

Abstract
Mutations in the high-affinity sulfonylurea receptor (SUR)-1 cause one of the severe recessively inherited diffuse forms of congenital hyperinsulinism or, when associated with loss of heterozygosity, focal adenomatosis. We hypothesized that SUR1 mutations would render the β-cell insensitive to sulfonylureas and to glucose. Stimulated insulin responses were compared among eight patients with diffuse hyperinsulinism (two mutations), six carrier parents, and ten normal adults. In the patients with diffuse hyperinsulinism, the acute insulin response to intravenous tolbutamide was absent and did not overlap with the responses seen in either adult group. There was positive, albeit significantly blunted, acute insulin response to intravenous dextrose in the patients with diffuse hyperinsulinism. Graded infusions of glucose, to raise and then lower plasma glucose concentrations over 4 h, caused similar rises in blood glucose but lower peak insulin levels in the hyperinsulinemic patients. Loss of acute insulin response to tolbutamide can identify children with diffuse SUR1 defects. The greater response to glucose than to tolbutamide indicates that ATP-sensitive potassium (KATP) channel–independent pathways are involved in glucose-mediated insulin release in patients with diffuse SUR1 defects. The diminished glucose responsiveness suggests that SUR1 mutations and lack of KATP channel activity may contribute to the late development of diabetes in patients with hyperinsulinism independently of subtotal pancreatectomy.
Several distinct forms of congenital hyperinsulinism have been identified in recent years (1). Sporadic nongenetic cases of transient hyperinsulinism can be associated with maternal diabetes and with perinatal stresses such as birth asphyxia or small-for-dates birth weight (2). Dominant genetic forms include activating glucokinase mutations that lower the glucose threshold for insulin release (3), gain-of-function mutations in glutamate dehydrogenase that cause both hyperinsulinism and hyperammonemia (4), and other types whose genetic bases have not yet been identified (5,6). However, mutations in the ATP-sensitive potassium (KATP) channel complex of the pancreatic β-cell plasma membrane cause some of the most severe clinical disease (7-11). Encoded by two adjacent genes on chromosome 11p, the sulfonylurea receptor (SUR)-1 regulates the channel activity, whereas the inwardly rectifying potassium channel (Kir6.2) constitutes the ion pore (12,13). Patients with KATP channel mutations who present the severe form of the disease are clinically diazoxide unresponsive, and many require 95% subtotal pancreatectomy to prevent recurrent hypoglycemia. They also exhibit a high risk of later developing diabetes, which is often attributed to their surgical treatment (14-16). SUR1 and Kir6.2 mutations can be expressed in two ways: autosomal recessive inheritance of two abnormal SUR1 or Kir6.2 alleles results in diffuse hyperinsulinism (formerly called nesidioblastosis), whereas inheritance of an abnormal paternal SUR1 allele with somatic loss of the maternal chromosome 11p15 leads to focal adenomatosis (17-19).
The KATP channel complex transduces the metabolic status of the β-cell into cell membrane electrical activity and thereby links insulin release with metabolic demands. SUR1, a member of the ATP-binding cassette superfamily, forms a hetero-octamer with Kir6.2 (20,21). Glucose entry and metabolism increase the ratio of ATP to ADP within the β-cell. At very high concentrations, ATP binding to Kir6.2 inhibits the channel activity, whereas magnesium nucleotides can antagonize this inhibition through interactions with the nucleotide binding folds of the SUR1 (22). The increased ATP-to-ADP ratio leads to closure of the KATP channel and hence depolarization of the β-cell membrane. The depolarization opens voltage-gated calcium channels, and the resultant elevation of the intracellular calcium concentration triggers exocytosis of insulin granules (23,24). Sulfonylureas modulate insulin secretion by binding to SUR1; some, like tolbutamide, stimulate insulin secretion, whereas others, like diazoxide, inhibit it.
The present study was undertaken to test the hypothesis that mutations in SUR1, the metabolic transducer, would render the β-cell unresponsive to sulfonylureas and insensitive to high or rising glucose levels as well as to falling glucose levels. Insulin responses to tolbutamide, a sulfonylurea that stimulates insulin release, and to both acute and prolonged graded glucose stimulation were measured in children with diffuse SUR1−/− hyperinsulinism. The effects of heterozygous SUR1 mutations were investigated by evaluating insulin secretion in the heterozygous parents of these children.
RESEARCH DESIGN AND METHODS
Subject characteristics
The clinical characteristics of the eight children with diffuse SUR1−/− hyperinsulinism who were studied are shown in Table 1. They were aged 2–20 years. Patients 1 and 2 were sisters, and patients 5, 6, and 8 were brothers. All eight children were unresponsive to treatment with diazoxide, a drug that inhibits insulin secretion by opening the potassium channel through its effect on SUR1. Five of the children had undergone subtotal pancreatectomy before this study, yet continued to experience episodes of hypoglycemia with fasting for <12 h. The other three had similarly severe hypoglycemia and were controlled with frequent feedings (one via gastrostomy tube). All eight hyperinsulinemic patients had 3992 –9 g → a and DelF 1388, the two most commonly found SUR1 mutations in the Ashkenazi Jewish population (25,26); four of the children studied were homozygous for 3992 –9 g → a, and four were compound heterozygous for DelF 1388 and 3992 –9 g → a.
TABLE 1.
