20/80 (25) | 1.00 | – | ||
| Endometrial cancer | 42/113 (37.2) | 1.78 | 0.94 | 3.34 | 0.075 |
| BOV | 34/124 (27.4) | 1.13 | 0.60 | 2.15 | 0.702 |
| LMP | 29/96 (44.6) | 1.30 | 0.67 | 2.53 | 0.443 |
| TOV | 77/264 (29.2) | 1.24 | 0.70 | 2.19 | 0.468 |
| EOC (LMP + TOV) | 106/360 (29.4) | 1.25 | 0.72 | 2.18 | 0.426 |
NOV: normal ovary; BOV: benign tumor of the ovary; LMP: low malignant potential; TOV: invasive tumor of the ovary; EOC: epithelial ovarian cancer.
Number of carriers includes heterozygotes (WT/Y or WT/D), homozygotes (Y/Y or D/D) and compound heterozygotes (Y/D).
Chi-square.
Impact of C282Y and H63D on overall survival of TOV patients
Kaplan-Meier analyses coupled to log-rank tests were used to estimate the association between C282Y and H63D mutations and overall survival for 246 TOV patients that had a minimum follow-up of 18 months or until death due to the disease. As shown in Figure 1, Kaplan-Meier analyses revealed that the C282Y mutation, but not the H63D mutation, was associated with a decreased survival of TOV patients. In fact, TOV patients with either a WT/Y or Y/Y genotype had decreased overall survival compared to TOV patients with a WT/WT genotype (log-rank = 22.49, p = 0.001, Fig. 1a). Comparatively, there was no significant overall survival difference for TOV patients with a WT/D or D/D genotype compared to WT/WT patients (log-rank = 4.86, p = 0.1821, Fig. 1b).
Figure 1.
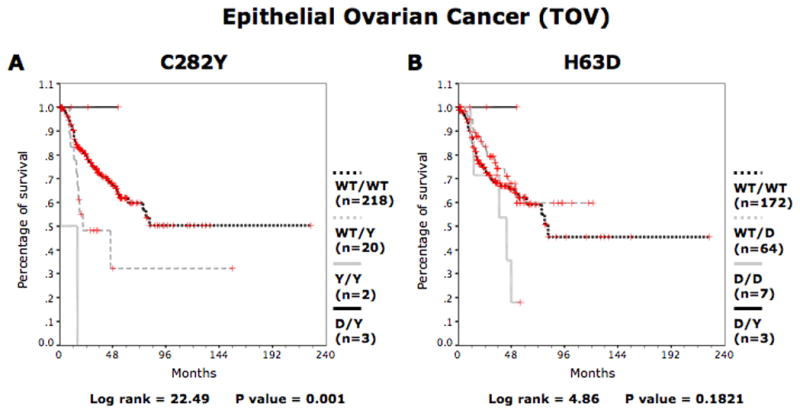
Kaplan-Meier survival curves for the entire group of TOV patients according to HFE genotype. For patients with invasive epithelial ovarian cancer overall survival curves are shown for (a) WT/WT, WT/Y, Y/Y and D/Y patients and (b) WT/WT, WT/D, D/D and D/Y patients. D represents H63D allele. Y represents the C282Y allele. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Impact of C282Y and H63D on overall survival of serous TOV patients
Since the serous TOV disease accounts for the majority of EOC cases in our cohort and is associated with a relatively poor patients outcome (77.6% mortality, Table 1), we determined the effect of the HFE mutations in this specific group, which was further restricted to patients with high-grade serous tumors, also known as type II EOC (grade 3). As shown in Figure 2a, high-grade serous patients with the WT/Y or Y/Y genotype were associated with significantly shorter survival time (log-rank = 12.34, p = 0.006, Kaplan-Meier). The median survival time of WT/WT patients was 33.7 months as compared to 19.4 months for WT/Y patients. In contrast to the C282Y mutation, the H63D mutation was not associated with overall survival in high-grade serous EOC patients (Fig. 2b). These results are consistent with the previous finding obtained when all TOV subtypes were considered together.
Figure 2.
