

Abstract
Eukaryotic mitochondria resulted from symbiotic incorporation of α-proteobacteria into ancient archaea species. During evolution, mitochondria lost most of the prokaryotic bacterial genes and only conserved a small fraction including those encoding 13 proteins of the respiratory chain. In this process, many functions were transferred to the host cells, but mitochondria gained a central role in the regulation of cell proliferation and apoptosis, and in the modulation of metabolism; accordingly, defective organelles contribute to cell transformation and cancer, diabetes, and neurodegenerative diseases. Most cell and transcriptional effects of mitochondria depend on the modulation of respiratory rate and on the production of hydrogen peroxide released into the cytosol. The mitochondrial oxidative rate has to remain depressed for cell proliferation; even in the presence of O2, energy is preferentially obtained from increased glycolysis (Warburg effect). In response to stress signals, traffic of pro- and antiapoptotic mitochondrial proteins in the intermembrane space (B-cell lymphoma-extra large, Bcl-2-associated death promoter, Bcl-2 associated X-protein and cytochrome c) is modulated by the redox condition determined by mitochondrial O2 utilization and mitochondrial nitric oxide metabolism. In this article, we highlight the traffic of the different canonical signaling pathways to mitochondria and the contributions of organelles to redox regulation of kinases. Finally, we analyze the dynamics of the mitochondrial population in cell cycle and apoptosis. Antioxid. Redox Signal. 16, 1150–1180.
VIII. Mitochondrial Biogenesis, Mitochondrial Dynamics, and Cell Cycle
I. Introduction
Mitochondria are the powerhouse of the cells providing energy for ionic pumps, muscular work, hormone secretion, and anabolic processes. Energy is mostly produced by sequential oxidoreductive reactions where electrons are transferred from NADH to oxygen and protons are extruded, and energy stored as an inner membrane potential, finally dissipated and accumulated as ATP by ATP synthase (Fig. 1). Considering the very low Km for oxygen of cytochrome oxidase (COX) (10−7 M), the terminal enzyme that transfer electrons to O2 to form H2O, it was previously stated that mitochondria follow the law of “all-nothing” and consume unrestrictedly all supplied O2. However, this assumption is more adequate for isolated mitochondria than for the organelles in vivo; indeed, mitochondria are able to adapt O2 uptake to different physiological or pathological situations. This notion is relevant, because the production of reactive oxygen species (ROS) is a consequence of monovalent reduction of molecular oxygen, and depends on the electron flow rate. In the last years, we and others demonstrated that nitric oxide (NO) is a powerful modulator of oxygen uptake by reversible binding to COX and thus, it reduces O2 utilization and increases ROS production. These two effects proved the regulatory adaptability of mitochondria; many cell functions are related to the transition between high and low oxidative rate and among them those normal pathways that drive cell fate: proliferation, cell cycle arrest, and apoptosis. The regulation of mitochondrial oxidative rate and the production of hydrogen peroxide (H2O2) are also related to pathological processes such as cell transformation and cancer, hypoxia, diabetes, and neurodegeneration. In thisarticle, we analyze redox effects and the function and dynamics of mitochondria in the signaling for cell proliferation and death.
FIG. 1.

General organization of mitochondrial electron transfer chain and the formation of O2 species are modulated by NO. Electrons from reduced metabolites from the intermediary metabolism and tricarboxylic cycle enter the respiratory chain as NADH (to NADH dehydrogenase at complex I) or from succinate (to succinate dehydrogenase at complex II) and lead to a two-step reduction of reduced ubiquinol (to semiubiquinone, and ubiquinone). This sequential pathway finally reduces O2 to water and, depending on electron entrance, extrudes two or three protons that creates an inner membrane potential and re-enter by ATP synthase with dissipation of energy and formation of ATP. From a low to high concentration, NO progressively inhibits cytochrome oxidase, complex II-III, and complex I. Myx, myxothiazole; AA, antimycin A; FeS, Fe sulfur complex; Cyt, cytochrome; SUCC, succinate; FUM, fumarate.
II. Introduction to Mitochondrial Biology
A. The physiology of mitochondria and redox biology
Mitochondria are organelles derived from the primitive symbiosis of archeon ancestors with the prokaryotic α-proteobacteria species (142). α-proteobacteria similar to Ricketsia prowasecki or Bartonella henselae has DNA homologous to mitochondrial DNA. However, in the evolution process leading to modern eukaryotic cells, mitochondria lost the ability to synthesize most of the proteins encoded by the primitive bacterial DNA, and only conserve a small circular polycystronic 16 Kb mtDNA controlling the synthesis of about 67 proteins, including 13 polypeptides of the electron transfer chain; the rest of the bacterial genes were transferred to the nuclear genome. It is noteworthy that relatively small DNA from B. henselae encodes for more than 1600 proteins (128).
Along evolution, mitochondria conserved some bacterial phenotypic characteristics while acquired new exciting functions given by complex regulation of energy production, the orchestration of intermediary metabolism, and, importantly, the control of cell proliferation and programmed cell death. The most striking fact is that during the transition to modern organisms, mitochondria incorporated different cell signaling pathways to become a central modulator of cell fate.
In 1950, Gerschman et al. proposed univalent reduction of O2 as causative of deleterious effects of radiation (84). The putative formation of superoxide anion (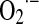 ) was later confirmed by McCord and Fridovich, who recognized cerebrocuprein as superoxide dismutase (SOD), the enzyme that catalyzes dismutation of superoxide to nonradical H2O2 (148). Several years later, Boveris, Cadenas, Turrens, and Chance detected the production of
) was later confirmed by McCord and Fridovich, who recognized cerebrocuprein as superoxide dismutase (SOD), the enzyme that catalyzes dismutation of superoxide to nonradical H2O2 (148). Several years later, Boveris, Cadenas, Turrens, and Chance detected the production of 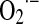 and H2O2 within mitochondria (18, 19, 221). At first glance, production of ROS was considered a toxic effect in the active oxygen metabolizing organelles. Mitochondria contain highly efficient enzymes to detoxify ROS, such as Mn2+-superoxide dismutase (SOD2), glutathione peroxidase 1 (GPx1), and members of the thioredoxin (Trx2) superfamily that may be included in the nucleoid structure (120). Nucleoids harbor 2–8 mtDNA copies and the mitochondrial single-stranded DNA binding protein and mitochondrial transcription factor A (TFAM) are major constituents of nucleoids. Packaging of mtDNA by TFAM is likely to be important for transcription and replication, similar to the regulation of nuclear genes by histones, which are themselves regulated by protein modification (82).
and H2O2 within mitochondria (18, 19, 221). At first glance, production of ROS was considered a toxic effect in the active oxygen metabolizing organelles. Mitochondria contain highly efficient enzymes to detoxify ROS, such as Mn2+-superoxide dismutase (SOD2), glutathione peroxidase 1 (GPx1), and members of the thioredoxin (Trx2) superfamily that may be included in the nucleoid structure (120). Nucleoids harbor 2–8 mtDNA copies and the mitochondrial single-stranded DNA binding protein and mitochondrial transcription factor A (TFAM) are major constituents of nucleoids. Packaging of mtDNA by TFAM is likely to be important for transcription and replication, similar to the regulation of nuclear genes by histones, which are themselves regulated by protein modification (82).
The existence of Mn2+-superoxide dismutase (SOD2) consuming the produced superoxide yield, and the further diffusion of formed H2O2 to cytosol and even outside cells (154) protects mitochondria from undesirable oxidative effects. However, repeated exposure to oxygen species accumulates oxidative damage that alters mitochondrial lipids and proteins through oxidation of cysteine and the genome through oxidation of nuclear and mitochondrial DNA. The importance of mitochondrial 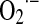 formation is best demonstrated by manganese superoxide dismutase (MnSOD)−/− mice, which die postnatally due to dilated cardiomyopathy or neurodegenerative processes (231). Major alterations were found in mitochondria such as reduced antioxidant capacity, increased mtDNA damage, and reduced activities of enzymes of the respiratory chain and citric acid cycle. Almost 20 years ago, Boveris et al. (19) discovered that the mitochondrial production of ROS depends on the partial reduction of membrane ubiquinone to intermediary ubisemiquinone (UQ–.; reaction 1), a transitional redox status that undergoes auto-oxidation by one-electron reduction of a small quantity of utilized O2 (2%–3%) to
formation is best demonstrated by manganese superoxide dismutase (MnSOD)−/− mice, which die postnatally due to dilated cardiomyopathy or neurodegenerative processes (231). Major alterations were found in mitochondria such as reduced antioxidant capacity, increased mtDNA damage, and reduced activities of enzymes of the respiratory chain and citric acid cycle. Almost 20 years ago, Boveris et al. (19) discovered that the mitochondrial production of ROS depends on the partial reduction of membrane ubiquinone to intermediary ubisemiquinone (UQ–.; reaction 1), a transitional redox status that undergoes auto-oxidation by one-electron reduction of a small quantity of utilized O2 (2%–3%) to 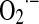 (reaction 2) further dismutated to H2O2 (reaction 3), which is freely diffusible to cytosol. In the absence of mitochondrial inhibitors, the rate of this nonenzymatic compulsive monovalent reduction of O2 depends on the rate of redox electron transfer to molecular O2.
(reaction 2) further dismutated to H2O2 (reaction 3), which is freely diffusible to cytosol. In the absence of mitochondrial inhibitors, the rate of this nonenzymatic compulsive monovalent reduction of O2 depends on the rate of redox electron transfer to molecular O2.
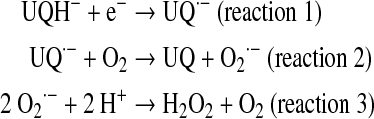 |
Otherwise, a maximal rate of superoxide formation is experimentally achieved by exposing mitochondria to inhibitors of mitochondrial complexes. The typical inhibitors rotenone and antimycin, respectively, block electron transfer through complexes I and III (Fig. 1). Diverse conditions from genetic mutations (in ND1-5 and NBD4L genes in mtDNA, i.e., myoclonic epilepsy with ragged red fibers and mitochondrial miopathy, encephalopathy, lactic acidosis, and stroke) to post-translational changes of mitochondrial components (acetylation, nitration of complex I, or mtDNA heteroplasmy [rho-0 cells]) markedly slow electron flow between the mitochondrial complexes and increase the 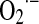 yield. In these cases, the
yield. In these cases, the 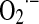 production rate in mitochondria correlates linearly and negatively with selective inhibition of electron transfer rate at complexes I and III, whereas in the absence of such inhibition, superoxide yield is directly dependent on the electron transfer rate (Fig. 2A).
production rate in mitochondria correlates linearly and negatively with selective inhibition of electron transfer rate at complexes I and III, whereas in the absence of such inhibition, superoxide yield is directly dependent on the electron transfer rate (Fig. 2A).
FIG. 2.

Production of superoxide anion and hydrogen peroxide by the inhibition of mitochondrial oxygen uptake. (A) Inverse relationship between oxygen utilization and superoxide formation in mitochondria. The inverse relationship between superoxide production and residual Complex I activity is shown in fibroblasts of patients with isolated Complex I deficiency—Measurement of superoxide production was performed with hydroethydine in an inverted fluorescence microscope. Fluorescence intensity in the indicated compartment (left y-axis) is expressed as percentage of vehicle-treated control (CT). Closed and open symbols represent patient cell lines with a known (13 patients) and hitherto unknown (8 patients) as mutation, respectively. Linear regression analysis reveals an inverse correlation between superoxide production and residual CI activity for the whole cohort of patient cell lines. Reprinted from Verkaart et al. (224). © 2007 by Elsevier. (B) Effects of NO on electron transfer rate and hydrogen peroxide production. The effects of NO result in the inhibition of mitochondrial respiratory rate with an inverse increase of mitochondrial H2O2 yield, the product of dismutation of 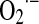 . The trace was obtained by simultaneous polarographic determinations of O2 utilization and fluorometric detection of H2O2, in rat heart submitochondrial particles (reproduced from Poderoso et al.) (182). © Elsevier, 1996. CI, confidence interval.
. The trace was obtained by simultaneous polarographic determinations of O2 utilization and fluorometric detection of H2O2, in rat heart submitochondrial particles (reproduced from Poderoso et al.) (182). © Elsevier, 1996. CI, confidence interval.
Direct demonstration of superoxide production by complex II was obtained by Zhang et al. (244) in purified succinate-CoQ reductase and succinate dehydrogenase; the enzymes were found to generate superoxide by autooxidation of flavin; and reconstitution of complex II with the bc1 complex to yield an active succinate-cytochrome (cyt) c reductase inhibited superoxide formation.
B. NO and mitochondrial redox metabolism
Mitochondrial NO utilization involves modulatory aspects on O2 consumption and 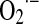 and H2O2 production. It is worth noting that NO also slows down the electron transfer between complexes. The components of the electron transfer chain have different sensitivity to inhibition by NO (Fig. 1) (184). Below 0.2 μM, NO reversibly inhibits COX and controls mitochondrial respiration; at 0.3–0.5 μM, it inhibits electron transfer between cyts b and c1 (182, 184), whereas relatively prolonged 0.5–1 μM NO exposure selectively inhibits the NADH dehydrogenase activity at mitochondrial complex I in intact cells (169) or isolated mitochondria (194), a hallmark of aging, sepsis, and neurodegenerative entities, such as Parkinson disease. Our studies revealed that the resultant segmental inhibition of the electron transfer chain at complexes I-III by NO is also followed by a very high burst of
and H2O2 production. It is worth noting that NO also slows down the electron transfer between complexes. The components of the electron transfer chain have different sensitivity to inhibition by NO (Fig. 1) (184). Below 0.2 μM, NO reversibly inhibits COX and controls mitochondrial respiration; at 0.3–0.5 μM, it inhibits electron transfer between cyts b and c1 (182, 184), whereas relatively prolonged 0.5–1 μM NO exposure selectively inhibits the NADH dehydrogenase activity at mitochondrial complex I in intact cells (169) or isolated mitochondria (194), a hallmark of aging, sepsis, and neurodegenerative entities, such as Parkinson disease. Our studies revealed that the resultant segmental inhibition of the electron transfer chain at complexes I-III by NO is also followed by a very high burst of 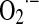 production rate (Fig. 2B). As a consequence of inhibitory NO effects, the reduction level of the mitochondrial components favors additional reactions of NO with ubiquinol (UQ) (183) and complex I components, and the formation of peroxynitrite (ONOO−) (reaction 4).
production rate (Fig. 2B). As a consequence of inhibitory NO effects, the reduction level of the mitochondrial components favors additional reactions of NO with ubiquinol (UQ) (183) and complex I components, and the formation of peroxynitrite (ONOO−) (reaction 4).
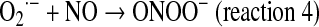 |
The equimolecular formation of superoxide with regard to NO utilization represents an accurate mechanism to control mitochondrial NO level and O2 uptake. Further, the modulation of NO utilization pathways and mitochondrial NO, 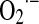 , H2O2, and ONOO− generation participate significantly in life processes (33, 34). In thisarticle, we show that H2O2, and the related level of oxidative stress play a significant role in the activation of signaling pathways central to the control of mitochondrial dynamics, energy balance and cell proliferation, differentiation, apoptosis, and senescence. Moreover, redox status is clearly related to the activity of growth factors and to cell transformation and cancer. Our proposal is that grading production and actions of matrix NO and concomitant changes in MnSOD modulate H2O2 and oxidative stress and set the platform for cell transformation (33), through opportune signals for the related physiological or pathological responses.
, H2O2, and ONOO− generation participate significantly in life processes (33, 34). In thisarticle, we show that H2O2, and the related level of oxidative stress play a significant role in the activation of signaling pathways central to the control of mitochondrial dynamics, energy balance and cell proliferation, differentiation, apoptosis, and senescence. Moreover, redox status is clearly related to the activity of growth factors and to cell transformation and cancer. Our proposal is that grading production and actions of matrix NO and concomitant changes in MnSOD modulate H2O2 and oxidative stress and set the platform for cell transformation (33), through opportune signals for the related physiological or pathological responses.
C. H2O2 and antagonistic antioxidant enzymes
H2O2 freely diffuses through cell membranes, and, thus, a similar mitochondrial and cytosolic H2O2 steady-state concentration ([H2O2]ss) should be expected. In agreement, [H2O2]ss has been calculated as ∼10–8 to 10–9 M in rat liver cytosol, rat liver mitochondria, and stimulated perfused liver (25), as well as after diffusion in hepatocytes (221). In this context, cell or tissue [H2O2]ss could be calculated as the ratio between H2O2 mitochondrial yield (d[H2O2]/dt) and the H2O2 catabolizing enzyme activities (catalase, GPx, and Trx, Eq. 1).
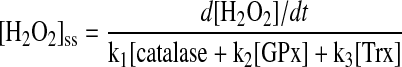 |
(Equation 1) |
More recently, Trx has emerged as a very important antioxidant system. Trx is oxidized during degradation of H2O2 to a disulfide intermediary that can be regenerated by peroxiredoxins (Prxs) and Trx reductase (98). Therefore, the actual function of Trx system relies on its capability to metabolize H2O2 in mitochondria (Trx2) and outside mitochondria in cytosol and nucleus (Trx1). It is worth noting that the increase in atmospheric oxygen a million years ago induced cell proliferation and pluricellularity; about 2 billion years ago, the ambient oxygen tension of the Earth's atmosphere increased rapidly from 1% to more than 15% of present levels within less than 200 million years (96). The development of antioxidant systems similar to Trx1/2 sustained a discrete cell redox status with low [H2O2]ss (10−11M). Activation of the extracellular signal-regulated kinase (ERK) cascade by ROS is consistent with the observation that relatively low levels of ROS are mitogenic (77). In addition, ROS may promote tumorigenesis by oxidizing DNA leading to proto-oncogenic mutations. The expression of Trx system proteins has been found to be changed in many diseases including cancer, diabetes, cardiovascular and neurodegenerative diseases, or rheumatoid arthritis. In particular, Trx is overexpressed in aggressive tumors that have reduced mitochondrial activity and electron transfer rate, associated to poor NO and H2O2 yields. In contrast, increase in oxygen during evolution accompanied a higher redox status with high [H2O2]ss (10−8–10−9M) that signals for cell cycle arrest, aging, senescence, and limits organ growth and body size (33). A discrete increase of H2O2 triggers most of the proliferation signaling pathways by activation of transcription factors such as nuclear factor-κB (NF-κB), tumor protein (p53), V-raf-1 murine leukemia viral oncogene homolog (RAF), hypoxia inducible factor-α (HIF-α), adaptor-related protein complex 1 (AP-1), and glucocorticoid receptor, etc. Therefore, the maintainance of cell proliferation rhythm depends on the redox balance between ROS and antioxidants (i.e., SOD, Trx, catalase, GPx, and nonenzymatic antioxidants such as gluthatione) (126).
D. The intermembrane space and the redox status
The intermembrane space (IMS) was initially described as a simple space between the inner and the outer mitochondrial membranes (OMMs) (∼10-15Å). Translocation of nuclear-encoded proteins by the translocases of the outer and inner membrane TOM-TIM, the assembling machinery complex, ADP-ATP translocation, and ion fluxes through specific channels are examples of an intense IMS physiological activity. As an important part of this article, we and others recently reported activation of kinases in the IMS as a part of canonical signaling pathways (79). A common feature in this case, and in the different mitochondrial IMS pathways, is the dependence on the redox status as determined by the H2O2 released by the organelles in the electron transfer chain. Recently, Iñarrea et al. (108) found that  reaches also the IMS, and that Cu-Zn SOD (SOD I) translocates into this compartment, likely to metabolize superoxide outside the mitochondrial matrix (223). In the presence of ROS, diverse oxidoreductive reactions occur in the IMS (102). In first place, electron transference between some of the components of the mitochondrial electron transfer chain proceeds in the IMS, similar to reduction and release of cyt c or phosphorylation of NDUFA1 gene product component of Complex I (41). In this context, the IMS contains a system to refold proteins by selective oxidation and retains them in mitochondria. An example is the disulfide relay integrated by two coupled proteins: mitochondrial intermembrane space assembly machinery (Mia40) and the sulfihydryl oxidase endogenous retroviral sequence 1 (ERV1) (207). Mia40 contains reactive cysteines forming a disulfide bridge; one of these cysteines is reduced by thiols of the protein to be imported after entering the TOM channel in the outer membrane. The two protein thiols form a new intramolecular disulfide, and reduced Mia40 is re-oxidized to its dithiol variant by complementary ERV1 protein, thus closing the cycle; ERV1 is reoxidized by cyt c (Fig. 3). By this means, the precursor protein can be oxidized and reduced by determined n times, during its refolding in the IMS (153). The Mia40 disulfide bridge has been recognized as necessary for the import of small Tim chaperones (for instance Tim 8 and Tim 13) that act as chaperones in the traffic of proteins to the inner membrane and to the mitochondrial matrix (159). As shown in this article, activation and phosphorylation of cytosolic kinases can be accomplished in mitochondria. Phosphorylation of kinases as ERK1/2 and protein kinase B (Akt) 1 and 2 depends on the presence of their upstream kinases mitogen protein kinase kinase 1 and 2 (MEK1/2) and PI-3K-dependent kinase 1 (PDK1) that are constitutively active in the IMS (10). Considering that this activation requires transitional binding between kinases and activators and that oxidation of thiols directs the protein interactions, it will be necessary to study the role of Mia40 disulfide bridge in the contributions of mitochondria to cell signaling pathways.
reaches also the IMS, and that Cu-Zn SOD (SOD I) translocates into this compartment, likely to metabolize superoxide outside the mitochondrial matrix (223). In the presence of ROS, diverse oxidoreductive reactions occur in the IMS (102). In first place, electron transference between some of the components of the mitochondrial electron transfer chain proceeds in the IMS, similar to reduction and release of cyt c or phosphorylation of NDUFA1 gene product component of Complex I (41). In this context, the IMS contains a system to refold proteins by selective oxidation and retains them in mitochondria. An example is the disulfide relay integrated by two coupled proteins: mitochondrial intermembrane space assembly machinery (Mia40) and the sulfihydryl oxidase endogenous retroviral sequence 1 (ERV1) (207). Mia40 contains reactive cysteines forming a disulfide bridge; one of these cysteines is reduced by thiols of the protein to be imported after entering the TOM channel in the outer membrane. The two protein thiols form a new intramolecular disulfide, and reduced Mia40 is re-oxidized to its dithiol variant by complementary ERV1 protein, thus closing the cycle; ERV1 is reoxidized by cyt c (Fig. 3). By this means, the precursor protein can be oxidized and reduced by determined n times, during its refolding in the IMS (153). The Mia40 disulfide bridge has been recognized as necessary for the import of small Tim chaperones (for instance Tim 8 and Tim 13) that act as chaperones in the traffic of proteins to the inner membrane and to the mitochondrial matrix (159). As shown in this article, activation and phosphorylation of cytosolic kinases can be accomplished in mitochondria. Phosphorylation of kinases as ERK1/2 and protein kinase B (Akt) 1 and 2 depends on the presence of their upstream kinases mitogen protein kinase kinase 1 and 2 (MEK1/2) and PI-3K-dependent kinase 1 (PDK1) that are constitutively active in the IMS (10). Considering that this activation requires transitional binding between kinases and activators and that oxidation of thiols directs the protein interactions, it will be necessary to study the role of Mia40 disulfide bridge in the contributions of mitochondria to cell signaling pathways.
