

Abstract
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare syndrome conferring predisposition to clinical disease caused by weakly virulent mycobacteria, such as Mycobacterium bovis Bacille Calmette Guérin (BCG) vaccines and nontuberculous, environmental mycobacteria (EM). Since 1996, MSMD-causing mutations have been found in six autosomal genes involved in IL-12/23-dependent, IFN-γ-mediated immunity. The aim of this review is to provide the description of the two described forms of X-linked recessive (XR) MSMD. Germline mutations in two genes, NEMO and CYBB, have long been known to cause other human diseases—incontinentia pigmenti (IP) and anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) (NEMO/IKKG), and X-linked chronic granulomatous disease (CGD) (CYBB)—but specific mutations in either of these two genes have recently been shown to cause XR-MSMD. NEMO is an essential component of several NF-κB-dependent signaling pathways. The MSMD-causing mutations in NEMO selectively affect the CD40-dependent induction of IL-12 in mononuclear cells. CYBB encoded for gp91phox, which is an essential component of the NADPH oxidase in phagocytes. The MSMD-causing mutation in CYBB selectively affects the respiratory burst in macrophages. Mutations in NEMO and CYBB may therefore cause MSMD by selectively exerting their deleterious impact on a single signaling pathway (CD40–IL-12, NEMO) or a single cell type (macrophages, CYBB). These experiments illustrate how specific germline mutations in pleiotropic genes can dissociate signalling pathways or cell lineages, thereby resulting in surprisingly narrow clinical phenotypes.
Keywords: Mycobacteria, X-linked primary immunodeficiency, NEMO, CYBB, interleukin-12, interferon-γ, monocytes, macrophages
Introduction
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare syndrome conferring predisposition to clinical disease caused by weakly virulent mycobacteria, such as Mycobacterium bovis Bacille Calmette Guérin (BCG) vaccines and nontuberculous, environmental mycobacteria (EM) (OMIM 209950). 1–3 It was recently found, as expected, that patients are also vulnerable to the more virulent Mycobacterium tuberculosis.4–6 The syndrome was probably first reported in 1951, in an Algerian child with disseminated BCG disease,7 and cases of ”idiopathic” infections with BCG have since been reported worldwide.8 Children with unexplained EM disease were also identified, once EM species had been described, with opportunistic EM, such as Mycobacterium avium, implicated most frequently. Typically, unlike patients with most conventional primary immunodeficiencies,9 patients with MSMD are otherwise healthy and are not prone to other unusually severe infections, with the notable exception of systemic nontyphoidal salmonellosis, which is documented in about half the patients, including some without mycobacterial diseases.10, 11 However, other severe infectious diseases, as diverse as cytomegalovirus and varicella zoster virus diseases,12 human herpes virus-8–associated Kaposi’s sarcoma,13 listeriosis,14 klebsiellosis,15 nocardiosis, 16,17 histoplasmosis,18 paracoccidioidomycosis,19 coccidioidomycosis,20,21 and leishmaniasis, 22 have been reported, mostly in single patients, raising the possibility that the clinical phenotype of MSMD may well extend beyond diseases caused by Mycobacterium or Salmonella. The original denomination of MSMD may therefore not accurately describe all patients, particularly if genetic etiologies common to various infectious phenotypes are described.
The syndrome of MSMD was long thought to be Mendelian, based on the large number of consanguineous and/or multiplex kindreds identified. The occurrence of MSMD in patients born to consanguineous parents and in siblings strongly suggested that most cases followed an autosomal recessive (AR) mode of inheritance.2 Consistent with this hypothesis, six MSMD-causing genes to be identified were all autosomal.3, 23 Since 1996, MSMD-causing mutations have been found in six autosomal genes—IFNGR1, IFNGR2, STAT1, IL12B, IL12RB1, and IRF8—all involved in IL-12/23-dependent, IFN-γ–mediated immunity. Mutations in IFNGR1 (encoding the ubiquitously expressed IFN-γ receptor ligand-binding chain, IFN-γR1),24–29 IFNGR2 (IFN-γR2),30–34 and STAT1 (STAT-1)35, 36 impair cellular responses to IFN-γ, a cytokine produced by NK and T lymphocytes. Mutations in IL12B 16, 37 and IL12RB138–40 impair the production of IFN-γ, by blocking the production of the p40 subunit of IL-12 and IL-23 (encoded by IL12B) or the response to IL-12 and IL-23 (IL12RB1, encoding the β1 chain of the IL-12 and IL-23 receptors) in phagocytes and dendritic cells (Fig. 1). IL-12 is a relatively specific, potent inducer of IFN-γ, whereas the newly described IL-23 is involved in the induction of IL-17 cytokines. Mutations in IFR8, an interferon regulatory factor inducible by IFN-γ, impair IL-12 secretion by monocytes and dendritic cells. 23 ”MSMD” may therefore be described as disorders of the IL-12–IFN-γ circuit, at least in patients bearing these defects. Interestingly, the genetic investigation of MSMD from this perspective has led to the description of the related, but different disorder of AR, complete STAT1 deficiency41,42 and partial recessive STAT1 deficiency43–45 in patients highly vulnerable to both mycobacteria and viruses. These patients have impaired responses to IFN-γ, accounting for mycobacterial diseases, and impaired responses to IFN-α/β and IFN-λ, accounting for overwhelming viral diseases.46, 47
Figure 1.