Clinical characteristics of the children with congenital hyperinsulinism
| SUR1 genotype | Patient number | Sex | Age (years) | Age at presentation | Age at pancreatectomy | Current treatment |
|---|---|---|---|---|---|---|
| Diffuse hyperinsulinism | ||||||
| DelF 1388/3992 –9 g → a | 1 | F | 20 | 4 h | 2 months | Frequent feedings |
| DelF 1388/3992 –9 g → a | 2 | F | 14 | Newborn | 8 days | None |
| DelF 1388/3992 –9 g → a | 3 | F | 13 | 11 months | ND* | None since age 8 years |
| DelF 1388/3992 –9 g → a | 4 | M | 2 | 3 days | 1, 2, 30 months* | Glucagon-octreotide infusion |
| 3992 –9 g → a/3992 –9 g → a | 5 | M | 13 | 10 days | ND | Frequent feedings |
| 3992 –9 g → a/3992 –9 g → a | 6 | M | 11 | 2 days | ND | Frequent gastrostomy feedings |
| 3992 –9 g → a/3992 –9 g → a | 7 | F | 6 | 1–2 days | 1 month | None |
| 3992 –9 g → a/3992 –9 g → a | 8 | M | 4 | At birth | 1 month | None |
Surgery done after the time of the study; ND, no surgery.
Control subjects included four female and two male SUR1+/− heterozygous carrier parents aged 36–48 years, and six female and four male normal adults aged 19–48 years. All control subjects were normoglycemic, and none had a history of diabetes. All had normal fasting blood glucose and insulin levels.
The study was reviewed and approved by the Children’s Hospital of Philadelphia Institutional Review Board. Written informed consent was obtained from the subjects or their parents before participation in the study. Verbal assent was obtained from the older children.
Acute insulin responses
Subjects were admitted to the Children’s Hospital of Philadelphia General Clinical Research Center. Octreotide or glucagon therapy was withdrawn for at least 24 h before study, and dextrose was infused intravenously as necessary to prevent hypoglycemia. Studies were carried out after an overnight fast, and dextrose was infused as needed to maintain the blood glucose concentration between 60 and 80 mg/dl. Two peripheral venous catheters were inserted: one for infusion and the other for blood sampling. Glucose (0.5 g/kg to a maximum of 20 g) was administered intravenously over 2 min. Twenty minutes later, tolbutamide (25 mg/kg to a maximum of 1 g) was administered intravenously over 1 min. Blood samples for glucose and insulin concentrations were obtained at −10, −5, and 0 min before the glucose bolus and at 1, 3, 5, 10, and 20 min afterwards. After the tolbutamide injection, blood samples were collected at 1, 3, 5, 10, 20, 30, 40, and 60 min. Whole blood glucose was measured by the glucose oxidase method (Glucose Analyzer; Yellow Springs Instruments, Yellow Springs, OH). Plasma insulin concentrations were measured by microenzyme immunoparticle assay with a sensitivity of 1.0 μU/ml (Abbott IMx, Abbott Park, IL). Acute insulin responses (AIRs) were calculated as the mean of the increment in insulin concentration at 1 and 3 min after stimulation. Glucose disposal rate was calculated from the glucose decay curve after intravenous glucose as the percent decline per minute.
Graded glucose infusion studies
Studies were performed after an overnight fast, and intravenous dextrose was administered as needed to maintain the blood glucose concentration between 60 and 80 mg/dl. Glucose was infused for a total of 4 h. Every 40 min, the rate of glucose infusion was increased from 0 to 4, 8, and 16 mg · kg−1 · min−1 and then decreased again to 8, 4, and 0 mg · kg−1 · min−1. Blood glucose and plasma insulin concentrations were measured at baseline and every 10 min throughout the infusion. Insulin concentrations were plotted against glucose levels, resulting in two curves for each subject: the up curve corresponding to the escalating rate of dextrose infusion and the down curve to the diminishing rate of dextrose infusion that followed. Glucose sensitivity was quantified as the slope of the up curve (μU · mg−1 · 10−2). Differences between the up and down curves were analyzed two ways. The insulin displacement was calculated as the difference between the up- and down-curve insulin concentrations at the approximate midpoint value between the peak and basal glucose concentrations (blood glucose of 150 mg/dl), expressed as a percent of the up-curve value. The second analysis used a comparison of the linear phases of the up and down curves.
Statistical analysis
All data are presented as means ± SE. An alternate Welch t test was used to compare the results between the different groups.
RESULTS
AIRs
The AIRs to glucose and tolbutamide in a normal adult and those in child number 6 with SUR1−/− hyperinsulinism are shown in Fig. 1A and B, respectively. The normal adult responded briskly to both stimuli, with AIRs of similar magnitude (45 μU/ml to glucose and 34 μU/ml to tolbutamide). In contrast, the SUR1−/− child had a smaller AIR to glucose (31 μU/ml), despite an even greater elevation in blood glucose concentration (345 vs. 290 mg/dl in the normal control), and no AIR to tolbutamide (4 μU/ml).
FIG. 1. AIRs to glucose and tolbutamide in children with diffuse SUR1−/− hyperinsulinism.
