

Abstract
While the pathologies associated with in utero smoke exposure are well established, their underlying molecular mechanisms are incompletely understood. We differentiated human embryonic stem cells in the presence of physiological concentrations of tobacco smoke and nicotine. Using post hoc microarray analysis, quantitative PCR, and immunoblot analysis, we demonstrated that tobacco smoke has lineage- and stage-specific effects on human embryonic stem cell differentiation, through both nicotine-dependent and -independent pathways. We show that three major stem cell pluripotency/differentiation pathways, Notch, canonical Wnt, and transforming growth factor-β, are affected by smoke exposure, and that Nodal signaling through SMAD2 is specifically impacted by effects on Lefty1, Nodal, and FoxH1. These events are associated with upregulation of microRNA-302a, a post-transcriptional silencer of Lefty1. The described studies provide insight into the mechanisms by which tobacco smoke influences fetal development at the cellular level, and identify specific transcriptional, post-transcriptional, and signaling pathways by which this likely occurs.
Keywords: human embryonic stem cell, differentiation, mesoderm, microRNA, tobacco, nicotine
Introduction
The human fetus is susceptible to molecular and biological teratogens throughout gestation. The human placenta is particularly permeable to many of the compounds in tobacco smoke such as nicotine, carbon monoxide, and polyaromatic hydrocarbons (Longo, 1976; Myers et al., 1996; Pastrakuljic et al., 1998; Whyatt et al., 2001). Fetuses exposed to tobacco smoke in utero are more likely to be born pre-term and underweight, both of which are associated with an increased risk of numerous pathologies including respiratory distress syndrome, cardiovascular defects, cleft lip and palate, immunodeficiency, and an increased risk of Sudden Infant Death Syndrome (Andres and Day, 2000; Hackshaw et al., 2011; Higgins, 2002). In utero tobacco smoke exposure has also been linked to an increased risk of pediatric hematological malignancies (John et al., 1991; Magnani et al., 1990). Later in life, children who were exposed to tobacco smoke in utero have been shown to be at increased risk of developing attention deficit and hyperactivity disorders, as well as other behavioral and psychological problems (Indredavik et al., 2007).
Although the clinical manifestations of in utero tobacco smoke exposure are well documented, the underlying mechanisms for these pathologies are not completely understood. Recent analysis of microarray profiles of fetal tissues exposed to tobacco smoke in utero has demonstrated global changes in mRNA expression (Hussain et al., 2008), however, the link between these large molecular perturbations and their effects on developmental mechanisms are for the most part unknown. Stem cell models have been used to investigate the effects of tobacco on cellular development (Zdravkovic et al., 2008; Zhang et al., 2005), however, these studies were confined to examining the effects of nicotine only, and relied on models of brief exposure that may not reflect the long-term exposure experienced by fetal tissue. Additionally, the singular evaluation of nicotine in these studies excluded consideration of the effects of other components of tobacco smoke, such as carbon monoxide and polyaromatic hydrocarbons, which are known to have physiological and potential developmental effects (Longo, 1976; Whyatt et al., 2001).
To determine the effects of tobacco smoke and nicotine on human embryonic development, we developed a human embryonic stem cell (hESC) culture model of tobacco smoke and nicotine exposure. We exposed hESCs to tobacco smoke-infused and nicotine-supplemented medium during spontaneous hESC differentiation through embryoid body formation. We examined whether tobacco smoke affects the formation of embryonic germ layers and impacts the pluripotent state in hESCs. Our results also led us to examine the role of the Nodal signaling pathway in mediating the effects of tobacco smoke on human embryonic development.
Materials and Methods
Post hoc expression profiling analysis
Expression profiling of cord tissue from smoking and non-smoking mothers was performed as previously described (Hussain et al., 2008) with approval from the Institutional Review Board at the University of Connecticut. All data were previously deposited to Gene Expression Omnibus (GSE11798; http://www.ncbi.nlm.nih.gov/geo/). Anonymous raw microarray data were formatted in Excel (Microsoft), and the data uploaded into the Core Analysis function of Ingenuity Pathway Analysis (IPA; Ingenuity Systems) using the following settings: all databases were selected to maximize the identification of microarray probes; the Ingenuity Knowledge Base (genes only) reference set was used to investigate experimentally observed direct relationships only; duplicate probes were resolved using the average fold change. Parameters were set to p<0.05 and a fold-change of at least 1.5. Data of affected biological pathways were taken from the Top Functions tab of the analyzed data set in IPA.
hESC culture and differentiation
Undifferentiated GFP H9 stem cell lines were maintained in culture and differentiated by human embryoid body (hEB) formation as previously described (Gaur et al., 2010; King et al., 2009; Ritner et al., 2011). At day four of hEB formation, equal numbers of differentiating hEBs were plated onto gelatin-coated plates in the presence of differentiation medium (DM; Knockout DMEM (Invitrogen), 20% Defined Fetal Bovine Serum (Hyclone), 2 mM glutamine, 0.1 mM non-essential amino acids, 0.1 mM 2-mercaptoethanol), DM infused with tobacco smoke as described below, or DM with 3.30 μM l-nicotine (Acros Organics). Tobacco smoke and nicotine concentrations were chosen as discussed in Results. Medium was exchanged every 48 hours with freshly prepared medium.
Media preparation and analysis
Research-grade cigarettes (3R4F) were purchased from the Reference Cigarette Program at the University of Kentucky maintained by the U.S. Department of Agriculture. Cigarettes were smoked using a closed extraction system previously described (Carp and Janoff, 1978). One cigarette was smoked into 20 mL pre-warmed DM, and 10 mL of smoke-infused DM was filtered through a 0.22 μM syringe filter. A 125 μL aliquot of filtered, smoke-infused medium was added to 2.875 mL pre-warmed DM. The nicotine concentrations of smoke-infused medium and control DM were determined by gas chromatography at the Tobacco Biomarkers Core Facility of the Helen Diller Family Comprehensive Cancer Center of the University of California San Francisco. The described dilution yielded an average nicotine concentration of 3.39 μM, approximating fetal tissue exposure to the smoke of one cigarette as explained in Results.
TUNEL staining and flow cytometry
Apoptosis was assayed in differentiating hESCs at days 8, 14, and 21 by scraping cells into cold phosphate-buffered saline (PBS), disaggregating cells in 0.05% trypsin-EDTA for 5 minutes at 37C, resuspending cells in cold PBS with 20% horse serum, and terminal deoxynucleotidyl transferase dUTP nick end-labeling using the TMR TUNEL Staining kit (Roche) according to the manufacturer’s instructions. Fluorescein isothiocyanate signals were collected for ≥50,000 cells using a BD LSR II flow cytometer, and data were analyzed using FlowJo software (Tree Star, Inc.) on a Macintosh platform.
Quantitative real-time PCR
Total RNA from control, tobacco smoke-exposed, and nicotine treated hEBs was obtained using TRIZOL reagent (Invitrogen). For quantitative real-time PCR (qPCR) analysis of mRNA expression, cDNA was synthesized from total RNA using SuperScript III (Invitrogen) following the manufacturer’s instructions, and cDNA was pre-amplified and qPCR performed as previously described (Ritner et al., 2011). For qPCR analysis of microRNA expression, cDNA was transcribed from total RNA using the TaqMan microRNA Reverse Transcription kit (Applied Biosystems). Relative expression was determined using the TaqMan Assay (Applied Biosystems) on an Applied Biosystems 7300 Real-Time PCR system with the following primer pairs: Oct4 (Hs00742896_s1), Nanog (Hs02387400_g1), Nestin (Hs00707120_s1), βIII tubulin (Hs00964965_m1), Brachyury (Hs00610080_m1), Tal1 (Hs00268434_m1), Nkx2-5 (Hs00231763_m1), α-fetoprotein (Hs00173490_m1), FoxA2 (Hs00232764_m1), Hes1 (Hs00172878_m1), Nodal (Hs01086749_m1), Lef1 (Hs01547250_m1), Lefty1 (Hs00764128_s1), FoxH1 (Hs00182690_m1). All assays were normalized to the signal for GAPDH (4326317E). MicroRNA-302a expression was quantified using hsa-miR-302a primer pairs (ABI 000529) normalized against RNU44 (001094) and RNU48 (001006) controls.