FIG. 3.

The IMS, a redox compartment with different functions. Many unfolded proteins traverse the IMS to reach the inner membrane or to exit mitochondria. The graphic shows the disulfide bridge relay given by the two complementary intermediaries Mia40 and Erv1 in the IMS. The two components compose a cycle by which an unfolded protein with thiol groups forms a disulfide bridge with oxidized Mia40 (Mia40ox) that allows the protein to be refolded with intermolecular disulfide formation and Mia40 reduction. Mia40red returns to its oxidized state by reduction of Erv1, subsequently recovered by given electrons to Cyt c, and ultimately to cytochrome oxidase. MIA40, mitochondrial intermembrane space assembly machinery; ERV1, endogenous retroviral sequence; Cyt c, cytochrome c; COX, cytochrome oxidase; IMM, inner mitochondrial membrane; IMS, intermembrane space; OMM, outer mitochondrial membrane.
III. Mitochondrial Metabolism and Cell Proliferation
A. The Warburg effect: The mitochondrial control of proliferation
Many years ago, the German scientist Otto Warburg revealed that tumors have a reduction of aerobic metabolism and a parallel increase of glycolysis rate (228, 229). Decreased aerobic respiration in the presence of available oxygen was then referred to as the Warburg effect (196). For many years, this effect was considered a particular feature of cancer tissues, but nowadays, it is considered a mandatory metabolic change to allow cells to divide and proliferate (83, 209). In this context, and considering the clinical significance of tumor growth and proliferation, two questions arise: first, what are the mitochondrial signaling mechanisms for the control of respiration rate and second, what is the physiological meaning and the ultimate reason for reduced O2 utilization during cell division and its contribution to tumor biology? Moreover, recent studies have shown that the Warburg effect not only results in intrinsic biochemical modifications in the organelles, but also depends on selective accumulation of glycolytic intermediaries. A reduction of the respiratory rate results from mutations in COX in tumors, reduced mitochondrial biogenesis, excessive mitochondrial fission, deficit of proapoptotic proteins such as p53 and antioxidants such as Trx, excess of mitochondrial inhibitors such as myelocytomatosis viral oncogene homolog (cMyc) or Akt, or reduced Akt catabolism by deletion or suppression of phosphatase antensin homolog (PTEN). We will consider in this article the mechanisms that promote the Warburg effect and the platform of tumorigenesis (Fig. 4).
FIG. 4.

Interaction between ROS and p53. An equilibrated production of ROS and p53 in normal cells (left) could be disrupted by a modest increase of p53 that increases discretely the ROS production, which increases the activation of pro-proliferative kinases such as Akt and ERK1/2. High p53 arrest cells and increases apoptosis, with low Akt and high respiratory activity and very high ROS. In contrast lack of p53, reduces the activity of cytochrome oxidase, lowers ROS, and provides the platform for transformation. In the end, NO lowers electron transfer rate but differentially with the Warburg condition is not proliferative because it increases ROS and decreases Akt activity; whereas it increases the activity of pro-apoptotic kinases, such as p38 and mitogen-activated protein kinase. Akt, protein kinase B; ERK, extracellular signal-regulated kinase; ETC, electron transfer chain; p53, tumor protein; ROS, reactive oxygen species.
B. Mitochondria and redox control in normal and tumor cells
The Warburg effect underlies the simultaneous increase in glycolytic rate with a reduced mitochondrial respiratory rate; a kind of mitochondrial dysfunction that is found in tumor metabolic reprogramming pathways. We previously reported a significant decrease in respiratory activities of lung and mammary tumor cell lines, and only a third of the mitochondrial electron transfer rate of normal organelles could be detected in the cancer cells (78). Notably, although tumor mitochondria were more bizarre and with less cristae (Fig. 5), functional respiratory status was similar to those of embryonic proliferating liver or the pregnant mammary gland, thus indicating that mitochondrial reprogramming is an essential requisite for both normal or tumor cell proliferation and organ growth.
FIG. 5.

Electron microscopy of normal and tumor mitochondria. In the upper panel, mitochondria from LM3 mammary tumor cells (right) are compared with those from normal mammary cells of pregnant mice. In the lower panel, a similar comparation is made between mitochondria isolated from lung tumor P07 cell line (right) and organelles from normal lung cells. Bar=0.33 μM. Reproduced according to American Association for Cancer Research (AACR) from Galli et al. (78).
Multiple causes participate in the genesis of alterations in tumor mitochondria. Recent studies revealed that cancer cells of various tissue origins exhibit frequent mutations in their mtDNA (52). Deletion of in-frame mtDNA genes has been documented in human renal, gastric, and colon carcinoma (188). Since the mtDNA encodes for 13 protein components of the mitochondrial respiratory chain, it is likely that specific mtDNA mutations may cause malfunction of the respiratory chain, thus forcing the cells to increase glycolysis to maintain their ATP supply. Surprisingly, this condition increases the activity of proliferative kinases such as Akt, probably by NADH accumulation at low mitochondrial respiratory rate that suppresses PTEN, the phosphatase that inactivates Akt (177). However, although mtDNA mutations are associated to high ROS and dysfunctional mitochondria, and cancer, the existence of similar mitochondrial features in genetic diseases lacks any particular clinical association with neoplasia, which suggests that cancer mutations are not the cause of, but a consequence of disease.
C. Stem cells, mitochondrial ROS metabolism, and differentiation
The pluripotent embryo cells embryonic stem cells (ESCs) have a low mitochondrial population. Mitochondria are large, with low energy potential; most of the energy becomes from glycolysis, which is limited only by a low ATP reservoir that precludes glucose phosphorylation to glucose 6-phosphate, required for the entrance to the cells. Instead, cells differentiated into trophectoderm of mice and rats, have more elongated mitochondria, with higher membrane potential and more O2 utilization (100). This behavior is reciprocal to that described in tumor cells. The tumor cells have a decreased respiratory rate associated with an enhancement of anaerobic glycolysis. The reduction of the respiratory rate depends on a uniform transcriptional reduction of mitochondrial components. It is clear that the most important effect of decreasing the mitochondrial respiratory rate is related to a marked decrease of ROS up to a level that allows a maximal activity of the proliferative kinases, such as ERK1/2 and Akt, which impedes the triggering of the apoptotic machinery by exerting the antiapoptotic control and by non-activation of proapoptotic kinases, such as c-Jun-NH2-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK). Conversely, differential activation of JNK and p38 MAPK conducts to cell differentiation and finally to cell cycle arrest and apoptosis. Differentiating effects of ROS produced during various electron transfer reactions in vivo have been reported in different species. In the mammalian hematopoietic system, hematopoietic stem cells contain low levels of ROS. However and unexpectedly, the common myeloid progenitors produce significantly increased levels of ROS. The functional significance of this difference in ROS level in the two progenitor types remains unresolved. Owusu-Ansah and Banerjee (170) showed that Drosophila multipotent hematopoietic progenitors, which are largely similar to the mammalian myeloid progenitors, display increased levels of ROS under in vivo physiological conditions, which are downregulated on differentiation. Scavenging ROS from these hematopoietic progenitors by using in vivo genetic tools, such as overexpression of GPx or catalase, retards their differentiation into mature blood cells while, conversely, increasing the hematopoietic progenitor ROS triggers precocious differentiation into all three mature blood cell types found in Drosophila; suggesting that the rise in ROS primed the relatively quiescent stem-like progenitor cells for differentiation and that this signaling pathway involves JNK and forkhead O-box (FOXO) activation.
D. ROS and mitochondrial malignancy: The example of p53
It was understood that ROS regulates the activation and duration of signaling through redox-dependent signal transduction pathways involving the cyclic oxidation/reduction of cysteine residues in kinases, phosphatases, and other regulatory factors (24). Burhans and Heintz noted that “signaling circuits may be segregated in organelles or other subcellular domains with distinct redox states, permitting them to respond independently to changes in the oxidation state of two major thiol reductants, glutathione and thioredoxin” (24). In the previous years, we emphasized the notion that grading redox status and steady-state concentration of oxidants, such as H2O2, bring up differential cysteine oxidations in the signaling circuits and, therefore, different, even opposite responses on cell proliferation, senescence, or arrest (137). As just stated, most of the cell H2O2 comes from mitochondria, a subcellular domain with redox variations (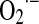 , NO, H2O2, and ONOO−).
, NO, H2O2, and ONOO−).
The tumor suppressor protein, p53 is a well-known transcription factor modulator of the regulation of cell cycle entry; p53 is frequently deleted or mutated in cancer cells. In the case of p53, there exist cross-synergistic effects that exemplify the extreme complexity of the redox regulation of cell cycle. Overexpression of p53 induces the activation of several pro-oxidant enzymes such as ROS-generating proline oxidase and genes such as p66 that interact with cyt c to increase H2O2 in mitochondria (137). On the other hand, evidence indicates that redox activation of human p53 involves post-translational modifications such as thiol redox modulation of critical cysteine residues (Cys174, Cys238) in its DNA binding domains. These post-translational modifications, likewise, explain the actions of p53 on the cell cycle without modifying nontranscriptional mitochondrial effects (Fig. 4). In addition, p53 counteracts the transition to a warburgian condition by increasing mitochondrial respiration at the COX level due the transactivation of SCO2 (synthesis of cyt c oxidase 2) gene (146). This effect increases the respective percentage of 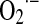 and H2O2 formed in mitochondria where there exist important targets of transcription-dependent and -independent actions of p53 required to trigger apoptosis. The effect of p53 in mitochondrial ROS homeostasis is evidenced by its participation in maintaining mtDNA copy number; p53 null mice and p53 knockdown human primary fibroblasts exhibit mtDNA depletion and decreased mitochondrial mass under normal growth conditions of cell culture (130). In this situation, the p53-depleted cells exhibited significant disruption of cellular ROS homeostasis, characterized by reduced mitochondrial and cellular levels of
and H2O2 formed in mitochondria where there exist important targets of transcription-dependent and -independent actions of p53 required to trigger apoptosis. The effect of p53 in mitochondrial ROS homeostasis is evidenced by its participation in maintaining mtDNA copy number; p53 null mice and p53 knockdown human primary fibroblasts exhibit mtDNA depletion and decreased mitochondrial mass under normal growth conditions of cell culture (130). In this situation, the p53-depleted cells exhibited significant disruption of cellular ROS homeostasis, characterized by reduced mitochondrial and cellular levels of 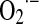 . Thus, tumors associated with loss of p53 function involve the simultaneous decrease of mitochondrial biogenesis with lowering of mitochondrial respiration, two conditions that increase glycolytic activity, and allow cells to grow by resetting energy production and ROS generation. In addition, mutant p53 enhances the mitochondrial effects of the oncoproteins rat sarcoma small GTPase (RAS) and cMyc, which contributes to the mitochondrial platform of malignancy (178).
. Thus, tumors associated with loss of p53 function involve the simultaneous decrease of mitochondrial biogenesis with lowering of mitochondrial respiration, two conditions that increase glycolytic activity, and allow cells to grow by resetting energy production and ROS generation. In addition, mutant p53 enhances the mitochondrial effects of the oncoproteins rat sarcoma small GTPase (RAS) and cMyc, which contributes to the mitochondrial platform of malignancy (178).
It is known that p53 is able to modulate the mitochondrial ROS production unless it is not clear whether this effect leads to cell death or promotes malignancy (129, 168). It would also be noted that the amount of ROS can be determinative for cell outcome mediated by p53. It is remarkable that low levels of ROS may result in activation of signaling pathways such as proliferation and motility and p53 antioxidant functions, and that in contrast, high ROS levels may result in activation of p53-mediated apoptosis. These important roles of p53 are exerted through the shuttling and balance between nucleus and mitochondria, especially in the context of ROS and antioxidants (90). In this context, nuclear tumor suppressor p53 can transactivate proapoptotic genes or antioxidant genes depending on stress severity, whereas cytoplasmic p53 induces mitochondrial-dependent apoptosis without gene transactivation. Although SIRT1, a p53 deacetylase, inhibits p53-mediated transactivation, how SIRT1 regulates these p53 multifunctions is unclear. Han et al. demonstrated that SIRT1 blocks nuclear translocation of cytoplasmic p53 in response to endogenous ROS and triggers mitochondrial dependent apoptosis in mouse embryonic stem (mES) cells. ROS generated by antioxidant free culture caused p53 translocation into mitochondria in wild-type mES cells, but induced p53 translocation into the nucleus in SIRT1−/− mES cells. Endogenous ROS triggered apoptosis of wild-type mES through mitochondrial translocation of p53 and Bcl-2, B-cell lymphoma 2 (BAX), but inhibited NANOG expression of SIRT1−/− mES, thus indicating that SIRT1 makes mES cells sensitive to ROS and inhibits p53-mediated suppression of NANOG expression. These results showed that endogenous ROS control is important for mES cell maintenance in culture.
Pro-oxidant effects of p53 and ROS increase after p53 translocation into mitochondria can be related to a general increase of the respiratory electron transfer rate. In addition to activation of SCO2, p53 is linked to AMP-activated protein kinase (AMPK). We recently demonstrated that phosphorylated AMPK enters to adipose and muscle mitochondria and inhibits NOS, thus increasing oxygen uptake (70). pAMPK activates p53 by phosphorylation at Ser15 and in contrast, p53 promotes transcriptional activation of AMPK gene. Since AMPK increases energy waste by inhibiting energy storing as fat (i.e., AMPK phophorylates and inhibits acetylCoA carboxylase, the first enzyme in fatty acid synthesis), cooperative effects between AMPK and p53 in mitochondria increase respiration and contribute to inhibit the Warburg effect and cell proliferation. This mechanism is useful for critical cell conditions with death expectancy.
Among the identified targets of p53, there are several genes with apparent antioxidant function. These are microsomal glutathione transferase homolog PIG12, aldehyde dehydrogenase ALDH4A1, GPx1, Mn superoxide dismutase SOD2, and catalase. In addition, two members of the sestrin family SESN1 (PA26) and SESN2 (Hi95) were also found to be regulated by p53 (168). Sestrins act as components of the Prx regeneration system. In tight cooperation with sulfiredoxin (Srx), the sestrins act as subunits of cysteinic sulfinyl reductase by regenerating inactive Prxs that overoxidize in response to massive bursts of H2O2 occurring during signal transduction. The contribution of these antioxidant products to p53 functions was elusive until it was found that in unstressed cells a p53 function is required for reducing the intracellular ROS levels. Abrogation of p53 functions by means of RNAi, or by overexpression of dominant negative p53, Mdm2, or papilloma virus E6 gene product results in a substantial increase in intracellular ROS. Similar increases in ROS were observed in tissues of the p53−/− mice. The increases in ROS in the p53-deficient cells correlated with substantial downregulation of the p53 regulated genes GPx1, SESN1, and SESN2, thus suggesting that basal physiological levels of p53 are sufficient for maintaining functional state of the antioxidant genes. Basal levels of p53 were also found sufficient for maintaining the expression of catalase and TIGAR (168).
E. The glycolytic effects for mitochondrial oxidative rate
Sinthupibulyakit et al. (208) demonstrated that the inhibition of glycolysis by 2-deoxyglucose (2-DG) is not enough in wild-type A549 cells to abolish the Warburg effect and set up the platform for persistent cell proliferation. However, a cytotoxic effect of 2-DG was evident when cells were knock-down for p53, thus indicating that glycolysis takes part in the Warburg effect only when mitochondria are disabled (208). However, it was also evidenced that ATP synthesis in selected tumors is not dependent exclusively on glycolysis but still on mitochondrial oxidative phosphorylation (OXPHOS). In this case, the driving force for enhanced glycolysis would not be the absolute reduction of mitochondrial respiratory rate, but a relative decrease of both glycolytic and mitochondrial respiration is likewise imposed by uncontrolled tumor cell proliferation (20, 158).
Constitutive levels of p53 are coupled to normal mitochondrial respiration through its target gene, SCO2, which has a critical function in maintaining the cyt c oxidase complex, the major site of O2 utilization (137). Increased O2 utilization is related to the increase of mitochondrial ROS. Reciprocally, diffusion of H2O2 activates p53 expression, and the antitumor and pro-warburgian action of p53 is mediated by inducing transactivation of genes related to ROS production. Up-regulation of these pro-oxidant enzymes leads to oxidative stress and consequently to apoptosis. This was the first clear connection between p53 and ROS generation. More candidates have been added to the list of p53-induced pro-oxidant genes, which include BAX, p53-up-regulated mediator of apoptosis (PUMA), and p66shc. However, p53 also mediates the modulation of ROS effects and prevention of apoptotic cell death by stimulating the expression of antioxidant enzymes (197).
F. Mitochondrial signaling in hypoxia
The main transcription factor in hypoxic signaling is the HIF. HIF is stabilized and reacts with hypoxia-responsive elements in DNA activating gene promoters to activate proliferative genes (Fig. 6). Moreover, hypoxia impedes the oxidation of prolyl HIF residues by prolyl hydroxylase and allows HIF to react with pVHL (the von Hippel Lindau protein), to be ubiquitinated, and degraded in the proteasome (49), a process activated in the presence of O2. On the contrary, more stable HIF-1 in anaerobiosis activates the transcriptional machinery for the glycolytic enzymes in the tumor cells. HIF also promotes glycolysis by stimulating expression pf PDH kinase I and decreasing oxidation by pyruvate (158).
FIG. 6.

Hypoxia signaling and HIF. The scheme shows the pathways for HIF degradation and proliferation in accord to oxygen levels. HIF, hypoxia inducible factor.
During hypoxia, HIF is expressed and integrated as HIF-1α–cMyc axis. Cells with activation of this axis display persistent DNA damage and mutations that sustain malignant proliferation (239). In addition, hypoxia stimulates mitochondrial NO synthase (mtNOS) and, thus, depresses the electron transfer rate (243).
Mechanisms of HIF in hypoxia rely on deactivation of genes related to cell proliferation such as vascular growth factor or insulin growth factor (IGF) (Fig. 6). In addition, HIF2-α stimulates the expression of one of the genes related to pluripotency transcription factors, OCT-4, without varying NANOG or SOXS2 (53). By this modulation, hypoxia sustains proliferation in embryos and tumoral tissues that share a hypoxic milieu (1.5%–5% O2). In those disorders associated to hypoxia, proliferative signals could be confined to peripheral tissues, as “clubbing” in the fingers of patients with lung diseases. The unsolved question is whether hypoxia increases the production of ROS in mitochondria and contributes to HIF stabilization. Klimova and Chandel (122) proposed that HIF is stabilized by ROS production at mitochondrial complex III. In this case, HIF was not stabilized by deleting cyt b. However, ROS were not completely abolished when cyt b was deleted (mutant cybrids), whereas the use of mitochondria-targeted antioxidants prevent HIF stabilization (112). In fact, we observed many years ago that the complete extraction of mitochondrial UQ abrogates the production of superoxide and H2O2 by the organelles (184). Moreover, ROS production by complex III requires a selective inhibition of electron transfer, whereas hypoxia decreases electron transfer more uniformly at all complexes (including I and III ones) by reducing the redox pairs on the side of substrate. In addition, a blockade at the level of cyt b in the presence of ATP (likewise glycolytic in the presence of HIF) could induce retrograde formation of 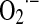 at complex I that would preferably be inhibited in this experimental condition. In contrast, Vieira et al. (225) studied the role of O2 and derived ROS in the development of neural tissues. In this case, the authors highlighted that culturing human ESCs (hESCs) from blastocysts in 4% O2 (instead 20% at normoxia) impeded differentiation and favored cell self-renewal (64). Accordingly, low (but not high) O2 tension increases the amount of inner mass cells in the blastocyst (94). Consequently, Vieira et al. remarked on the importance of using appropriate (low) levels of oxygen in the environment of hESCs to maintain pluripotency. Westfall et al. (233) characterized oxygen-sensitive transcriptional programs and gene expression profiles in two distinct hESC lines cultured in 4% versus 20% O2. At O2 tension of 20%, multiple HIF-controlled genes as well as genes coding for glycolytic enzymes and pentose phosphate enzymes were downregulated in the hESCs. In this context, the transition from pluripotency to differentiation is accepted as a process that increases O2 and switches glycolytic to oxidative metabolism with 2.5% higher production of mitochondrial ROS (47). In contrast, low oxygen levels were required for neuroblast proliferation. In agreement, Carreras et al. reported (33) that low ROS production associates to maximal hepatoblast proliferation during development. However, discrete ROS production can stimulate, by oxidation of critical cysteines, proliferative kinases as Akt (10) and, thus, very low ROS yield could collaborate in stabilizing HIF, an effect rapidly lost at crescent ROS levels.
at complex I that would preferably be inhibited in this experimental condition. In contrast, Vieira et al. (225) studied the role of O2 and derived ROS in the development of neural tissues. In this case, the authors highlighted that culturing human ESCs (hESCs) from blastocysts in 4% O2 (instead 20% at normoxia) impeded differentiation and favored cell self-renewal (64). Accordingly, low (but not high) O2 tension increases the amount of inner mass cells in the blastocyst (94). Consequently, Vieira et al. remarked on the importance of using appropriate (low) levels of oxygen in the environment of hESCs to maintain pluripotency. Westfall et al. (233) characterized oxygen-sensitive transcriptional programs and gene expression profiles in two distinct hESC lines cultured in 4% versus 20% O2. At O2 tension of 20%, multiple HIF-controlled genes as well as genes coding for glycolytic enzymes and pentose phosphate enzymes were downregulated in the hESCs. In this context, the transition from pluripotency to differentiation is accepted as a process that increases O2 and switches glycolytic to oxidative metabolism with 2.5% higher production of mitochondrial ROS (47). In contrast, low oxygen levels were required for neuroblast proliferation. In agreement, Carreras et al. reported (33) that low ROS production associates to maximal hepatoblast proliferation during development. However, discrete ROS production can stimulate, by oxidation of critical cysteines, proliferative kinases as Akt (10) and, thus, very low ROS yield could collaborate in stabilizing HIF, an effect rapidly lost at crescent ROS levels.