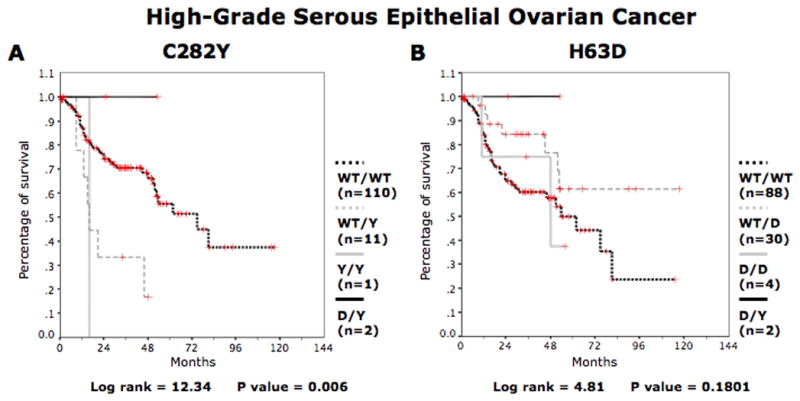
Kaplan-Meier survival curves for high-grade serous subtype of ovarian cancer patients according to HFE genotype. For patients with high-grade serous subtype ovarian cancer overall survival curves are shown for (a) WT/WT, WT/Y, Y/Y and D/Y patients and (b) WT/WT, WT/D, D/D and D/Y. D represents H63D allele. Y represents the C282Y allele. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The C282Y mutation is an independent predictor of overall survival in TOV patients
Finally, we compared the predictive strength of the C282Y and H63D mutations to clinical parameters (age, stage and grade) known to be associated with overall survival of ovarian cancer patients. Univariate COX regression analyses in TOV patients revealed that the C282Y mutation, age, stage and grade were all associated with reduced survival (Table 4). Not surprisingly, the H63D mutation was not associated with survival. In a multivariate analysis, the C282Y mutation was shown to be an independent predictor of overall survival (HR = 0.102 (WT/Y) and 0.278 (Y/Y), p = 0.002 (WT/Y) and 0.101 (Y/Y)), along with stage (HR = 2.263, p < 0.001) and age (HR = 1.029, p < 0.006).
Table 4.
Multivariate COX regression analyses for overall survival in invasive epithelial ovarian cancer (TOV) patients
| B | Wald | p-value | HR | 95% CI | ||
|---|---|---|---|---|---|---|
| Lower | Upper | |||||
| Univariate analyses1 | ||||||
| C282Y mutation (WT/WT)2 | – | 15.442 | 0.001 | – | – | – |
| C282Y mutation (WT/Y)2 | −2.378 | 10.677 | 0.001 | 0.093 | 0.022 | 0.386 |
| C282Y mutation (Y/Y)2 | −1.544 | 3.926 | 0.048 | 0.213 | 0.046 | 0.983 |
| H63D mutation (WT/WT)2 | – | 3.681 | 0.159 | – | – | – |
| H63D mutation (WT/D)2 | −0.712 | 2.307 | 0.129 | 0.491 | 0.196 | 1.230 |
| H63D mutation (D/D)2 | −0.975 | 3.653 | 0.056 | 0.377 | 0.139 | 1.025 |
| Age | 0.026 | 6.413 | 0.011 | 1.026 | 1.006 | 1.047 |
| Grade | 0.468 | 6.258 | 0.012 | 1.597 | 1.107 | 2.306 |
| Stage | 0.827 | 16.738 | <0.001 | 2.287 | 1.539 | 3.399 |
| Multivariate analyses3 | ||||||
| Age | 0.028 | 7.469 | 0.006 | 1.029 | 1.008 | 1.050 |
| Stage | 0.817 | 15.862 | <0.001 | 2.263 | 1.514 | 3.384 |
| C282Y mutation (WT/WT)2 | – | 16.149 | <0.001 | – | – | – |
| C282Y mutation (WT/Y) | −2.279 | 9.603 | 0.002 | 0.102 | 0.024 | 0.433 |
| C282Y mutation (Y/Y) | −1.281 | 2.687 | 0.101 | 0.278 | 0.060 | 1.285 |
HR: hazard ratio.
Univariate COX regression analyses using the ENTER model.