Immunoblot analysis
Whole cell lysates from hESC cultures differentiated for 14 days were prepared in 50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% deoxycholate, 1% NP-40, 0.1% SDS, and Protease Inhibitor Cocktail (Roche), separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and analyzed by immunoblot as previously described (Epting et al., 2004). Undifferentiated hESCs treated with 50 ng/ml human Activin A (Humanzyme; HZ-1137) for 4 hr at 37°C were used as a positive control for phosphorylated SMAD2. Primary antibodies included monoclonal rabbit anti-SMAD2 (Cell Signaling; 3122), monoclonal mouse anti-phospho(Ser465/467)-SMAD2 (Cell Signaling; 3103), and polyclonal goat anti-actin (Santa Cruz Biotechnology; SC-1616). Immunoblot signals were quantified with ImageJ64 (NIH).
Statistical analysis
Differences between two groups (qPCR) were tested for significance by unpaired t-test, and for three or more groups (TUNEL) by one-way analysis of variance, followed by Bonferroni’s post-test to isolate specific differences. A value of p<0.05 was considered significant. All analyses were performed using SPSS (IBM) for Macintosh.
Results
Tobacco smoke exposure alters fetal gene expression
Previous analysis of umbilical cord tissue from fetuses exposed to tobacco smoke in utero demonstrated upregulation of genes important for fetal growth, angiogenesis, and development of connective tissue (Hussain et al., 2008). To determine whether these findings extended to a sensitivity of specific embryonic germ layers to tobacco smoke exposure, we performed further post hoc pathway analysis of this data set. Using cutoffs for fold-change in gene expression greater than 1.5, and significance of p<0.05, we identified genes that could be ascribed to the development of specific physiological systems (Table 1). This identified gene products important in the development of tissues arising from mesoderm (e.g., AKT2, annexin A1, BCL3, interleukin-23, PI3 kinase, cyclooxygenase-2, SOCS1, and STAT5B (Boniface et al., 2008; Chen et al., 2003; Damazo et al., 2007; Diao et al., 2009; Palmer and Chen, 2008; Peng et al., 2003; Ramjaun and Downward, 2007; Trenerry et al., 2011)), as well as transcripts fundamental to cellular development across germ layers (e.g., CREB1, integrin β1, focal adhesion kinase, neurofibromin 1, palladin (Chatzizacharias et al., 2010; Diwakar et al., 2008; Faraldo et al., 2000; Ingram et al., 2002; Jin, 2011; Kossler et al., 2011; Leone et al., 2011; Luo et al., 2005; Potocnik, 2000; Sengbusch et al., 2002; Wang and Moser, 2008; Yashpal et al., 2005; Zubenko and Hughes, 2010)). The systems with the highest numbers of differentially expressed genes in tobacco-exposed tissues included the hematological, musculoskeletal, and cardiovascular systems, all of which have mesodermal origins (Fig. 1).
Table 1.
Fold-change >1.5 (p<0.05) in system-specific genes with in utero tobacco exposure
| ID | Gene | Fold- change | p-value |
|---|---|---|---|
| Hematological development | |||
| SOCS1 | suppressor of cytokine signaling 1 | −2.10 | 0.05 |
| AP3D1 | AP-3 complex subunit δ1 | −2.01 | 0.00 |
| ANXA1 | annexin A1 | −1.94 | 0.04 |
| CSF2RA | GMCSF receptor/CD116 | −1.94 | 0.03 |
| SYK | spleen tyrosine kinase | −1.86 | 0.04 |
| CST3 | cystatin 3 | −1.79 | 0.04 |
| KLF13 | Kruppel-like factor 13 | −1.77 | 0.02 |
| CDK6 | cyclin-dependent kinase 6 | −1.71 | 0.03 |
| WASL | Wiskott-Aldrich syndrome gene-like | −1.67 | 0.00 |
| MLLT10 | leukemia AF-10 protein | −1.64 | 0.02 |
| LMAN1 | lectin mannose-binding 1 | −1.63 | 0.05 |
| TOP2A | DNA topoisomerase 2α | −1.63 | 0.02 |
| NF1 | neurofibromin 1 | −1.62 | 0.01 |
| AKT2 | RACβ serine/threonine-protein kinase | −1.60 | 0.01 |
| PTGS2 | cyclooxygenase-2 | −1.59 | 0.04 |
| CD8B | cluster of differentiation 8B | −1.58 | 0.02 |
| KLK3 | prostate-specific antigen | −1.57 | 0.05 |
| IFNK | interferon κ | −1.56 | 0.01 |
| SLC19A1 | folate transporter 1 | −1.55 | 0.04 |
| TSC2 3 | TSC22 domain family protein 3 | −1.55 | 0.04 |
| PIK3R1 | phosphatidylinositol 3-kinase regulatory subunit α | −1.53 | 0.01 |
| BCL3 | B-cell lymphoma 3-encoded protein | −1.51 | 0.01 |
| CREB1 | cAMP responsive element binding protein 1 | −1.51 | 0.02 |
| PALLD | palladin | −1.51 | 0.03 |
| IL9R | interleukin 9 receptor | 1.51 | 0.02 |
| CSNK1A1 | casein kinase I isoform α | 1.53 | 0.01 |
| IL23A | interleukin-23 subunit α | 1.53 | 0.00 |
| PTK2 | focal adhesion kinase | 1.62 | 0.02 |
| ITGB1 | integrin β1 | 1.67 | 0.02 |
| SMARCC1 | SWI/SNF complex subunit/BAF155 | 1.67 | 0.04 |
| PR47 | 47 kDa platelet receptor for type III collagen | 1.69 | 0.04 |
| EBI3 | Epstein-Barr virus induced gene 3 | 1.72 | 0.05 |
| PDPN | podoplanin | 1.76 | 0.01 |
| STAT5B | signal transducer and activator of transcription 5B | 1.94 | 0.04 |
| LCAT | lecithin-cholesterol acyltransferase | 1.96 | 0.03 |
| Musculoskeletal development | |||
| SOCS1 | suppressor of cytokine signaling 1 | −2.10 | 0.05 |
| ANXA1 | annexin A1 | −1.94 | 0.04 |
| NOX1 | NADPH oxidase 1 | −1.76 | 0.02 |
| FOSL2 | Fos-related antigen 2 | −1.64 | 0.00 |
| IGFBP5 | insulin-like growth factor-binding protein 5 | −1.64 | 0.01 |
| NF1 | neurofibromin 1 | −1.62 | 0.01 |
| ADRA1A | α1A adrenergic receptor | −1.60 | 0.05 |
| AKT2 | RACβ serine/threonine-protein kinase | −1.60 | 0.01 |
| PTGS2 | cyclooxygenase-2 | −1.59 | 0.04 |
| ABCC4 | ATP-binding cassette sub-family C member 4 | −1.58 | 0.00 |
| KLK3 | prostate-specific antigen | −1.57 | 0.05 |
| PIK3R1 | phosphatidylinositol 3-kinase regulatory subunit α | −1.53 | 0.01 |
| BCL3 | B-cell lymphoma 3-encoded protein | −1.51 | 0.01 |
| CREB1 | cAMP responsive element binding protein 1 | −1.51 | 0.02 |
| IL23A | interleukin-23 subunit alpha | 1.53 | 0.00 |
| PTK2 | focal adhesion kinase | 1.62 | 0.02 |
| ITGB1 | integrin β1 | 1.67 | 0.02 |
| STAT5B | signal transducer and activator of transcription 5B | 1.94 | 0.04 |
| Cardiovascular development | |||
| ANXA1 | annexin A1 | −1.94 | 0.04 |
| NOX1 | NADPH oxidase 1 | −1.76 | 0.02 |
| ADRA1A | α1A adrenergic receptor | −1.60 | 0.05 |
| PTGS2 | cyclooxygenase-2 | −1.59 | 0.04 |
| KLK3 | prostate-specific antigen | −1.57 | 0.05 |
| GLRX3 | glutaredoxin-3 | −1.53 | 0.03 |
| PTK2 | focal adhesion kinase | 1.62 | 0.02 |
| ITGB1 | integrin β1 | 1.67 | 0.02 |
| Neurological development | |||
| FKBP5 | FK506-binding protein 5 | −3.86 | 0.00 |
| ARHGAP32 | Rho GTPase-activating protein 32 | −1.95 | 0.04 |
| ANK2 | ankyrin 2, neuronal | −1.94 | 0.04 |
| DOCK1 | dedicator of cytokinesis, 180 kDa | −1.90 | 0.02 |
| FAM126A | family with sequence similarity 126, member A | −1.90 | 0.02 |
| NTRK2 | TrkB receptor | −1.84 | 0.04 |
| STX3 | syntaxin 3 | −1.81 | 0.03 |
| MAP6 | microtubule-associated protein 6 | −1.75 | 0.05 |
| FKBP4 | FK506-binding protein 4 | −1.70 | 0.01 |
| UNC13A | unc-13 homolog A | −1.66 | 0.03 |
| KCNJ5 | G protein-activated inward rectifier K+ channel 4/5 | −1.64 | 0.03 |
| NF1 | neurofibromin 1 | −1.62 | 0.01 |
| CREB1 | cAMP responsive element binding protein 1 | −1.51 | 0.02 |
| PALLD | palladin | −1.51 | 0.03 |
| LAMA5 | laminin subunit α5 | 1.56 | 0.04 |
| PTK2 | focal adhesion kinase | 1.62 | 0.02 |
| ITGB1 | integrin β1 | 1.67 | 0.02 |
| ARHGEF10 | Rho guanine nucleotide exchange factor 10 | 1.78 | 0.02 |
| CDC42BPA | serine/threonine-protein kinase MRCKα | 1.79 | 0.00 |
Fig. 1. Tobacco smoke exposure has a widespread effect on fetal tissue development.