G. Mechanistic target of rapamycin (serine/threonine kinase)/Akt pathways
The mechanistic target of rapamycin (serine/threonine kinase) (mTOR)/Akt pathway plays an important role in the regulation of cell proliferation, mitochondrial biogenesis, and energy balance. The complete picture of the different mechanisms leading to reprogramming of mitochondrial function and energy balance are not yet clear. For instance, mTORC1 phosphorylates S6K, and this stimulates mitochondrial biogenesis. Otherwise, mTORC2 increases phosphorylation of Akt at Ser476; this phosphorylation, and a second one dependent on PDK1 occurring on Thr308 are as well stimulated by the IGF-1/phosphoinositide 3-kinase (PI3K) pathway that promotes cell growth and survival (10).
Robey and Hay considered Akt to be the Warburg kinase (196). In this context, Akt, which is expressed in most tumors, is accompanied by two- to threefold increase in intracellular ATP content. The change in energy metabolism induced by Akt seems to rely on the activation of glycolysis (Fig. 7). Concerning the specific connections between RTK/PI3K/Akt/mTOR pathway and the Warburg effect, it is noteworthy that the rate-limiting glycolytic enzyme pyruvate kinase M2 (PKM2) isoform is exclusively expressed in embryonic, proliferating, and tumor cells, and plays an essential role in tumor metabolism and growth (147). In addition, Sun et al. (216) identified mTOR as a central activator of the Warburg effect by inducing PKM2 and other glycolytic enzymes (hexokinase [HK], phosphofructokinase [PFK]) under normoxic conditions. The authors suggested that the levels of PKM2 were augmented in mouse kidney tumors due to the deficiency of tuberous sclerosis complex 2 and the consequent mTOR activation, and was decreased in human cancer cells by mTOR suppression. As an integrated tumorigenic response, mTOR up-regulation of PKM2 expression occurs through HIF1α-mediated transcription activation, and c-Myc-heterogeneous nuclear ribonucleoproteins-dependent regulation of PKM2 gene splicing (216); disruption of PKM2 suppresses oncogenic mTOR-mediated tumorigenesis. However, how can increased respiration by Akt with Warburg's depression of oxidative metabolism and mitochondrial impairment be reconciled? Recently, we reported that Akt2 decreases mitochondrial oxidative metabolism through phosphorylation of nNOS present in mitochondria at Ser1412 (68). Akt2-dependent nNOS phosphorylation produces a positive allosteric change resulting in a decrease of Km from 20 to 12 μM L-arginine, and high-matrix NO levels that inhibit mitochondrial respiration. It is not clear whether the same effects on nNOS are equally produced by Akt1 and 2; it can be surmised that Akt1 is activated predominantly by the glycolytic pathway, and Akt2 depresses mitochondria, favors displacement of acetyl-CoA to fat synthesis for cell proliferation, and limits glucose complete oxidation to be utilized in glycolysis.
FIG. 7.

Transition from high to low respiratory rate: The Warburg requirement for cell proliferation. Different mechanisms underlie the Warburg effect characterized by low O2 utilization in the presence of enough available oxygen. This effect is given by increased glycolysis associated to high expression and activation of glycolytic enzymes (PK, GAPDH, PFK, HK, and LDH) and reduced mitochondrial respiration. Low respiratory rate depends on several factors such as mitochondrial fission, and defective combined effects of p53, c-myc, and Akt that sustain high oxidative rate or promote adhesion of HK II. GAPDH, glyceraldehide 3-phosphate dehydrogenase; HK, hexokinase; LDH, lactate dehydrogenase; PFK, phosphofructokinase; PK, pyruvate kinase.
H. Hexokinase
HK catalyzes the first step of the glycolytic pathway where glucose is phosphorylated to glucose-6-phosphate (G-6-P) by phosphate transfer from ATP. By this means, G-6-P remains inside the cells and cannot be further released to the extracellular milieu except by hepatocytes that possess glucose-6-phosphatase to regulate glycemia. Most G-6-P is utilized through the glycolytic pathway to provide a discrete 2–3 mol ATP per mol of glucose and more importantly, to allow pyruvate to be decarboxylated in mitochondria to acetyl-CoA and enter into the tricarboxylic acid. There exist four different HK (1–4) in mammals (235). From a kinetics perspective, HK1-3 have a very low Km for glucose (∼0.02 mM), whereas HK4 or glucokinase has a high Km (∼5 mM). HK-1 and HK-2 are localized predominantly on the OMM (Fig. 7), HK-3 in a perinuclear compartment (235), and HK-4 is located in the cytosol (liver and pancreas).
Different studies have confirmed that the HK-2 isoform is predominantly expressed in mitochondria from malignant tumors with high glycolytic rate (26, 27). HK-2 is bound to the mitochondrial anion channel voltage-dependent anion channel (VDAC) and to the ADP/ATP translocator (ANT) that is linked to ATP synthase. On the bases of kinetic behavior and subcellular localization, Mathupala et al. (145) proposed three reasons by which HK-2 increases the glycolytic rate. First, in its localization, HK-2 has preferential access to ATP released by ANT. Second, binding to mitochondria protects HK-2 from the potent inhibition exerted by the product of the catalyzed reaction, G-6-P. Finally, the very low Km for glucose of HK-2 assures a high G-6-P flow rate to the glycolytic pathway. In this context, it is clear that HK-2 anchorage to the mitochondrial outer membrane is required for increase of glycolysis in the tumor tissues. It is accepted that pro-proliferative and anti-apoptotic Akt contributes to cancer metabolic reprogramming by increasing HK-2 bound to VDAC (145).
I. The regulation of glycolysis and proliferation by the ubiquitination system
ATP-mediated inhibition of PFK1 is reverted by increasing the amount of the enzyme up to physiological levels. As just mentioned, inhibition of mitochondrial ATP synthesis with NO (and potassium cyanide or oligomycin) triggers a rapid PFK1 activation in intact, but not in disrupted astrocytes, which indicates that the effect of NO on PFK1 does require an endogenous allosteric activator, such as fructose-2,6-bisphosphate (F2,6P2). F2,6P2 is the most potent physiological allosteric activator of PFK1. In agreement with this, NO promotes a rapid and persistent accumulation of F2,6P2 in astrocytes, but not in neurons. Further, the inhibition of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3), that is, the bifunctional enzyme responsible for F2,6P2 formation and degradation, rendered astrocytes unable to increase F2,6P2 levels, activate PFK1 and up-regulate the glycolytic rate. Further, Almeida, et al. (4) recently discovered that the bifunctional glycolysis-promoting enzyme PFKFB3 is degraded by the E3 ubiquitin ligase APC/C-Cdh1, which also degrades cell-cycle proteins. Therefore, both proliferation and aerobic glycolysis were prevented by overexpression of Cdh1 and enhanced by its silencing. In this study, they also demonstrated that although glycolysis is essential for cell proliferation, its initiation in the presence of active Cdh1 does not result in proliferation. The authors linked the notion that the proliferative response, in normal or neoplastic cells, is dependent on degradation of allosteric effectors of regulatory PFK1 with implications for cell proliferation, neoplastic transformation, and the prevention and treatment of cancer. Accordingly, the presence of PFKFB3 is tightly controlled to ensure the up-regulation of glycolysis at a specific point in G1. On this basis, Tudzarovaa et al. (220) suggested that this up-regulation of glycolysis and its associated events represent the nutrient-sensitive restriction point in mammalian cells. As mentioned earlier, other glycolytic enzymes, such as HK(s), pyruvate kinase, and glyceraldehyde-3-phosphate dehydrogenase, are potential targets of NO-mediated glycolysis activation.
IV. ROS: From Proliferation to Cell Death
It has been more than 30 years since it was proposed that higher organisms can achieve programmed cell death, which is illustrated by a common set of morphological features, coining the term “apoptosis” for cells that display these characteristics. Apoptosis is central to mammalian development and also plays a critical role in cellular homeostasis. Since apoptosis is crucial for maintaining cell number in the adult stage, deregulation of this process may contribute to the development of neurodegenerative disorders, immunodeficiency, and cancer (65).
At a molecular level, apoptosis is regulated by two protein families: the Bcl-2 family which is involved in the initiation phase, and the caspase family that is responsible for the execution part (217). The pathways that lead to caspase activation during apoptosis have been widely described. These are the extrinsic and intrinsic pathways that ultimately result in the activation of caspases-3 and -7, promoting proteolysis of typical substrates (1). The extrinsic pathway involves binding of tumor necrosis factor α (TNF-α) and TNF receptor superfamily member 6 (FAS) ligand to membrane receptors leading to caspase-8 activation, whereas the intrinsic pathway entails mitochondrial oxidative stress, damage, and mitochondrial cyt c release. Released cyt c triggers the assembly of the apoptosome complex with apoptotic protease-activating factor-1 (APAF-1) and procaspase-9, which induces activation of caspase-9. Both pathways converge on caspase-3 activation, thus resulting in nuclear degradation and cellular morphological changes (191).
The Bcl-2 family constitutes a key regulator in the intrinsic pathway of apoptosis controlling mitochondrial outer membrane permeabilization and the consequent release of apoptogenic factors (44, 206). Selective protein interactions among different pro- and antiapoptotic members of this family are crucial to apoptosis regulation. These interactions are mostly influenced by α-helical segments known as Bcl-2 homology (BH) domains and comprise both cytosolic and membrane conformers of select family members (131). Bcl-2 family members have typically been grouped into three classes. One group inhibits apoptosis (Bcl-2, B-cell lymphoma-extra large [Bcl-xL], Bcl-W, MCL1, Bcl-B and A1), whereas a second class promotes apoptosis (BAX, Bcl-2 homologous antagonist killer [BAK] and BOK). A third different class (Bcl-2-associated death promoter [BAD], Bcl-2 interacting killer, BID, BIM, BH3-only protein, and PUMA) has a conserved BH3 domain that can bind and regulate the anti-apoptotic Bcl-2 proteins to promote apoptosis. It appears that the pro-apoptotic family members BAX and BAK are crucial for inducing permeabilization of the OMM and the subsequent release of apoptogenic molecules (such as cyt c and direct IAP-binding protein with low pl (DIABLO) [also known as second mitochondria-derived activator of caspases (SMAC)]), which leads to caspase activation. The anti-apoptotic family members, such as Bcl-2 and Bcl-xL, inhibit BAX and BAK. Recent evidence indicates that BH3-only proteins de-repress BAX and BAK by direct binding and inhibiting of Bcl-2 and other anti-apoptotic family members (234). Contrary, a different model proposes direct activation of BAX and BAK by some BH3-only proteins (similar to BIM, truncated BID, and PUMA) (240). BAX and BAK promote caspase activation by their effects on mitochondria. These two pro-apoptotic Bcl-2 family members induce the release of proteins from the IMS (162). Mitochondrial outer membrane permeabilization results in the release of cyt c and other soluble proteins into the cytosol. At the same time as cyt c releases, BAX and BAK induce mitochondria to fragment into smaller units, which suggest a link between mitochondrial division progression and the role of the Bcl-2 proteins (144). It has been described that lymphocytes can probably use alternative APAF1-, caspase-9-, and cyt c-independent, but pro-apoptotic Bcl-2-dependent, pathways for caspase activation and cell killing (91). The action of IAPs (inhibitor of apoptosis proteins) that bind and neutralize certain caspases can be antagonized by the binding of SMAC/DIABLO, which is released from mitochondria after the activation of BAX and/or BAK.
Over the past decade, several investigators have provided new insights into how the plasticity of the mitochondria plays a central role in relaying diverse cellular signals. Together, these studies revealed that H2O2 and NO can regulate cell proliferation (22, 32, 33). Transient production of H2O2 is considered an intracellular signal for cell growth and transformation triggered by surface receptor activation (193) as well as determined by mitochondrial metabolic status (28). ERK1/2 activation is a H2O2- dependent process, and it translocates into the mitochondria during brain development (5). In vitro, ERK1/2 activation in mitochondria was maximal at 10−6 M H2O2, an effect also observed in embryonic hepatoblasts and isolated postnatal P2 hepatocytes, whereas a decreased phosphorylation concomitant with p38 MAPK activation was observed in the quiescent adult cells (33). The regulation of MAPKs cascades was associated to the modulation of mtNOS in the progression of proliferating to quiescent cell stages. Proliferating phenotypes are characterized by low levels of mtNOS expression and activity, with a resulting NO-dependent [H2O2]ss yield of 10−11 to 10−12 M, and high cyclin D1 expression. In contrast, quiescent phenotypes presented an opposite pattern with NO-mediated H2O2 levels of 10−9 M. In agreement, increases of mtNOS and [H2O2]ss are parallel during rat brain and cerebellum development at the stage of synaptic plasticity (195).
Distinctive effects of H2O2 are illustrated in transformed cells; increased proliferation in P07 tumor lung cells and mammary MM3 cell lines was observed at 1 μM H2O2, whereas cells became arrested at 50 μM H2O2 (78). It is remarkable that mtNOS expression is reduced in some tumor cell lines such as M3 and MM3; whereas in others such as P07, it is high but with an activity consistently lower than in normal tissues. Accordingly, mitochondrial H2O2 production is significantly lower in mitochondria from tumor cells compared with normal mitochondria. Redox status also determines cell fate in the NIH/3T3 cell line. At low H2O2 concentrations, cell proliferation increased, and cyclin D1 expression was up-regulated (Fig. 8A). Conversely, high H2O2 concentration caused a decrease in cell proliferation and cyclin D1 expression and triggered apoptosis (Fig. 8A and B). Apoptosis was achieved on activation of the mitochondrial-caspase-3 dependent pathway resulting in the release of cyt c to cytosol and retention of Bcl-xL in mitochondria (Fig. 8C). In this context, a low functional level of OXPHOS, decreased mtNOS, and low NO-dependent H2O2 production represent a common pattern of the active tumor growth and of embryonic tissues.
FIG. 8.

The fate of NIH/3T3 cells depends on the redox status. (A) Cyclin D1 expression increased on low H2O2 stimuli, whereas it decreased at a high H2O2 concentration. (B) High redox status triggered apoptosis by 10-fold as determined by flow cytometry with Annexin V staining (upper panel) and propidium iodide (lower panel). (C) Translocation of Bcl-xL and Cyt c from mitochondria to cytosol was determined 24 and 48 h after H2O2 stimuli. *represents p<0.05 vs control. From Antico et al., according to Creative Commons Attribution License (11). Bcl-xL, B-cell lymphoma-extra large. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
The antiproliferative effects of NO have been demonstrated in a variety of cell types from normal tissues and diverse tumors (140, 226). On exposure to NO from different sources, either NOS or NO-donors, cells stop growth at G1 or G2 phase, or show a delay in S phase progression. Up-regulation of endogenous NO production, by L-arginine supplementation or by expression of the inducible NO synthase (iNOS), inhibited proliferation of lymphocytes, vascular smooth muscle cells, and pancreatic tumor cells (89). Conversely, the treatment of hematopoietic progenitor cells with an NO scavenger increased cell proliferation (118).
The action of NO on the cell cycle elicits a series of molecular events. A well-characterized effect is the up-regulation of the cyclin-dependent kinase inhibitor p21Cip1/Waf1 (109) in a process mediated by MAPK. Another remarkable action of NO on cell cycle is the regulation of cyclin expression. Exogenous NO decreases the synthesis of cyclin D1 but not of cyclin E in breast cancer cells, and this down-regulation is accomplished without an increase in the degradation of Cdk4/6 regulatory protein (180).
It has been stated that NO directly induces cyt c release from the mitochondria through mitochondrial potential loss (190) or by tyrosine nitration of cyt c (99). High concentrations of NO and ONOO− were reported to cause DNA damage and lead to p53-mediated growth arrest and apoptosis in tumor cells (7). NFκB plays a protective role against apoptosis through the up-regulation of genes encoding anti-apoptotic proteins (38). NO inhibits NFκB activation by inducing the expression of the NFκB inhibitor IκBα and by stabilization of the NFκB/IκBα complex (179). In contrast to NO, oxidative stress activates IκB kinase (IKK), which leads to the phosphorylation of IκBα and activation of NFκB. The activation of IKK and phosphorylation of IκBα is blocked by antioxidants and NO (38). Inappropriate activation of NFκB has been linked to inflammatory events associated with autoimmune arthritis, asthma, septic shock, lung fibrosis, glomerulonephritis, atherosclerosis, and acquired immunodeficiency syndrome. In contrast, complete and persistent inhibition of NFκB has been linked to apoptosis, inappropriate immune cell development, and delayed cell growth (12).
Although NO promotes apoptosis in some cells, it also displays antiapoptotic properties in other cell types. It has been claimed that the anti-apoptotic mechanism involves the gene transcription of protective proteins, such as heat shock proteins, hemeoxygenase and cyclooxygenase-2, and the direct inhibition of the apoptotic activators of proteases of the caspase family by S-nitrosylation of the cysteine thiol group in the catalytic site in a cell specific way (48).
V. Kinases, Mitochondria, and Cell Cycle
Cell cycle stages include a long growth phase (G1), a DNA replicating phase (S), a short growth phase (G2), and cell division (mitosis, M). Transition from one phase of the cycle to the next is coordinated by specific cyclins and the sequential activation and inactivation of cyclin-dependent protein kinases (39, 95, 60).
A. The MAPK cascade
ERK1/2 (also known as p42/p44MAPK, respectively) belongs to the family of MAPKs, which include ERK5, the JNK1/2/3, and the p38 MAP kinases (α, β, γ, δ). These kinases are activated through a sequential phosphorylation cascade that amplifies and transduces signals from the cell membrane to the nucleus and, as recently described, to the mitochondria (79, 10). On receptor activation, membrane-bound GTP-loaded RAS recruit RAF kinases (A, B or C) and becomes activated. Raf phosphorylates two serine residues on the kinase MEK1/2, which, in turn, phosphorylates ERK1/2 on threonine and tyrosine residues. As a result, active ERK1/2 regulates many cytoplasmatic, nuclear, and mitochondrial substrates that perform key biological functions. Depending on the cell type, duration, and magnitude of the stimulus and its subcellular localization, ERK activation controls a wide range of cell responses, such as proliferation, migration, differentiation, and death (175, 160).
Mitogens induce a biphasic activation of ERK1/2, with a prompt burst of kinase activity peaking at 5–10 min followed by a second wave of lower but sustained activity that persists throughout the G1 phase for up to 6 h (115). Nuclear translocation of ERK1/2 occurs within 15 min of activation, persists during the entire G1 phase, and can be reversed on removing the mitogenic stimulus. ERK1/2 activation should be sustained until late G1 for successful S phase entry and ERK1/2 translocation to the nucleus is essential for G1 to S phase progression (238). On translocation to the nucleus, activated ERK1/2 phosphorylates the factors Elk-1, Sap-1a, and TIF-IA (42, 245). The ERK1/2 signaling pathway promotes cell survival by a dual mechanism comprising the posttranslational modification and inactivation of cell death machinery components, and the increased transcription of pro-survival genes (8, 125). ERK1/2 can influence the FOXO transcription factors that activate multiple target genes involved in tumor suppression including BIM and FASL for inducing apoptosis (23) and p27kip1 and cyclin D for cell cycle regulation (63). FOXO3a expression is associated with suppression of tumor progression, and inhibiting its expression promotes cell transformation, tumor progression, and angiogenesis (87). Recently, evidences have indicated that FOXO3 is placed in mitochondria where it interacts with Sirtuin3 (SirT3). SirT3 is a member of the class III histone deacetylases named sirtuins. The sirtuins (SirT 1–7) are a conserved family of proteins possessing NAD+-dependent deacetylase activity, distinct from class I and II histone deacetylases. SirT3 is the major mitochondrial deacetylase. SirT3 activates FOXO3 by deacetylation and increases its effects on the suppression of proliferation (110). ERK activity can also mediate antiproliferative events, such as apoptosis, autophagy, and senescence in vitro and in vivo. A common hallmark of this response is the sustained activation of ERK, which contrasts with the transient nature of ERK stimulation characteristic of cases in which ERK regulates progression of the cell cycle (29).