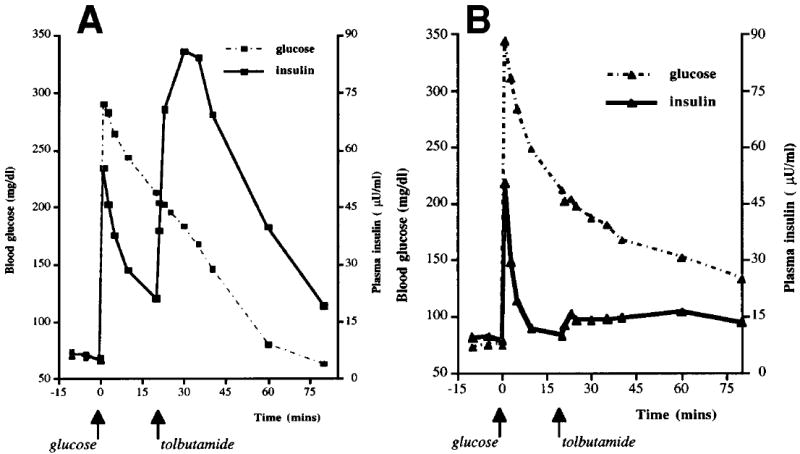
A: Normal adult control. B: Patient 6 with diffuse SUR1−/− hyperinsulinism
Table 2 compares the results of the AIR tests in the group of eight children with diffuse SUR1−/− hyperinsulinism, their carrier parents, and normal adults. Baseline insulin levels in the SUR1−/− group were comparable to those in the normal adults. AIR to tolbutamide was absent in the children with diffuse hyperinsulinism (P < 0.005). The 95% CI of the AIR to tolbutamide in the diffuse hyperinsulinemic patients (−2.3 to 3.7 μU/ml) did not overlap with the normal control subjects (26.4–79.7 μU/ml). Glucose stimulation provoked a positive but smaller AIR in the diffuse hyperinsulinemic patients (P < 0.05). The glucose disposal rates among the SUR1−/− children and the normal adult groups were similar. Mean insulin responses to the two AIR tests did not differ among the four children with homozygous 3992 –9 g → a mutations and the four children with compound heterozygous DelF 1388/3992 –9 g → a mutations (AIR tolbutamide −0.5 ± 2 vs. 2 ± 2 μU/ml and AIR glucose 8 ± 3 vs. 20 ± 6 μU/ml, respectively). There was also no difference between the five SUR1−/− children who had undergone subtotal pancreatectomy and the three without surgery (AIR tolbutamide −0.1 ± 2 vs. 2 ± 1 μU/ml and AIR glucose 13 ± 4 vs. 17 ± 8 μU/ml, respectively).
TABLE 2.
AIRs to glucose and tolbutamide in children with congenital hyperinsulinism
| Subject group | n | Baseline plasma insulin concentration (μU/ml) | Peak blood glucose concentration (mg/dl) | AIR to glucose (μU/ml) | Glucose disposal rate (%/min) | AIR to tolbutamide (μU/ml) |
|---|---|---|---|---|---|---|
| Diffuse SUR1−/− hyperinsulinism | 8 | 11 ± 2 (NS*) | 293 ± 17 (NS) | 14 ± 4 (<0.05) | 2 ± 0.3 (NS) | 0.7 ± 1 (<0.005) |
| SUR1+/− carriers | 6 | 10 ± 2 (NS) | 198 ± 12 (NS) | 37 ± 13 (NS) | 2 ± 0.2 (NS) | 31 ± 8 (NS) |
| Normal adults | 10 | 8 ± 1 | 262 ± 16 | 42 ± 9 | 2 ± 0.5 | 53 ± 12 |
Data are means ± SE (P) or means ± SE.
Significance values are presented for the difference between each group and the normal adults.
As shown in Table 2, AIRs to both glucose and tolbutamide were similar in the SUR1+/− carriers and the normal adults. The 95% CI of the AIR to tolbutamide in the diffuse hyperinsulinemic patients did not overlap with that of the carriers (9.7–52.3 μU/ml). The glucose disposal rates of the carriers were also similar to those of the normal adults.
Insulin response to graded glucose infusion
Graded glucose infusion studies were performed in three children with SUR1−/− diffuse hyperinsulinism who had not undergone subtotal pancreatectomy. Surgery-naive patients were chosen for this study to eliminate subtotal pancreatectomy as a potential confounding variable that could cause impaired glucose responsiveness. Figure 2 shows the blood glucose and plasma insulin concentrations in the graded glucose infusion studies of the same SUR1−/− child (number 6) and normal adult control as in Fig. 1. In the normal adult (Fig. 2A), the rise and subsequent fall in blood glucose was associated with a parallel, though slightly delayed, rise and fall in insulin concentration (peak 87 μU/ml). As shown in Fig. 2B, child number 6 with SUR1−/− hyperinsulinism had a similar rise and fall in blood glucose but achieved a peak insulin concentration of only 29 μU/ml despite a higher peak blood glucose concentration. Figure 3 shows the insulin concentrations plotted against blood glucose concentrations from the studies of Fig. 2. The slope of the up curve was steeper in the normal adult (37 μU · mg−1 · 10−2) than in the child with SUR1−/− hyperinsulinism (4 μU · mg−1 · 10−2), indicating diminished sensitivity to glucose for insulin secretion in the patient.
FIG. 2. Insulin response to graded glucose infusion in diffuse SUR1−/− hyperinsulinism.
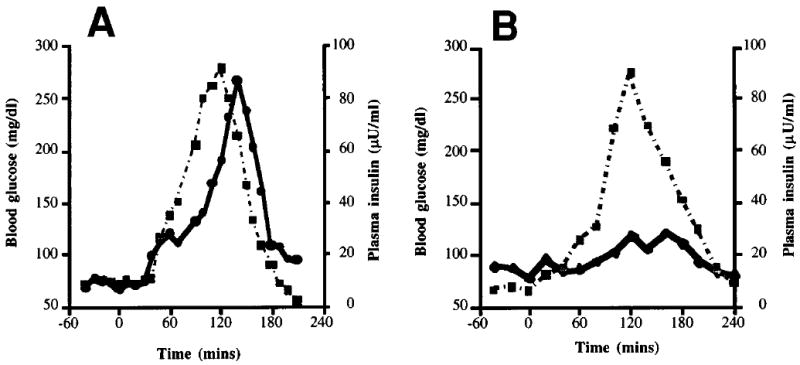
A: The same normal adult as in Fig. 1A. B: Patient 6 with diffuse SUR1−/− hyperinsulinism who had never undergone subtotal pancreatectomy.
 ■, Glucose;
■, Glucose;
 ●, insulin.
●, insulin.
FIG. 3. Relationship of plasma insulin to blood glucose during graded glucose infusion.