A previously described data set (Hussain et al., 2008) was analyzed using Ingenuity Pathway Analysis with cutoffs of fold-change >1.5 and significance at p<0.05. The top four tissue- or organ-specific biological functions were identified. Three of the four systems identified (hematological, musculoskeletal, cardiovascular) derive from embryonic mesoderm, while the remaining system, neurological, arises from ectoderm.
Development of a cell culture model of tobacco smoke exposure
To further explore the mechanisms responsible for tobacco-related alterations in the human developmental program, we developed a cell culture model of tobacco smoke exposure in differentiating human embryonic stem cells (hESCs). Nicotine concentrations in fetal serum have been demonstrated to be 1.12 times higher than in maternal serum (Luck et al., 1985). In adults, one cigarette causes blood nicotine concentrations from 0.03 μM to 0.45 μM with an average of 0.20 μM (Russell et al., 1980). Other studies have demonstrated that there is a considerable amount of variability in the blood nicotine levels between individuals who smoke the same number of cigarettes (Benowitz et al., 1982). In fetal tissues, the concentration of nicotine is considerably higher, with reported concentrations of 0.9–14.2 μM in amniotic fluid and 0.3–15.4 μM in fetal serum (Luck et al., 1985). Based on these reports, and the concentrations studied in other investigations of nicotine effect on hESCs (0.1–6.0 μM) (Zdravkovic et al., 2008), we targeted a nicotine concentration of 3–5 μM in our experiments.
We smoked cigarettes into a closed system containing hESC differentiation medium, and determined the concentration of nicotine in the smoked medium using gas chromatography. The mean nicotine concentration achieved by smoking one research grade cigarette into 20 mL DM followed by 1:24 dilution into DM was 3.4±1.9 μM, compared to control (no smoke) samples with nicotine concentrations <0.06 μM, the minimum concentration that could be detected (N=5, p<0.01).
Nicotine exists in two isomers: physiologically active l-nicotine (S-(−)-nicotine), which is present in tobacco, and less active d-nicotine (R-(+)-nicotine), which is absent from tobacco. In the process of smoking, approximately 2.6% of all l-nicotine in tobacco is converted to d-nicotine (Liu et al., 2008). Since gas chromatography was unable to resolve the relative percentages of l- and d-nicotine, we assumed that our smoke-exposed medium contained 3.30 μM l-nicotine. Based on these calculations, we performed all subsequent experiments with either smoke-exposed medium containing an estimated active nicotine concentration of 3.30 μM, or medium with 3.30 μM l-nicotine added, to distinguish between the nicotine-dependent and -independent effects of tobacco smoke exposure.
Since hESCs require fastidious culture conditions (King et al., 2009), we wanted to assess the effect of tobacco smoke and nicotine exposure on their survival. We assayed apoptosis by TUNEL staining in tobacco- and nicotine-exposed cultures compared to untreated controls (Fig. 2), and found that while there was a trend toward increasing apoptosis (% TUNEL+ cells) in differentiating cultures over time (7.8±1.6 versus 17.5±1.5%, p=0.03), there were no significant differences between treatment groups and control.
Fig. 2. Tobacco smoke exposure does not affect apoptosis in differentiating human embryonic stem cells.

hESCs were cultured in differentiation conditions for 8, 14, or 21 days in control, nicotine-supplemented, or tobacco smoke-exposed media, and apoptosis was quantified by flow cytometric analysis of TUNEL staining. Data shown are mean±SD (N=3). Apoptosis levels were similar in all conditions at each time point, and an increase in apoptosis was observed with progress of differentiation.
Tobacco smoke- and nicotine-exposed hESCs demonstrate a delay in differentiation
Octamer-binding transcription factor 4 (Oct4/POU5F1) and Nanog are required for maintenance of pluripotency (Kashyap et al., 2009), and their expression is used as a marker of undifferentiated hESCs (King et al., 2011). To determine whether tobacco smoke exposure affects the differentiation of hESCs, we assayed Oct4 and Nanog mRNA expression by qPCR in differentiating hESCs treated with tobacco smoke containing nicotine, or nicotine alone (Fig. 3). Compared to untreated differentiating cells, both Oct4 (Tobacco: 1.0±0.3 versus 2.9±0.2 at day 14, p<0.001; 1.0±0.3 versus 2.7±0.2 at day 21, p=0.001; Nicotine: 1.0±0.3 versus 2.7±0.2 at day 14, p=0.001; 1.0±0.3 versus 2.6±0.1 at day 21, p=0.001) and Nanog (Tobacco: 1.0±0.1 versus 2.5±0.5 at day 14, p=0.007; 1.0±0.2 versus 2.5±0.3 at day 21, p=0.002; Nicotine: 1.0±0.1 versus 2.8±0.2 at day 14, p<0.001; 1.0±0.2 versus 2.5±0.1 at day 21, p<0.001) expression was upregulated in hESCs cultured in differentiation medium for 14 and 21 days. These time points were specifically chosen as they represent stages at which differentiating hESC cultures are comprised mostly of differentiated cells alongside a small population of residual pluripotent Oct4+Nanog+ cells (day 14), or manifest tissue-specific cells from all three germ layers with few if any Oct4+Nanog+ cells (day 21) (King et al., 2011; King et al., 2009; Ritner et al., 2011). There was no significant difference in the levels of these two transcripts between hESCs exposed to tobacco smoke-infused medium or medium with nicotine alone. This suggests that the tobacco exposure causes a delay in differentiation, and that the effects of tobacco smoke on hESC pluripotency, as measured by Oct4 and Nanog, may be primarily due to the effects of nicotine.
Fig. 3. Tobacco smoke exposure causes persistence of human embryonic stem cell pluripotency.

hESCs were cultured in differentiation conditions for 14 or 21 days in control, nicotine-supplemented, or tobacco smoke-exposed media, and expression of Oct4 and Nanog was evaluated by quantitative real-time PCR. Data shown are mean±SD (N=4). Significant increases in expression of both pluripotency genes were observed at both time points. This effect was similar for both tobacco smoke exposure and nicotine alone.