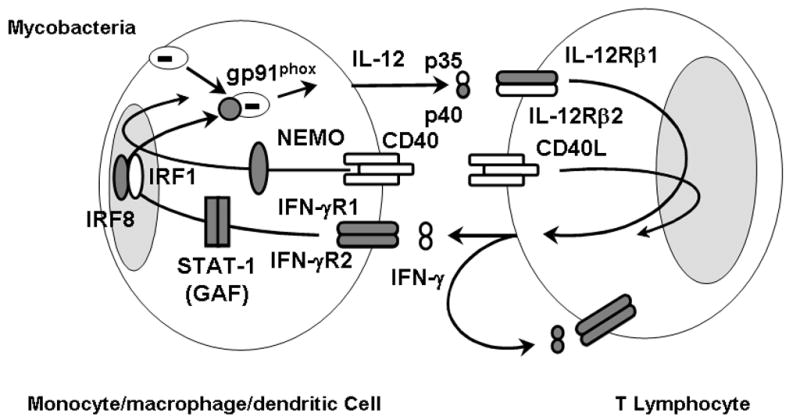
MSMD-causing gene products in the IL-12/23–IFN-γ circuit. Schematic representation of cytokine production and cooperation between monocytes-macrophages-dendritic cells and NKT cells. Mutant molecules in patients with MSMD are indicated in gray. Allelic heterogeneity of the eight genes results in the definition of 15 genetic disorders. The IL-12–IFN-γ loop and the CD40L-activated CD40 pathway, mediating cooperation between T cells and monocyte/cells, are crucial for protective immunity to mycobacterial infection in humans. IRF8 is an IFN-γ–inducible transcription factor required for the induction of various target genes, including IL-12. The NEMO mutations in the LZ domain mostly impair CD40-NEMO–dependent pathways. The Q231P and T178P gp91phox mutations specifically abolish the respiratory burst in monocyte-derived macrophages; gp91phox induction might led to release of IL-12 (for which, any evidence have been demonstrated).
Moreover, the high level of allelic heterogeneity at these six loci accounted for the existence of 13 known distinct genetic disorders, depending on whether the alleles were null or hypomorphic, associated with a lack of protein expression or the expression of an abnormal protein, in which case, depending on the molecular mechanism of disease, and more surprisingly, whether the alleles conferred recessive or dominant MSMD (Table 1). Indeed, whereas MSMD was first noticed to be inherited as an AR and its first genetic etiologies were actually found to be AR, autosomal dominant (AD) MSMD, with both sporadic cases and multiplex kindreds with cases in several generations, has been found to be associated with specific IFNGR1, STAT1, and IRF8 alleles .23, 26, 35, 36 A mutation in IFNGR2 has even been found to be dominant in cells from a healthy individual, raising the possibility that there might be MSMD patients with dominant IFN-γR2 deficiency.32 Four of the 13 known autosomal genetic etiologies of MSMD are AD, the others being AR. However, these known genetic etiologies account for no more than half the patients with MSMD, and probably even fewer of the patients with other unexplained infectious diseases, such as nontyphoidal salmonellosis. Interestingly, MSMD may also segregate as an X-linked recessive (XR) trait. We will herein review the two known forms of XR MSMD, due to mutations in NEMO and CYBB.
Table 1.
Genetic aetiologies of MSMD
| Gene | Inheritance | Defect | Protein |
|---|---|---|---|
| IL12B | AR | C | E− |
| IL12RB1 | AR | C | E− |
| AR | C | E+ | |
| IFNGR1 | AR | C | E− |
| AR | C | E+ | |
| AR | P | E+ | |
| AD | P | E+ | |
| IFNGR2 | AR | C | E− |
| AR | C | E+ | |
| AR | P | E+ | |
| STAT1 | AD | P | E+P− |
| AD | P | E+P+B− | |
| IRF8 | AD | P | E+ |
| NEMO | XR | P | E+ |
| CYBB | XR | P | E+ |
Modes of inheritance are either autosomal dominant (AD), autosomal recessive (AR) or X-linked recessive (XR). The functional defects are either complete (C) or partial (P). The mutant proteins are either expressed (E+) or not (E−), being not phosphorylated (P−) or not binding DNA (P+ B−) upon IFN-γ stimulation.
X-linked recessive MSMD type 1
The occurrence of mycobacterial disease in multiple, maternally related males in other kindreds suggested, as early as 1996, that there might be at least one XR form of MSMD. 48, 49 Two patients from a kindred with X-linked MSMD were described clinically in 1991,50 another patient related to these cases was identified in 1994,51 and a fourth case relative with the same underlying genetic etiology was reported in 2006 (OMIM 300636).52 Clinically, four maternally related males developed disseminated Mycobacterium avium complex infection but presented no other severe infectious diseases, with the exception of invasive Haemophilus influenzae type b infection in one patient and culture-proven, miliary tuberculosis in another. The known autosomal defects of the IL-12–IFN-γ circuit were excluded in these kindreds. However, a related immunological phenotype was identified by Frucht and Holland in 1996.48 They reported low levels of IFN-γ and IL-12 production by the patients’ mononuclear cells upon activation with phytohemaggutinin (PHA) or CD3-specific antibodies. The production of IL-12 in response to microbial stimulation, in the form of BCG, BCG plus IFN-γ and LPS, was normal. Impaired IL-12 production by the patients’ monocytes was shown to be the primary defect in assays in which autologous and heterologous monocytes and T cells were cocultured in the presence of PHA, resulting in a secondary defect in IFN-γ production. The monocyte defect was cell-autonomous, but dependent on the presence of T cells, indicating a defect in the T cell-dependent monocyte activation pathway.