Categorical variable.
Multivariate COX regression analyses using the Forward-Wald model.
Discussion
Our study analyzed 677 women that presented at our center with gynecological disorders, for the two most common HFE mutations (C282Y and H63D). An interesting observation of our study is that the frequency of the C282Y mutation was markedly higher and statistically significant in patients with ovarian tumors when compared with two control groups, namely NOV (control, tumor-free cases) and endometrial cancers. Statistical analysis showed that the presence of C282Y mutation increased the risk of TOV by 4.63 times and of LMP by 5.57 times compared to NOV.
Another important finding is that the C282Y mutation was found to be associated with shorter survival of EOC patients. This decrease in survival time was observed both when all EOC subtypes were considered together and when only high-grade (grade 3) serous subtype cases were evaluated. This is important because ovarian cancer subtypes (serous, endometrioid, mucinous and clear cell) are increasingly being considered different diseases, with marked higher mortality being consistently observed in the high-grade serous subtype.28
What would be the mechanism(s) by which the C282Y mutation predisposes to and influences the outcomes of ovarian cancer? Iron levels should be considered since iron is a potential carcinogen based on the capability of this metal to induce oxidative stress by catalyzing hydroxyl radical formation through the Fenton reaction.2 Iron-catalyzed oxidative stress causes lipid peroxidation and protein modification, DNA damage with consequent promotion of mutagenesis, and leads to the depletion of antioxidant defenses. In addition to its contribution to a higher incidence of cancer via oxidative-stress pathways, iron may further promote cancer growth as an essential cofactor for cell proliferation, since iron is a co-factor for the ribonucleotide reductase enzyme, which is essential for DNA synthesis. In this aspect, it is noteworthy that the incidence of ovarian cancer is higher in postmenopausal than in premenopausal women, stages that are characterized by profound changes in body iron levels. In fact, as a result of menstrual cessation, body iron levels tend to increase sharply after menopause.29–31 Unfortunately, we were unable to determine whether the associations with genotype were mediated through body iron stores because traditional measurements of iron, such as serum iron, ferritin and transferrin saturation might have been confounded by anemia associated with their cancer.32,33 However, in our cohort, we found only two patients that were homozygous for the C282Y mutation and developed an ovarian carcinoma with very short survival time after diagnosis. These are the patients that would have been at risk to develop significant iron overload compared to women presenting with any other HFE genotype.34–36 In fact, while the C282Y heterozygotes have marginally increased serum ferritin and transferrin levels, unless combined with H63D mutations (compound heterozygotes), these individuals are not at increased risk of developing iron overload.34 Still, it is possible that the sharp accumulation of iron that occurs after menopause may be accelerated and aggravated in women carrying a single copy of the C282Y mutation, thereby increasing the chances of developing an ovarian cancer. Moreover, there is some evidence that HFE-related increased expression of TfR and consequent effect on tumor cellular homeostasis may modulate drug-induced apoptosis and ensure the survival of cancer cells,37,38 which ultimately would affect patient outcomes.
There are other mechanisms that may also explain the association between HFE genotype, ovarian cancer incidence and outcomes. HFE, as a nonclassical MHC class-like molecule, has been proposed to have an immunological function. In fact, the C282Y mutation interferes with the normal expression of MHC-I molecules and impairs the MHC-I presentation pathways,39 and there is some evidence indicating that HFE molecules may be directly recognized by a subset of T cells.40 MHC-I and T cell recognition are fundamental in the shaping of the T cell repertoire and, intriguingly, altered CD4/CD8 T cells ratios have been observed in HH patients.41 In turn, evidence that immune responses are important in ovarian cancer has been provided by a study revealing that immune responses and, more specifically, the presence of intratumoral T cells are associated with clinical outcomes in advanced ovarian cancer.42
On the other hand, the C282Y mutation, by preventing the correct association of HFE with β2m,8,43 causes the retention of misfolded HFE molecules in the endoplasmic reticulum (ER) and can trigger the unfolded protein response (UPR).44 Importantly, there is increasing evidence that the UPR response is associated with tumor progression and therapy resistance.45 In fact, cancer cells may adapt to ER stress and evade stress-induced apoptotic pathways by activating UPR branches.46,47 The HFE C282Y mutation could contribute to this process either by accelerating or differentially activating UPR branches linked to tumor cell resistance.48
Finally, we cannot completely rule out that other mechanisms, such as linkage disequilibrium with other genetic variants at risk on chromosome 6p,49 were the HFE gene is located, might explain the association between C282Y heterozygosis, risk of ovarian cancer and survival.