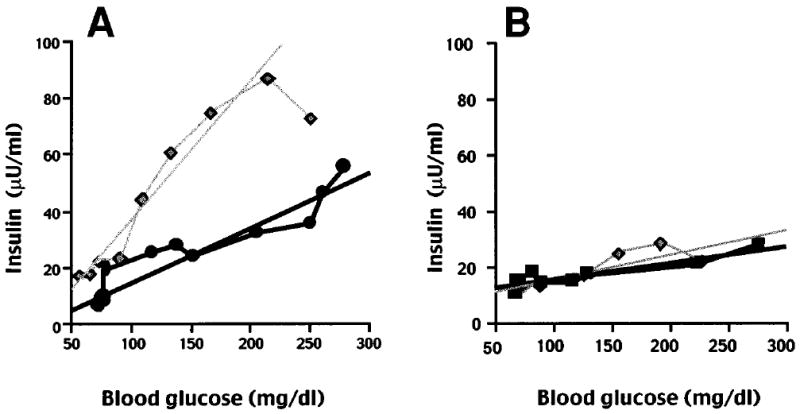
A: The same normal adult as in Fig. 2A. B: Patient 6 with diffuse SUR1−/− hyperinsulinism. The up curve for each subject corresponded to the escalating dextrose infusion, and the down curve corresponded to the subsequent diminishing dextrose infusion.
●,
■, Up curve;
 , down curve.
, down curve.
Table 3 summarizes the results of the graded glucose infusion studies in the three children with SUR1−/− diffuse hyperinsulinism, five SUR+/− carriers, and four normal adults. Because there was no difference between the carriers and normal control subjects on either AIR or on any of the graded glucose infusion parameters assessed, the two adult groups were combined for data analysis. Peak blood glucose concentration in the SUR1−/− children (298 ± 50 mg/dl) was equivalent to that in the adults. The peak plasma insulin concentrations in the SUR1−/− children overlapped the lower end of values seen in the adults, but the mean insulin concentration was significantly less in the former group (P < 0.05). Glucose sensitivity in the SUR1−/− children also was less than that in the adults (P < 0.01). There was no insulin displacement at 150 mg/dl glucose in the children with SUR1−/− hyperinsulinism. When insulin levels were plotted against blood glucose concentrations, the control subjects showed a hysteresis loop that resulted in a greater slope for the down curve compared with the up curve (124 ± 43 vs. 57 ± 13 μU · mg−1 · 10−2; P < 0.05 by a paired non-parametric test). The SUR1−/− children had loss of the hysteresis loop, with down slopes that were similar to the up slopes (11 ± 3 vs. 4 ± 2 μU · mg−1 · 10−2; NS).
TABLE 3.
Insulin response to graded glucose infusion in unoperated children with diffuse SUR1−/− hyperinsulinism
| Peak blood glucose (mg/dl) | Peak plasma insulin (μU/ml) | Glucose sensitivity (μU · mg−1 · 10−2) | Insulin displacement at 150 mg/dl glucose (%) | |
|---|---|---|---|---|
| SUR1−/− children | ||||
| Patient 3 | 350 | 56 | 0 | 0 |
| Patient 5 | 198 | 18 | 6 | 0 |
| Patient 6 | 345 | 51 | 4 | 0 |
| Mean ± SE | 298 ± 50 (NS*) | 42 ± 12 (P < 0.05) | 4 ± 2 (P < 0.01) | 0 ± 0 (P = 0.0001) |
| Adults (n = 9) | ||||
| Mean ± SE | 257 ± 15 | 120 ± 31 | 57 ± 13 | 47 ± 7 |
| 95% CI | 223–292 | 48–191 | 27–86 | 32–63 |
Significance of the difference between the mean values of the children and adults by the alternate Welch t test.
DISCUSSION
The results of these studies show that children with SUR1−/− hyperinsulinism do not respond to the insulin secretagogue, tolbutamide, and have a diminished, although positive, AIR to intravenous glucose compared with control subjects. AIR testing did not differentiate children with diffuse SUR1−/− hyperinsulinism homozygous for the 3992 –9 g → a mutation from those compound heterozygous for DelF 1388/3992 –9 g → a mutations. During graded glucose infusion studies, children with diffuse SUR1−/− hyperinsulinism who had never undergone subtotal pancreatectomy exhibited reduced β-cell glucose sensitivity. Thus, SUR1−/− hyperinsulinism causes blunted but not absent glucose responsiveness. The abnormalities found on AIRs and graded glucose infusion studies of these patients were not age related because the SUR1−/− children were as old as 20 years and the adults as young as 19 years.
Patients with SUR1−/− hyperinsulinism had no AIR to tolbutamide. A previous report by Ehrlich and Martin (27) found excessive insulin release in response to intravenous tolbutamide in 15 children with idiopathic hypoglycemia. The earlier study, however, measured glucose and insulin responses from 15 to 120 min after tolbutamide and did not assess the immediate response. Furthermore, the cause(s) of hypoglycemia in these children was not known and likely represented a heterogeneous group of disorders (not necessarily diffuse SUR1−/− hyperinsulinism). Intravenous tolbutamide tests have also been used in the assessment of patients with insulinomas, again looking only at delayed (120–180 min) responses (28).
The loss of AIR to tolbutamide in children with SUR1−/− hyperinsulinism is not surprising given the fact that tolbutamide binds to SUR1 and these children have a lack of KATP channel activity due to defective SUR1 proteins (29). Sharma et al. (30) have reported that the truncated and misfolded mutant SUR1 proteins do not transit to the plasma membrane. Similarly, these children are clinically unresponsive to treatment with diazoxide, which inhibits insulin secretion by binding to SUR1. Loss of AIR to tolbutamide can therefore serve as a diagnostic marker for children who will not benefit from diazoxide therapy, although it is remotely possible that an SUR1 mutation different from those studied here may allow the molecule to respond to one or the other ligand. Patients with mutations in Kir6.2 would be expected to have the same responses as those with SUR1 mutations, since both components of the KATP channel are required for normal channel activity.