It was only later recognized that one of these four American patients had sparse teeth, leading to suspicion of a disorder related to anhidrotic ectodermal dysplasia (EDA) with immunodeficiency (EDA-ID). EDA-ID is a complex developmental and immunological syndrome caused by hypomorphic mutations in NEMO/IKKG, encoding a regulatory component of the IKK complex.53–55 In its classical form, the EDA developmental syndrome is characterized by the lack of skin appendages, resulting in anodontia or hypodontia with conical incisors (decidual or permanent teeth), atrichosis or hypotrichosis (sparse hair, no eyebrows and eyelashes), and a lack of sweat glands (with heat intolerance). Most children with EDA do not suffer from any detectable immunodeficiency and carry mutations in the ectodysplasin pathway.56 However, some children with EDA have been found to display severe infectious diseases and an impaired antibody response to glycan antigens—pneumococcal capsular antigens in particular—leading Abinun to coin the term EDA-ID.57–59 No overt immunological abnormalities other than a poor inflammatory response have been identified in these patients. The range of infections varied considerably from case to case, with viral, bacterial, and fungal diseases observed, but most children suffered from invasive pneumococcal disease, which was by far the most frequent type of infection in these children. Some children have been found to have a much milder developmental phenotype, with conical incisors as the sole developmental abnormality.60, 61 Some children with hypomorphic mutations in NEMO have even been found to have no detectable developmental phenotype.62–64
The underlying genetic mutations in NEMO in children with EDA-ID were identified by serendipity, when two mothers with IP surprisingly gave birth at term to boys with EDA-ID,65, 66 after it had been shown that IP, an X-linked dominant disorder lethal in utero in boys, was associated with loss-of-function mutations in NEMO.67, 68 The two unrelated mothers were found to carry the same NEMO mutation, which, unlike the other IP-causing null mutations, was hypomorphic, indicating that there was a genotype-phenotype correlation in males, but not in females. Since 2001, up to 80 patients with hypomorphic mutations in NEMO have been reported53–55, 59–64, 66, 67, 69–80 and many more have been diagnosed worldwide. For all mutations found (nonsense, frameshift, and missense), no obvious correlation between a given genotype and a particular infectious or immunological phenotype has been identified. However, the W420X mutation has been found to cosegregate with a severe phenotype of EDA-ID, with osteopetrosis and lymphedema.55, 65, 81, 82
Patients from the American kindred with XR-MSMD and impaired IL-12 production by monocytes upon stimulation by PHA-activated T cells were found to have the novel E315A NEMO mutation.52 One boy in each of two other unrelated families, one from France and the other from Germany, developed infectious disease diagnosed as probable tuberculosis. The French patient presented very mild signs of EDA, limited to conical decidual incisors; he was vaccinated with BCG and, at the age of two years, had cervical lymphadenitis, fever, and a positive tuberculin skin test (TST). He was diagnosed with tuberculosis and treated for this disease. The German patient was not vaccinated with BCG, and at nine years of age, he was hospitalized for persistent fever and a strongly positive TST, suggestive of mycobacterial disease. This patient displayed no developmental abnormality. The same novel R319Q hemizygous mutation in NEMO was identified in these two kindreds.
The E315A and R319Q mutations were not mere polymorphisms, and were found to affect residues conserved in the NEMO genes of nine and six species (of nine studied). Moreover, residues E315 and R319 disrupt the formation of the salt bridge in the leucine zipper domain (LZD) of NEMO, suggesting that mutations in either of the amino acids may disturb the plasticity of the LZD-helix of NEMO, interfering with the CD40-NEMO-NF-κB signaling pathway. The folding defect of the E315A mutant is responsible for the defect in binding to ubiquitin chains.83 These hypomorphic mutations in NEMO are associated with impaired NF-κB activation of c-Rel containing proteins in response to CD40. Not all CD40-dependent pathways are affected, as the induction of IL-12 in monocytes and dendritic cells is impaired, whereas that of the co-stimulatory molecules CD80 and CD86 is affected. The CD40–NF-κB pathway in B cells has also been shown to be intact. Moreover, these mutations do not affect NF-κB activation in response to classical activators, such as TNF-α, IL-1β, or lipopolysaccharide (LPS). The mutant NEMO protein was normally expressed in the hematopoietic and non hematopoietic cells tested. The identification of these mutations therefore provided a molecular basis for XR-MSMD in the absence of other infections. Nonetheless, impairment of the CD40–IL-12 pathway cannot itself provide a full explanation for the predisposition to mycobacterial diseases, as CD40- and CD40L-deficient patients are prone to tuberculosis and regional ”BCG-it is,” but none has yet been shown to be particularly susceptible to M. avium disease or disseminated ”BCG-osis.” Other pathways affected by the NEMO mutations may also be involved. The key genetic feature of these two related mutations, found in six patients from three kindreds, is that they dissociate the multiple NEMO-dependent, NF-κB signalling pathways. By affecting the CD40–IL-12 pathway, while maintaining at least the other pathways tested, these two mutations result in a very narrow clinical phenotype, apparently restricted to MSMD, unlike other NEMO mutations, resulting in a much broader immunological and clinical phenotype.