Tobacco and nicotine have widespread and distinct effects on embryonic germ layer development
To further delineate the impact of tobacco smoke on hESC differentiation, we examined the effects of exposure on the expression of genes that identify the three embryonic germ layers, ectoderm, mesoderm and endoderm (Fig. 4). We used qPCR to determine the expression of βIII tubulin and Nestin (ectoderm), Brachyury, Tal1 and Nkx2-5 (mesoderm), and α-fetoprotein and FoxA2 (endoderm) in tobacco smoke- and nicotine-exposed hESCs at 14 and 21 days in differentiation medium. At day 14, a nearly 10-fold increase in the early mesodermal marker, Brachyury, was observed in tobacco smoke-exposed cells (1.0±0.1 versus 9.1±0.7, p<0.001), as well as a roughly 3-fold increase in expression of the later ectodermal marker, βIII tubulin (1.0±0.1 versus 3.1±0.2, p<0.001). At day 21, an increase in βIII tubulin (Tobacco: 1.0±0.2 versus 33.9±1.9, p<0.001; Nicotine: 1.0±0.2 versus 26.1±6.1, p<0.001), Brachyury (Tobacco: 1.0±0.1 versus 64.6±4.0, p<0.001; Nicotine: 1.0±0.1 versus 44.3±4.5, p<0.001), and the definitive endodermal marker, FoxA2 (Tobacco: 1.0±0.3 versus 30.9±4.2, p<0.001; Nicotine: 1.0±0.3 versus 27.3±2.8, p<0.001), were observed in both tobacco smoke- and nicotine-exposed cells. Ordinarily, these early germ layer markers are no longer expressed at such high levels by day 14 of differentiation in culture (King et al., 2011), and their upregulation at this later time point suggests a delay in germ layer specification. The increase in these genes was greater in tobacco-exposed cells compared to cells treated with nicotine alone (βIII tubulin: 33.9±1.9 versus 26.1±6.1, p=0.10; Brachyury: 64.6±4.0 versus 44.3±4.5, p=0.004; FoxA2: 30.9±4.2 versus 27.3±2.8, p=0.28). While the difference in gene expression was only statistically significant for Brachyury, these trends suggest that other components of tobacco smoke impact germ layer development as well.
Fig. 4. Effects of tobacco smoke exposure on embryonic germ layer development.

hESCs were cultured in differentiation conditions for 14 (upper panel) or 21 (lower panel) days in control, nicotine-supplemented, or tobacco smoke-exposed media, and expression of markers for embryonic ectoderm (Ecto), mesoderm (Meso) and endoderm (Endo) was evaluated by quantitative real-time PCR. Data shown are mean±SD (N=4). Persistent expression of the early mesodermal marker, Brachyury, and ectodermal marker, βIII tubulin, was seen despite culture under differentiation conditions at both time points. In addition, persistently elevated expression of markers for all three germ layers was observed at 21 days, suggesting a widespread delay in differentiation. In some cases, the changes in expression were similar with both tobacco smoke and nicotine treatment (i.e., nestin, Nkx2-5, α-fetoprotein), suggesting these effects may be predominantly due to nicotine, while for other markers, the effects were greater for tobacco smoke (i.e., βIII tubulin, Brachyury, Tal1, FoxA2), consistent with additive effects from other smoke components.
Nearly identical increases in expression between tobacco smoke and nicotine exposures were observed in the early endodermal and ectodermal markers, 3-fetoprotein (2.9±0.3 versus 2.8±0.2, p=0.66) and Nestin (12.4±1.2 versus 13.8±1.1, p=0.21), respectively, consistent with a predominant role for nicotine in early germ layer-specific developmental events. Nkx2-5, a late mesodermal marker of the cardiac lineage, increased in both tobacco smoke- (1.0±0.3 versus 16.3±2.0, p<0.001) and nicotine-treated (1.0±0.3 versus 18.9±3.2, p<0.001) cells to comparable levels (18.9±3.2 versus 16.3±2.0, p=0.30). In contrast, Tal1, a late mesodermal marker of the hematopoietic lineage, increased seven-fold in tobacco-exposed cells over untreated cells (1.0±0.3 versus 7.0±1.1, p<0.001), while expression in cells treated with nicotine alone increased only slightly over the untreated cells (1.0±0.3 versus 1.3±0.2, p=0.22).
Taken together, these data suggest that tobacco smoke, as a multicomponent bioactive agent, has a widespread effect on development of all three embryonic germ layers, consistent with persistent expression of the pluripotency markers, Oct4 and Nanog (Fig. 3). Specifically, it appears that the nicotine component of tobacco smoke has a significant impact on cardiac specification (Nkx2-5) within mesoderm, while other components of tobacco smoke likely have a greater impact on hematopoietic differentiation (Tal1) and earlier mesodermal development (Brachyury). In contrast, nicotine appears to have a dominant effect on the early development of ectoderm (Nestin) and endoderm (α-fetoprotein), whereas other tobacco smoke components may target later developmental events in these tissues (βIII tubulin and FoxA2, respectively).
Nodal expression is altered with tobacco smoke exposure
Given the widespread developmental effects of tobacco smoke exposure, we examined the expression of sentinel genes for the Notch, TGFβ and canonical Wnt signaling pathways in differentiating hESCs exposed to tobacco smoke and nicotine (Fig. 5A). The Notch pathway controls multiple cell differentiation pathways during embryonic and adult life (Liu et al., 2010). Wnt signaling through its canonical pathway has been implicated in germ layer specification (Wang and Wynshaw-Boris, 2004). Signaling through TGFβ regulates multiple effectors of embryonic stem cell pluripotency and differentiation (Ogawa et al., 2007). We assessed the expression of Hes1, a downstream effector of Notch, Nodal, a TGFβ ligand specifically implicated in cell differentiation (Watabe and Miyazono, 2009), and Lef1, a transcription factor that reports canonical Wnt activity (Ling et al., 2009), in tobacco smoke- and nicotine-treated differentiating hESCs at 14 days. Expression levels of all three sentinel genes were upregulated with both tobacco smoke and nicotine exposure, however, the most prominent impact was seen with Nodal expression (Tobacco: 1.0±0.2 versus 72.4±5.3, p=0.0001; Nicotine: 1.0±0.2 versus 48.3±3.4, p=0.0001). In addition, Nodal expression in response to tobacco smoke was 50% higher compared to nicotine exposure alone (72.4±5.3 versus 48.3±3.4, p=0.003), suggesting both nicotine-dependent and -independent effects of tobacco smoke on Nodal-mediated TGFβ signaling.
Fig. 5. Nodal signaling mediates the effects of tobacco smoke on differentiation.

hESCs were cultured in differentiation conditions for 14 days in control, nicotine-supplemented, or tobacco smoke-exposed media. A) Expression of genes associated with Notch (Hes1), TGFβ (Nodal) and canonical Wnt (Lef1) signaling pathways was evaluated by quantitative real-time PCR. Data shown are mean±SD (N=3). While all three developmentally important pathways were affected by tobacco smoke exposure, the greatest effect was seen with Nodal. In addition, the significant difference between Nodal upregulation with tobacco smoke and nicotine alone suggested that the effects of tobacco smoke on Nodal occur through nicotine-dependent and -independent mechanisms. B) Expression of Nodal, the Nodal inhibitor, Lefty1, and the Nodal effector, FoxH1, was evaluated by quantitative real-time PCR. Data shown are mean±SD (N=3). While the Nodal effector, FoxH1, was upregulated with tobacco smoke and nicotine exposure, Lefty1, a Nodal inhibitor, was downregulated, implicated the Nodal pathway in mediating tobacco smoke related effects. C) Whole cell lysate from Activin A-stimulated, undifferentiated hESCs (C; positive control), and hESCs differentiated for 14 days and treated with tobacco smoke- (T) or nicotine-supplemented medium (N), or untreated medium (U), were analyzed by immunoblot for total SMAD2 and phosphorylated SMAD2 (P-SMAD2) expression (left panel). Actin expression was used as a loading control. Immunoblot signals for SMAD2 and P-SMAD2 were quantified, normalized to Actin signals, and expressed as a ratio of P-SMAD2:SMAD2 (right panel). Typical results are shown. There was no difference in SMAD2 expression between samples, however, P-SMAD2 was upregulated with tobacco smoke exposure and more so with nicotine treatment, consistent with a delay in differentiation. The increase in SMAD2 phosphorylation with nicotine compared to tobacco smoke exposure suggests that nicotine may be the predominant effector of Nodal signaling through SMAD2, even though Nodal expression was higher with total tobacco smoke.