The complete loss of AIR to tolbutamide but only partial blunting of AIR to glucose provides evidence that KATP channel–independent pathways are involved in glucose-mediated insulin secretion in children with SUR1−/− hyperinsulinism. The existence of KATP channel–independent pathways has been suggested by studies of islets from rats and mice (31-33) and was recently demonstrated in human islets in vitro (34). The precise KATP channel–independent pathways are still unclear (35). Proposed mechanisms include glucose-mediated elevations in intracellular calcium concentrations via mobilization of calcium sequestered in the endoplasmic reticulum (36-38). Intracellular calcium release may involve inositol-1,4,5-triphosphate (39) or the calcium release–activated nonselective cation channel (iCRAN) (40,41). Furthermore, a non–calcium-dependent mechanism involving protein kinases A and C, ATP, and GTP has been suggested (42).
The reduced AIR to glucose and the diminished glucose sensitivity on graded glucose infusion studies suggest that SUR1−/− β-cells are less glucose responsive. Additionally, the insulin displacement at 150 mg/dl glucose is lost in children with SUR1−/− hyperinsulinism. Both factors may lead to post-prandial hyperglycemia in children with SUR1−/− hyperinsulinism. These findings corroborate the clinical observation that hyperinsulinemic patients can exhibit episodes of both hypoglycemia and hyperglycemia, sometimes even in the same day. Recent studies in SUR1−/− knockout (and Kir6.2 knockout) mice did not have the severe hypoglycemia seen in the human disease but demonstrated a loss of first-phase and attenuated second-phase glucose-stimulated insulin secretion, consistent with the impairments in the AIR to glucose and glucose sensitivity observed in the present studies (43). Insulin secretory defects on intravenous glucose tolerance testing and graded glucose infusions have been reported in different types of maturity-onset diabetes of the young (MODY) and autosomal-dominantly inherited forms of diabetes caused by non-SUR1 single-gene mutations (44-46). In one MODY family studied, reductions in insulin secretory oscillations during prolonged glucose infusion were also detected in genetic marker–positive family members who were not yet diabetic (47). This closely resembles our children with SUR1−/− hyperinsulinism who, because of their single gene defect, exhibited abnormal insulin release on graded glucose infusion, but who were not diabetic. The finding of normal glucose disposal rates in the children with diffuse SUR1−/− hyperinsulinism suggests that they may have adapted to impaired insulin release by increasing peripheral sensitivity to insulin. The AIR to glucose did not differ between the children with diffuse SUR1−/− hyperinsulinism who had and those who had not undergone subtotal pancre-atectomy, and the abnormal response to graded glucose infusion was found in our SUR1−/− hyperinsulinemic patients without surgical intervention. This finding suggests that SUR1−/− β-cells may contribute, independently of subtotal pancreatectomy, to the increased risk of diabetes seen in diffuse SUR1−/− hyperinsulinemic patients.
The children with SUR1−/− hyperinsulinism had no insulin displacement at 150 mg/dl glucose. It is unclear whether this signifies loss of the normal glucose potentiation of insulin release or loss of the normal lag in the suppression of the insulin secretory response to a falling glucose. It is also possible that the flat glucose sensitivity curve of the patients with diffuse SUR1−/− hyperinsulinism made it difficult to detect any glucose potentiation. In any case, the hysteresis loop formed by the difference in insulin levels along the up and down curves in the control subjects was not apparent in the children with SUR1−/− hyperinsulinism. Presumably, this observation is due to the loss of KATP channel activity and provides further evidence of the glucose blindness of SUR1−/− β-cells. How this effect relates to the glucose potentiation of insulin secretion (48-51) is unknown. Further studies of glucose responsiveness in children with hyperinsulinism are needed to better understand how this phenomenon is altered by SUR1 mutations.
The heterozygous SUR1+/− carrier parents demonstrated normal insulin release on both AIRs and graded glucose infusion studies. Thus, performance of AIR testing and graded glucose infusion studies on parents of children with hyperinsulinism cannot serve as a simple clinical way of identifying heterozygous carriers and thereby predicting which families potentially transmit paternal-only or autosomal recessive SUR1 mutations. The normal responses seen in the heterozygous SUR1+/− parents also suggest that in nonobese individuals without a history of diabetes, carrying one mutated SUR1 allele does not increase the risk of hypoglycemia or diabetes.
Because patients with the different types of hyperinsulinism follow markedly different clinical courses, including responsiveness to medical therapies such as diazoxide, identifying the type of hyperinsulinism in any given patient allows individualized tailoring of the therapeutic plan for that patient. Furthermore, the diagnosis of diffuse versus focal hyperinsulinism is currently based on histopathologic examination of subtotal pancreatectomy specimens; genetic analysis of the SUR1 mutations is still available only through research laboratories. This distinction is important because local excision in focal hyperinsulinism is far preferable to blind 95% subtotal pancreatectomy; local excision carries a lower risk of complications and is potentially curative. Complete loss of AIR to tolbutamide constitutes a potentially valuable clinical method for preoperatively identifying children with diffuse hyperinsulinism. Because children with focal hyperinsulinism also have a subpopulation of normal β-cells outside their focal lesion, they would be expected to retain an AIR to tolbutamide and have an AIR to glucose intermediate between that seen in the patients with diffuse hyperinsulinism and that seen in normal individuals. Further studies of patients with congenital hyperinsulinism at the time of diagnosis (both diffuse and focal) are needed to confirm the predictive value of the AIR to tolbutamide as a clinical marker for diffuse disease.