It has been recently shown that on proliferative stimulus, hERK1 translocates to mitochondria in HeLa cells where it associates with transport proteins such as VDAC1, as well as with proteins related to signaling, metabolism in histones H2A and H4 providing a new perspective for assessing ERK function in the regulation of cell signaling and trafficking (80). Several studies have indicated that ERK can modulate mitochondrial functions, predominantly those associated with cell death. For instance, ERK signaling appears to promote mitochondrial ATP synthase function in glucose-deprived astrocytes (242), to maintain mitochondrial membrane potential and prevent cyt c release (132), and to inactivate BAD (113). Mitochondrial fractions of normal rat brain homogenates show 10-fold lower levels of ERK1/2 than those observed in crude homogenates (5). However, the presence of a mitochondrial pool of ERK1/2 in normal as well as stressed tissues supports a potential physiological role for ERK in mitochondrial regulation. Our group has previously demonstrated the presence of ERK1/2, p38, and JNK1/2 within the mitochondrion (79). ERK1/2 translocation to brain mitochondria triggers a developmental pattern that is maximal between E19-P2 stages and afterward declines at P3, just before maximal translocation to nucleus, and up to adulthood. These results suggest that developmental mitochondrial activation of ERK1/2 cascade contributes to its nuclear translocation effects, and further enlightening mitochondrial energetic and redox status to the proliferating/differentiating nuclear pathways. Interestingly, both ultrastructural and biochemical subfractionation studies showed ERK localized within the mitochondrion in association with the outer membrane/IMS fraction (5). Consequently, it is clear that ERK is positioned in an ideal location for modulating mitochondrial death mediators and respiratory or metabolic processes.
The effects of NO and H2O2 outcome in the modulation of MAPKs and cyclin D1 are noteworthy. ERK stimulates cell proliferation and induction of active cyclin D1 by numerous mechanisms including the enhancement of AP-1 activity. It has been stated that NO induces a gradual elevation of intracellular [Ca2+] that leads to activation of ERK and enhances cell division. The functional blockade of Ca2+ and the inhibition of calmodulin prevent ERK activation and antagonize the mitogenic effect of NO (152).
Many studies point out a role for p38 MAPK signaling in regulating cell death events, including translocation of BAX from cytosolic to mitochondrial compartments (173), caspase-independent potassium efflux (17), and transcriptional regulation of TR3, a steroid receptor-like protein that translocates from the nucleus to the mitochondria to initiate the intrinsic pathway of apoptosis (97). p38 MAPK translationally downregulates cyclin D1. This effect is due to phosphorylation of cyclin D1 at Thr286, which leads to the ubiquitination of the protein. Similarly, NO activates p38 and suppresses proliferation through the activation of JAK2- signal transducer and activator of transcription protein 5 and cyclin D1/cdk4 (103); also, mitochondrial NO activates p38 and promotes cell cycle arrest in normal development and hypothyroidism (33, 72, 69).
JNKs play a critical role in death receptor-initiated extrinsec as well as mitochondrial intrinsic apoptotic pathways. JNKs activate apoptotic signaling by the upregulation of pro-apoptotic genes through the transactivation of specific transcription factors or by directly modulating the activities of mitochondrial pro- and anti-apoptotic proteins through distinct phosphorylation events. This is achieved not only through the activation of intermediates such as BAX (241), but also by many subcellular fractionation studies showing localization of activated JNK to the mitochondria. In addition, recent detection of mitochondrially targeted scaffold proteins provides compelling evidence for the biological relevance of mitochondrial JNK. For example, Sab (SH3BP5), which is a JNK-binding protein, that co- localizes with mitochondria (236) may serve a function analogous to that of certain A-kinase-anchor-proteins (AKAPs) in the localization of kinase activity to the mitochondria. The effects of JNK on the mitochondria often entail stimulation of apoptosis. Treatment of isolated rat brain mitochondria with active JNK causes the inhibition of anti-apoptotic BCL-2 and Bcl-xL, thus promoting the release of cyt c and a decrease in mitochondrial membrane potential (203).
Kamata et al. (116) showed that TNFα-induced ROS cause oxidation and inhibition of JNK-inactivating phosphatases by converting their catalytic cysteine to sulfenic acid. This results in sustained JNK activation, which is required for cyt c release and caspase 3 cleavage, as well as necrotic cell death.
B. Akt/protein kinase B
The serine/threonine kinase Akt plays a major role in cell proliferation, survival, adhesion, migration, metabolism, and tumorigenesis. The critical effects of Akt activation are determined by the phosphorylation of its downstream effectors located in the cytoplasm, nucleus, and, recently described, mitochondria (16, 9). Mammals have three closely related PKB genes, encoding the isoforms Akt1/PKBα, Akt2/PKBβ, and Akt3/PKBγ. Although the Akt isoforms are ubiquitously expressed, evidence suggests that the relative isoform expression levels differ between tissues. Akt1 is the mainly expressed isoform in most tissues, whereas Akt2 is highly enriched in insulin target tissues (36). Akt1 deficient mice show normal glucose tolerance and insulin-stimulated glucose clearance from blood but display severe growth retardation (46). It has also been demonstrated that cells derived from Akt1 deficient mouse embryos are also more susceptible to pro-apoptotic stimuli (43).
Akt kinases are typically activated by engagement of receptor tyrosine kinases by peptide growth factors and cytokines, as well as oxidative stress and heat shock. Akt activation relies on PtdIns-3,4,5-P3, and, to a lesser extent, on PtdIns-3,4-P2, which are products of PI3K (74). The interaction of PtdIns-3,4,5-P3 with the Pleckstrin homology domain of Akt1 favors the binding with their upstream activators, and it undergoes phosphorylation at two residues, one in the activation loop (Thr308) and the other in the C-terminal tail (Ser473). Phosphorylation at Ser473 appears to precede and facilitate phosphorylation at Thr308 (199, 10). Akt1 is phosphorylated at Ser473 by mTORC2 (107), whereas PDK1 accounts for the phosphorylation at Thr308 (36). Once activated, Akt1 not only phosphorylates an ever-increasing list of substrates in the cytosol but also translocates to the nucleus and mitochondria (16, 10).
Akt traffic to mitochondria seems to be important for the second phophorylation at Thr308. The oxidation of Cys310 that is adjacent to Thr308 to sulfinic acid favors interactions between constitutive mitochondrial PDK1 and pAktSer476 and likewise, a transient phosphoryl-thiol compound (10).
Inhibition of apoptosis by Akt1 can be mediated by multiple mechanisms, some of them tissue specific. Akt inactivates the proapoptotic factor BAD (at Ser136), procaspase-9, and the transcription factor family Forkhead that induces the expression of FASL, BIM, and PUMA (Fig. 9). Forkhead phosphorylation leads to its cytoplasmatic localization and binding to 14-3-3 proteins, thus rendering it unable to mediate nuclear transcription. Further, Akt activates the transcription factor, cAMP response element-binding protein as well as IKK, a positive regulator of NF-κB, both of which are regulators of gene expression with anti-apoptotic activity (185). Regulation of NF-κB activity occurs mainly by modulation of its associated inhibitory molecule IκB. When IκB is phosphorylated by the kinase complex IKKα and -β, it undergoes ubiquitination and degradation, thus releasing the active transcription factor. IKKα can be activated by multiple kinases, such as Akt. As a result, NF-κB positively regulates the expression of Bcl-2, Bcl-xL, and other antiapoptotic proteins as though IAP.
FIG. 9.

Akt phosphorylates and inactivates the FOXO family of transcription factors. In lower metazoans, FOXO proteins promote the expression of pro-apoptotic genes, such as BIM and FAS. In mammalian cells, FOXO also promotes expression of p27KIP1 and p21CIP1 to inhibit cell cycle entry. The CDK inhibitor p27KIP1 and p21CIP1 proteins can also be phosphorylated by Akt, leading to their accumulation in the cytoplasm. Akt increases the translation of cyclin D mRNA through phosphorylation of GSK3, and Akt phosphorylation of Myt1 drives the cell cycle to M phase. A prominent function of Akt on cell survival is mediated by its direct phosphorylation and negative regulation on caspases 3 and 9 as well as BAD. Phosphorylation of MDM2 by Akt leads to its nuclear translocation and, thus, promoting p53 degradation and finally leading to a reduction in transcription of p21CIP1 mRNA. CDK, cyclin-dependent kinase; FOXO, forkhead O-box; GSK3, glycogen synthase kinase-3; BIM, Bcl-2-like protein 11; MDM2, mouse double minute 2; Myt1, myelin transcription factor; UB, ubiquitin; FAS, TNF receptor superfamily member 6.
Once inserted in the mitochondrial membrane, pro-apoptotic factor BAX is a key regulator of mitochondrial permeability that mediates apoptosis. BAX is phosphorylated by Akt in its inhibitory site (Ser184) close to the C-terminus domain that contributes to cell death suppression mediated by BAX in neutrophils (81). BAX forms a dimer with Bcl-2 and prevents death repressor activity, thus determining cell sensitivity to apoptosis in response to specific stimuli. On the other hand, Akt activation induces Bcl-2 expression in BAF/3 cells (3). Later studies have demonstrated that neuronal apoptosis induced by NO is accompanied by a Bcl-2 decrease and a BAX synthesis increase. In addition, Akt1 activation can cause a protective effect in hippocampal primary neurons through association with the scaffold protein JIP1 (JNK interacting protein 1) that acts, thus avoiding recruiting and assembly of JNK and delaying apoptosis start in response to toxic agents (121).
Akt inhibits apoptosis not only in culture cells but also in in vivo models. Homozygous PTEN mutant mice develop an abnormal patterning and over-growth of the cephalic and caudal regions. Analysis of PTEN mutant embryos reveals regions of increased proliferation. In addition, immortalized mouse embryonic fibroblast cell lines from PTEN homozygous mutant embryos show decreased sensitivity to various apoptotic stimuli, including UV irradiation, heat shock, osmotic stress, and tumor necrosis factor α stimulation (211). Apoptotic prevention is also observed in freshly isolated MyrAkt thymocytes, which express high levels of cyclins D2 and E (141).
Cell cycle control by Akt resides in the subcellular localization regulation of FOXO members, cdk inhibitors (p21CIP1 and p27KIP1), and β-catenin as well as the p53 intracellular concentration (via Mdm2 stabilization) (Fig. 8). Glycogen synthase kinase 3 (GSK3) inhibition by Akt prevents the phosphorylation of β-catenin, which impedes its degradation; hence, it is translocated to the nucleus. Once in the nucleus, β-catenin interacts with different transcription factors, such as human T-cell factor-1 (TCF)/mouse lymphoid enhancer factor (LEF-1) family, to induce the expression of several genes, such as Cyclin D1, which induces cell cycle progression via regulation of RB hyperphosphorylation and inactivation. In a similar way, decreased Cyclin D1 phosphorylation by GSK3 promotes the stabilization of this protein (62). Akt phosphorylates p21 and inhibits its antiproliferative effects by retaining it within the cytoplasm (246). A similar mechanism has been described for p27 (135). After stimulation with growth factors, Mdm2 is phosphorylated by Akt and enters the nucleus, wherein it induces a decrease in both p53 levels and its transactivation. In the absence of the tumor suppressor p19/p14ARF, the complex Mdm2-p53 leaves the nucleus and enters the cytoplasm, where p53 becomes degraded through an ubiquitin/proteasome-mediated process (205).
G1/S transition by Akt takes place through the blockage of cell cycle inhibitors p21CIP and p27KIP as a result of phosphorylation, which mediates their cytoplasmic localization and binding to 14-3-3 proteins (55). Moreover, p21CIP and p27KIP levels decrease through phosphorylation of FOXO members, which once in the cytoplasm become unable to promote the synthesis of the inhibitors (150) (Fig. 8). Further, although genetic ablation of one Cdkn1b allele accelerated the development of neoplastic lesions in thyroid glands from PTEN−/− mice, it also abolished the gender differences in survival and reduced the difference in neoplastic lesion development rate, underlining a key role of p27 in mediating estrogen action in the thyroid follicular cells (11).
Translation inhibition is achieved when 4E-BP1 protein binds to initiation factor eIF-4E. Akt phosphorylates 4E-BP1, thus preventing the binding to initiation factor removing the inhibitory effect on protein synthesis (85). Protein synthesis stimulation by Akt can favor the expression of a vast gene number related to growth such as c-Myc, c-Fos and cyclins G1, D1, and D3.
C. Protein kinase C
The protein kinase C (PKC) family of Ser/Thr kinases comprises 10 members in the AGC kinase branch of the kinome (163). PKCs have traditionally been recognized as lipid-sensitive enzymes that are activated by growth factor receptors that stimulate phospholipase C, which, in turn, produces diacylglycerol (DAG) to activate PKC. Recent studies have shown that PKCs can also be controlled through phosphorylations on both serine/threonine and tyrosine residues that influence the stability, protease/phosphatase resistance, protein-protein interactions, subcellular localization, and substrate specificity of the enzyme. What is more, they can be activated by less conventional lipid cofactors (such as ceramide or arachidonic acid) or through lipid-independent mechanisms (such as oxidative modifications or tyrosine nitration) that allow for PKC signaling at all other sites within the cell (212).
Immunoelectron microscopy studies have shown that α and β isoforms of PKC are localized in the mitochondrial inner membrane of carp retinal Müller cells (67). In agreement, considerable emerging evidence indicates that PKC isoforms play a direct role in regulating mitochondrial function. PKCδ activation induces and enhances the apoptotic events that occur during ischemia–reperfusion and malignant progression of cancer cells, whereas activation of PKCɛ inhibits and reduces these events (50). Renal proximal tubular cells react to oxidative stress by trafficking activated PKCɛ to the mitochondria, which inhibits the electron transport chain, ATP production, and Na+ transport, probably in part through direct phosphorylation of Na+-K+-ATPase (166). Overexpression of-wild type PKCɛ in MCF-7 cells inhibits activation of caspases-8 and -9 and decreases tumor necrosis factor-induced mitochondrial depolarization, which normally leads to the release of mitochondrial cyt c and cell death triggered by TNF-related apoptosis-inducing ligand (14). Further studies emphasized that the Bcl-2 level increases, whereas that of the apoptotic protein BID decreases by PKCɛ at both the protein and mRNA levels. Additionally, PKCɛ depletion or overexpression of a dominant negative PKCɛ is associated with a decrease in Bcl-2 protein levels. These findings present clear evidence that PKCɛ mediates its antiapoptotic effect via the mitochondria by regulating both the activities of proapoptotic and antiapoptotic proteins as well as the translocation of these proteins to the organelle (139).
A connection between PKC and MAPK pathway has been demonstrated in mammary epithelial cells. PKC zeta overexpression markedly altered the adhesive, spreading, and migratory abilities of immortalized mammary epithelial cells (222). Taken together, these results suggest that in these cells PKC zeta modulates several of the critical events involved in tumor development and dissemination through the activation of ERK pathway.
D. Protein kinase A
Protein kinase A (PKA) is considered the most important effector of cAMP action (174, 210). Eukaryotic cells express multiple forms of PKA regulatory and catalytic subunits, which assemble as different holoenzyme isoforms. Although many other AGC kinases possess canonical protein–protein or protein–lipid interaction modules regulating their targeting, these are absent in PKA, with the exception of the dimerization and docking (D/D) domain. This explains and underlines the need for AKAPs to control the localization of PKA (210). By these means, PKA is concentrated in particulate membranes and cellular organelles through interactions with AKAPs. Different AKAPs bind specific cellular components, thereby placing PKA to particular cellular localizations and enhancing the efficiency of signal-transducing pathways by concentrating PKA near sites of cAMP generation or at PKA targets (66). Phosphorylation of nuclear and cytoplasmic substrates mediated by PKA is critical for multiple cell functions, including metabolism, differentiation, synaptic transmission, ion channel activity, growth, and development (66, 58).
Similar to other kinases, it has been demonstrated that PKA plays a critical role in mammalian mitochondrial physiology, thus suggesting that activated PKA can efficiently phosphorylate mitochondrial substrates and, therefore, alter their function. Several mitochondrial proteins are substrates of PKA, including the nuclear-encoded 18-kDa subunit of complex I (NDUFS4), BAD and steroidogenic acute regulatory (StAR) protein. Phosphorylation of NDUFS4 by PKA enhances the activity of mitochondrial respiratory complexes (57). This would increase ATP synthesis in response to conditions that induce cAMP signal transduction.
Activation of PKA by extracellular ligands or cAMP analogs leads to the inhibition of apoptosis since PKA phosphorylates and inactivates BAD. cAMP signaling to mitochondria depends essentially on the localization of PKA to the OMM. Delocalization of PKA from mitochondria induced by dominant negative AKAP121 mutants reduces phosphorylation of BAD and causes apoptosis (92). On the contrary, overexpression of AKAP121 increases BAD phosphorylation and protects cells against proapoptotic stimuli (2).
Transport of cholesterol from cytosol to mitochondria is a highly regulated process that is stimulated by PKA-dependent phosphorylation and activation of StAR. Mitochondrial PKA along with constitutive MEK1/2 drives ERK1/2 phosphorylation in this organelle. As a consequence, active ERK1/2 interacts with StAR protein, leading to the phosphorylation at Ser232 in the presence of cholesterol, thus regulating its transportation (181).
AKAP121, which tethers PKAII to the OMM, includes a K homology (KH) RNA-binding motif. This domain binds the 3′ untranslated regions (3′UTRs) of transcripts encoding the Fo-f subunit of mitochondrial ATP synthase and MnSOD. Binding requires a structural motif in the 3′UTR that is stimulated by PKA phosphorylation (86). AKAP121 expressed in HeLa cells promotes the translocation of MnSOD mRNA from cytosol to mitochondria and an increase in mitochondrial MnSOD content. Therefore, AKAP121 can facilitate import of mitochondrial proteins in response to cAMP stimulation (86).
It has also been demonstrated that Rab32, a member of the Ras superfamily of small-molecular-weight G-proteins, interacts directly with type II regulatory subunit of PKA functioning as an AKAP (6). Rab32 and a proportion of the cellular PKA pool are associated with mitochondria. Transient transfection of a GTP binding-deficient mutant of Rab32 promotes aberrant accumulation of elongated mitochondria at the microtubule-organizing center. This implicates Rab32 as a key factor in synchronization of mitochondrial fission. Thus, Rab32 is a dual function protein that participates in both mitochondrial anchoring of PKA and mitochondrial dynamics (6).
Brennan et al. showed that the R1 regulatory subunits of PKA form interprotein disulphide dimers during cardiac oxidative stress (21). In cardiac tissues, this redox change induces a subcellular translocation and kinase activation, thus resulting in phosphorylation of multiple PKA substrates, which increases the amplitude of myocyte contraction.
VI. Mitochondrial Biogenesis
A. Transcriptional control of mitochondrial biogenesis
Normal mitochondrial number, structure, and function are supported by mitochondrial biogenesis, a cellular program that adjusts energy production by synthesis of new organelles and organelle components, and mediates inter-organelle interactions. Mitochondria cannot be generated de novo; instead, they proliferate by growth and division of pre-existing organelles (232). Mitochondria divide during mitosis, providing daughter cells with a normal complement of mitochondria. There are, however, instances in which mitochondrial divisions are not tied to cell cycle. For example, muscle mitochondria will proliferate during myogenesis, but also after exercise (164). Mitochondrial biogenesis can also be stimulated by other pathways such as ROS (189), NO (165), calcium fluxes (61), and hypoxia (149).
Since the mitochondrial genome encodes only a fraction of the mitochondrial proteins, mitochondrial biogenesis requires communication between mitochondria and the nucleus (119). Further, the large network of enzymes involved in generating bioenergy by the tricarboxylic acid cycle and fatty acid oxidation pathway are encoded by nuclear genes. In this way, mitochondrial biogenesis involves the import of nucleus-encoded proteins from the cytosol, the incorporation of mitochondrion synthesized and imported membrane lipids, the amplification of the mitochondrial genome, and the translation of mitochondrion-encoded proteins.
The most prevalent transcription factors activating promoters of mitochondrial genes are the nuclear respiratory factor 1 and 2 (NRF-1 and NRF-2), and the estrogen-related receptor α (ERRα) that work in concert with transcriptional activators of the peroxisome proliferator-activated receptor-γ-coactivator-1 (PGC-1) family: PGC-1α, PGC-1β, and PRC (PGC-1-related coactivator) (201). This family of coactivators has been identified as a crucial factor linking external stimuli to mitochondrial biogenesis (132) (Fig. 10). The differential expression of PGC-1 family coactivators is controlled by an array of environmental signals including temperature, energy deprivation, and availability of nutrients and growth factors (202). The PGC-1 family of coactivators regulates several metabolic pathways such as cellular respiration, thermogenesis, and hepatic glucose metabolism, thus demonstrating the ability of PGC-1α to alter the metabolic state of the cell in response to changes in the cellular or extracellular environment (61, 189).
FIG. 10.

Regulation of mitochondrial biogenesis machinery by calcium and kinases. Extracellular stimuli increase intracellular calcium levels resulting in kinases activation by transcriptional pathways. Some kinases activate PGC-1α coactivator by transcriptional activation of its gene or by direct phosphorylation, thus resulting in the activation of nuclear respiratory factors NRF-1 and NRF-2 that mediate the transcription of several mitochondrial genes that will promote mitochondrial biogenesis. CaMK, calcium/calmodulin-dependent protein kinase; CREB, cAMP response element-binding protein; ER, endoplasmic reticulum; NRF, nuclear respiratory factor; PGC-1, peroxisome proliferator-activated receptor-γ-coactivator-1; TFAM, mitochondrial transcription factor A.