Nodal is essential for left-right axis symmetry and its expression contributes to the maintenance of stem cell pluripotency (Beddington and Robertson, 1999; Lee et al., 2011; Ogawa et al., 2007). Upon binding to its receptor, Nodal stimulates a signaling cascade that leads to the phosphorylation and binding of SMAD2/3 (Besser, 2004). The phospho-SMAD2/3 complex enters the nucleus and forms a transcriptional complex with SMAD4 and FoxH1 to control the expression of Nodal and other targets that promote pluripotency, including Oct4 (Lee et al., 2011) and Lefty, a negative regulator of Nodal (Pogoda et al., 2000; Saijoh et al., 2000).
Using qPCR, we assessed the expression of Lefty1, Nodal, and FoxH1 in differentiating hESCs exposed to tobacco smoke and nicotine (Fig. 5B). At day 14, Nodal expression was 50–75-fold higher in nicotine- and tobacco smoke-exposed cells, respectively, compared to untreated cells (Nicotine: 1.0±0.2 versus 48.3±3.4, p=0.0001; Tobacco: 1.0±0.2 versus 72.4±5.3, p=0.0001), and FoxH1 expression was also upregulated in exposed cells (Nicotine: 1.0±0.2 versus 23.9±2.6, p=0.0001; Tobacco: 1.0±0.2 versus 19.4±1.3, p=0.0001). Lefty1 expression was depressed, more so in tobacco smoke-exposed cells (Nicotine: 1.0±0.1 versus 0.6±0.1, p=0.008; Tobacco: 1.0±0.1 versus 0.3±0.0, p=0.0003). These data suggest that Nodal signaling may be responsible for persistence of the pluripotent state observed with tobacco smoke exposure (Fig. 3). In addition, the increased expression of Nodal in response to tobacco smoke compared to nicotine alone (72.4±5.3 versus 48.3±3.4, p=0.003) supports both nicotine-dependent and -independent effects of tobacco smoke on Nodal signaling.
Nodal signaling through SMAD2 mediates the delay in differentiation seen with tobacco smoke exposure
To confirm that increased Nodal expression results in increased Nodal activity in tobacco smoke-exposed hESCs, we evaluated SMAD2 and phosph-SMAD2 levels in differentiating hESCs exposed to tobacco smoke and nicotine by immunoblot (Fig. 5C). Activin A stimulation of undifferentiated hESCs was used as a positive control for SMAD2 phosphorylation. We observed an increase in phosphorylated SMAD2 relative to total SMAD2 in tobacco smoke- and nicotine-exposed cells at 14 days of differentiation, consistent with an increase in signaling through Nodal. This provided biochemical confirmation that Nodal signaling likely causes the delay in hESC differentiation seen with tobacco exposure.
MicroRNA-302a, a positive regulator of Nodal signaling, is upregulated with tobacco smoke exposure
MicroRNAs (miRNAs) are small RNAs thought to control the post-transcriptional expression of >30% of all coding genes, and influence numerous biological processes, including stem cell pluripotency (Mallanna and Rizzino, 2010). miRNAs primarily support post-transcriptional gene silencing by targeting specific mRNA transcripts for degradation or by inhibiting their translation (Mallanna and Rizzino, 2010). Oct4 and Nanog have been shown to promote pluripotency by activating the transcription of the miRNA-302 cluster. The miRNA-302 cluster is only expressed in early stages of embryogenesis and has been shown to promote pluripotency (Card et al., 2008). miRNA-302a specifically has been demonstrated to inhibit translation of Lefty1 (Rosa et al., 2009), a negative regulator of Nodal (Hamada et al., 2002). Because of the effect of tobacco smoke exposure on Nodal expression and signaling through SMAD2, we assessed miRNA-302a expression in differentiating hESCs exposed to tobacco smoke by qPCR (Fig. 6). We observed an increase in miRNA-302a expression at day 14 of differentiation in tobacco smoke-exposed cells compared to untreated cells (2.3±0.6 versus 1.0±0.2, p=0.02). This is consistent with the persistence in pluripotency observed, as evidenced by the increase in Oct4 and Nanog expression (Fig. 3). This also suggests a post-transcriptional regulatory process to explain the observed activation of Nodal signaling, through miR-302a-mediated downregulation of Lefty1 (Fig. 5B).
Fig. 6. MicroRNA-302a is upregulated with tobacco smoke exposure.
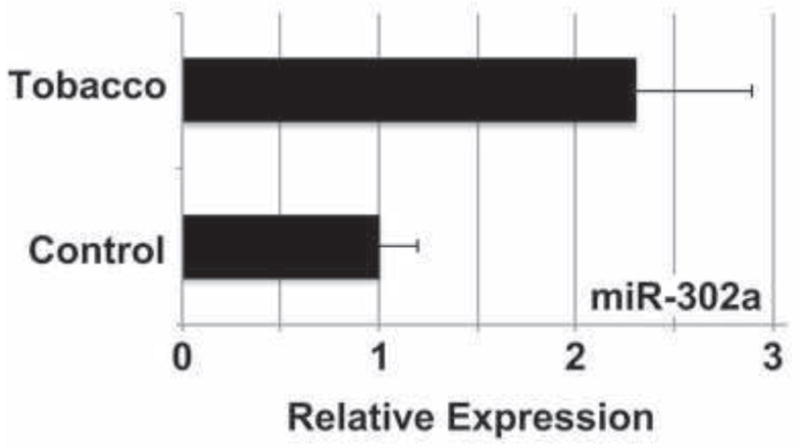
hESCs were cultured in differentiation conditions for 14 days in control or tobacco smoke-exposed medium, and miRNA-302a expression was evaluated by quantitative real-time PCR. Data shown are mean±SD (N=3). Tobacco smoke induced a >2-fold increase in miR-302a expression, consistent with the observed downregulation of the Nodal inhibitor, Lefty1 (Fig. 5B).
Discussion
We found that in vivo exposure to tobacco smoke influenced gene expression in cord tissue obtained from smoking versus non-smoking mothers, and that gene expression patterns indicated predominant effects on organ systems derived from mesoderm (hematological, musculoskeletal, reproductive, cardiovascular). Others have recently shown that graded Nodal signaling titrates the balance between pluripotency and specifically mesoendodermal differentiation in embryonic stem cells (Lee et al., 2011), supporting our studies implicating Nodal signaling in tobacco smoke-mediated developmental patterns.
Studies in differentiating hESCs in culture showed sustained expression levels of the pluripotency markers, Oct4 and Nanog, as well as the early mesodermal marker, Brachyury, in both tobacco and nicotine treated cells, suggesting a nicotine-dependent delay in differentiation. However, the delayed up-regulation of the hematopoietic lineage marker, Tal1, in day 21 exposed cells suggests a nicotine-independent delay in the differentiation of the hematopoietic lineage. These results indicate that tobacco smoke can perturb normal development independently of nicotine, and that modeling tobacco smoke exposure in vitro with nicotine exposure alone fails to account for the myriad other toxins in tobacco smoke that can affect differentiation.
hESC have proven to be a versatile system for modeling fetal toxicology and development (Pal et al., 2011). We have shown that with gas chromatography, it is possible to deliver physiological concentrations of tobacco smoke components to hESCs. Our approach contrasts that of other groups who have exposed cell cultures to tobacco smoke without quantifying the concentration of nicotine or other components that were being delivered (Crane-Godreau et al., 2009; Nana-Sinkam et al., 2007). Although other studies have provided results indicating the potential effects of tobacco components on cells, it is quite possible that those studies introduced toxins at concentrations significantly above physiological levels. Subtle changes in key signaling pathways may be overlooked or even exaggerated under these circumstances.
Genome-wide examination of the effects of tobacco smoke on fetal tissues suggests that tobacco smoke results in perturbed development of several distinct tissues and organ systems, particularly those of mesodermal origin. Using titered tobacco smoke and nicotine exposure models, we demonstrated elevated expression of pluripotency markers, Oct4 and Nanog, at days 14 and 21 of differentiation with exposure to tobacco smoke and nicotine. Nearly identical expression of pluripotency markers in both total smoke and nicotine treated cells suggests that the nicotine component in tobacco smoke has a widespread effect on stem cell pluripotency and differentiation.