In summary, the loss of AIR to tolbutamide may be a useful clinical identifier of children with congenital hyperinsulinism due to diffuse SUR1−/− mutations. The reduced glucose responsiveness of SUR1−/− also has ramifications for the care of children with congenital hyperinsulinism. The primary glucose-sensing defect in diffuse SUR1−/− hyperinsulinism may directly contribute, independently from surgical treatment, to the increased risk of diabetes or impaired glucose tolerance in these patients. Furthermore, investigations of the insulin secretory dynamics in patients with diffuse SUR1−/− mutations may shed light on normal β-cell signaling. Our study provides the first in vivo evidence of the involvement of KATP channel–independent pathways in glucose-mediated insulin secretion in humans.
Acknowledgments
This work was partially supported by grants from the National Institutes of Health (MO1 RR00240, RO1 DK53012, and RO1 DK56268 [C.A.S.]; T32 DK07314 [A.G., R.J.F.]; and DK16746 and DK20579 [M.A.P.]), grants from the American Diabetes Association (C.A.S.), the Lawson Wilkins Pediatric Endocrine Society Award (A.G., R.J.F.), and fellowship grants from Eli Lilly and Pharmacia-Upjohn (A.G., R.J.F., A.K). The Diabetes Research and Training Center (DK20595) at the University of Chicago supported the development of the graded glucose infusion studies.
The authors wish to thank the nursing staff of the Children’s Hospital of Philadelphia and the staff of the General Clinical Research Center for their expert assistance in carrying out these studies.
Glossary
- AIR
acute insulin response
- KATP channel
ATP-sensitive potassium channel
- Kir6.2
inwardly rectifying potassium channel
- MODY
maturity-onset diabetes of the young
- SUR
sulfonylurea receptor
Footnotes
This study was presented in part at the Second International Conference on ATP Sensitive Potassium Channels and Disease, St. Charles, IL, September 1998, and at the 81st Annual Meeting of the Endocrine Society, San Diego, CA, June 1999.
References
- 1.Glaser B, Thornton P, Otonkoski T, Junien C. Genetics of neonatal hyperinsulinism. Arch Dis Child Fetal Neonatal Ed. 2000;82:F79–F86. doi: 10.1136/fn.82.2.F79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanley CA. Hyperinsulinism in infants and children. Pediatr Clin North Am. 1997;44:363–374. doi: 10.1016/s0031-3955(05)70481-8. [DOI] [PubMed] [Google Scholar]
- 3.Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, Stanley CA, Thornton PS, Permutt MA, Matchinsky FM, Herold KC. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338:226–230. doi: 10.1056/NEJM199801223380404. [DOI] [PubMed] [Google Scholar]
- 4.Stanley CA, Lieu YK, Hsu BYL, Burlina AB, Greenberg CR, Hopwood NJ, Perlman K, Rich BH, Zammarchi E, Poncz M. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. 1998;338:1352–1357. doi: 10.1056/NEJM199805073381904. [DOI] [PubMed] [Google Scholar]
- 5.Thornton PS, Satin-Smith MS, Herold K, Glaser B, Chiu KC, Nestorowicz A, Permutt MA, Baker L, Stanley CA. Familial hyperinsulinism with apparent autosomal dominant inheritance: clinical and genetic differences from the autosomal recessive variant. J Pediatr. 1998;132:9–14. doi: 10.1016/s0022-3476(98)70477-9. [DOI] [PubMed] [Google Scholar]
- 6.Kukuvitis A, Deal C, Arbour L, Polychronakos C. An autosomal dominant form of familial persistent hyperinsulinemic hypoglycemia of infancy, not linked to the sulfonylurea receptor locus. J Clin Endocrinol Metab. 1997;82:1192–1194. doi: 10.1210/jcem.82.4.3904. [DOI] [PubMed] [Google Scholar]
- 7.Thomas PM, Cote GJ, Hallman DM, Mathew PM. Homozygosity mapping to chromosome 11p of the gene for familial persistent hyperinsulinemic hypoglycemia of infancy. Am J Hum Genet. 1995;56:416–421. [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas PM, Cote GJ, Wohllk N, Mathew PM, Gagel RF. The molecular basis for familial persistent hyperinsulinemic hypoglycemia of infancy. Proc Assoc Am Physicians. 1996;108:14–19. [PubMed] [Google Scholar]
- 9.Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, Aguilar-Bryan L, Gagel RF, Bryan J. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. 1995;268:426–429. doi: 10.1126/science.7716548. [DOI] [PubMed] [Google Scholar]
- 10.Thomas P, Ye Y, Lightner E. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Human Mol Genet. 1996;5:1809–1812. doi: 10.1093/hmg/5.11.1809. [DOI] [PubMed] [Google Scholar]
- 11.Nestorowicz A, Inagaki N, Gonoi T, Schoor KP, Wilson BA, Glaser B, Landau H, Stanley CA, Thornton PS, Seino S, Permutt MA. A nonsense mutation in the inward rectifier potassium channel gene, Kir6.2, is associated with familial hyperinsulinism. Diabetes. 1997;46:1743–1748. doi: 10.2337/diab.46.11.1743. [DOI] [PubMed] [Google Scholar]
- 12.Inagaki N, Gonoi T, Clement JP, 4th, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- 13.Dukes ID, Philipson LH. K+ channels: generating excitement in pancreatic beta-cells. Diabetes. 1996;45:845–853. doi: 10.2337/diab.45.7.845. [DOI] [PubMed] [Google Scholar]