In summary, we identified the C282Y mutation in the HFE gene as a possible risk factor to develop epithelial ovarian cancer. We also observed, for the first time, an association between the presence of the C282Y mutation and a shorter survival in ovarian cancer patients, which indicates that the HFE C282Y mutation may act as a modifier of ovarian cancer disease progression. Whether this is due to iron dysregulation or some other function of the HFE gene remains to be determined. Additional studies including larger cohorts of patients would be needed to reinforce these results and understand the biologic role of HFE in ovarian cancer occurrence and progression, which may very well be also relevant to other cancers.
Acknowledgments
The authors are very grateful to the staff and patients at the Gynecologic Oncology Service at the Hôpital Notre-Dame for providing the blood samples. The authors wish to thank Manon de Ladurantaye, Laura Montermini, Hortence Makui, Lise Portelance, Louise Champoux, Marie-Line Puiffe and Antonio Layoun for their technical assistance. P.O.G. is a recipient of a Ph.D. studentship from the Fonds de la Recherche en Santé du Québec (FRSQ) and received additional support from the Institut du cancer de Montréal/Canderel scholarship and the Molecular Biology Program of the Université de Montréal. S.M. was supported by studentships from Canderel funds of the Institut du cancer de Montréal. M.M.S. is the recipient of a Research Scholarship—Junior 2 award from the FRSQ (Fonds de la recherche en santé du Québec). Blood samples were obtained through the Banque de tissus et de données of the Réseau de recherche sur le cancer of the Fonds de la Recherche en Santé du Québec (FRSQ), affiliated with the Canadian Tumor Repository Network (CTRNet).
Grant sponsor: Canadian Institutes of Health Research; Grant number: MOP44045; Grant sponsors: The Cancer Research Society, Institut du Cancer de Montréal (Initiative René Malo)
Abbreviations
- BOV
benign tumor of the ovary
- EOC
epithelial ovarian cancer
- HH
hereditary hemochromatosis
- HR
hazard ratio
- LMP
low malignant potential
- MHC-I
major histocompatibility class I
- NOV
normal ovary
- OR
odds ratio
- TfR
transferrin receptor
- TOV
invasive tumor of the ovary
- UPR
unfolded protein response
References
- 1.Kwok JC, Richardson DR. The iron metabolism of neoplastic cells: alterations that facilitate proliferation? Crit Rev Oncol Hematol. 2002;42:65–78. doi: 10.1016/s1040-8428(01)00213-x. [DOI] [PubMed] [Google Scholar]
- 2.Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82–83:969–74. doi: 10.1016/0378-4274(95)03532-x. [DOI] [PubMed] [Google Scholar]
- 3.Porto G, De Sousa M. Iron overload and immunity. World J Gastroenterol. 2007;13:4707–15. doi: 10.3748/wjg.v13.i35.4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos M, de Sousa M. In vitro modulation of T-cell surface molecules by iron. Cell Immunol. 1994;154:498–506. doi: 10.1006/cimm.1994.1094. [DOI] [PubMed] [Google Scholar]
- 5.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–97. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 6.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R, Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 7.Nichols GM, Bacon BR. Hereditary hemochromatosis: pathogenesis and clinical features of a common disease. Am J Gastroenterol. 1989;84:851–62. [PubMed] [Google Scholar]
- 8.Feder JN, Tsuchihashi Z, Irrinki A, Lee VK, Mapa FA, Morikang E, Prass CE, Starnes SM, Wolff RK, Parkkila S, Sly WS, Schatzman RC. The hemochromatosis founder mutation in HLA-H disrupts b2-microglobulin interaction and cell surface expression. J Biol Chem. 1997;272:14025–8. doi: 10.1074/jbc.272.22.14025. [DOI] [PubMed] [Google Scholar]