Our assessment of embryonic germ layer development suggests a nicotine-dependent delay specifically in the mesodermal lineage and a minor delay in endodermal differentiation. Interestingly, the delay in differentiation is more pronounced in the tobacco smoke exposed cells, compared to treatment with nicotine alone. The more pronounced upregulation of Brachyury in tobacco smoke-treated cells than with nicotine exposure at day 14, with subsequent similar alterations in both tobacco smoke- and nicotine-treated cells at day 21, suggest that various components of tobacco smoke, including nicotine, may play important roles in the tempo of cell differentiation in a stage-specific manner. The absence of significant changes in expression of the primitive endodermal marker, α-fetoprotein, with either tobacco smoke or nicotine treatment at either time point suggests that there is very little effect of tobacco components on the development of early endoderm. In contrast, a significant increase in FoxA2 expression, a hallmark of definitive endoderm, was observed in both total smoke and nicotine exposed cells, suggesting a nicotine-dependent delay later in endodermal differentiation.
In mesoderm, the effects of tobacco smoke and nicotine vary between lineages. High expression of Brachyury at day 14 in smoke treated cells relative to control and nicotine treatment suggests a nicotine-independent delay of early mesodermal differentiation. Nkx2-5, an early marker of cardiac mesoderm, is upregulated in nicotine and tobacco treated cells at both 14 and 21 days. However, Tal1, a marker of the hematopoietic lineage, is upregulated at the later time point only in tobacco smoke-exposed cells. These observations suggest a nicotine-dependent effect on cardiac mesoderm as distinct from the nicotine-independent effects on hematopoietic development.
In addition to their effects on both germ layer development and lineage determination, tobacco smoke and nicotine had distinct effects on major signaling pathways that regulate stem cell pluripotency and differentiation. Three of the most important pathways implicated in stem cell activity, Notch, canonical Wnt, and TGFβ, were altered by tobacco exposure, with the most prominent change in expression seen with the TGFβ-associated cytokine, Nodal. Nodal signals through SMAD2, and we observed upregulation of SMAD2 phosphorylation that paralleled upregulation of Nodal expression. Interestingly, while total smoke had a greater effect on Nodal expression than nicotine, the level of SMAD2 phosphorylation was higher in nicotine-treated cells than in those exposed to total tobacco smoke. A possible explanation is that while nicotine may be only one component of tobacco smoke that impacts Nodal expression, nicotine may affect other Nodal-independent pathways that converge on SMAD2 signaling as well.
Until recently, previous investigations into the role of Nodal signaling in ESC fate decisions have been difficult to reconcile. There has been evidence that Nodal both maintains self-renewal and plutipotency (Mavrakis et al., 2007; Vincent et al., 2003), and that it promotes differentiation (Beattie et al., 2005; James et al., 2005). A recent study, however, sheds light on the mechanism by which Nodal signaling participates in both processes (Lee et al., 2011). Lee et al. showed that any interference with endogenous Nodal signaling levels impacts Oct4 expression in a graded manner through SMAD2 binding to specific sequences in its promoter (Lee et al., 2011). In their studies, high-level Nodal signaling with Activin induced differentiation toward mesoendoderm, while low-level signaling promoted SMAD2 binding to and positive regulation of the Oct4 promoter. Repression with the Nodal inhibitor, SB431542, induced trophectodermal differentiation. This work provides mechanistic support for our observation that while tobacco/nicotine-induced Nodal signaling through SMAD2 resulted in persistence of pluripotency (i.e., expression of Oct4 and Nanog) and delayed differentiation, its effects on differentiation across germ layers were stage-dependent and favored mesoendoderm. It is likely that various components of tobacco smoke, including nicotine, are disrupting the graded Nodal signaling elucidated by Lee et al. (Lee et al., 2011).
We also observed suppressed expression of the competitive Nodal inhibitor, Lefty1, and upregulated expression of the downstream Nodal effector, FoxH1, with both tobacco smoke and nicotine, consistent with the observations made for Nodal expression and signaling. In addition, we found that miR-302a, a member of a cluster of short regulatory RNAs known to play a role in stem cell differentiation, was upregulated with tobacco smoke exposure. Lefty1 is a known target of miR-302a, and likely mediates the effects of tobacco smoke on Lefty1 expression leading to Nodal activation. These studies also provide the first evidence of which we are aware that tobacco smoke exposure impacts the post-transcriptional regulation of cellular activity through miRNAs.
In summary, we have employed a controlled system for delivering physiological concentrations of nicotine and tobacco smoke to differentiating hESCs. We have demonstrated that hESCs differentiated in the presence of tobacco smoke and nicotine display widespread alterations in pluripotency and differentiation potential. We also provide evidence that nicotine is not the sole component of tobacco smoke responsible for developmental effects, and that both nicotine and other tobacco smoke components impact cellular differentiation in a stage- and lineage-dependent manner. Beyond these observations, we have shown that at least some of the effects of tobacco smoke on stem cell pluripotency are mediated through Nodal signaling through SMAD2 to FoxH1, as influenced by the Nodal inhibitory protein, Lefty1, and its regulatory miRNA, miR-302a. These studies provide important insight into both the global dysregulation and specific molecular mechanisms of development that may be responsible for the adverse effects of cigarette smoke on the embryo, fetus, infant, and child through exposure in utero. They also confirm that exposure to tobacco smoke, as well as nicotine in various forms, may have profoundly deleterious effects on a wide range of tissues in the developing embryo.
Highlights.
Pathologies associated with in utero smoke exposure are incompletely understood.
We used human embryonic stem cells as a model of tissue differentiation.
Notch, canonical Wnt, and TGFβ pathways were affected by smoke exposure.
Nodal signaling through SMAD2 was mediated by miR-302a, Lefty1, and FoxH1.