- 14.Shilyansky J, Cutz E, Filler RM. Endogenous hyperinsulinism: diagnosis, management, and long-term follow-up. Semin Pediatr Surg. 1997;6:115–120. [PubMed] [Google Scholar]
- 15.Liebowitz B, Glaser B, Higazi AA, Salameh M, Cerasi E, Landau H. Hyperinsulinemic hypoglycemia of infancy (nesidioblastosis) in clinical remission: high incidence of diabetes mellitus and persistent beta-cell dysfunction at long-term follow-up. J Clin Endocrinol Metab. 1995;80:386–392. doi: 10.1210/jcem.80.2.7852494. [DOI] [PubMed] [Google Scholar]
- 16.Lovvorn HN, 3rd, Nance ML, Ferry RJ, Jr, Stolte L, Baker L, O’Neill JA, Jr, Schnaufer L, Stanley CA, Adzick NS. Congenital hyperinsulinism and the surgeon: lessons learned over 35 years. J Pediatr Surg. 1999;34:786–793. doi: 10.1016/s0022-3468(99)90374-3. [DOI] [PubMed] [Google Scholar]
- 17.Verkarre V, Fournet JC, de Lonlay P, Gross-Morand MS, Devillers M, Rahier J, Brunelle F, Robert JJ, Nihoul-Fekete C, Saudubray JM, Junien C. Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinism in focal adenomatous hyperplasia. J Clin Invest. 1998;102:1286–1291. doi: 10.1172/JCI4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryan F, Devaney D, Joyce C, Nestorowicz A, Permutt MA, Glaser B, Barton DE, Thornton PS. Hyperinsulinism: molecular aetiology of focal disease. Arch Dis Child. 1998;79:445–447. doi: 10.1136/adc.79.5.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glaser B, Ryan F, Donath M, Landau H, Stanley CA, Baker L, Barton DE, Thornton PS. Hyperinsulinism caused by paternal-specific inheritance of a recessive mutation in the sulfonylurea-receptor gene. Diabetes. 1999;48:1652–1657. doi: 10.2337/diabetes.48.8.1652. [DOI] [PubMed] [Google Scholar]
- 20.Aguilar-Bryan L, Clement JP, IV, Gonzalez G, Kunjilwar K, Babenko A, Bryan J. Toward understanding the assembly and structure of KATP channels. Physiol Rev. 1998;78:227–245. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- 21.Babenko AP, Aguilar-Bryan L, Bryan J. A view of sur/KIR6.X, KATP channels. Annu Rev Physiol. 1998;60:667–687. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]
- 22.Bryan J, Aguilar-Bryan L. Sulfonylurea receptors: ABC transporters that regulate ATP-sensitive K(+) channels. Biochim Biophys Acta. 1999;1461:285–303. doi: 10.1016/s0005-2736(99)00164-9. [DOI] [PubMed] [Google Scholar]
- 23.Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, IV, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphatase as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- 24.Aguilar-Bryan L, Bryan J. ATP-sensitive potassium channels, sulfonylurea receptors, and persistent hyperinsulinemic hypoglycemia of infancy. Diabetes Rev. 1996;4:336–343. [Google Scholar]
- 25.Nestorowicz A, Wilson BA, Schoor KP, Inoue H, Glaser B, Landau H, Stanley CA, Thornton PS, Clement JP, 4th, Bryan J, Aguilar-Bryan L, Permutt MA. Mutations in the sulfonlurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews. Human Mol Genet. 1996;5:1813–1822. doi: 10.1093/hmg/5.11.1813. [DOI] [PubMed] [Google Scholar]
- 26.Glaser B, Chiu KC, Anker R, Nestorowicz A, Landau H, Ben-Bassat H, Shlomai Z, Kaiser N, Thornton PS, Stanley CA, Spielman RS, Goglin-Ewens K, Cerasi E, Baker L, Rice J, Donis-Keller H, Permutt MA. Familial hyperinsulinism maps to chromosome 11p14-15.1, 30 cM centromeric to the insulin gene. Nat Genet. 1994;7:185–188. doi: 10.1038/ng0694-185. [DOI] [PubMed] [Google Scholar]
- 27.Ehrlich RM, Martin JM. Tolbutamide tolerance test and plasma-insulin response in children with idiopathic hypoglycemia. J Pediatr. 1967;71:485–493. doi: 10.1016/s0022-3476(67)80098-2. [DOI] [PubMed] [Google Scholar]
- 28.McMahon MM, O’Brien PC, Service FJ. Diagnostic interpretation of the intravenous tolbutamide test for insulinoma. Mayo Clin Proc. 1989;64:1481–1488. doi: 10.1016/s0025-6196(12)65703-6. [DOI] [PubMed] [Google Scholar]
- 29.Dunne MJ, Kane C, Shepherd RM, Sanchez JA, James RF, Johnson PR, Aynsley-Green A, Lu S, Clement JP, 4th, Lindley KJ, Seino S, Aguilar-Bryan L. Familial persistent hyperinsulinemic hypoglycemia of infancy and mutations in the sulfonylurea receptor. N Engl J Med. 1997;336:703–706. doi: 10.1056/NEJM199703063361005. [DOI] [PubMed] [Google Scholar]
- 30.Sharma N, Crane A, Clement JP, 4th, Gonzalez G, Babenko AP, Bryan J, Aguilar-Bryan L. The C terminus of SUR1 is required for trafficking of KATP channels. J Biol Chem. 1999;274:20628–20632. doi: 10.1074/jbc.274.29.20628. [DOI] [PubMed] [Google Scholar]
- 31.Sato Y, Aizawa T, Komatsu M, Okada N, Yamada T. Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic B-cell. Diabetes. 1992;41:438–443. doi: 10.2337/diab.41.4.438. [DOI] [PubMed] [Google Scholar]
- 32.Gembal M, Gilon P, Henquin JC. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J Clin Invest. 1992;89:1288–1295. doi: 10.1172/JCI115714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Best L, Yates AP, Tomlinson S. Stimulation of insulin secretion by glucose in the absence of diminished (86Rb+) permeability. Biochem Pharmacol. 1992;43:2483–2485. doi: 10.1016/0006-2952(92)90330-l. [DOI] [PubMed] [Google Scholar]