X-linked recessive MSMD type 2
A second form of X-linked recessive MSMD has recently been reported (OMIM 3000645).84 Four maternally related men from a large French kindred were identified as suffering from mycobacterial diseases.85 The first patient, now aged 58 years, was not vaccinated with BCG in infancy but developed a clinical disease diagnosed as disseminated tuberculosis at the age of 34. The other three men, now aged 61, 56, and 37 years, suffered from disseminated BCG disease (BCG-osis) with lymph node involvement, or from recurrent regional BCG-itis, shortly after vaccination. Two of these men suffered relapses years later. Moreover, an obligate carrier from first kindred developed tuberculous salpingitis at the age of 29 years. Three maternally related men from another unrelated French kindred also displayed MSMD. The patients are aged 37, 40, and 37 suffered from BCG-osis.85 The seven patients did not suffer from any other infectious diseases. Other members of the two families were vaccinated with BCG, with no complications. The production of IL-12 and IFN-γ by the patients’ cells in response to BCG was normal and, unlike patients with NEMO mutations (E315A and R319Q) these patients had an intact T cell-dependent pathway of monocyte IL-12 production. Moreover, CD40-dependent IL-12 production by monocytes and dendritic cells was normal. The involvement of NEMO was further excluded in theses kindreds by genetic means, including sequencing of the coding region and genetic linkage analysis. This linkage analysis identified two regions with a maximum LOD score of 2.29, on the short and long arms of the X chromosome. The patients from the two kindreds therefore clearly displayed a new form of XR-MSMD.85
The sequencing of candidate genes in the intervals to which genes known to be associated with primary immunodeficiencies, including CD40LG, have been mapped led to a specific mutation (Q231P) being identified in CYBB in the four male patients from the first family and another specific mutation (T178P) has been identified in CYBB in the three male patients from the second family. This gene encodes gp91phox, is located on the short arm of the X chromosome, and contains 13 exons. CYBB is expressed strongly in all phagocytic cells (including granulocytes, monocytes, and macrophages) and, to a lesser extent, in B cells. Germline mutations in CYBB are responsible for the most common form of chronic granulomatous disease (CGD) (OMIM 306400), a primary immunodeficiency disease in which phagocytic cells display little or no nicotinamide adenine dinucleotide phosphatase (NADPH) oxidase activity.86–88 CGD patients suffer from recurrent life-threatening infections caused by multiple bacteria and fungi, Staphylococcus and Aspergillus in particular.89 Mycobacterial diseases, caused by BCG and M. tuberculosis, are seen in about 30 to 40% of these patients, with a higher prevalence of tuberculosis in endemic countries.90, 91 A few rare cases have been reported of isolated mycobacterial disease, albeit in patients under the age of 17 years.90 Patients also present steroid-sensitive granulomas, apparently not triggered by infectious agents. Three forms of X-linked CGD have been distinguished, based on X91 protein levels—X910 (no protein), X91− (low levels), and X91+ (normal levels)—but there is no strict correlation with mutant gp91 function and the resulting clinical features.89, 92,93 Unlike the cells of CGD patients, neutrophils and monocytes from our seven patients displayed a perfectly functional respiratory burst, in terms of both superoxide production and hydroxide peroxide release after phorbol ester induction and the physiological stimuli.85 Moreover, their granulocytes killed Sthaphylococcus aureus correctly, unlike granulocytes from CGD patients. This explains the lack of any of the common clinical features of CGD, whether infectious or granulomatous, in these seven adult patients, in the absence of any anti-infectious or immunosuppressive prophylaxis.
However, the hemizygous Q231P and T178P mutations were shown to affect respiratory burst function in monocyte-derived macrophages (MDMs) and EBV-transformed B cells. Indeed, when macrophages were activated with BCG, PPD, or IFN-γ and triggered with phorbol ester, the respiratory burst function was completely abolished.85 Morever, the growth of BCG in the patients’ MDMs in vitro was enhaced. These alterations were documented in vitro and probably reflect the respiratory burst activity in tissue-resident macrophages in vivo. Indeed, B cells are known to play no substantial role in protective immunity to mycobacteria, as attested by the lack of mycobacterial diseases in B cell-deficient children.94 This experiment of nature therefore demonstrates that the respiratory burst in macrophages is essential for protective immunity to mycobacteria. These patients provide a cellular basis for the susceptibility to mycobacteria observed in CGD patients. The lack of environmental mycobacterial disease in patients with these mutations and the extreme rarity of such disease in CGD patients also indicate that the macrophage respiratory burst is not critical for the control of EM. The respiratory burst in granulocytes and monocytes also seems to be dispensable for protective immunity to mycobacteria. However, all six BCG-vaccinated patients in these kindreds had BCG disease (regional or disseminated forms), as do more than the 30 to 50% of vaccinated CGD patients. This may reflect the impact of modifier genes, or other unknown environmental factors. In any event, these data also strongly suggest that the macrophage respiratory burst is not involved in immunity to other bacteria and to fungi, consistent with the results obtained for patients with congenital or acquired neutropenia.95 Immunity to these microbes principally involves the respiratory burst in granulocytes and monocytes.96
Why is the respiratory burst defective in macrophages (and B cells) but not in granulocytes or monocytes? The CYBB mutations are the germline mutations and were found in all cell types tested, including granulocytes and monocytes. However, the gp91phox expression is different in all phagocytic cells, particularly in MDMs and EBV-B cells, correlating with the defect in the NADPH activity. Moreover, gp65phox, a precursor of the mature gp91phox, has been detected in the macrophages and EBV-B cells, indicating the impaired maturation of gp65phox to gp91phox associated with impaired formation of the flavocytochrome b558, the assembly and activation of which depend on a cell-specific threshold. Smaller amounts of gp91phox protein can support flavocytochrome b558 assembly and substantial oxidase activity in granulocytes and monocytes but not in B cells and macrophages. Additional experiments using Chinese hamster ovary (CHO) epithelial cell line and PLB-985 cell line found a small amounts of gp91phox and the presence of gp65 precursor by these two CYBB alleles.85 The detailed biochemical mechanism underlying the selective, cell-specific impact of these germline mutations, which result in impaired flavocytochrome b558 complex assembly required for NADPH activity, remains unknown.85 The NADPH oxidase launches a series of biochemical reactions resulting in the successive production of multiple, short-lived reactive oxygen species (ROS).96 In the in vitro and ex vivo assays, we determined the levels of only some of the ROS, such as superoxide and hydrogen peroxide, produced by cells extracted from their natural, physiological environment, and stimulated with artificial stimuli. For example, we determined hydrogen peroxide release by monocyte-derived macrophages. It is possible, although unlikely, that tissue macrophages in vivo, and perhaps even granulocytes or monocytes in vivo, have a phenotype different from that described in our study. Despite these hypothetical reservations, which it is not yet possible to tackle experimentally in humans, these data strongly suggest that the Q231P and T178P CYBB mutations confers XR-MSMD because they selectively affect macrophages.