PGC-1α was originally cloned as a cold-inducible coactivator of peroxisome proliferator-activated receptor-γ (PPAR-γ) in brown adipose tissue (187), but it has emerged as a potent coactivator of a plethora of transcription factors impacting on whole-body energy expenditure (54). In order to exert such a wide array of functions, PGC-1α directly coactivates multiple transcription factors, including nuclear receptors—such as the PPARs, or the thyroid hormone receptor, glucocorticoid receptors, estrogen receptors, and ERRs among others—and nonnuclear receptor transcription factors such as myocyte enhancer factor-2 and the family of FOXO transcription factors (30). PGC-1α is highly expressed in tissues with high metabolic demands, such as heart, skeletal muscle, and kidney. A variety of signaling mechanisms have been proposed to regulate the expression of PGC-1α and mitochondrial biogenesis, including NO-soluble guanylate cyclase (166), β-adrenergic/cAMP, calcineurin A, calcium/calmodulin-dependent protein kinase, and AMPK (189) (Fig. 10). The p38 MAPK is also thought to regulate PGC-1α through phosphorylation of Thr262, Ser265, and Thr298 within the repressor domain of PGC-1α, thus stabilizing the protein and increasing its ability to function as a coactivator. Interestingly, there is a strong overlap in the genes transcriptionally regulated by AMPK and those by PGC-1α, thus suggesting that PGC-1α might be an important mediator of AMPK-induced gene expression. Supporting this hypothesis, AMPK activation leads to increased PGC-1α expression, and AMPK requires PGC-1α activity to modulate the expression of several key players in mitochondrial and glucose metabolism (30). Ectopic expression of PGC-1α was later shown to stimulate the biogenesis of mitochondria by increasing the expression of NRF-1 and 2 and enhancing the transcriptional activity of NRF-1 on the promoter for TFAM (186).
NRF-1 is a 68 kDa polypeptide with the presence of a C-terminal transcriptional activation domain. Serine phosphorylation of the N-terminal domain enhances both its DNA-binding and trans-activation functions (106). NRF-1 has been linked to the transcriptional control of genes involved in mitochondrial function and biogenesis. NRF-1 regulates the expression of many genes required for mitochondrial respiratory function, including the vast majority of nuclear genes that encode subunits of the five respiratory complexes. NRF-1 also mediates mtDNA transcription and replication through TFAM. NRF-1 occupancy of the TFAM promoter increases under pro-oxidant conditions, and the control of TFAM expression by NRF-1 is blocked by inhibition of NRF-1 phosphorylation by Akt (202).
Characterization of COX genes led to the identification of a second regulatory factor designated as NRF-2 (201). Notably, most of the known COX subunit genes as well as individual subunit genes from complexes I, II, and V contain NRF-2 sites. NRF-2 is a regulator of both cytosolic and mitochondrial antioxidant programs. NRF-2 binds to the antioxidant response element to regulate the transcription of antioxidant enzymes including heme oxygenase-1, NAD(P)H quinone oxidoreductase-1, glutathione-S-transferases, the glutathione synthetic enzyme glutamate-cysteine ligase, GPx, glutathione reductase, and the Trx and Prx families (51) (Fig. 11).
FIG. 11.

NO, oxidative stress, antioxidant enzymes, and mitochondrial biogenesis. Increased mitochondrial NO mostly by increased mtNOS activity derives in increased production of 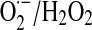 by mitochondrial respiratory complex inhibition. NO is an activator of PGC-1α and subsequently of mitochondrial biogenesis, and increased production of ROS activates the nuclear transcription factor NRF2 that regulates antioxidant enzymes genes transcription and mitochondrial biogenesis. SOD, superoxide dismutase; Prx, peroxiredoxin; HO-1, heme oxigenase 1; Trx, thioredoxin; GPx, glutathione peroxidase.
by mitochondrial respiratory complex inhibition. NO is an activator of PGC-1α and subsequently of mitochondrial biogenesis, and increased production of ROS activates the nuclear transcription factor NRF2 that regulates antioxidant enzymes genes transcription and mitochondrial biogenesis. SOD, superoxide dismutase; Prx, peroxiredoxin; HO-1, heme oxigenase 1; Trx, thioredoxin; GPx, glutathione peroxidase.
B. Mitochondrial biogenesis, NO, and ROS
Mitochondria are crucial to cellular health, so a high level of mitochondrial biogenesis is considered indicative of intact metabolic and bioenergetic functionality and cellular well-being (124). Treatment of various cells with small-to-moderate levels of NO increases their mtDNA content, and, consequently, mitochondrial biogenesis (165). This effect is sensitive to removal of NO by the NO scavenger, oxyhemoglobin, and is dependent on the induction of PGC-1α by cyclic guanosine 3′,5′-monophosphate (cGMP)-dependent signal transduction pathway(s).
Increased mitochondrial biogenesis is a part of the cellular response to oxidative stress (Fig. 11). Mitochondrial biogenesis attenuates oxidative stress by increasing mitochondrial capacity to metabolize reducing equivalents. Cells undergoing mitochondrial biogenesis consume less oxygen, maintain mitochondrial membrane potential, and produce fewer ROS, which together decrease cellular damage and increase survival (189). Neurons transfected with PGC-1α are protected against H2O2-induced oxidative stress (213).
PGC-1α stimulates mSirT3 gene expression, and SirT3 mediates the effects of PGC-1α on decreasing intracellular ROS levels and increasing mitochondrial biogenesis, in response to rising ROS in muscle cells (127). Since mitochondria are a major source for ROS, mtDNA is particularly vulnerable to ROS-induced mutations, and lesions and ROS-related damage have been implicated in cancer, aging, diabetes, and neurodegenerative diseases. The interaction between mitochondrial biogenesis and cell cycle arrest could be the strategy used for sensing and restoring integrity to damaged mtDNA and for protecting cells from mitochondrial metabolic disorders.
VII. Mitochondrial Dynamics
In recent years, it has been realized that mitochondria are not small round organelles, as seen after isolation from various tissues, but constitute in young and healthy cells a network of elongated filamentous structures. They are in constant movement and change shape and size through dynamic processes of fission and fusion, conducted by a number of dynamin-like GTPases (88). Fusion and fission events facilitate inner and outer membrane fusion and the exchange of organelle contents such as solutes, proteins, and mitochondrial DNA (136). The processes of fusion and fission are necessary for many cell functions including mitosis, fuel sensing, ATP production, autophagy, and apoptosis (106) (Fig. 12).
FIG. 12.

Mitochondrial dynamics and biological functions. Mitochondrial biogenesis starts the mitochondrial cycle by division of pre-existing organelles, and mitophagy ends it by degradation of impaired mitochondria. In between, mitochondria undergo several cycles of fission and fusion that generate multiple heterogeneous mitochondria or interconnected mitochondrial networks depending on the physiological conditions. Fused mitochondrial networks are important for the dissipation of metabolic energy and for complementation of mtDNA gene products to counteract the decline of respiratory functions in aging in heteroplasmic cells.
Mitochondrial functions are coordinated to their dynamic behavior. Mitochondrial morphology is tightly regulated by balanced fusion and fission events. A shift of this balance causes excessive fragmentation or elongation of mitochondria. These processes require several factors, many of them conserved during evolution. Disturbances in mitochondrial dynamics in humans cause severe neurodegenerative diseases, indicating that neurons appear to be strongly susceptible to mitochondrial dysfunction (157). The most prominent examples of these disorders are dominant optic atrophy (DOA) (218), peripheral axonal neuropathies (known as Charcot- Marie Tooth disease, [Züchner (250)]), and also hearing loss (deafness-dystonia-optic neuropathy) (156), thus demonstrating the importance of these morphology-modulating mechanisms for normal cell physiology.
Mitochondrial mobility is thought to be especially important in neuronal cells to deliver mitochondria to the site of synapses where they not only provide ATP for sustained synaptic transmission but also probably help buffer calcium changes.
Recent reports demonstrated that cells lacking mitochondrial fusion are respiration defective, and that mitochondrial dynamics participates in regulating mitochondrial electron transport activity, thus indicating the presence of an intricate form-function relationship (104).
A. Mitochondrial fusion
Mitochondrial fusion is a mechanistically complex event that involves the fusion of two lipid membranes (the inner mitochondrial membrane [IMM] and the OMM). This process is dependent on both mitochondrial membrane potential and GTP. It is a two-step process, where the outer and the IMM fuse in separable steps. The pro-fusion protein located in the IMM (optic atrophy gene 1 [OPA1]) and the pro-fusion proteins located in the OMM (mitofusins [Mfns]) are detected in the same complex, thereby suggesting that the two steps are co-regulated or coordinated. The disruption of one of them is sufficient to stop the whole mitochondrial fusion process (247).
It was previously demonstrated by cell-free fusion assays that Mfn 1 and Mfn2 mediate OMM fusion, whereas OPA1 mediates IMM fusion (151). Fusion events can be transient (outer membrane only) or complete (inner and outer membrane), probably having alternate stimuli and purpose; however both play an important role in mitochondrial function (106).
OPA1 was discovered as the gene whose mutations were responsible for the autosomal dominant optic atrophy (ADOA) (111). OPA1 is encoded by a single gene with eight transcript variants resulting from alternative splicing. These variants are differentially proteolyzed into long and short forms yielding different OPA1 isoforms in the mitochondria that exert various activities in promoting fusion and shaping mitochondrial cristae. Indeed, the control of mitochondrial cristae morphology seems to be a specific feature of OPA1 activity, because Mfn1 and Mfn2 disruption does not induce aberrant cristae structures (214). Proteolytic processing of OPA1 is regulated by four different proteases, and is considered a potential regulatory mechanism for the inactivation of fusion, thereby resulting in fragmentation and even apoptosis (157).
Mfns 1 and 2 integrate into the OMM with two transmembrane regions separated by 2–3 aminoacids facing the IMS orienting the GTPase N-terminal domain and the C-terminal coiled-coil regions toward the cytosol. The C-terminal coiled-coil region of Mfn1 and Mfn2 mediates tethering between mitochondria through homo- or heterotype complexes formed between adjacent mitochondria. Mfn2 presents a lower GTPase activity and a higher affinity for GTP, compared with Mfn1 (247). Mfn 1 and 2 knock-out mice die in gestation due a placental defect.
Mutations in Mfn2 gene cause the autosomal dominant neurodegenerative disease Charcot-Marie Tooth type 2A (250). A reduced expression of Mfn2 has been found in obese and diabetic subjects (248).
Loss of Mfn2 causes a specific alteration in the expression of subunits that participate in respiratory complexes which leads to reduced activity. Overexpression of Mfn2 in cultured cells causes the formation of a perinuclear tightly cluster mitochondrial mass, a marked enhancement of mitochondrial membrane potential, and increased glucose oxidation. Mfn2-induced mitochondrial cluster is an aggregation of small fragmented mitochondria (104). Small mitochondria in the cluster are functionally impaired and release cyt c to the cytosol, thus leading to apoptotic cell death. The identical phenotype induced by overexpression of wild-type and the GTPase-defective mutant of Mfn2 indicates that formation of mitochondrial clusters is not the result of mitochondrial fusion.
Proteins that participate in mitochondrial fusion or fission also play a regulatory role in mitochondrial metabolism. In this sense, Mfn2 ablation causes alterations in mitochondrial metabolism characterized by reduced mitochondrial membrane potential and cellular oxygen consumption, and depressed substrate oxidation. Under these conditions, the energetic needs of the cell are balanced by a higher rate of glucose uptake and glycolysis and a lower rate of glycogen synthesis. A cell with low Mfn 2 activity relies on the use of anaerobic glycolysis to generate energy (249).
De Brito and Scorrano found that Mfn2 tethers the endoplasmic reticulum (ER) to mitochondria (56). Crosstalk between the ER and mitochondria is a good example of local Ca2+ signaling. The second messenger InsP3 can induce the release of Ca2+ from the ER, which opens high conductance Ca2+-permeable ion channels, and Ca2+ is taken up by mitochondria through a Ca2+-selective uniporter channel in the IMM (172). In fibroblasts lacking Mfn2, the distance between ER and mitochondria increased and both organelles had altered shapes. The ER was interconnected and manifest as a reticular pattern in either wild-type or Mfn 1-deficient cells, whereas the organelle was swollen and aggregated with reduced luminal continuity in cells lacking Mfn2. Mfn2 is, therefore, needed to maintain an interconnected and tubular ER (56) and to stabilize interactions between mitochondria, and mitochondria-ER.
B. Mitochondrial fusion machinery and apoptosis
Mitochondrial fragmentation during apoptosis may result from insufficient fusion. Silencing of Mfn1 and Mfn2 results in mitochondrial fragmentation and increased sensitivity to apoptotic stimuli (215). Further, overexpression of Mfn1 or Mfn2, in addition to increasing mitochondrial connectivity, results in delayed BAX activation, cyt c release, and apoptotic death, thus suggesting a role for Mfns in cell death (214).
Mfns has also been shown to inhibit the ERK/MAPK signaling pathway (40). An Mfn2 mutant lacking the C-terminal coiled-coil domain, which is required for mitochondrial fusion activity, still inhibits ERK 1/2 activation, thus indicating that Mfn2 regulation of ERK 1/2 signaling is independent of its role in mitochondrial fusion. Overexpression of Mfn2 in heart muscle cells or vascular smooth muscle cells induces apoptosis through inhibition of RAS-PI3K-Akt signaling. Thus, Mfn2 could act in a pro-apoptotic manner (204).
Loss of OPA1 induces spontaneous apoptosis of cells, thus providing another important link between apoptosis and mitochondrial morphogenesis (167). OPA1 haplo-insufficiency is responsible for the most common form of ADOA, a neuropathy resulting from degeneration of the retinal ganglion cells and optic nerve atrophy. In-vitro studies showed that down-regulation of OPA1 in HeLa cells using specific small interfering RNA led to fragmentation of the mitochondrial network concomitant with the dissipation of the mitochondrial membrane potential and a drastic disorganization of the cristae. These events are followed by cyt c release and caspase-dependent apoptotic nuclear events. Conversely, overexpression of OPA1 prevents mitochondrial fission and protects cells from apoptotic death occurring by the intrinsic pathway, but not apoptosis induced by the extrinsic pathway circumventing the mitochondria (75). OPA1 protects against apoptosis by preventing cyt c release independently from mitochondrial fusion. OPA1 does not interfere with activation of the mitochondrial “gatekeepers” BAX and BAK, but it controls the shape of mitochondrial cristae, thereby keeping their junctions tight during apoptosis. Tightness of cristae junctions correlates with oligomerization of two forms of OPA1, a soluble, IMS and an integral inner membrane one (75). The pro-apoptotic Bcl-2 family member BID, which widens cristae junctions, also disrupts OPA1 oligomers (237).
C. Mitochondrial fission
The most relevant genes identified up-to-date that directly mediate mitochondrial fission are dynamin related protein 1 (Drp1)/dynamin-like gene and fission 1 homolog protein (hFis1). Drp1 is a large GTPase that mediates mitochondrial fission in mammalian cells (214). Most Drp1 is found soluble in the cytosol of cells where it shuttles onto and off mitochondria (230). Drp1 assembles into spirals at division sites around the OMM to drive the fission process. Mutations that block Drp1 GTPase activity yield dominant negative mutants that inhibit mitochondrial fission, thus resulting in excessively tubular mitochondrial networks due to ongoing fusion.
Drp1 appears to constrict mitochondria due to the assembly of the spiral or to subsequent changes in helical diameter. The recruitment of Drp1 to the mitochondria in mammals is still unclear (230). The outer membrane protein hFis1 has been proposed to fulfill this function but given the uniform distribution of hFis1 on the mitochondrial surface and the fact that its removal does not alter the recruitment of Drp1 to mitochondria, it is likely that other factors contribute to the assembly of Drp1 and its function. In mammalian cells, actin filaments and microtubules also appear to function in recruitment of Drp1 to mitochondria (59).
The association of Drp1 with mitochondria and its activity are regulated by phosphorylation, S-nitrosylation, ubiquitination, and sumoylation (37). During mitosis, Drp1 is activated by cyclin-dependent kinases (CDKs)/cyclin B phosphorylation of Ser618 (human), resulting in mitochondrial fragmentation. Conversely, Drp1 GTPase activity is inactivated by phosphorylation at Ser637 (human) mediated by PKA resulting in mitochondrial fusion. Thus, Drp1 can be positively or negatively regulated by kinases. The phosphatase calcineurin regulates mitochondrial morphology by de-phosphorylating Drp1 at Ser637 in response to Ca2+ signals (232). During physiological (i.e., agonist evoked) Ca2+ signaling, activation of PKA can prevail over calcineurin-mediated dephosphorylation of Drp1 (35). Moreover, compartmentalization of both Ca2+ and cAMP signals could play a role in the local regulation of mitochondrial shape. Conversely, long-lasting Ca2+ plateaus in the cytosol are linked to full activation of calcineurin and to generalized fragmentation that could account for the apoptotic mitochondrial fission.
S-nitrosylation is a redox-related modification of thiols (such as in cysteine residues) by NO, which transduces NO activity and affects a variety of proteins involved in a number of cellular processes (71). A recent study has suggested that NO produced in response to β-amyloid protein, a key mediator of Alzheimer disease, triggers mitochondrial fission and subsequent synaptic loss and neuronal damage, in part via S-nitrosylation of Drp1 at Cys644 (45). S-Nitrosylation of Drp1 induces formation of Drp1 dimers, which function as building blocks for tetramers and higher order structures of Drp1, and activates Drp1 GTPase activity (161).
Sumoylation by SUMO1 protects Drp1 from degradation and positively regulates its activity in mitochondrial fission (93). MARCH5, a ubiquitin ligase in the outer membrane, associates with and ubiquitylates Drp1, Fis1, and Mfns. Mitochondrial protein MTP18 and ganglioside-induced differentiation associated protein (GDAP1) also influence mitochondrial fission (219). MTP18 is a downstream effector of PI3K signaling, is localized in the IMS, and it has been proposed to regulate mitochondrial fission through recruitment of Drp1 to the OMM. Its absence leads to cyt c release from mitochondria.
GDAP1 is a mitochondrial outer membrane protein that is mutated in Charcot- MarieTooth disease type 4A (176). Overexpression of GDAP1 causes fragmentation of mitochondria with no effect on cell death, whereas down-regulation of GDAP1 or the expression of certain patient mutant forms, which are truncated and no longer localized to mitochondria, tends to elongate mitochondria.
D. Mitochondrial fission machinery and apoptosis
Mitochondrial fission is an early event during apoptosis, occurring within the same time as activation of the pro-apoptotic Bcl-2 family member BAX and permeabilization of the OMM that leads to the release of multiple IMS proteins, before caspase activity and membrane bubbling (230). The process occurs very close in time to cyt c release, either just before or simultaneously.
Excessive mitochondrial fission can occur in the absence of apoptosis when mitochondria are exposed to uncoupling agents that disrupt IMM electrochemical potential (214). Carbonyl cyanide p-trifluoromethoxy phenyl-hydrazone-induced mitochondrial fragmentation is reversible. Viral infection also can induce mitochondrial fragmentation that is not linked to apoptosis. However, apoptosis is invariably associated with mitochondrial fragmentation. Moreover, the excessive mitochondrial fission appears to be a requisite step in intrinsic apoptosis pathways, at least for the normal rate of cyt c release and caspase activation. On apoptotic stimulation, Drp1 is recruited to the OMM where it localizes with BAX and Mfn2 at fission sites. During apoptosis, Drp1 sumoylation increases, along with increased Drp1 association with the OMM and increased mitochondrial fission. Down-regulation of Drp1 expression by RNAi or overexpression of a dominant-negative mutant Drp1 K38A delays but does not block BAX recruitment and activation on the mitochondrial membranes and inhibits cyt c release from mitochondria (73). This implies that both BAX and Drp1 function upstream cyt c release in the apoptotic cascade of events with Drp1 potentially participating in efficient BAX activation. The overexpression of Drp1 K38E blocks remodeling of the mitochondrial cristae, an event that allows for the translocation of cyt c from the cristae stores to the IMS, from where it is released.
The link between the fusion machinery and BAX was recently highlighted by the discovery that BAX is required for the regulation of Mfn2 activity and lateral assembly into foci along the mitochondrial tubules (117).
E. Mitochondrial dynamics, NO and ROS
Oxidative stress elicited by NO and/or 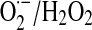 results in mitochondrial fission. The dynamic process is thought to function as a rescue mechanism for dysfunctional/damaged mitochondria by fusing with healthy ones or alternatively, when damage is excessive by isolating mitochondria for subsequent engulfment into autophagosomes (mitophagy), in order to rescue the cell (88). If this is not possible, then apoptosis or necrosis signal take over and execute cell death.
results in mitochondrial fission. The dynamic process is thought to function as a rescue mechanism for dysfunctional/damaged mitochondria by fusing with healthy ones or alternatively, when damage is excessive by isolating mitochondria for subsequent engulfment into autophagosomes (mitophagy), in order to rescue the cell (88). If this is not possible, then apoptosis or necrosis signal take over and execute cell death.
Since mitochondria are a major source of ROS, mtDNA is particularly vulnerable to ROS-induced mutations and lesions. Gradual and progressive accumulation of mtDNA mutations leads to a loss of functional respiratory chain complexes, resulting in a decline of bioenergetic capacity. When two solitary mitochondria carrying mutations in two different genes fuse, each one will contribute an intact allele and synthesize a functional gene product, thus restoring respiratory activity (232). On the other hand, mitophagy constitutes a mechanism to remove dysfunctional mitochondria from the cell and thereby preventing proliferation of mutated mtDNA. Remarkably, inhibition of fission decreases mitophagy and results in a decline of respiratory capacity, whereas arrest of autophagy leads to the accumulation of mitochondria with low membrane potential and low OPA1, that are less likely to be engaged in subsequent fusion events (232).