- 9.Feder JN, Penny DM, Irrinki A, Lee VK, Lebron JA, Watson N, Tsuchihashi Z, Sigal E, Bjorkman PJ, Schatzman RC. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA. 1998;95:1472–7. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gross CN, Irrinki A, Feder JN, Enns CA. Co-trafficking of HFE, a nonclassical major histocompatibility complex class I protein, with the transferrin receptor implies a role in intracellular iron regulation. J Biol Chem. 1998;273:22068–74. doi: 10.1074/jbc.273.34.22068. [DOI] [PubMed] [Google Scholar]
- 11.Salter-Cid L, Brunmark A, Li Y, Leturcq D, Peterson PA, Jackson MR, Yang Y. Transferrin receptor is negatively modulated by the hemochromatosis protein HFE: implications for cellular iron homeostasis. Proc Natl Acad Sci USA. 1999;96:5434–9. doi: 10.1073/pnas.96.10.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roy CN, Carlson EJ, Anderson EL, Basava A, Starnes SM, Feder JN, Enns CA. Interactions of the ectodomain of HFE with the transferrin receptor are critical for iron homeostasis in cells. FEBS Lett. 2000;484:271–4. doi: 10.1016/s0014-5793(00)02173-6. [DOI] [PubMed] [Google Scholar]
- 13.Roy CN, Penny DM, Feder JN, Enns CA. The hereditary hemochromatosis protein, HFE, specifically regulates transferrin-mediated iron uptake in HeLa cells. J Biol Chem. 1999;274:9022–8. doi: 10.1074/jbc.274.13.9022. [DOI] [PubMed] [Google Scholar]
- 14.Corsi B, Levi S, Cozzi A, Corti A, Altimare D, Albertini A, Arosio P. Overexpression of the hereditary hemochromatosis protein, HFE, in HeLa cells induces and iron-deficient phenotype. FEBS Lett. 1999;460:149–52. doi: 10.1016/s0014-5793(99)01330-7. [DOI] [PubMed] [Google Scholar]
- 15.Riedel HD, Muckenthaler MU, Gehrke SG, Mohr I, Brennan K, Herrmann T, Fitscher BA, Hentze MW, Stremmel W. HFE downregulates iron uptake from transferrin and induces iron-regulatory protein activity in stably transfected cells. Blood. 1999;94:3915–21. [PubMed] [Google Scholar]
- 16.Wang J, Chen G, Pantopoulos K. The haemochromatosis protein HFE induces an apparent iron-deficient phenotype in H1299 cells that is not corrected by co-expression of beta 2-microglobulin. Biochem J. 2003;370:891–9. doi: 10.1042/BJ20021607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy D, Cai Q, Felty Q, Narayan S. Estrogen-induced generation of reactive oxygen and nitrogen species, gene damage, and estrogen-dependent cancers. J Toxicol Environ Health Part B: Crit Rev. 2007;10:235–57. doi: 10.1080/15287390600974924. [DOI] [PubMed] [Google Scholar]
- 18.Kabat G, Rohan T. Does excess iron play a role in breast carcinogenesis? an unresolved hypothesis. Cancer Causes Control. 2007;18:1047–53. doi: 10.1007/s10552-007-9058-9. [DOI] [PubMed] [Google Scholar]
- 19.Beckman LE, Van Landeghem GF, Sikstrom C, Wahlin A, Markevarn B, Hallmans G, Lenner P, Athlin L, Stenling R, Beckman L. Interaction between haemochromatosis and transferrin receptor genes in different neoplastic disorders. Carcinogenesis. 1999;20:1231–3. doi: 10.1093/carcin/20.7.1231. [DOI] [PubMed] [Google Scholar]
- 20.Kallianpur AR, Hall LD, Yadav M, Christman BW, Dittus RS, Haines JL, Parl FF, Summar ML. Increased prevalence of the HFE C282Y hemochromatosis allele in women with breast cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:205–12. doi: 10.1158/1055-9965.epi-03-0188. [DOI] [PubMed] [Google Scholar]