Conclusion
Elucidation of the molecular basis of the two forms of XR-MSMD was of immunological interest, as it indicated the role of the CD40–IL-12 pathway in monocyte-macrophages and dendritic cells (XR-MSMD type 1), and of the CYBB-dependent NADPH oxidase assembly and respiratory burst in macrophages (XR-MSMD type 2), in protective immunity to mycobacteria in humans. Mutations in NEMO are clearly connected with the IL-12–IFN-γ circuit, as CD40 stimulation leads to the induction of IL-12. The connection of the CYBB mutation with the previously identified mutations in autosomal genes controlling the IL-12–IFN-γ circuit is less clear. It may be that CYBB is controlled by IFN-γ, and the respiratory burst acts as an effector mechanism to destroy mycobacteria in macrophages. Alternatively, CYBB and the respiratory burst may be required for the optimal production of IL-12 by macrophages infected with mycobacteria. It is also possible that gp91phox and the other MSMD-causing gene products are not directly connected but work independently to eliminate mycobacteria. These hypotheses are currently being tested in vitro. In any event, both NEMO and CYBB may be considered bona fide MSMD-causing genes, and MSMD is therefore allelic with two other XR primary immunodeficiencies: EDA-ID (NEMO) and CGD (CYBB) (Fig. 1).
These experiments occuring in nature are also of interest to the genetic community. Indeed, the two forms of XR-MSMD illustrate how subtle germline mutations in pleiotropic genes, involved in multiple signaling pathways (NEMO) and multiple cell types (CYBB), can impact on one pathway or one cell type, respectively. The XR-MSMD NEMO mutations do not impair NEMO-dependent NF-κB activation in response to most stimuli tested, accounting for the lack of associated EDA-ID developmental and infectious diseases—pyogenic bacterial diseases in particular. The XR-MSMD CYBB mutation does not impair the respiratory burst in granulocytes and monocytes, accounting for the lack of granulomas and infectious diseases seen in CGD—and of bacterial and fungal infections in particular. The MSMD-causing NEMO mutations thus seem to impairing one pathway selectively, whereas the MSMD-causing CYBB mutation appears to impair one cell type selectively. From a genetic standpoint, these findings indicate that subtle mutations associated with a narrow phenotype may be found in genes, null mutations of which result in a broad phenotype, whether in mice or in humans.97–100 Candidate genes should not be excluded a priori based solely on the phenotype associated with null alleles. The NEMO mutation, in particular, is reminiscent of the STAT1 mutations previously found to affect one of the two signaling pathways involving STAT1.35, 36 As for CYBB, it now appears that germline mutations mimicking ”conditional knockouts” in mice can be seen in humans. Candidate genes for subtle, narrow phenotypes should therefore not be excluded based solely on their involvement in multiple pathways or on the basis of their involvement in multiple cell types.
Acknowledgments
We thank all members of the two branches of the laboratory of Human Genetics of Infectious Diseases for discussions. This work was supported by the ANR, The Rockefeller University Center for Clinical and Translational Science Grant number 5UL1RR024143, The Rockefeller University, EU Grants HOMITB HEALTH-F3-2008-200732 and NEOTIM EEA05095KKA.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Hamosh A, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–7. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 3.Filipe-Santos O, et al. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18:347–61. doi: 10.1016/j.smim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Alcais A, et al. Tuberculosis in children and adults: two distinct genetic diseases. J Exp Med. 2005;202:1617–21. doi: 10.1084/jem.20052302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boisson-Dupuis S, et al. IL-12Rbeta1 deficiency in two of fifty children with severe tuberculosis from Iran, Morocco, and Turkey. PLoS One. 2011;6:e18524. doi: 10.1371/journal.pone.0018524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabarsi P, et al. Lethal Tuberculosis in a Previously Healthy Adult with IL-12 Receptor Deficiency. J Clin Immunol. 2011 April 13; doi: 10.1007/s10875-011-9523-9. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.Mimouni J. Our experiences in three years of BCG vaccination at the center of the O.P.H.S. at Constantine; study of observed cases (25 cases of complications from BCG vaccination) Alger Medicale. 1951;55:1138–47. [PubMed] [Google Scholar]
- 8.Casanova JL, et al. Idiopathic disseminated bacillus Calmette-Guerin infection: a French national retrospective study. Pediatrics. 1996;98:774–8. [PubMed] [Google Scholar]
- 9.Picard C, Casanova JL, Abel L. Mendelian traits that confer predisposition or resistance to specific infections in humans. Curr Opin Immunol. 2006;18:383–90. doi: 10.1016/j.coi.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Bustamante J, et al. Novel primary immunodeficiencies revealed by the investigation of paediatric infectious diseases. Curr Opin Immunol. 2008;20:39–48. doi: 10.1016/j.coi.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Patel SY, et al. Genetically determined susceptibility to mycobacterial infection. J Clin Pathol. 2008;61:1006–12. doi: 10.1136/jcp.2007.051201. [DOI] [PubMed] [Google Scholar]