Acknowledgments
We thank Melanie Bedolli and Meenakshi Gaur for experimental advice and helpful discussions. This work was supported in part by funds from the Connecticut Department of Health and the University of Connecticut General Clinical Research Center to C.O., and a grant from the National Heart, Lung and Blood Institutes (HL085377), a gift from the Polin Foundation, and funds from the Department of Pediatrics, University California, San Francisco to H.S.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andres RL, Day MC. Perinatal complications associated with maternal tobacco use. Semin Neonatol. 2000;5:231–241. doi: 10.1053/siny.2000.0025. [DOI] [PubMed] [Google Scholar]
- Beattie GM, Lopez AD, Bucay N, Hinton A, Firpo MT, King CC, Hayek A. Activin A maintains pluripotency of human embryonic stem cells in the absence of feeder layers. Stem Cells. 2005;23:489–495. doi: 10.1634/stemcells.2004-0279. [DOI] [PubMed] [Google Scholar]
- Beddington RS, Robertson EJ. Axis development and early asymmetry in mammals. Cell. 1999;96:195–209. doi: 10.1016/s0092-8674(00)80560-7. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Kuyt F, Jacob P., 3rd Circadian blood nicotine concentrations during cigarette smoking. Clin Pharmacol Ther. 1982;32:758–764. doi: 10.1038/clpt.1982.233. [DOI] [PubMed] [Google Scholar]
- Besser D. Expression of nodal, lefty-a, and lefty-B in undifferentiated human embryonic stem cells requires activation of SMAD2/3. J Biol Chem. 2004;279:45076–45084. doi: 10.1074/jbc.M404979200. [DOI] [PubMed] [Google Scholar]
- Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. 2008;226:132–146. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card DA, Hebbar PB, Li L, Trotter KW, Komatsu Y, Mishina Y, Archer TK. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol Cell Biol. 2008;28:6426–6438. doi: 10.1128/MCB.00359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carp H, Janoff A. Possible mechanisms of emphysema in smokers. In vitro suppression of serum elastase-inhibitory capacity by fresh cigarette smoke and its prevention by antioxidants. Am Rev Respir Dis. 1978;118:617–621. doi: 10.1164/arrd.1978.118.3.617. [DOI] [PubMed] [Google Scholar]
- Chatzizacharias NA, Kouraklis GP, Theocharis SE. The role of focal adhesion kinase in early development. Histol Histopathol. 2010;25:1039–1055. doi: 10.14670/HH-25.1039. [DOI] [PubMed] [Google Scholar]
- Chen Q, Rho JY, Fan Z, Laulederkind SJ, Raghow R. Congenital lack of COX-2 affects mechanical and geometric properties of bone in mice. Calcif Tissue Int. 2003;73:387–392. doi: 10.1007/s00223-002-0009-x. [DOI] [PubMed] [Google Scholar]
- Crane-Godreau MA, Maccani MA, Eszterhas SK, Warner SL, Jukosky JA, Fiering S. Exposure to Cigarette Smoke Disrupts CCL20-Mediated Antimicrobial Activity in Respiratory Epithelial Cells. Open Immunol J. 2009;2:86–93. doi: 10.2174/1874226200902010086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damazo AS, Moradi-Bidhendi N, Oliani SM, Flower RJ. Role of annexin 1 gene expression in mouse craniofacial bone development. Birth Defects Res A Clin Mol Teratol. 2007;79:524–532. doi: 10.1002/bdra.20368. [DOI] [PubMed] [Google Scholar]
- Diao Y, Wang X, Wu Z. SOCS1, SOCS3, and PIAS1 promote myogenic differentiation by inhibiting the leukemia inhibitory factor-induced JAK1/STAT1/STAT3 pathway. Mol Cell Biol. 2009;29:5084–5093. doi: 10.1128/MCB.00267-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwakar G, Zhang D, Jiang S, Hornyak TJ. Neurofibromin as a regulator of melanocyte development and differentiation. J Cell Sci. 2008;121:167–177. doi: 10.1242/jcs.013912. [DOI] [PubMed] [Google Scholar]
- Epting CL, Lopez JE, Shen X, Liu L, Bristow J, Bernstein HS. Stem cell antigen-1 is necessary for cell-cycle withdrawal and myoblast differentiation in C2C12 cells. J Cell Sci. 2004;117:6185–6195. doi: 10.1242/jcs.01548. [DOI] [PubMed] [Google Scholar]
- Faraldo MM, Deugnier MA, Thiery JP, Glukhova MA. Development of mammary gland requires normal beta 1-integrin function. Adv Exp Med Biol. 2000;480:169–174. doi: 10.1007/0-306-46832-8_21. [DOI] [PubMed] [Google Scholar]
- Gaur M, Ritner C, Sievers R, Pedersen A, Prasad M, Bernstein HS, Yeghiazarians Y. Timed inhibition of p38MAPK directs accelerated differentiation of human embryonic stem cells into cardiomyocytes. Cytotherapy. 2010;12:807–817. doi: 10.3109/14653249.2010.491821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackshaw A, Rodeck C, Boniface S. Maternal smoking in pregnancy and birth defects: a systematic review based on 173 687 malformed cases and 11.7 million controls. Hum Reprod Update. 2011;17:589–604. doi: 10.1093/humupd/dmr022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada H, Meno C, Watanabe D, Saijoh Y. Establishment of vertebrate left-right asymmetry. Nat Rev Genet. 2002;3:103–113. doi: 10.1038/nrg732. [DOI] [PubMed] [Google Scholar]
- Higgins S. Smoking in pregnancy. Curr Opin Obstet Gynecol. 2002;14:145–151. doi: 10.1097/00001703-200204000-00007. [DOI] [PubMed] [Google Scholar]
- Hussain N, Krueger W, Covault J, Walsh S, Kranzler HR, Oncken C. Effects of prenatal tobacco exposure on gene expression profiling in umbilical cord tissue. Pediatr Res. 2008;64:147–153. doi: 10.1203/PDR.0b013e31817c5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indredavik MS, Brubakk AM, Romundstad P, Vik T. Prenatal smoking exposure and psychiatric symptoms in adolescence. Acta Paediatr. 2007;96:377–382. doi: 10.1111/j.1651-2227.2006.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram DA, Zhang L, McCarthy J, Wenning MJ, Fisher L, Yang FC, Clapp DW, Kapur R. Lymphoproliferative defects in mice lacking the expression of neurofibromin: functional and biochemical consequences of Nf1 deficiency in T-cell development and function. Blood. 2002;100:3656–3662. doi: 10.1182/blood-2002-03-0734. [DOI] [PubMed] [Google Scholar]
- James D, Levine AJ, Besser D, Hemmati-Brivanlou A. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005;132:1273–1282. doi: 10.1242/dev.01706. [DOI] [PubMed] [Google Scholar]
- Jin L. The actin associated protein palladin in smooth muscle and in the development of diseases of the cardiovasculature and in cancer. J Muscle Res Cell Motil. 2011;32:7–17. doi: 10.1007/s10974-011-9246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John EM, Savitz DA, Sandler DP. Prenatal exposure to parents’ smoking and childhood cancer. Am J Epidemiol. 1991;133:123–132. doi: 10.1093/oxfordjournals.aje.a115851. [DOI] [PubMed] [Google Scholar]
- Kashyap V, Rezende NC, Scotland KB, Shaffer SM, Persson JL, Gudas LJ, Mongan NP. Regulation of stem cell pluripotency and differentiation involves a mutual regulatory circuit of the NANOG, OCT4, and SOX2 pluripotency transcription factors with polycomb repressive complexes and stem cell microRNAs. Stem Cells Dev. 2009;18:1093–1108. doi: 10.1089/scd.2009.0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King FW, Liszewski W, Ritner C, Bernstein HS. High-Throughput Tracking of Pluripotent Human Embryonic Stem Cells with Dual Fluorescence Resonance Energy Transfer Molecular Beacons. Stem Cells Dev. 2011;20:475–484. doi: 10.1089/scd.2010.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King FW, Ritner C, Liszewski W, Kwan HC, Pedersen A, Leavitt AD, Bernstein HS. Subpopulations of human embryonic stem cells with distinct tissue-specific fates can be selected from pluripotent cultures. Stem Cells Dev. 2009;18:1441–1450. doi: 10.1089/scd.2009.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossler N, Stricker S, Rodelsperger C, Robinson PN, Kim J, Dietrich C, Osswald M, Kuhnisch J, Stevenson DA, Braun T, Mundlos S, Kolanczyk M. Neurofibromin (Nf1) is required for skeletal muscle development. Hum Mol Genet. 2011;20:2697–2709. doi: 10.1093/hmg/ddr149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaras C, Mandala E, Saratzis A, Platogiannis D, Barbetakis N, Papoti S, Christopoulou M, Ilonidis G, Bischiniotis T. Pro-brain natriuretic peptide is a sensitive marker for detecting cardiac metastases in patients with non-small cell lung cancer. Onkologie. 2009;32:389–392. doi: 10.1159/000218368. [DOI] [PubMed] [Google Scholar]