Barsoum et al., showed that nitrosative stress triggers persistent mitochondrial fragmentation prior to neuronal cell death in isolated neurons in vitro and in an experimental mouse model of ischemic stroke injury in vivo (13). NO-induced fragmentation results in ultrastructural damage of mitochondria, increased ROS, and reduced ATP. The decline in ATP synthesis and subsequent mitochondrial fragmentation is due to nitrosative stress-dependent respiratory chain complexes (I and IV) inhibition. Thus, persistent mitochondrial fission is accompanied by bioenergetic compromise. Free radical scavengers such as reduced glutathione prevent NO-mediated mitochondrial fragmentation and cell death. In the past few years, multiple findings have suggested that disruptions of mitochondrial function and dynamics contribute to neurodegenerative diseases (123). Overactivation of the NMDA-subtype of glutamate receptor triggers excessive calcium influx, contributing to neurodegenerative conditions. Such dysregulation of calcium signaling results in generation of excessive free radicals, including reactive oxygen and nitrogen species. Excessive calcium-induced NO production can contribute to the accumulation of misfolded proteins, specifically by S-nitrosylation of the ubiquitin E3 ligase, parkin, and the formation of S-nitrosylated dynamin-related protein 1, which causes abnormal mitochondrial fragmentation and resultant synaptic damage (161).
Mitochondrial fission allows for mitochondrial renewal, redistribution, and proliferation into synapses the competing process; whereas mitochondrial fusion allows mitochondria to interact and communicate with each other, facilitating mitochondrial movement and distribution across long distances and to the synapses. Probably, mitochondrial fusion serves as a protective mechanism by preventing deficiencies of individual injured and dysfunctional mitochondria from damaging the entire neuron while maintaining an adequate bioenergetic capacity. However, mitochondrial fission could also be protective in facilitating autophagic clearance of damaged and inactive mitochondria.
VIII. Mitochondrial Biogenesis, Mitochondrial Dynamics, and Cell Cycle
Cellular proliferation is an energy-consuming activity that is controlled by checkpoints of the cell cycle (114). Cells that make the decision to divide should be metabolically prepared to support the energetic demand imposed by proliferation. Essentially, the “‘metabolic cycle”’ during cellular proliferation alternates between a first oxidative phase that is characterized by the biosynthesis of many cellular components (G1 phase) and is supported by the energy derived from mitochondrial activity followed by a reductive phase where the replication of DNA and the biosynthesis of mitochondria (S/G2/M phases) is supported by nonrespiratory modes of energy generation (143, 192).
Alternatively, the cells can become reversibly arrested at the G1/S boundary (restriction point) of the cell cycle. The G1/S arrest is triggered by a metabolic stress checkpoint of the cycle that is controlled by the activation of AMPK, which is a metabolic sensor of the energy charge in higher eukaryotic cells (31). The activation of AMPK promotes the phosphorylation of p53 at Ser15 (114), a modification that prevents its degradation and results in the cellular accumulation of p53 and induces the expression of its target gene, p21. P21 is an inhibitor of cyclin-dependent kinases, which promotes a cell-cycle arrest at the level of G1 and G2 (138). Owusu-Ansah et al. (170) showed that mutations in genes encoding components of complex IV and complex I activate the G1-S checkpoint using distinct mechanisms that result in cyclin E down-regulation or inactivation of Cyclin E-CDK2 restricting entry into S phase. These mechanisms involved the reduction of ATP and activation of AMPK by the rise of AMP or the increase in ROS production and the activation of JNK and FOXO.
Recent findings in mammalian cells showed that cyclin D1, which is involved in the phosphorylation and inactivation of the retinoblastoma protein, marking the entry of cells into the S phase of the cycle, inhibits mitochondrial function (198) and represses the activity of NRF-1 (227, 200).
The control of mitochondrial biogenesis during the cell cycle in mammalian cells remains unknown and controversial. Recently, Lee et al. showed in synchronized cultures of HeLa cells that mitochondrial mass and membrane potential increased during the progression of G1 to mitosis and after cell division, these parameters were reduced again (133). In addition, mitochondrial DNA contents increased from G(1)/S to G(2) phase concomitant with a consistent increase of NRF-1 level, although the levels of TFAM and PRC were not changed. The strongest in vivo link between NRF-1 and the control of mitochondrial function comes from the results of targeted disruption of the NRF-1 gene in mice (105). Homozygosity of the null allele results in lethality between embryonic days 3.5 and 6.5 (E3.5 and E6.5). The null blastocysts fail to grow in culture despite having a normal morphology. Homozygous null blastocysts are defective in maintaining a mitochondrial membrane potential and have severely reduced mtDNA levels (119).
PGC-1α and PGC-1β stimulate Mfn2 expression through a transcriptional mechanism involving the activation of the Mfn2 promoter, which requires the integrity of an ERRα-binding element. PGC-1β also alters mitochondrial morphology. Under conditions of enhanced energy expenditure, PGC-1α is induced and stimulates mitochondrial biogenesis, Mfn2 expression, and mitochondrial function.
Morphological changes of mitochondria during the cell cycle were also reported (133). Individual mitochondria act in a concerted manner at different stages of the cell cycle (106) (Fig. 13). In mitosis, fragmented mitochondria localize to opposite telomeres of daughter mitochondria in M phase, thus indicating concerted movement and dispersion between daughter cells. In G0, both filamentous and fragmented mitochondria occur. In G1–S, most surprisingly, mitochondria form a giant tubular network with tubular elements undergoing fission and fusion. The giant voluminous mitochondrial network at G1-S could serve several cellular functions such as efficient homogeneization and complementation of mitochondrial DNA in the continuous matrix, enhanced mitochondrial ATP to compensate for reduced glycolytic ATP during G1-S, and cell protection against apoptosis at this crucial cell cycle stage (155). Inducing mitochondrial hyperfusion by acute inhibition of Drp1 leads to buildup of cyclin E and initiation of replication in serum starved cells at G0. Regulation of cyclin E levels by hyperfused mitochondria at G1-S could explain why knockouts of Mfns phenocopy the effects of cyclin E knockouts. Several lines of evidence support the view that Mfn2 exerts a regulatory role on the cell cycle. Mfn2 overexpression in vascular smooth muscle cells (VSMCs) causes growth arrest characterized by an increased number of cells in G0/G1 stage and a reduction of cells in S or G2/M phases. Whether the effects of Mfn2 are universal in different cell types and species is unknown. (136).
FIG. 13.

Mitochondrial dynamics and cell cycle. During the cell cycle, rapid changes in mitochondrial morphology and dynamics accompany mitosis, and allow the appropriate segregation of the organelles and mtDNA into daughter cells.
The rapid changes in mitochondrial morphology and dynamics that accompany mitosis might be of relevance for the appropriate segregation of the organelles and mtDNA into daughter cells during proliferation (143). Fission of mitochondria is an early event of mitosis that might be triggered in response to the signaling cascades that accompany progression through the cell cycle. Fission of the organelle is likely to be triggered by proteasomal degradation of Mfns. Functionally, fission of mitochondria assures a stochastic distribution of the organelles within the two daughter cells by a process that might be actively controlled and mediated by microtubules. However, it has been reported that after mitochondrial fission 25%–40% of the organelles lack mtDNA (134). This situation could contribute to the asymmetric segregation of mtDNA into daughter cells explaining certain mtDNA depletion syndromes. However, the fusion of mitochondria in late telophase, just before cytokinesis, a process that is controlled by Mfns and the development of ΔΨ, is likely to ameliorate the possible unequal distribution of mtDNA during cellular proliferation. Mitochondrial biogenesis increases mitochondrial mass, whereas mitochondrial fission increases the actual number of mitochondria. Berman et al. (15) showed that Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. The authors found that the rate of fission was markedly higher than the rate of fusion in healthy neuronal processes, and that this imbalance was compensated by growth of mitochondrial organelles mediated by Bcl-xL. Thus, fission and fusion are integrated with the control of mitochondrial mass (biogenesis and degradation) to determine mitochondrial morphology.
IX. Concluding Remarks
Endosymbiosis leads ancient bacteria to be transformed into modern mitochondria. During evolution, the organelles lost many bacterial functions, but they gained two important new functions. First, to release ATP to the cytosol via the ATP/ADP translocase that is not present in bacteria. Second, to modulate cell fate by the adjustment of cell cycle for development and growth, or alternatively for aging, involution, or senescence. These actions depend on the regulation of electron transfer rate and on the production of ROS that are controlled in vivo by NO and by the combined transcriptional activity on nuclear and mtDNAs. Although bacterial NOS are present in mitochondria from proteobacteria, they require a complementary oxidative domain to provide electrons for NO synthesis. Modern mitochondria utilize NO from canonical cytosolic NOS or from NOS translocated into mitochondria to reversibly inhibit COX or to release H2O2. We and others have previously shown that the level of H2O2 determines cell fate by acting on transcription factors, signaling pathways, and cell cycle regulators. It is highlighted here that kinases participating in cell cycle and cell protection, such as Akt and ERK1/2 and PKA/PKC, are trafficked to mitochondria where they are activated by redox modifications through specific cysteine oxidations.
The involvement of cell-cycle regulators and oncogenic proteins in cancer development extend beyond the control of cell proliferation, to new functions that may be implicated in metabolic alterations related to the pathogenesis of human cancers. This is consistent with the observation that changes in metabolism are part of a coordinated response of the cell to distinct physiological and pathological conditions. In mammalian cells, the metabolic response entails a permanent coordination of cell activity, including cell proliferation with nutrient availability, hormonal and stress signaling, and with regulation of energy homeostasis (101). This links external signaling events with the activation or inhibition of particular metabolic pathways, such as oxidative or glycolytic metabolism, and biosynthetic routes. The clinical relevance of the mitochondrial regulation of the cell cycle is already given by the changes in proliferation and apoptosis, and in intermediary metabolism and synthesis of macromolecules as reported in cancer with low mitochondrial production of H2O2 and activation of ERK1/2 and Akt, or in neurodegenerative disorders, hypothyroidism, and diabetes with high mitochondrial H2O2, and activation of JNK and p38MAPK. It is then remarkable that the proliferative effects of Warburg effect are linked to low mitochondrial ROS levels rather than to low respiration rate and high glucose consumption; high NO levels are associated to high H2O2 and cell cycle arrest in spite of the low oxygen utilization.
The mitochondrial morphology is continuously modified by functional requirements to adapt to different cell demands. Mitochondria can exhibit continuous shape changes such as branching, bending, and retractions, an increase in the number of cristae or change in their shape and may fuse or increase in size to form giant mitochondria. The mitochondrial respiration and metabolism may be spatially and temporally regulated by the architecture and positioning of the organelle. The orchestrated synthesis of mitochondrial components and mitochondrial biogenesis and the transitional dynamics variations of the organelles are as well regulated by H2O2 and NO and contribute to set the respiratory rate, the redox status, and, ultimately, cell behavior.
The structural mitochondrial alterations in human tumors are heterogeneous and not specific for any neoplasm. Mitochondrial changes are associated with mitochondrial-DNA mutations, tumoral microenvironment conditions, and mitochondrial fusion–fission disequilibrium. Functionally, the structural alterations suppose the presence of hypoxia-tolerant and hypoxia-sensitive cancer cells. Pharmacological approaches designed to act on both glycolysis and OXPHOS can be considered as a new approach to selectively kill cancer cells.
On the other hand, the study of the pathogenesis of the neurodegenerative disorders appears terribly complex; nevertheless, the common role of mitochondria dysfunction (metabolic, morphologic, or dynamic) appears very clear, and provides not only a leading theme for further studies, but also a promising pharmacological target in these devastating diseases.
Abbreviations Used
- 2-DG
2-deoxyglucose
- 3′UTRs
3′ untranslated regions
- ADOA
autosomal dominant optic atrophy
- AKAP
A-kinase-anchor-proteins
- Akt
protein kinase B
- AMPK
AMP-activated protein kinase
- ANT
ADP/ATP translocator
- AP-1
adaptor-related protein complex 1
- APAF-1
apoptotic protease-activating factor-1
- BAD
Bcl-2-associated death promoter
- BAK
Bcl-2 homologous antagonist killer
- BAX
Bcl-2, B-cell lymphoma 2
- Bcl-xL
B-cell lymphoma-extra large
- BH
Bcl-2 homology
- BIM
Bcl-2 interacting mediator of cell death
- CaMK
calcium/calmodulin-dependent protein kinase
- CDKs
cyclin-dependent kinases
- COX
cytochrome oxidase
- CREB
cAMP response element-binding protein
- cMyc
myelocytomatosis viral oncogene homology
- cyt
cytochrome
- DIABLO
direct IAP-binding protein with low pl
- Drp
dynamin related protein
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- ERRα
estrogen-related receptor α
- ERV1
endogenous retroviral sequence 1
- ESCs
embryonic stem cells
- FAS
TNF receptor superfamily member 6
- FOXO
forkhead O-box
- F2,6P2
fructose-2,6-bisphosphate
- G-6-P
glucose-6-phosphate
- GDAP1
ganglioside-induced differentiation associated protein
- GPx
glutathione peroxidase
- GSK3
glycogen synthase kinase 3
- hESCs
human ESCs
- hFis1
fission 1 homologue protein
- HIF
hypoxia inducible factor
- HK
hexokinase
- [H2O2]ss
H2O2 steady state concentration
- IKK
IκB kinase
- IMM
inner mitochondrial membrane
- IMS
intermembrane space
- JNK
c-Jun-NH2-terminal kinase
- MAPK
mitogen-activated protein kinases
- MEK
mitogen-activated protein kinase kinase
- mES
mouse embryonic stem
- Mfn
mitofusin
- Mia40
mitochondrial intermembrane space assembly machinery
- mtNOS
mitochondrial NO synthase
- mTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NF-κB
nuclear factor-κB
- NO
nitric oxide
- NRF
nuclear respiratory factor
-
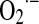 −
−
superoxide anion
- OMM
outer mitochondrial membrane
- ONOO−
peroxynitrite
- OPA1
optic atrophy gene 1
- OXPHOS
oxidative phosphorylation
- p53
tumor protein p53
- PDK1
PI-3K-dependent kinase 1
- PFK
phosphofructokinase
- PFKFB3
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase
- PGC-1
peroxisome proliferator-activated receptor-γ-coactivator-1
- PI3K
phosphoinositide 3-kinase
- PKA
protein kinase A
- PKC
protein kinase C
- PKM2
pyruvate kinase M2
- PPAR-γ
peroxisome proliferator-activated receptor
- Prx
peroxiredoxin
- PTEN
phosphatase antensin homolog
- PUMA
p53-up-regulated mediator of apoptosis
- RAS
rat sarcoma small GTPase
- RAF
V-raf-1 murine leukemia viral oncogene homolog
- ROS
reactive oxygen species
- SirT3
Sirtuin3
- SMAC
second mitochondria-derived activator of caspases
- SOD
superoxide dismutase
- StAR
steroidogenic acute regulatory
- TFAM
mitochondrial transcription factor A
- TIM
translocase of the inner membrane
- TNF-α
tumor necrosis factor α
- TOM
translocase of the outer membrane
- Trx
thioredoxin reductases
- UQ
ubiquinol
- VDAC
voltage-dependent anion channel
Acknowledgments
The authors thank Soledad Galli and Yael Alippe for electron microphotographs and studies on ERK1/2. This work was supported by the Agencia Nacional de Promoción Científica y Tecnológica (Foncyt) grants PICT 1625, PICT 21461, and PICT 34785 (to M.C.C. and J.J.P.), University of Buenos Aires (Ubacyt M058), Conicet (PIP 5495 to M.C.C.), and Fundación Pérez Companc, Buenos Aires, Argentina.