- 12.Dorman SE, et al. Viral infections in interferon-gamma receptor deficiency. J Pediatr. 1999;135:640–3. doi: 10.1016/S0022-3476(99)70064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camcioglu Y, et al. HHV-8-associated Kaposi sarcoma in a child with IFNgammaR1 deficiency. J Pediatr. 2004;144:519–23. doi: 10.1016/j.jpeds.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Roesler J, et al. Listeria monocytogenes and recurrent mycobacterial infections in a child with complete interferon-gamma-receptor (IFNgammaR1) deficiency: mutational analysis and evaluation of therapeutic options. Exp Hematol. 1999;27:1368–74. doi: 10.1016/s0301-472x(99)00077-6. [DOI] [PubMed] [Google Scholar]
- 15.Pedraza S, et al. Clinical disease caused by Klebsiella in 2 unrelated patients with interleukin 12 receptor beta1 deficiency. Pediatrics. 2010;126:e971–6. doi: 10.1542/peds.2009-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Picard C, et al. Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am J Hum Genet. 2002;70:336–48. doi: 10.1086/338625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luangwedchakarn V, et al. A novel mutation of the IL12RB1 gene in a child with nocardiosis, recurrent salmonellosis and neurofibromatosis type I: first case report from Thailand. Asian Pac J Allergy Immunol. 2009;27:161–5. [PubMed] [Google Scholar]
- 18.Zerbe CS, Holland SM. Disseminated histoplasmosis in persons with interferon-gamma receptor 1 deficiency. Clin Infect Dis. 2005;41:e38–41. doi: 10.1086/432120. [DOI] [PubMed] [Google Scholar]
- 19.Moraes-Vasconcelos D, et al. Paracoccidioides brasiliensis disseminated disease in a patient with inherited deficiency in the beta1 subunit of the interleukin (IL)-12/IL-23 receptor. Clin Infect Dis. 2005;41:e31–7. doi: 10.1086/432119. [DOI] [PubMed] [Google Scholar]
- 20.Vinh DC, et al. Refractory disseminated coccidioidomycosis and mycobacteriosis in interferon-gamma receptor 1 deficiency. Clin Infect Dis. 2009;49:e62–5. doi: 10.1086/605532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vinh DC, et al. Interleukin-12 receptor beta1 deficiency predisposing to disseminated Coccidioidomycosis. Clin Infect Dis. 2011;52:e99–e102. doi: 10.1093/cid/ciq215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanal O, et al. A case of interleukin-12 receptor beta-1 deficiency with recurrent leishmaniasis. Pediatr Infect Dis J. 2007;26:366–8. doi: 10.1097/01.inf.0000258696.64507.0f. [DOI] [PubMed] [Google Scholar]
- 23.Hambleton S, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365:127–38. doi: 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newport MJ, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med. 1996;335:1941–9. doi: 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- 25.Jouanguy E, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. N Engl J Med. 1996;335:1956–61. doi: 10.1056/NEJM199612263352604. [DOI] [PubMed] [Google Scholar]
- 26.Jouanguy E, et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet. 1999;21:370–8. doi: 10.1038/7701. [DOI] [PubMed] [Google Scholar]
- 27.Jouanguy E, et al. In a novel form of IFN-gamma receptor 1 deficiency, cell surface receptors fail to bind IFN-gamma. J Clin Invest. 2000;105:1429–36. doi: 10.1172/JCI9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorman SE, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364:2113–21. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 29.Kong XF, et al. A novel form of cell type-specific partial IFN-gammaR1 deficiency caused by a germ line mutation of the IFNGR1 initiation codon. Hum Mol Genet. 2010;19:434–44. doi: 10.1093/hmg/ddp507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101:2364–9. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doffinger R, et al. Partial interferon-gamma receptor signaling chain deficiency in a patient with bacille Calmette-Guerin and Mycobacterium abscessus infection. J Infect Dis. 2000;181:379–84. doi: 10.1086/315197. [DOI] [PubMed] [Google Scholar]
- 32.Rosenzweig, et al. A novel mutation in IFN-gamma receptor 2 with dominant negative activity: biological consequences of homozygous and heterozygous states. J Immunol. 2004;173:4000–8. doi: 10.4049/jimmunol.173.6.4000. [DOI] [PubMed] [Google Scholar]
- 33.Vogt G, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. 2005;37:692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 34.Vogt G, et al. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med. 2008;205:1729–37. doi: 10.1084/jem.20071987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dupuis S, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293:300–3. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- 36.Chapgier A, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006;2:e131. doi: 10.1371/journal.pgen.0020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altare F, et al. Inherited interleukin 12 deficiency in a child with bacille Calmette-Guerin and Salmonella enteritidis disseminated infection. J Clin Invest. 1998;102:2035–40. doi: 10.1172/JCI4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fieschi C, et al. Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor beta1 deficiency: medical and immunological implications. J Exp Med. 2003;197:527–35. doi: 10.1084/jem.20021769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fieschi C, et al. A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors. Blood. 2004;104:2095–101. doi: 10.1182/blood-2004-02-0584. [DOI] [PubMed] [Google Scholar]
- 40.de Beaucoudrey L, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore) 2010;89:381–402. doi: 10.1097/MD.0b013e3181fdd832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dupuis S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–91. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 42.Chapgier A, et al. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol. 2006;176:5078–83. doi: 10.4049/jimmunol.176.8.5078. [DOI] [PubMed] [Google Scholar]