- Lee KL, Lim SK, Orlov YL, Yit le Y, Yang H, Ang LT, Poellinger L, Lim B. Graded Nodal/Activin signaling titrates conversion of quantitative phospho-SMAD2 levels into qualitative embryonic stem cell fate decisions. PLoS Genet. 2011;7:e1002130. doi: 10.1371/journal.pgen.1002130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone V, D’Angelo D, Ferraro A, Pallante P, Rubio I, Santoro M, Croce CM, Fusco A. A TSH-CREB1-microRNA Loop Is Required for Thyroid Cell Growth. Mol Endocrinol. 2011 doi: 10.1210/me.2011-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene. 2009;433:1–7. doi: 10.1016/j.gene.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Liu B, Yao W, Su Q. Racemization of S-(−)-nicotine during smoking and its relationship with pyrolysis process. J Analytic App Pyrolysis. 2008;81:157–161. [Google Scholar]
- Liu J, Sato C, Cerletti M, Wagers A. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr Top Dev Biol. 2010;92:367–409. doi: 10.1016/S0070-2153(10)92012-7. [DOI] [PubMed] [Google Scholar]
- Longo LD. Carbon monoxide: effects on oxygenation of the fetus in utero. Science. 1976;194:523–525. doi: 10.1126/science.973133. [DOI] [PubMed] [Google Scholar]
- Luck W, Nau H, Hansen R, Steldinger R. Extent of nicotine and cotinine transfer to the human fetus, placenta and amniotic fluid of smoking mothers. Dev Pharmacol Ther. 1985;8:384–395. doi: 10.1159/000457063. [DOI] [PubMed] [Google Scholar]
- Luo H, Liu X, Wang F, Huang Q, Shen S, Wang L, Xu G, Sun X, Kong H, Gu M, Chen S, Chen Z, Wang Z. Disruption of palladin results in neural tube closure defects in mice. Mol Cell Neurosci. 2005;29:507–515. doi: 10.1016/j.mcn.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Magnani C, Pastore G, Luzzatto L, Terracini B. Parental occupation and other environmental factors in the etiology of leukemias and non-Hodgkin’s lymphomas in childhood: a case-control study. Tumori. 1990;76:413–419. doi: 10.1177/030089169007600501. [DOI] [PubMed] [Google Scholar]
- Mallanna SK, Rizzino A. Emerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cells. Dev Biol. 2010;344:16–25. doi: 10.1016/j.ydbio.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis KJ, Andrew RL, Lee KL, Petropoulou C, Dixon JE, Navaratnam N, Norris DP, Episkopou V. Arkadia enhances Nodal/TGF-beta signaling by coupling phospho-SMAD2/3 activity and turnover. PLoS Biol. 2007;5:e67. doi: 10.1371/journal.pbio.0050067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SR, Spinnato JA, Pinorini-Godly MT, Cook C, Boles B, Rodgers GC. Characterization of 4-aminobiphenyl-hemoglobin adducts in maternal and fetal blood-samples. J Toxicol Environ Health. 1996;47:553–566. doi: 10.1080/009841096161537. [DOI] [PubMed] [Google Scholar]
- Nana-Sinkam SP, Lee JD, Sotto-Santiago S, Stearman RS, Keith RL, Choudhury Q, Cool C, Parr J, Moore MD, Bull TM, Voelkel NF, Geraci MW. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med. 2007;175:676–685. doi: 10.1164/rccm.200605-724OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Saito A, Matsui H, Suzuki H, Ohtsuka S, Shimosato D, Morishita Y, Watabe T, Niwa H, Miyazono K. Activin-Nodal signaling is involved in propagation of mouse embryonic stem cells. J Cell Sci. 2007;120:55–65. doi: 10.1242/jcs.03296. [DOI] [PubMed] [Google Scholar]
- Pal R, Mamidi MK, Das AK, Bhonde R. Human embryonic stem cell proliferation and differentiation as parameters to evaluate developmental toxicity. J Cell Physiol. 2011;226:1583–1595. doi: 10.1002/jcp.22484. [DOI] [PubMed] [Google Scholar]
- Palmer S, Chen YH. Bcl-3, a multifaceted modulator of NF-kappaB-mediated gene transcription. Immunol Res. 2008;42:210–218. doi: 10.1007/s12026-008-8075-4. [DOI] [PubMed] [Google Scholar]
- Pastrakuljic A, Schwartz R, Simone C, Derewlany LO, Knie B, Koren G. Transplacental transfer and biotransformation studies of nicotine in the human placental cotyledon perfused in vitro. Life Sci. 1998;63:2333–2342. doi: 10.1016/s0024-3205(98)00522-0. [DOI] [PubMed] [Google Scholar]
- Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, Sundararajan D, Chen WS, Crawford SE, Coleman KG, Hay N. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoda HM, Solnica-Krezel L, Driever W, Meyer D. The zebrafish forkhead transcription factor FoxH1/Fast1 is a modulator of nodal signaling required for organizer formation. Curr Biol. 2000;10:1041–1049. doi: 10.1016/s0960-9822(00)00669-2. [DOI] [PubMed] [Google Scholar]
- Potocnik AJ. Role of beta 1 integrin for hemato-lymphopoiesis in mouse development. Curr Top Microbiol Immunol. 2000;251:43–50. doi: 10.1007/978-3-642-57276-0_6. [DOI] [PubMed] [Google Scholar]
- Ramjaun AR, Downward J. Ras and phosphoinositide 3-kinase: partners in development and tumorigenesis. Cell Cycle. 2007;6:2902–2905. doi: 10.4161/cc.6.23.4996. [DOI] [PubMed] [Google Scholar]
- Ritner C, Wong SS, King FW, Mihardja SS, Liszewski W, Erle DJ, Lee RJ, Bernstein HS. An engineered cardiac reporter cell line identifies human embryonic stem cell-derived myocardial precursors. PLoS One. 2011;6:e16004. doi: 10.1371/journal.pone.0016004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa A, Spagnoli FM, Brivanlou AH. The miR-430/427/302 family controls mesendodermal fate specification via species-specific target selection. Dev Cell. 2009;16:517–527. doi: 10.1016/j.devcel.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Russell MA, Jarvis M, Iyer R, Feyerabend C. Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. Br Med J. 1980;280:972–976. doi: 10.1136/bmj.280.6219.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijoh Y, Adachi H, Sakuma R, Yeo CY, Yashiro K, Watanabe M, Hashiguchi H, Mochida K, Ohishi S, Kawabata M, Miyazono K, Whitman M, Hamada H. Left-right asymmetric expression of lefty2 and nodal is induced by a signaling pathway that includes the transcription factor FAST2. Mol Cell. 2000;5:35–47. doi: 10.1016/s1097-2765(00)80401-3. [DOI] [PubMed] [Google Scholar]
- Sengbusch JK, He W, Pinco KA, Yang JT. Dual functions of [alpha]4[beta]1 integrin in epicardial development: initial migration and long-term attachment. J Cell Biol. 2002;157:873–882. doi: 10.1083/jcb.200203075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenerry MK, Gatta PA, Cameron-Smith D. JAK/STAT signaling and human in vitro myogenesis. BMC Physiol. 2011;11:6. doi: 10.1186/1472-6793-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent SD, Dunn NR, Hayashi S, Norris DP, Robertson EJ. Cell fate decisions within the mouse organizer are governed by graded Nodal signals. Genes Dev. 2003;17:1646–1662. doi: 10.1101/gad.1100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HV, Moser M. Comparative expression analysis of the murine palladin isoforms. Dev Dyn. 2008;237:3342–3351. doi: 10.1002/dvdy.21755. [DOI] [PubMed] [Google Scholar]
- Wang J, Wynshaw-Boris A. The canonical Wnt pathway in early mammalian embryogenesis and stem cell maintenance/differentiation. Curr Opin Genet Dev. 2004;14:533–539. doi: 10.1016/j.gde.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Watabe T, Miyazono K. Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Res. 2009;19:103–115. doi: 10.1038/cr.2008.323. [DOI] [PubMed] [Google Scholar]
- Whyatt RM, Jedrychowski W, Hemminki K, Santella RM, Tsai WY, Yang K, Perera FP. Biomarkers of polycyclic aromatic hydrocarbon-DNA damage and cigarette smoke exposures in paired maternal and newborn blood samples as a measure of differential susceptibility. Cancer Epidemiol Biomarkers Prev. 2001;10:581–588. [PubMed] [Google Scholar]
- Yashpal NK, Li J, Wheeler MB, Wang R. Expression of (Lafaras et al.)1 integrin receptors during rat pancreas development--sites and dynamics. Endocrinology. 2005;146:1798–1807. doi: 10.1210/en.2004-1292. [DOI] [PubMed] [Google Scholar]
- Zdravkovic T, Genbacev O, LaRocque N, McMaster M, Fisher S. Human embryonic stem cells as a model system for studying the effects of smoke exposure on the embryo. Reprod Toxicol. 2008;26:86–93. doi: 10.1016/j.reprotox.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Zhang H, Guo D, Wang L, Zhao Y, Cheng Y, Qiao Z. Effect of nicotine on Oct-4 and Rex-1 expression of mouse embryonic stem cells. Reprod Toxicol. 2005;19:473–478. doi: 10.1016/j.reprotox.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Zubenko GS, Hughes HB., 3rd Effects of the A(-115)G variant on CREB1 promoter activity in two brain cell lines: Interactions with gonadal steroids. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:1365–1372. doi: 10.1002/ajmg.b.31133. [DOI] [PMC free article] [PubMed] [Google Scholar]