- 21.Osborne NJ, Gurrin LC, Allen KJ, Constantine CC, Delatycki MB, McLaren CE, Gertig DM, Anderson GJ, Southey MC, John KO, Powell LW, Hopper JL, et al. HFE C282Y homozygotes are at increased risk of breast and colorectal cancer. Hepatology. 2010;51:1311–8. doi: 10.1002/hep.23448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondrashova TV, Neriishi K, Ban S, Ivanova TI, Krikunova LI, Shentereva NI, Smirnova IA, Zharikova IA, Konova MV, Taira S, Tsyb AF. Frequency of hemochromatosis gene (HFE) mutations in Russian healthy women and patients with estrogen-dependent cancers. Biochim Biophys Acta. 2005;1762:59–65. doi: 10.1016/j.bbadis.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Gunel-Ozcan A, Alyilmaz-Bekmez S, Guler E, Guc D. HFE H63D mutation frequency shows an increase in Turkish women with breast cancer. BMC Cancer. 2006;6:37. doi: 10.1186/1471-2407-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abraham BK, Justenhoven C, Pesch B, Harth V, Weirich G, Baisch C, Rabstein S, Ko Y-D, Bruning T, Fischer H-P, Haas S, Brod S, et al. Investigation of genetic variants of genes of the hemochromatosis pathway and their role in breast cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1102–7. doi: 10.1158/1055-9965.EPI-05-0013. [DOI] [PubMed] [Google Scholar]
- 25.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 26.Mangasser-Stephan K, Tag C, Reiser A, Gressner AM. Rapid genotyping of hemochromatosis gene mutations on the lightcycler with fluorescent hybridization probes. Clin Chem. 1999;45:1875–8. [PubMed] [Google Scholar]
- 27.Girouard J, Giguere Y, Delage R, Rousseau F. Prevalence of HFE gene C282Y and H63D mutations in a French-Canadian population of neonates and in referred patients. Hum Mol Genet. 2002;11:185–9. doi: 10.1093/hmg/11.2.185. [DOI] [PubMed] [Google Scholar]
- 28.Köbel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C, Leung S, Bowen NJ, Ionescu DN, Rajput A, Prentice LM, Miller D, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 2008;5:e232. doi: 10.1371/journal.pmed.0050232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nils M, Keld-Erik B, Lars O, Marianne K, Kirsten SLJ. Iron status in Danish women, 1984–1994: a cohort comparison of changes in iron stores and the prevalence of iron deficiency and iron overload. Eur J Haematol. 2003;71:51–61. doi: 10.1034/j.1600-0609.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 30.Zacharski LR, Ornstein DL, Woloshin S, Schwartz LM. Association of age, sex, and race with body iron stores in adults: analysis of NHANES III data. Am Heart J. 2000;140:98–104. doi: 10.1067/mhj.2000.106646. [DOI] [PubMed] [Google Scholar]
- 31.Koziol JA, Ho NJ, Felitti VJ, Beutler E. Reference centiles for serum ferritin and percentage of transferrin saturation, with application to mutations of the HFE gene. Clin Chem. 2001;47:1804–10. [PubMed] [Google Scholar]
- 32.Maccio A, Madeddu C, Massa D, Mudu MC, Lusso MR, Gramignano G, Serpe R, Melis GB, Mantovani G. Hemoglobin levels correlate with interleukin-6 levels in patients with advanced untreated epithelial ovarian cancer: role of inflammation in cancer-related anemia. Blood. 2005;106:362–7. doi: 10.1182/blood-2005-01-0160. [DOI] [PubMed] [Google Scholar]
- 33.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005;352:1011–23. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]
- 34.Beutler E, Waalen J. Response to Moirand et al.-HFE based re-evaluation of heterozygous hemochromatosis. Am J Med Genet. 2004;124A:218–9. doi: 10.1002/ajmg.a.20358. [DOI] [PubMed] [Google Scholar]
- 35.Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, McLaren CE, Bahlo M, Nisselle AE, Vulpe CD, Anderson GJ, Southey MC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221–30. doi: 10.1056/NEJMoa073286. [DOI] [PubMed] [Google Scholar]