- 43.Kong XF, et al. A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood. 2010;116:5895–906. doi: 10.1182/blood-2010-04-280586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chapgier A, et al. A partial form of recessive STAT1 deficiency in humans. J Clin Invest. 2009;119:1502–14. doi: 10.1172/JCI37083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vairo D, et al. Severe impairment of IFN{gamma} and IFN{alpha} responses in cells of a patient with a novel STAT1 splicing mutation. Blood. 2011 Jul 19; doi: 10.1182/blood-2011-01-330571. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 46.Jouanguy E, et al. Human primary immunodeficiencies of type I interferons. Biochimie. 2007;89:878–83. doi: 10.1016/j.biochi.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 47.Zhang SY, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. 2008;226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x. [DOI] [PubMed] [Google Scholar]
- 48.Frucht DM, Holland SM. Defective monocyte costimulation for IFN-gamma production in familial disseminated Mycobacterium avium complex infection: abnormal IL-12 regulation. J Immunol. 1996;157:411–6. [PubMed] [Google Scholar]
- 49.Frucht DM, et al. IL-12-Independent costimulation pathways for interferon-gamma production in familial disseminated Mycobacterium avium complex infection. Clin Immunol. 1999;91:234–41. doi: 10.1006/clim.1999.4688. [DOI] [PubMed] [Google Scholar]
- 50.Nedorost ST, et al. Rosacea-like lesions due to familial Mycobacterium avium-intracellulare infection. Int J Dermatol. 1991;30:491–7. doi: 10.1111/j.1365-4362.1991.tb04869.x. [DOI] [PubMed] [Google Scholar]
- 51.Holland SM, et al. Treatment of refractory disseminated nontuberculous mycobacterial infection with interferon gamma. A preliminary report. N Engl J Med. 1994;330:1348–55. doi: 10.1056/NEJM199405123301904. [DOI] [PubMed] [Google Scholar]
- 52.Filipe-Santos O, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203:1745–59. doi: 10.1084/jem.20060085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zonana J, et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO) Am J Hum Genet. 2000;67:1555–62. doi: 10.1086/316914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain A, et al. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. 2001;2:223–8. doi: 10.1038/85277. [DOI] [PubMed] [Google Scholar]
- 55.Doffinger R, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–85. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 56.Priolo M, Lagana C. Ectodermal dysplasias: a new clinical-genetic classification. J Med Genet. 2001;38:579–85. doi: 10.1136/jmg.38.9.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abinun M. Ectodermal dysplasia and immunodeficiency. Arch Dis Child. 1995;73:185. doi: 10.1136/adc.73.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abinun M, et al. Anhidrotic ectodermal dysplasia associated with specific antibody deficiency. Eur J Pediatr. 1996;155:146–7. doi: 10.1007/BF02075774. [DOI] [PubMed] [Google Scholar]
- 59.Carrol ED, et al. Anhidrotic ectodermal dysplasia and immunodeficiency: the role of NEMO. Arch Dis Child. 2003;88:340–1. doi: 10.1136/adc.88.4.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ku CL, et al. NEMO mutations in 2 unrelated boys with severe infections and conical teeth. Pediatrics. 2005;115:e615–9. doi: 10.1542/peds.2004-1754. [DOI] [PubMed] [Google Scholar]
- 61.Hubeau M, et al. A new mechanism of X-linked anhidrotic ectodermal dysplasia with immunodeficiency: impairment of ubiquitin binding despite normal folding of NEMO protein. Blood. 2011;118:926–35. doi: 10.1182/blood-2010-10-315234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puel A, et al. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am J Hum Genet. 2006;78:691–701. doi: 10.1086/501532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orange JS, et al. Human nuclear factor kappa B essential modulator mutation can result in immunodeficiency without ectodermal dysplasia. J Allergy Clin Immunol. 2004;114:650–6. doi: 10.1016/j.jaci.2004.06.052. [DOI] [PubMed] [Google Scholar]
- 64.Mooster JL, et al. Immune deficiency caused by impaired expression of nuclear factor-kappaB essential modifier (NEMO) because of a mutation in the 5' untranslated region of the NEMO gene. J Allergy Clin Immunol. 2010;126:127–32. e7. doi: 10.1016/j.jaci.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mansour S, et al. Incontinentia pigmenti in a surviving male is accompanied by hypohidrotic ectodermal dysplasia and recurrent infection. Am J Med Genet. 2001;99:172–7. doi: 10.1002/1096-8628(2001)9999:9999<::aid-ajmg1155>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 66.Dupuis-Girod S, et al. Osteopetrosis, lymphedema, anhidrotic ectodermal dysplasia, and immunodeficiency in a boy and incontinentia pigmenti in his mother. Pediatrics. 2002;109:e97. doi: 10.1542/peds.109.6.e97. [DOI] [PubMed] [Google Scholar]
- 67.Smahi A, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature. 2000;405:466–72. doi: 10.1038/35013114. [DOI] [PubMed] [Google Scholar]
- 68.Smahi A, et al. The NF-kappaB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371–5. doi: 10.1093/hmg/11.20.2371. [DOI] [PubMed] [Google Scholar]
- 69.Aradhya S, et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma) Am J Hum Genet. 2001;68:765–71. doi: 10.1086/318806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kosaki K, et al. Female patient showing hypohidrotic ectodermal dysplasia and immunodeficiency (HED-ID) Am J Hum Genet. 2001;69:664–6. doi: 10.1086/323003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishikomori R, et al. X-linked ectodermal dysplasia and immunodeficiency caused by reversion mosaicism of NEMO reveals a critical role for NEMO in human T-cell development and/or survival. Blood. 2004;103:4565–72. doi: 10.1182/blood-2003-10-3655. [DOI] [PubMed] [Google Scholar]