- 34.Straub SG, James RFL, Dunne MJ, Sharp GWG. Glucose activates both KATP channel-dependent and KATP channel-independent signaling pathways in human islets. Diabetes. 1998;47:758–763. doi: 10.2337/diabetes.47.5.758. [DOI] [PubMed] [Google Scholar]
- 35.Shepherd RM, Cosgrove KE, O’Brien RE, Barnes PD, Ammala C, Dunne MJ. Hyperinsulinism of infancy: towards an understanding of unregulated insulin release. Arch Dis Child Fetal Neonatal Ed. 2000;82:F87–F97. doi: 10.1136/fn.82.2.F87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roe MW, Lancaster ME, Mertz RJ, Worley JF, III, Dukes ID. Thapsigargin inhibits the glucose-induced decrease of intracellular Ca2+ in mouse islets of Langerhans. Am J Physiol. 1994;266:E852–E862. doi: 10.1152/ajpendo.1994.266.6.E852. [DOI] [PubMed] [Google Scholar]
- 37.Hamakawa N, Tada T. Interplay of glucose-stimulated Ca2+ sequestration and acetylcholine-induced Ca2+ release at the endoplasmic reticulum in rat pancreatic β-cells. Cell Calcium. 1995;17:21–31. doi: 10.1016/0143-4160(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 38.Dukes ID. Calcium influx and efflux pathways in insulin-secreting cells. Biophys J. 1993;64:A127. Abstract. [Google Scholar]
- 39.Gromada J, Frokjaer-Jensen J, Disseng S. Glucose stimulates voltage- and calcium-dependent inositol triphosphate production and intracellular calcium mobilization in insulin-secreting BTC3 cells. Biochem J. 1996;314:339–345. doi: 10.1042/bj3140339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Worley JF, III, McIntyre MS, Spencer B, Dukes ID. Depletion of intracellular Ca2+ stores activates a maitotoxin-sensitive nonselective cationic current in β-cells. J Biol Chem. 1994;269:32055–32058. [PubMed] [Google Scholar]
- 41.Roe MW, Worley JF, III, Qian F, Mittal AA, Mertz RJ, Philipson LH, Dukes ID. Characterization of a Ca2+ release-activated nonselective cation current regulating membrane potential and [Ca2+]i oscillations in transgenic-derived β-cells. J Biol Chem. 1998;273:10402–10410. doi: 10.1074/jbc.273.17.10402. [DOI] [PubMed] [Google Scholar]
- 42.Yajima H, Komatsu M, Schermerhorn T, Aizawa T, Kaneko T, Nagai M, Sharp GW, Hashizume K. cAMP enhances insulin secretion by an action on the ATP-sensitive K+ channel-independent pathway of glucose signaling in rat pancreatic islets. Diabetes. 1999;48:1006–1012. doi: 10.2337/diabetes.48.5.1006. [DOI] [PubMed] [Google Scholar]
- 43.Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 knockout mice: a model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem. 2000;275:9270–9277. doi: 10.1074/jbc.275.13.9270. [DOI] [PubMed] [Google Scholar]
- 44.Byrne MM, Sturis J, Clement K, Vionnet N, Pueyo ME, Stoffel M, Takeda J, Passa P, Cohen D, Bell GI, Velho G, Froguel P, Polonsky KS. Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest. 1994;93:1120–1130. doi: 10.1172/JCI117064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byrne MM, Sturis J, Menzel S, Yamagata K, Fajans SS, Dronsfield MJ, Bain SC, Hattersley AT, Velho G, Froguel P, Bell GI, Polonsky KS. Altered insulin secretory responses to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY3 on chromosome 12. Diabetes. 1996;45:1503–1510. doi: 10.2337/diab.45.11.1503. [DOI] [PubMed] [Google Scholar]
- 46.Fajans SS, Bell GI, Bowden DW, Halter JB, Polonsky KS. Maturity-onset diabetes of the young. Life Sci. 1994;55:413–422. doi: 10.1016/0024-3205(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 47.Herman WH, Fajans SS, Ortiz FJ, Smith MJ, Sturis J, Bell GI, Polonsky KS, Halter JB. Abnormal insulin secretion, not insulin resistance, is the genetic or primary defect of MODY in the RW pedigree. Diabetes. 1994;43:40–46. doi: 10.2337/diab.43.1.40. [DOI] [PubMed] [Google Scholar]
- 48.Wajngot A, Alvarsson M, Glaser A, Efendic S, Luthman H, Grill V. Glucose potentiation of arginine-induced insulin secretion is impaired in subjects with a glucokinase Glu256Lys mutation. Diabetes. 1994;43:1402–1406. doi: 10.2337/diab.43.12.1402. [DOI] [PubMed] [Google Scholar]
- 49.McCulloch DK, Raghu PK, Johnston C, Klaff LJ, Kahn SE, Beard JC, Ward WK, Benson EA, Koerker DJ, Bergman RN, Palmer JP. Defects in beta-cell function and insulin sensitivity in normoglycemic streptozocin-treated baboons: a model of preclinical insulin-dependent diabetes. J Clin Endocrinol Metab. 1988;67:785–792. doi: 10.1210/jcem-67-4-785. [DOI] [PubMed] [Google Scholar]
- 50.McRae JR, Metz SA, Robertson RP. A role for endogenous prostaglandins in defective glucose potentiation of nonglucose insulin secretagogues in diabetics. Metabolism. 1981;30:1065–1075. doi: 10.1016/0026-0495(81)90049-4. [DOI] [PubMed] [Google Scholar]
- 51.Efendic S, Lins PE, Cerasi E. Potentiation and inhibition of insulin release in man following priming with glucose and with arginine: effect of somatostatin. Acta Endocrinol. 1979;90:259–271. doi: 10.1530/acta.0.0900259. [DOI] [PubMed] [Google Scholar]