References
- 1.Adams JM. Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19:488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Affaitati A. Cardone L. Carlucci A. de Cristofaro T. Ginsberg MD. Varrone S. Gottesman ME. Avvedimento VE. Feliciello A. Essential role of A-kinase anchor protein 121 for cAMP signaling to mitochondria. J Biol Chem. 2003;278:4286–4294. doi: 10.1074/jbc.M209941200. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed NN. Grimes HL. Bellacosa A. Chan TO. Tsichlis PN. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci U S A. 1997;94:3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almeida A. Bolaños JP. Moncada S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc Natl Acad Sci U S A. 2010;107:738–741. doi: 10.1073/pnas.0913668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alonso M. Melani M. Converso DP. Jaitovich A. Paz C. Carreras MC. Medina JH. Poderoso JJ. Mitochondrial extracellular signal-regulated kinases 1/2 (ERK1/2) are modulated during brain development. J Neurochem. 2004;89:248–256. doi: 10.1111/j.1471-4159.2004.02323.x. [DOI] [PubMed] [Google Scholar]
- 6.Alto NM. Soderling J. Scott JD. Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J Cell Biol. 2002;158:659–668. doi: 10.1083/jcb.200204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ambs S. Hussain SP. Harris CC. Interactive effects of nitric oxide and the p53 tumor suppressor gene in carcinogenesis and tumor progression. FASEB J. 1997;11:443–448. doi: 10.1096/fasebj.11.6.9194524. [DOI] [PubMed] [Google Scholar]
- 8.Anjum R. Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nature Rev. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- 9.Antico Arciuch VG. Alippe Y. Carreras MC. Poderoso JJ. Mitochondrial kinases in cell signaling: facts and perspectives. Adv Drug Deliv Rev. 2009;61:1234–1249. doi: 10.1016/j.addr.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 10.Antico Arciuch VG. Dima M. Liao XH. Refetoff S. Di Cristofano A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene. 2010;29:5678–5686. doi: 10.1038/onc.2010.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antico Arciuch VG. Galli S. Franco MC. Lam PY. Cadenas E. Carreras MC. Poderoso JJ. Akt1 intramitochondrial cycling is a crucial step in the redox modulation of cell cycle progression. PLoS One. 2009;4:e7523. doi: 10.1371/journal.pone.0007523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldwin AS., Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 13.Barsoum MJ. Yuan H. Gerencser AA. Liot G. Kushnareva Y. Gräber S. Kovacs I. Lee WD. Waggoner J. Cui J. White AD. Bossy B. Martinou JC. Youle RJ. Lipton SA. Ellisman MH. Perkins GA. Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. Embo J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basu A. Lu D. Sun B. Moor AN. Akkaraju GR. Huang J. Proteolytic activation of protein kinase C-epsilon by caspase-mediated processing and transduction of antiapoptotic signals. J Biol Chem. 2002;277:41850–41856. doi: 10.1074/jbc.M205997200. [DOI] [PubMed] [Google Scholar]
- 15.Berman SB. Chen YB. Qi B. McCaffery JM. Rucker EB., 3rd Goebbels S. Nave KA. Arnold BA. Jonas EA. Pineda FJ. Hardwick JM. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bijur GN. Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. 2003;87:1427–1435. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bossy-Wetzel E. Talantova MV. Lee WD. Scholzke MN. Harrop A. Mathews E. Gotz T. Han J. Ellisman MH. Perkins GA. Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- 18.Boveris A. Oshino N. Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boveris A. Cadenas E. Stoppani AO. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J. 1976;56:435–444. doi: 10.1042/bj1560435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brand A. Hermfisse U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J. 1997;11:388–395. doi: 10.1096/fasebj.11.5.9141507. [DOI] [PubMed] [Google Scholar]
- 21.Brennan JP. Bardswell SC. Burgoyne JR. Fuller W. Schröder E. Wait R. Begum S. Kentish JC. Eaton P. Oxidant–induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem. 2006;281:21827–21836. doi: 10.1074/jbc.M603952200. [DOI] [PubMed] [Google Scholar]
- 22.Burdon RH. Alliangana D. Gill V. Hydrogen peroxide and the proliferation of BHK-21 cells. Free Radic Res. 1995;23:471–486. doi: 10.3109/10715769509065268. [DOI] [PubMed] [Google Scholar]
- 23.Burgering BM. Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27:352–360. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- 24.Burhans WC. Heintz NH. The cell cycle is a redox cycle: Linking phase-specific targets to cell fate. Free Rad Biol Med. 2009;47:1282–1293. doi: 10.1016/j.freeradbiomed.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 25.Burk RF. Nishiki K. Lawrence RA. Chance B. Peroxide removal by selenium-dependent and selenium-independent gluthatione peroxidases in hemoglobin-free perfused rat liver. J Biol Chem. 1978;253:43–46. [PubMed] [Google Scholar]
- 26.Bustamante E. Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci U S A. 1977;74:3735–3739. doi: 10.1073/pnas.74.9.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bustamante E. Morris HP. Pedersen PL. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J Biol Chem. 1981;256:8699–8704. [PubMed] [Google Scholar]
- 28.Cadenas E. Davies KJ. Mitochondrial free radical generation, oxidative stress and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 29.Cagnol S. Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 30.Cantó C. Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cantó C. Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407–3423. doi: 10.1007/s00018-010-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carreras MC. Poderoso JJ. Mitochondrial nitric oxide in the signaling of cell integrated responses. Am J Physiol Cell Physiol. 2007;292:1569–1580. doi: 10.1152/ajpcell.00248.2006. [DOI] [PubMed] [Google Scholar]
- 33.Carreras MC. Converso DP. Lorente AS. Barbich MR. Levisman DM. Jaitovich A. Antico Arciuch VG. Galli S. Poderoso JJ. Mitochondrial nitric oxide synthase drives redox signals from proliferation and quiescence in rat liver development. Hepatology. 2004;40:157–166. doi: 10.1002/hep.20255. [DOI] [PubMed] [Google Scholar]
- 34.Carreras MC. Franco MC. Peralta JG. Poderoso JJ. Nitric oxide, complex I, and the modulation of mitochondrial reactive species in biology and disease. Mol Aspects Med. 2004;25:125–139. doi: 10.1016/j.mam.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 35.Cereghetti GM. Costa V. Scorrano L. Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010;17:1785–1794. doi: 10.1038/cdd.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan TO. Rittenhouse SE. Tsichlis PN. AKT/PKB and other D3 phosphoinositideregulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 37.Chang CR. Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. N Y Acad Sci. 2010;1201:34–39. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen H. Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. 2005;14:R283–R289. doi: 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- 39.Chen HZ. Tsai SY. Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen KH. Guo X. Ma D. Guo Y. Li Q. Yang D. Li P. Qiu X. Wen S. Xiao RP. Tang J. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. 2004;6:872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 41.Chen R. Fearnley IM. Peak-Chew SY. Walker JE. The phosphorylation of subunits of complex I from bovine heart mitochondria. J Biol Chem. 2004;279:26036–26045. doi: 10.1074/jbc.M402710200. [DOI] [PubMed] [Google Scholar]
- 42.Chen RH. Sarnecki C. Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen WS. Xu PZ. Gottlob K. Chen ML. Sokol K. Shiyanova T. Roninson I. Weng W. Suzuki R. Tobe K. Kadowaki T. Hay N. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chipuk JE. Bouchier-Hayes L. Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 45.Cho DH. Nakamura T. Fang J. Cieplak P. Godzik A. Gu Z. Lipton SA. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho H. Thorvaldsen JL. Chu Q. Feng F. Birnbaum MJ. Akt1/pkbalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 47.Cho YM. Kwon S. Pak YK. Seol HW. Choi YM. Park do J. Park KS. Lee HK. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun. 2006;348:1472–1478. doi: 10.1016/j.bbrc.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 48.Choi BM. Pae HO. Jang SI. Kim YM. Chung HT. Nitric oxide as a pro-apoptotic as well as anti-apoptotic modulator. J Biochem Mol Biol. 2002;35:116–126. doi: 10.5483/bmbrep.2002.35.1.116. [DOI] [PubMed] [Google Scholar]
- 49.Chowdhury R. McDonough MA. Mecinović J. Loenarz C. Flashman E. Hewitson KS. Domene C. Schofield CJ. Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure. 2009;17:981–989. doi: 10.1016/j.str.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 50.Churchill EN. Mochly-Rosen D. The roles of PKCdelta and epsilon isoenzymes in the regulation of myocardial ischaemia/reperfusion injury. Biochem Soc Trans. 2007;35:1040–1042. doi: 10.1042/BST0351040. [DOI] [PubMed] [Google Scholar]
- 51.Clark J. Simon DK. Transcribe to survive: transcriptional control of antioxidant defense programs for neuroprotection in Parkinson's disease. Antioxid Redox Signal. 2009;11:509–528. doi: 10.1089/ars.2008.2241. [DOI] [PubMed] [Google Scholar]
- 52.Copeland WC. Wachsman JT. Johnson FM. Penta JS. Mitochondrial DNA alterations in cancer. Cancer Invest. 2002;20:557–569. doi: 10.1081/cnv-120002155. [DOI] [PubMed] [Google Scholar]
- 53.Covello KL. Kehler J. Yu H. Gordan JD. Arsham AM. Hu CJ. Labosky PA. Simon MC. Keith B. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dali-Youcef N. Mataki C. Coste A. Messaddeq N. Giroud S. Blanc S. Koehl C. Champy MF. Chambon P. Fajas L. Metzger D. Schoonjans K. Auwerx J. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Acad Sci U S A. 2007;104:10703–10708. doi: 10.1073/pnas.0611568104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Datta SR. Brunet A. Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 56.de Brito OM. Scorrano L. Mitofusin 2 tethers endoplasmic reticulum (ER) to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 57.De Rasmo D. Panelli D. Sardanelli AM. Papa S. cAMP-dependent protein kinase regulates the mitochondrial import of the nuclear encoded NDUFS4 subunit of complex I. Cell Signal. 2008;20:989–997. doi: 10.1016/j.cellsig.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 58.de Rooij J. Zwartkruis FJ. Verheijen MH. Cool RH. Nijman SM. Wittinghofer A. Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 59.De Vos KJ. Allan VJ. Grierson AJ. Sheetz MP. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr Biol. 2005;15:678–683. doi: 10.1016/j.cub.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 60.Deshpande A. Sicinski P. Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene. 2005;24:2909–2915. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 61.Diaz F. Moraes CT. Mitochondrial biogenesis and turnover. Cell Calcium. 2008;44:24–35. doi: 10.1016/j.ceca.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Diehl JA. Cheng M. Roussel MF. Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dijkers PF. Medema RH. Pals C. Banerji L. Thomas NS. Lam EW. Burgering BM. Raaijmakers JA. Lammers JW. Koenderman L. Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ezashi T. Das P. Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci U S A. 2005;102:4783–4788. doi: 10.1073/pnas.0501283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fadeel B. Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 66.Feliciello A. Gottesman ME. Avvedimento EV. The biological functions of A-kinase anchor proteins. J Mol Biol. 2001;308:99–114. doi: 10.1006/jmbi.2001.4585. [DOI] [PubMed] [Google Scholar]
- 67.Fernandez E. Cuenca N. Garcia M. De Juan J. Two types of mitochondria are evidenced by protein kinase C immunoreactivity in the Muller cells of the carp retina. Neurosci Lett. 1995;183:202–205. doi: 10.1016/0304-3940(94)11151-8. [DOI] [PubMed] [Google Scholar]
- 68.Finocchietto PV. Barreyro F. Holod S. Peralta JG. Franco MC. Méndez C. Converso DP. Estévez AG. Carreras MC. Poderoso JJ. Control of Muscle Mitochondria by Insulin Entails Activation of Akt2-mtNOS Pathway: Implications for the Metabolic Syndrome. PLoS ONE. 2008;3:e1749. doi: 10.1371/journal.pone.0001749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finocchietto PV. Franco MC. Holod SA. Gonzalez AS. Converso DP. Antico Arciuch VG. Serra MP. Poderoso JJ. Carreras MC. Mitochondrial nitric oxide synthase: a master piece of metabolic adaptation, cell growth, transformation and death. Exp Biol Med (Maywood) 2009;234:1020–1028. doi: 10.3181/0902-MR-81. [DOI] [PubMed] [Google Scholar]
- 70.Finocchietto PV. Holod SA. Barreyro F. Peralta JG. Alippe Y. Giovambattista A. Carreras MC. Poderoso JJ. Defective leptin-AMPK pathway induces NO release and contributes to mitochondrial dysfunction and obesity in ob/ob Mice. Antioxid Redox Signal. 2011;15:2395–2406. doi: 10.1089/ars.2010.3857. [DOI] [PubMed] [Google Scholar]
- 71.Foster MW. Hess DT. Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Franco MC. Antico Arciuch VG. Peralta JG. Galli S. Levisman D. López LM. Romorini L. Poderoso JJ. Carreras MC. Hypothyroid phenotype is contributed by mitochondrial complex I inactivation due to translocated neuronal nitric oxide synthase. J Biol Chem. 2006;281:4779–4786. doi: 10.1074/jbc.M512080200. [DOI] [PubMed] [Google Scholar]
- 73.Frank S. Gaume B. Bergmann-Leitner ES. Leitner WW. Robert EG. Catez F. Smith CL. Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 74.Franke TF. Yang SI. Chan TO. Datta K. Kazlauskas A. Morrison DK. Kaplan DR. Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;2:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 75.Frezza C. Cipolat S. Martins de Brito O. Micaroni M. Beznoussenko GV. Rudka T. Bartoli D. Polishuck RS. Danial NN. De Strooper B. Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 76.Fritz V. Fajas L. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene. 2010;29:4369–4377. doi: 10.1038/onc.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fujinoa G. Noguchia T. Takedaa K. Ichijo H. Thioredoxin and protein kinases in redox signaling. Sem Cancer Biol. 2006;16:427–435. doi: 10.1016/j.semcancer.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 78.Galli S. Labato MI. Bal de Kier Joffé E. Carreras MC. Poderoso JJ. Decreased mitochondrial nitric oxide synthase activity and hydrogen peroxide relate persistent tumoral proliferation to embryonic behavior. Cancer Res. 2003;63:6370–6377. [PubMed] [Google Scholar]
- 79.Galli S. Antico Arciuch VG. Poderoso C. Converso DP. Zhou Q. Bal de Kier Joffé E. Cadenas E. Boczkowski J. Carreras MC. Poderoso JJ. Tumor cell phenotype is sustained by selective MAPK oxidation in mitochondria. PLoS One. 2008;3:e2379. doi: 10.1371/journal.pone.0002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Galli S. Jahn O. Hitt R. Hesse D. Opitz L. Plessmann U. Urlaub H. Poderoso JJ. Jares-Erijman EA. Jovin TM. A new paradigm for MAPK: structural interactions of hERK1 with mitochondria in HeLa cells. PLoS One. 2009;4:e7541. doi: 10.1371/journal.pone.0007541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gardai SJ. Hildeman DA. Frankel SK. Whitlock BB. Frasch SC. Borregaard N. Marrack P. Bratton DL. Henson PM. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem. 2004;279:21085–21095. doi: 10.1074/jbc.M400063200. [DOI] [PubMed] [Google Scholar]
- 82.Garrido N. Griparic L. Jokitalo E. Wartiovaara J. van der Bliek AM. Spelbrink JN. Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell. 2003;14:1583–1596. doi: 10.1091/mbc.E02-07-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gatenby RA. Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 84.Gerschman R. Gilbert DL. Nye SW. Dwyer P. Fenn W0. Oxygen poisoning and X-irradiation: a mechanism in common. Science. 1954;119:623–626. doi: 10.1126/science.119.3097.623. [DOI] [PubMed] [Google Scholar]
- 85.Gingras AC. Kennedy SG. O'Leary MA. Sonenberg N. Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt (PKB) signaling pathway. Genes Dev. 1998;12:502–513. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ginsberg MD. Feliciello A. Jones JK. Avvedimento EV. Gottesman ME. PKA-dependent binding of mRNA to the mitochondrial AKAP121 protein. J Mol Biol. 2003;327:885–897. doi: 10.1016/s0022-2836(03)00173-6. [DOI] [PubMed] [Google Scholar]
- 87.Greer EL. Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 88.Gregersen N. Bross P. Protein misfolding and cellular stress: an overview. Methods Mol Biol. 2010;648:3–23. doi: 10.1007/978-1-60761-756-3_1. [DOI] [PubMed] [Google Scholar]
- 89.Hajri A. Metzger E. Vallat F. Coffy S. Flatter E. Evrard S. Marescaux J. Aprahamian M. Role of nitric oxide in pancreatic tumour growth: in vivo and in vitro studies. Br J Cancer. 1998;78:841–849. doi: 10.1038/bjc.1998.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Han MK. Song EK. Guo Y. Ou X. Mantel C. Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241–251. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hao Z. Duncan GS. Chang CC. Elia A. Fang M. Wakeham A. Okada H. Calzascia T. Jang Y. You-Ten A. Yeh WC. Ohashi P. Wang X. Mak TW. Specific ablation of the apoptotic functions of cytochrome c reveals a differential requirement for cytochrome c and Apaf-1 in apoptosis. Cell. 2005;121:579–591. doi: 10.1016/j.cell.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 92.Harada H. Becknell B. Wilm M. Mann M. Huang LJ. Taylor SS. Scott JD. Korsmeyer S. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. J Mol Cell. 1999;4:413–422. doi: 10.1016/s1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- 93.Harder Z. Zunino R. McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 94.Harvey AJ. Kind KL. Pantaleon M. Armstrong DT. Thompson JG. Oxygen-regulated gene expression in bovine blastocysts. Biol Reprod. 2004;71:1108–1119. doi: 10.1095/biolreprod.104.028639. [DOI] [PubMed] [Google Scholar]
- 95.Helin K. Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev. 1998;8:28–35. doi: 10.1016/s0959-437x(98)80058-0. [DOI] [PubMed] [Google Scholar]
- 96.Holland HD. Early proterozoic atmospheric change. In: Bengtson S, editor. Early Life on Earth. New York: Columbia University Press; 1994. pp. 237–244. [Google Scholar]
- 97.Holmes WF. Soprano DR. Soprano KJ. Early events in the induction of apoptosis in ovarian carcinoma cells by CD437: activation of the p38 MAP kinase signal pathway. Oncogene. 2003;22:6377–6386. doi: 10.1038/sj.onc.1206694. [DOI] [PubMed] [Google Scholar]
- 98.Holmgren A. Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem Biophys Res Commun. 2010;396:120–124. doi: 10.1016/j.bbrc.2010.03.083. [DOI] [PubMed] [Google Scholar]
- 99.Hortelano S. Alvarez AM. Bosca L. Nitric oxide induces tyrosine nitration and release of cytochrome c preceding an increase of mitochondrial transmembrane potential in macrophages. FASEB J. 1999;13:2311–2317. doi: 10.1096/fasebj.13.15.2311. [DOI] [PubMed] [Google Scholar]
- 100.Houghton FD. Energy metabolism of the inner cell mass and trophectoderm of the mouse blastocyst. Differentiation. 2006;74:11–18. doi: 10.1111/j.1432-0436.2006.00052.x. [DOI] [PubMed] [Google Scholar]
- 101.Hsieh MC. Das D. Sambandam N. Zhang MQ. Nahle Z. Regulation of the PDK4 isozyme by the Rb–E2F1 complex. J Biol Chem. 2008;283:27410–27417. doi: 10.1074/jbc.M802418200. [DOI] [PubMed] [Google Scholar]
- 102.Hu J. Dong L. Outten CE. The redox environment in the mitochondrial intermembrane space is maintained separately from the cytosol and matrix. J Biol Chem. 2008;283:29126–29134. doi: 10.1074/jbc.M803028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang JS. Chuang LY. Guh JY. Chen CJ. Yang YL. Chiang TA. Hung MY. Liao TN. Effect of nitric oxide-cGMP-dependent protein kinase activation on advanced glycation end-product-induced proliferation in renal fibroblasts. J Am Soc Nephrol. 2005;16:231–239. doi: 10.1681/ASN.2005010030. [DOI] [PubMed] [Google Scholar]
- 104.Huang P. Yu T. Yoon Y. Mitochondrial clustering induced by overexpression of the mitochondrial fusion protein Mfn2 causes mitochondrial dysfunction and cell death. Eur J Cell Biol. 2007;86:289–302. doi: 10.1016/j.ejcb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 105.Huo L. Scarpulla RC. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol Cell Biol. 2001;21:644–654. doi: 10.1128/MCB.21.2.644-654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hyde BB. Twig G. Shirihai OS. Organellar vs cellular control of mitochondrial dynamics. Semin Cell Dev Biol. 2010;21:575–581. doi: 10.1016/j.semcdb.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ikenoue T. Inoki K. Yang Q. Zhou X. Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signaling. EMBO J. 2008;23:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Iñarrea P. Moini H. Han D. Rettori D. Aguiló I. Alava MA. Iturralde M. Cadenas E. Mitochondrial respiratory chain and thioredoxin reductase regulate intermembrane Cu,Zn-superoxide dismutase activity: implications for mitochondrial energy metabolism and apoptosis. Biochem J. 2007;405:173–179. doi: 10.1042/BJ20061809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ishida A. Sasaguri T. Kosaka C. Nojima H. Ogata J. Induction of the cyclindependent kinase inhibitor p21Sdi1Cip1/Waf1 by nitric oxide-generating vasodilator in vascular smooth muscle cells. J Biol Chem. 1999;272:10050–10057. doi: 10.1074/jbc.272.15.10050. [DOI] [PubMed] [Google Scholar]
- 110.Jacobs KM. Pennington JD. Bisht KS. Aykin-Burns N. Kim HS. Mishra M. Sun L. Nguyen P. Ahn BH. Leclerc J. Deng CX. Spitz DR. Gius D. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci. 2008;4:291–299. doi: 10.7150/ijbs.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jaeger W. Hereditary optic atrophy with dominant transmission; with special reference to the associated color-sense disorder. Albrecht Von Graefes Arch Ophthalmol. 1954;155:457–484. [PubMed] [Google Scholar]
- 112.Jezek P. Plecita-Hlavata L. Smolkova K. Rossignol R. Distinctions and similarities of cell bioenergetics and the role of mitochondria in hypoxia, cancer, and embryonic development. Int J Biochem Cell Biol. 2010;42:604–622. doi: 10.1016/j.biocel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 113.Jin K. Mao XO. Zhu Y. Greenberg DA. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J Neurochem. 2002;80:119–125. doi: 10.1046/j.0022-3042.2001.00678.x. [DOI] [PubMed] [Google Scholar]
- 114.Jones RG. Plas DR. Kubek S. Buzzai M. Mu J. Mu J. Xu Y. Birnbaum MJ. Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 115.Kahan C. Seuwen K. Meloche S. Pouysségur J. Coordinate, biphasic activation of p44 mitogen-activated protein kinase and S6 kinase by growth factors in hamster fibroblasts. Evidence for thrombin-induced signals different from phosphoinositide turnover and adenylylcyclase inhibition. J Biol Chem. 1992;267:13369–13375. [PubMed] [Google Scholar]
- 116.Kamata H. Honda S. Maeda S. Chang L. Hirata H. Karin M. Reactive oxygen species promote TNF-alpha-induced death and sustained JNK activation by inhibiting MAP kinase death. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 117.Karbowski M. Norris KL. Cleland MM. Jeong SY. Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- 118.Kashiwakura I. Kuwabara M. Murakami M. Hayase Y. Takagi Y. Effect of carboxy-PTIO, a nitric oxide scavenger, on the proliferation of murine hematopoietic progenitor cells in vitro. Res Commun Mol Pathol Pharmacol. 1998;99:329–337. [PubMed] [Google Scholar]
- 119.Kelly DP. Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 120.Kienhöfer J. Häussler DJ. Ruckelshausen F. Muessig E. Weber K. Pimentel D. Ullrich V. Bürkle A. Bachschmid MM. Association of mitochondrial antioxidant enzymes with DNA as integral nucleoid constituents. FASEB J. 2009;23:2034–2044. doi: 10.1096/fj.08-113571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim AH. Yano H. Cho H. Meyer D. Monks B. Margolis B. Birnbaum MJ. Chao MV. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron. 2002;35:697–709. doi: 10.1016/s0896-6273(02)00821-8. [DOI] [PubMed] [Google Scholar]