- 72.Jain A, et al. Specific NEMO mutations impair CD40-mediated c-Rel activation and B cell terminal differentiation. J Clin Invest. 2004;114:1593–602. doi: 10.1172/JCI21345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Orstavik KH, et al. Novel splicing mutation in the NEMO (IKK-gamma) gene with severe immunodeficiency and heterogeneity of X-chromosome inactivation. Am J Med Genet A. 2006;140:31–9. doi: 10.1002/ajmg.a.31026. [DOI] [PubMed] [Google Scholar]
- 74.Lee WI, et al. Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood. 2005;105:1881–90. doi: 10.1182/blood-2003-12-4420. [DOI] [PubMed] [Google Scholar]
- 75.Ku CL, et al. IRAK4 and NEMO mutations in otherwise healthy children with recurrent invasive pneumococcal disease. J Med Genet. 2007;44:16–23. doi: 10.1136/jmg.2006.044446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pachlopnik Schmid JM, et al. Transient hemophagocytosis with deficient cellular cytotoxicity, monoclonal immunoglobulin M gammopathy, increased T-cell numbers, and hypomorphic NEMO mutation. Pediatrics. 2006;117:e1049–56. doi: 10.1542/peds.2005-2062. [DOI] [PubMed] [Google Scholar]
- 77.Tono C, et al. Correction of immunodeficiency associated with NEMO mutation by umbilical cord blood transplantation using a reduced-intensity conditioning regimen. Bone Marrow Transplant. 2007;39:801–4. doi: 10.1038/sj.bmt.1705658. [DOI] [PubMed] [Google Scholar]
- 78.Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, Orange JS. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–1177. e16. doi: 10.1016/j.jaci.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Salt BH, Niemela JE, Pandey R, Hanson EP, Deering RP, Quinones R, Jain A, Orange JS, Gelfand EW. IKBKG (nuclear factor-kappa B essential modulator) mutation can be associated with opportunistic infection without impairing Toll-like receptor function. J Allergy Clin Immunol. 2008;121:976–82. doi: 10.1016/j.jaci.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mancini AJ, Lawley LP, Uzel G. X-linked ectodermal dysplasia with immunodeficiency caused by NEMO mutation: early recognition and diagnosis. Arch Dermatol. 2008;144:342–6. doi: 10.1001/archderm.144.3.342. [DOI] [PubMed] [Google Scholar]
- 81.Dupuis-Girod S, et al. Successful allogeneic hemopoietic stem cell transplantation in a child who had anhidrotic ectodermal dysplasia with immunodeficiency. Pediatrics. 2006;118:e205–11. doi: 10.1542/peds.2005-2661. [DOI] [PubMed] [Google Scholar]
- 82.Roberts CM, et al. A novel NEMO gene mutation causing osteopetrosis, lymphoedema, hypohidrotic ectodermal dysplasia and immunodeficiency (OL-HED-ID) Eur J Pediatr. 2010;169:1403–7. doi: 10.1007/s00431-010-1206-7. [DOI] [PubMed] [Google Scholar]
- 83.Lo YC, et al. Structural basis for recognition of diubiquitins by NEMO. Mol Cell. 2009;33:602–15. doi: 10.1016/j.molcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bustamante J, et al. A novel X-linked recessive form of Mendelian susceptibility to mycobaterial disease. J Med Genet. 2007;44:e65. doi: 10.1136/jmg.2006.043406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bustamante J, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12:213–21. doi: 10.1038/ni.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Royer-Pokora B, et al. Cloning the gene for an inherited human disorder--chronic granulomatous disease--on the basis of its chromosomal location. Nature. 1986;322:32–8. doi: 10.1038/322032a0. [DOI] [PubMed] [Google Scholar]
- 87.Dinauer MC, et al. The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature. 1987;327:717–20. doi: 10.1038/327717a0. [DOI] [PubMed] [Google Scholar]
- 88.Teahan C, et al. The X-linked chronic granulomatous disease gene codes for the beta-chain of cytochrome b-245. Nature. 1987;327:720–1. doi: 10.1038/327720a0. [DOI] [PubMed] [Google Scholar]
- 89.Roos D, Kuijpers TW, Curnutte JT. Chronic Granulomatous Disease. In: Ochs HD, Edvards Smith CI, Puch JM, editors. Primary Immunodeficiency Diseases. A molecular and genetic approach. Oxford University Press; New York: 2007. pp. 525–549. [Google Scholar]
- 90.Bustamante J, et al. BCG-osis and tuberculosis in a child with chronic granulomatous disease. J Allergy Clin Immunol. 2007;120:32–8. doi: 10.1016/j.jaci.2007.04.034. [DOI] [PubMed] [Google Scholar]
- 91.Lee PP, et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J. 2008;27:224–30. doi: 10.1097/INF.0b013e31815b494c. [DOI] [PubMed] [Google Scholar]
- 92.Holland SM. Chronic granulomatous disease. Clin Rev Allergy Immunol. 2010;38:3–10. doi: 10.1007/s12016-009-8136-z. [DOI] [PubMed] [Google Scholar]
- 93.Roos D, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update) Blood Cells Mol Dis. 2010;45:246–65. doi: 10.1016/j.bcmd.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Conley ME, et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199–227. doi: 10.1146/annurev.immunol.021908.132649. [DOI] [PubMed] [Google Scholar]
- 95.Boztug K, Klein C. Novel genetic etiologies of severe congenital neutropenia. Curr Opin Immunol. 2009;21:472–80. doi: 10.1016/j.coi.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 96.Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317:617–9. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- 98.Alcais A, Abel L, Casanova JL. Human genetics of infectious diseases: between proof of principle and paradigm. J Clin Invest. 2009;119:2506–14. doi: 10.1172/JCI38111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Notarangelo LD, Casanova JL. Primary immunodeficiencies: increasing market share. Curr Opin Immunol. 2009;21:461–5. doi: 10.1016/j.coi.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 100.Alcais A, et al. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Ann N Y Acad Sci. 2011;1214:18–33. doi: 10.1111/j.1749-6632.2010.05834.x. [DOI] [PubMed] [Google Scholar]