- 36.Gurrin LC, Osborne NJ, Constantine CC, McLaren CE, English DR, Gertig DM, Delatycki MB, Southey MC, Hopper JL, Giles GG, Anderson GJ, Olynyk JK, et al. The natural history of serum iron indices for HFE C282Y homozygosity associated with hereditary hemochromatosis. Gastroenterology. 2008;135:1945–52. doi: 10.1053/j.gastro.2008.08.056. [DOI] [PubMed] [Google Scholar]
- 37.Fassl S, Leisser C, Huettenbrenner S, Maier S, Rosenberger G, Strasser S, Grusch M, Fuhrmann G, Leuhuber K, Polgar D, Stani J, Tichy B, et al. Transferrin ensures survival of ovarian carcinoma cells when apoptosis is induced by TNFalpha, FasL, TRAIL, or Myc. Oncogene. 2003;22:8343–55. doi: 10.1038/sj.onc.1207047. [DOI] [PubMed] [Google Scholar]
- 38.Christopher RC, Srigiridhar K, Janine PW. Expression of the hemochromatosis gene modulates the cytotoxicity of doxorubicin in breast cancer cells. Int J Cancer. 2006;119:2200–4. doi: 10.1002/ijc.22079. [DOI] [PubMed] [Google Scholar]
- 39.de Almeida SF, Carvalho IF, Cardoso CS, Cordeiro JV, Azevedo JE, Neefjes J, de Sousa M. HFE cross-talks with the MHC class I antigen presentation pathway. Blood. 2005;106:971–7. doi: 10.1182/blood-2004-12-4640. [DOI] [PubMed] [Google Scholar]
- 40.Rohrlich PS, Fazilleau N, Ginhoux F, Firat H, Michel F, Cochet M, Laham N, Roth MP, Pascolo S, Nato F, Coppin H, Charneau P, et al. Direct recognition by alphabeta cytolytic T cells of Hfe, a MHC class Ib molecule without antigen-presenting function. Proc Natl Acad Sci USA. 2005;102:12855–60. doi: 10.1073/pnas.0502309102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porto G, Vicente C, Teixeira MA, Martins O, Cabeda JM, Lacerda R, Goncalves C, Fraga J, Macedo G, Silva BM, Alves H, Justica B, et al. Relative impact of HLA phenotype and CD4-CD8 ratios on the clinical expression of hemochromatosis. Hepatology. 1997;25:397–402. doi: 10.1002/hep.510250223. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 43.Waheed A, Parkkila S, Zhou XY, Tomatsu S, Tsuchihashi Z, Feder JN, Schatzman RC, Britton RS, Bacon BR, Sly WS. Hereditary hemochromatosis: effects of C282Y and H63D mutations on association with beta2-microglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7 cells. Proc Natl Acad Sci USA. 1997;94:12384–9. doi: 10.1073/pnas.94.23.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Almeida SF, Fleming JV, Azevedo JE, Carmo-Fonseca M, de Sousa M. Stimulation of an unfolded protein response impairs MHC class I expression. J Immunol. 2007;178:3612–9. doi: 10.4049/jimmunol.178.6.3612. [DOI] [PubMed] [Google Scholar]
- 45.Wang G, Yang ZQ, Zhang K. Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. Am J Transl Res. 2010;2:65–74. [PMC free article] [PubMed] [Google Scholar]
- 46.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32:469–76. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 47.Rabik CA, Fishel ML, Holleran JL, Kasza K, Kelley MR, Egorin MJ, Dolan ME. Enhancement of cisplatin [cis-diammine dichloroplatinum (II)] cytotoxicity by O6-benzylguanine involves endoplasmic reticulum stress. J Pharmacol Exp Ther. 2008;327:442–52. doi: 10.1124/jpet.108.141291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawless MW, Mankan AK, White M, O’Dwyer MJ, Norris S. Expression of hereditary hemochromatosis C282Y HFE protein in HEK293 cells activates specific endoplasmic reticulum stress responses. BMC Cell Biol. 2007;8:30. doi: 10.1186/1471-2121-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santos GC, Zielenska M, Prasad M, Squire JA. Chromosome 6p amplification and cancer progression. J Clin Pathol. 2007;60:1–7. doi: 10.1136/jcp.2005.034389. [DOI] [PMC free article] [PubMed] [Google Scholar]