- 122.Klimova T. Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–666. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- 123.Knott AB. Bossy-Wetzel E. Impact of nitric oxide on metabolism in health and age-related disease. Diabetes Obes Metabol. 2010;12:126–133. doi: 10.1111/j.1463-1326.2010.01267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Knott AB. Perkins G. Schwarzenbacher R. Bossy-Wetzel E. Mitocondrial fragmentation in neurodegeneration. Nature Rev. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nature Rev. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 126.Kondo N. Nakamura H. Masutani H. Yodoi J. Redox regulation of human thioredoxin network. Antiox Redox Signal. 2006;8:1881–1890. doi: 10.1089/ars.2006.8.1881. [DOI] [PubMed] [Google Scholar]
- 127.Kong X. Wang R. Xue Y. Liu X. Zhang H. Chen Y. Fang F. Chang Y. Sirtuin 3, a New Target of PGC-1a, Plays an Important Role in the Suppression of ROS and Mitochondrial Biogenesis. PLoS One. 2010;5:e11707. doi: 10.1371/journal.pone.0011707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kurland CG. Andersson SGE. Origin and evolution of the mitochondrial proteome. Microbiol Mol Biol Rev. 2000;64:786–820. doi: 10.1128/mmbr.64.4.786-820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lago CU. Sung HJ. Ma W. Wang PY. Hwang PM. p53, Aerobic metabolism, and cancer. Antioxid Redox Signal. 2011;15:1739–1748. doi: 10.1089/ars.2010.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lebedeva MA. Eaton JS. Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim Biophys Acta. 2009;1787:328–334. doi: 10.1016/j.bbabio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Leber B. Lin J. Andrews DW. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12:897–911. doi: 10.1007/s10495-007-0746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lee HJ. Bach JH. Chae HS. Lee SH. Joo WS. Choi SH. Kim KY. Lee WB. Kim SS. Mitogen-activated protein kinase/extracellular signal-regulated kinase attenuates 3-hydroxykynurenine-induced neuronal cell death. J Neurochem. 2004;88:647–656. doi: 10.1111/j.1471-4159.2004.02191.x. [DOI] [PubMed] [Google Scholar]
- 133.Lee S. Kim S. Sun X. Lee JH. Cho H. Cell cycle-dependent mitochondrial biogenesis and dynamics in mammalian cells. Biochem Biophys Res Commun. 2007;357:111–117. doi: 10.1016/j.bbrc.2007.03.091. [DOI] [PubMed] [Google Scholar]
- 134.Legros F. Malka F. Frachon P. Lombès A. Rojo M. Organization and dynamics of human mitochondrial DNA. J Cell Sci. 2004;117:2653–2662. doi: 10.1242/jcs.01134. [DOI] [PubMed] [Google Scholar]
- 135.Liang J. Zubovitz J. Petrocelli T. Kotchetkov R. Connor MK. Han K. Lee JH. Ciarallo S. Catzavelos C. Beniston R. Franssen E. Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 136.Liesa M. Palacín M. Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 137.Liu B. Chen Y. St. Clair DK. ROS and p53: versatile partnership. Free Rad Biol Med. 2008;44:1529–1535. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu G. Lozano G. p21 stability: linking chaperones to a cell cycle checkpoint. Cancer Cell. 2005;7:113–114. doi: 10.1016/j.ccr.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 139.Lu D. Sivaprasad U. Huang J. Shankar E. Morrow S. Basu A. Protein kinase C-epsilon protects MCF-7 cells from TNF-mediated cell death by inhibiting Bax translocation. Apoptosis. 2007;12:1893–1900. doi: 10.1007/s10495-007-0111-7. [DOI] [PubMed] [Google Scholar]
- 140.Lu Q. Jourd'Heuil FL. Jourd'Heuil D. Redox control ofG1/S cell cycle regulators during nitric oxide-mediated cell cycle arrest. J Cell Physiol. 2007;212:827–839. doi: 10.1002/jcp.21079. [DOI] [PubMed] [Google Scholar]
- 141.Malstrom S. Tili E. Kappes D. Ceci JD. Tsichlis PN. Tumor induction by an Lck-MyrAkt transgene is delayed by mechanisms controlling the size of the thymus. Proc Natl Acad Sci U S A. 2001;98:14967–14972. doi: 10.1073/pnas.231467698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Margulis L. Symbiotic theory of the origin of eukaryotic org anelles; criteria for proof. Symp Soc Exp Biol. 1975;29:21–38. [PubMed] [Google Scholar]
- 143.Martınez-Diez M. Santamarıa G. Ortega A'D. Cuezva JM. Biogenesis and Dynamics of Mitochondria during the Cell Cycle: Significance of 39UTRs. PLoS ONE. 2006;1:e107. doi: 10.1371/journal.pone.0000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Martinou JC. Youle RJ. Which came first, the cytochrome c release or the mitochondrial fission? Cell Death Differ. 2006;13:1291–1295. doi: 10.1038/sj.cdd.4401985. [DOI] [PubMed] [Google Scholar]
- 145.Mathupala SP. Koc YH. Perdersen PL. HHexokinase-2 bound to mitochondria: Cancer's stygian link to the “Warburg effect” and a pivotal target for effective therapy. Biochem Biophys Acta. 2010;1797:1225–1230. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Matoba S. Kang J-G. Patino WD. Wragg A. Boehm M. Gavrilova O. Hurley PJ. Bunz F. Hwang PM. P53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 147.Mazurek S. Eigenbrodt E. The tumor metabolome. Anticancer Res. 2003;23:1149–1154. [PubMed] [Google Scholar]
- 148.McCord JM. Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 149.McLeod CJ. Pagel I. Sack MN. The mitochondrial biogenesis regulatory program in cardiac adaptation to ischemia: a putative target for therapeutic intervention. Trends Cardiovasc Med. 2005;15:118–123. doi: 10.1016/j.tcm.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 150.Medema RH. Kops GJ. Bos JL. Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 151.Meeusen S. McCaffery JM. Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305:1747–1752. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- 152.Meini A. Garcia JB. Pessina GP. Aldinucci C. Frosin M. Palmi M. Role of intracellular Ca2+ and calmodulin/MAP kinase kinase/extracellular signal-regulated protein kinase signalling pathway in the mitogenic and antimitogenic effect of nitric oxide in glia- and neurone-derived cell lines. Eur J NeuroSci. 2006;23:1690–1700. doi: 10.1111/j.1460-9568.2006.04705.x. [DOI] [PubMed] [Google Scholar]
- 153.Mesecke N. Terziyska N. Kozany C. Baumann F. Neupert W. Hell K. Herrmann JM. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 154.Mishina NM. Tyurin-Kuzmin PA. Markvicheva KN. Vorotnikov AV. Tkachuk VA. Laketa V. Schultz C. Lukyanov S. Belousov VV. Does cellular hydrogen peroxide diffuse or act locally? Antioxid Redox Signal. 2011;14:1–7. doi: 10.1089/ars.2010.3539. [DOI] [PubMed] [Google Scholar]
- 155.Mitra K. Wunder C. Roysam B. Lin G. Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mohr J. Mageroy K. Sex-linked deafness of a possibly new type. Acta Genet Statist Med. 1960;10:54–62. doi: 10.1159/000151118. [DOI] [PubMed] [Google Scholar]
- 157.Möpert K. Hajek P. Frank S. Chen C. Kaufmann J. Santel A. Loss of Drp1 function alters OPA1 processing and changes mitochondrial membrane organization. Exp Cell Res. 2009;315:2165–2180. doi: 10.1016/j.yexcr.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 158.Moreno-Sánchez R. Rodríguez-Enríquez S. Marín-Hernández A. Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393–1418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 159.Müller JM. Milenkovic D. Guiard B. Pfanner N. Chacinska A. Precursor Oxidation by Mia40 and Erv1 Promotes Vectorial Transport of Proteins into the Mitochondrial Intermembrane Space. Mol Biol Cell. 2008;19:226–236. doi: 10.1091/mbc.E07-08-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Murphy LO. Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 161.Nakamura T. Lipton SA. Preventing Ca2+-mediated nitrosative stress in neurodegenerative diseases: Possible pharmacological strategies. Cell Calcium. 2010;47:190–197. doi: 10.1016/j.ceca.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Newmeyer DD. Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 163.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nisoli E. Clementi E. Moncada S. Carruba MO. Mitochondrial biogenesis as a cellular signaling framework. Biochem Pharmacol. 2004;67:1–15. doi: 10.1016/j.bcp.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 165.Nisoli E. Clementi E. Paolucci C. Cozzi V. Tonello C. Sciorati C. Bracale R. Valerio A. Francolini M. Moncada S. Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 166.Nowak G. Bakajsova D. Clifton GL. Protein kinase C-epsilon modulates mitochondrial function and active Na+ transport after oxidant injury in renal cells. Am J Physiol Renal Physiol. 2004;286:F307–F316. doi: 10.1152/ajprenal.00275.2003. [DOI] [PubMed] [Google Scholar]
- 167.Olichon A. Baricault L. Gas N. Guillou E. Valette A. Belenguer P. Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 168.Olovnikov IA. Kravchenko JE. Chumakov PM. Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense. Semin Cancer Biol. 2009;19:32–41. doi: 10.1016/j.semcancer.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Orsi A. Beltrán B. Clementi E. Hallén K. Feelisch M. Moncada S. Continuous exposure to high concentrations of nitric oxide leads to persistent inhibition of oxygen consumption by J774 cells as well as extraction of oxygen by the extracellular medium. Biochem J. 2000;346:407–412. [PMC free article] [PubMed] [Google Scholar]
- 170.Owusu-Ansah E. Yavari A. Mandal S. Banerjee U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet. 2008;40:356–361. doi: 10.1038/ng.2007.50. [DOI] [PubMed] [Google Scholar]
- 171.Owusu-Ansah E. Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Parekh A. Calcium signaling: mitofusins promote interorganellar crosstalk. Current Biol. 2009;19:R200–R203. doi: 10.1016/j.cub.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 173.Park MT. Choi JA. Kim MJ. Um HD. Bae S. Kang CM. Cho CK. Kang S. Chung HY. Lee YS. Lee SJ. Suppression of extracellular signal-related kinase and activation of p38 MAPK are two critical events leading to caspase-8- and mitochondria-mediated cell death in phytosphingosine-treated human cancer cells. J Biol Chem. 2003;278:50624–50634. doi: 10.1074/jbc.M309011200. [DOI] [PubMed] [Google Scholar]
- 174.Pearce LR. Komander D. Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 175.Pearson G. Robinson F. Beers Gibson T. Xu BE. Karandikar M. Berman K. Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 176.Pedrola L. Espert A. Wu X. Claramunt R. Shy ME. Palau F. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum Mol Genet. 2005;14:1087–1094. doi: 10.1093/hmg/ddi121. [DOI] [PubMed] [Google Scholar]
- 177.Pelicano H. Xu R-H. Du M. Feng L. Sasaki R. Carew JS. Hu Y. Ramdas L. Hu L. Keating MJ. Zhang W. Plunkett W. Huang P. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol. 2006;175:913–923. doi: 10.1083/jcb.200512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Peltenburg LT. de Bruin EC. Meersma D. Wilting S. Jürgensmeier JM. Schrier PI. C-Myc is able to sensitize human melanoma cells to diverse apoptotic triggers. Melanoma Res. 2004;14:3–12. doi: 10.1097/00008390-200402000-00002. [DOI] [PubMed] [Google Scholar]
- 179.Peng HB. Spiecker M. Liao JK. Inducible nitric oxide: an autoregulatory feedback inhibitor of vascular inflammation. J Immunol. 1998;161:1970–1976. [PubMed] [Google Scholar]
- 180.Pervin S. Singh R. Chaudhuri G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): potential role of cyclin D1. Proc Natl Acad Sci U S A. 2001;98:3583–3588. doi: 10.1073/pnas.041603998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Poderoso C. Maloberti P. Duarte A. Neuman I. Paz C. Maciel FC. Podesta EJ. Hormonal activation of a kinase cascade localized at the mitochondria is required for StAR protein activity. Mol Cell Endocrinol. 2009;300:37–42. doi: 10.1016/j.mce.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 182.Poderoso JJ. Carreras MC. Lisdero CL. Riobó NA. Schöpfer F. Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. [DOI] [PubMed] [Google Scholar]
- 183.Poderoso JJ. Carreras MC. Lisdero C. Schöpfer F. Riobó N. Giulivi C. Boveris A. Boveris AA. Cadenas E. The reaction of nitric oxide with ubiquinol: kinetic properties and biological significance. Free Rad Biol Med. 1999;26:925–935. doi: 10.1016/s0891-5849(98)00277-9. [DOI] [PubMed] [Google Scholar]
- 184.Poderoso JJ. Lisdero C. Schöpfer F. Riobó N. Carreras MC. Cadenas E. Boveris A. The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. J Biol Chem. 1999;274:37709–37716. doi: 10.1074/jbc.274.53.37709. [DOI] [PubMed] [Google Scholar]
- 185.Pugazhenthi S. Nesterova A. Sable C. Heidenreich KA. Boxer LM. Heasley LE. Reusch JE. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–10766. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- 186.Puigserver P. Spiegelman BM. Peroxisome proliferators-activated receptor γ-coactivator-1α (PGC-1α): Transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 187.Puigserver P. Wu Z. Park CW. Graves R. Wright M. Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 188.Radpour R. Fan AX. Kohler C. Holzgreve W. Zhong XY. Current understanding of mitochondrial DNA in breast cancer. Breast J. 2009;15:505–509. doi: 10.1111/j.1524-4741.2009.00767.x. [DOI] [PubMed] [Google Scholar]
- 189.Rasbach KA. Schnellmann RG. Signaling of mitochondrial biogenesis following oxidative injury. J Biol Chem. 2007;282:2355–2362. doi: 10.1074/jbc.M608009200. [DOI] [PubMed] [Google Scholar]
- 190.Reed CJ. Apoptosis and cancer: strategies for integrating programmed cell death. Semin Hematol. 2000;37:196–206. doi: 10.1016/s0037-1963(00)90055-6. [DOI] [PubMed] [Google Scholar]
- 191.Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999;17:2941–2953. doi: 10.1200/JCO.1999.17.9.2941. [DOI] [PubMed] [Google Scholar]
- 192.Reinke H. Gatfield D. Genome-wide oscillation of transcription in yeast. Trends Biochem Sci. 2006;31:189–191. doi: 10.1016/j.tibs.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 193.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 194.Riobó NA. Clementi E. Melani M. Boveris A. Cadenas E. Moncada S. Poderoso JJ. Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem J. 2001;359:139–145. doi: 10.1042/0264-6021:3590139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Riobó NA. Melani M. Sanjuan N. Fiszman ML. Gravielle MC. Carreras MC. Cadenas E. Poderoso JJ. The modulation of mitochondrial nitric-oxide synthase activity in rat brain development. J Biol Chem. 2002;277:42447–42455. doi: 10.1074/jbc.M204580200. [DOI] [PubMed] [Google Scholar]
- 196.Robey R. Hay N. Is Akt the Warburg kinase?-Akt-energy metabolism and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sablina AA. Budanov AV. Ilyinskaya GV. Agapova LS. Kravchenko JE. Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sakamaki T. Casimiro MC. Ju X. Quong AA. Katiyar S. Liu M. Jiao X. Li A. Zhang X. Lu Y. Wang C. Byers S. Nicholson R. Link T. Shemluck M. Yang J. Fricke ST. Novikoff PM. Papanikolaou A. Arnold A. Albanese C. Pestell R. Cyclin d1 determines mitochondrial function in vivo. Mol Cell Biol. 2006;26:5449–5469. doi: 10.1128/MCB.02074-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sarbassov DD. Guertin DA. Ali SM. Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;18:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 200.Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochem Biophys Acta. 2002;1576:1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- 201.Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 2002;286:81–89. doi: 10.1016/s0378-1119(01)00809-5. [DOI] [PubMed] [Google Scholar]
- 202.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 203.Schroeter H. Boyd CS. Ahmed R. Spencer JP. Duncan RF. Rice-Evans C. Cadenas E. c-Jun N-terminal kinase (JNK)-mediated modulation of brain mitochondria function: new target proteins for JNK signalling in mitochondrion-dependent apoptosis. Biochem J. 2003;372:359–369. doi: 10.1042/BJ20030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shen T. Zheng M. Cao C. Chen C. Tang J. Zhang W. Cheng H. Chen KH. Xiao RP. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem. 2007;282:23354–23361. doi: 10.1074/jbc.M702657200. [DOI] [PubMed] [Google Scholar]
- 205.Sherr CJ. Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev. 2000;10:94–99. doi: 10.1016/s0959-437x(99)00038-6. [DOI] [PubMed] [Google Scholar]
- 206.Shi Y. Mechanical aspects of apoptosome assembly. Curr Opin Cell Biol. 2006;18:677–684. doi: 10.1016/j.ceb.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 207.Sideris DP. Tokatlidis K. Oxidative protein folding in the intermembrane space. Antioxid Redox Signal. 2010;13:1189–1204. doi: 10.1089/ars.2010.3157. [DOI] [PubMed] [Google Scholar]
- 208.Sinthupibulyakit C. Ittarat W. St. Clair WH. St. Clair DK. p53 Protects lung cancer cells against metabolic stress. Int J Oncol. 2010;37:1575–1581. doi: 10.3892/ijo_00000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Skeen JE. Bhaskar PT. Chen CC. Chen WS. Nogueuira V. Hahn-Windgassen A. Kiyokawa H. Hay N. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTOR-dependent manner. Cancer Cell. 2006;10:269–280. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 210.Skroblin P. Grossmann S. Schäfer G. Rosenthal W. Klussmann E. Mechanisms of protein kinase a anchoring. Int Rev Cell Mol Biol. 2010;283:235–330. doi: 10.1016/S1937-6448(10)83005-9. [DOI] [PubMed] [Google Scholar]
- 211.Stambolic V. Suzuki A. de la Pompa JL. Brothers GM. Mirtsos C. Sasaki T. Ruland J. Penninger JM. Siderovski DP. Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 212.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.St. Pierre J. Drori S. Uldry M. Silvaggi JM. Rhee J. Jäger S. Handschin C. Zheng K. Lin J. Yang W. Simon DK. Bachoo R. Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 214.Suen DF. Norris KL. Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sugioka R. Shimizu S. Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem. 2004;279:52726–52734. doi: 10.1074/jbc.M408910200. [DOI] [PubMed] [Google Scholar]
- 216.Sun Q. Chen X. Ma J. Peng H. Wang F. Zha X. Wang Y. Jing Y. Yang H. Chen R. Chang L. Zhang Y. Goto J. Onda H. Chen T. Wang MR. Lu Y. You H. Kwiatkowski D. Zhang H. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Nat Acad Sci U S A. 2011;108:4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Taylor RC. Cullen SP. Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 218.Thiselton DL. Alexander C. Morris A. Brooks S. Rosenberg T. Eiberg H. Kjer B. Kjer P. Bhattacharya SS. Votruba M. A frameshift mutation in exon 28 of the OPA1 gene explains the high prevalence of dominant optic atrophy in the Danish population: evidence for a founder effect. Human Genet. 2001;109:498–502. doi: 10.1007/s004390100600. [DOI] [PubMed] [Google Scholar]
- 219.Tondera D. Czauderna F. Paulick K. Schwarzer R. Kaufmann J. Santel A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci. 2005;118:3049–3059. doi: 10.1242/jcs.02415. [DOI] [PubMed] [Google Scholar]
- 220.Tudzarovaa S. Colomboa SL. Stoebera K. Carcamoa S. Williamsa GH. Moncada S. Two ubiquitin ligases, APC/C-Cdh1 and SKP1-CUL1-F (SCF)-β-TrCP, sequentially regulate glycolysis during the cell cycle. Proc Nat Acad Sci USA. 2011;108:5278–5283. doi: 10.1073/pnas.1102247108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Turrens JF. Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Urtreger AJ. Grossoni VC. Falbo KB. Kazanietz MG. Bal de Kier Joffé ED. Atypical protein kinase C-zeta modulates clonogenicity, motility, and secretion of proteolytic enzymes in murine mammary cells. Mol Carcinog. 2005;2:29–39. doi: 10.1002/mc.20066. [DOI] [PubMed] [Google Scholar]
- 223.van der Loo B. Bachschmid M. Skepper JN. Labugger R. Schildknecht S. Hahn R. Müssig E. Gygi D. Lüscher TF. Age-associated cellular relocation of Sod 1 as a self-defense is a futile mechanism to prevent vascular aging. Biochem Biophys Res Commun. 2006;344:972–980. doi: 10.1016/j.bbrc.2006.03.224. [DOI] [PubMed] [Google Scholar]
- 224.Verkaart S. Koopm WJ. van Emrst-de Vries SE. Nijtmans LG. van den Heuvel LW. Smeitink JA. Willems PH. Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim Biophys Acta. 2007;1772:373–381. doi: 10.1016/j.bbadis.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 225.Vieira HL. Alves PM. Vercelli A. Modulation of neuronal stem cell differentiation by hypoxia and reactive oxygen species. Progress Neurobiol. 2011;93:444–455. doi: 10.1016/j.pneurobio.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 226.Villalobo A. Nitric oxide and cell proliferation. FEBS J. 2006;273:2329–2344. doi: 10.1111/j.1742-4658.2006.05250.x. [DOI] [PubMed] [Google Scholar]
- 227.Wang C. Li Z. Lu Y. Du R. Katiyar S. Yang J. Fu M. Leader JE. Quong A. Novikoff PM. Pestell RG. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Nat Acad Sci U S A. 2006;103:11567–11572. doi: 10.1073/pnas.0603363103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Warburg O. Wind F. Negelein E. The Metabolism of Tumors: Investigations from the Kaiser Wilheim Institute for Biology. Berlin-Dahlen, London: Constable & Co., Ltd.; 1930. On the metabolism of tumors in the body. [Google Scholar]
- 229.Warburg O. Wind F. Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Wasiak S. Zunino R. McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wenzel P. Schuhmacher S. Kienhöfer J. Müller J. Hortmann M. Oelze M. Schulz E. Treiber N. Kawamoto T. Scharffetter-Kochanek K. Münzel T. Bürkle A. Bachschmid MM. Daiber A. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280–289. doi: 10.1093/cvr/cvn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Westermann B. Mitochondrial fusion and fission in cell life and death. Nature Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 233.Westfall SD. Sachdev S. Das P. Hearne LB. Hannink M. Roberts RM. Ezashi T. Identification of oxygen-sensitive transcriptional programs in human embryonic stem cells. Stem Cells Dev. 2008;17:869–881. doi: 10.1089/scd.2007.0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Willis SN. Fletcher JI. Kaufmann T. van Delft MF. Chen L. Czabotar PE. Ierino H. Lee EF. Fairlie WD. Bouillet P. Strasser A. Kluck RM. Adams JM. Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 235.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 236.Wiltshire C. Matsushita M. Tsukada S. Gillespie DA. May GH. A new c-Jun N-terminal kinase (JNK)-interacting protein, Sab (SH3BP5), associates with mitochondria. Biochem J. 2002;367:577–585. doi: 10.1042/BJ20020553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Yamaguchi R. Lartigue L. Perkins G. Scott RT. Dixit A. Kushnareva Y. Kuwana T. Ellisman MH. Newmeyer DD. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. 2008;31:557–569. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Yamamoto T. Ebisuya M. Ashida F. Okamoto K. Yonehara S. Nishida E. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell- cycle progression. Curr Biol. 2006;16:1171–82. doi: 10.1016/j.cub.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 239.Yoo Y-G. Hayashi M. Christensen J. Huang LH. An essential role of the HIF-1α–cMyc axis in malignant progression. Ann N Y Acad Sci. 2009;1177:108–204. doi: 10.1111/j.1749-6632.2009.05043.x. [DOI] [PubMed] [Google Scholar]
- 240.Youle RJ. Cell biology. Cellular demolition and the rules of engagement. Science. 2007;315:776–777. doi: 10.1126/science.1138870. [DOI] [PubMed] [Google Scholar]
- 241.Yu W. Sanders BG. Kline K. RRR-a-tocopheryl succinate induced apoptosis of human breast cancer cells involves Bax translocation to mitochondria. Cancer Res. 2003;63:2483–2491. [PubMed] [Google Scholar]
- 242.Yung HW. Wyttenbach A. Tolkovsky AM. Aggravation of necrotic death of glucose-deprived cells by the MEK1 inhibitors U0126 and PD184161 through depletion of ATP. Biochem Pharmacol. 2004;68:351–360. doi: 10.1016/j.bcp.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 243.Zaobornij T. Valdez LB. La Padula P. Costa LE. Boveris A. Effect of sustained hypobaric hypoxia during maturation and aging on rat myocardium. II. mtNOS activity. J Appl Physiol. 2005;98:2370–2375. doi: 10.1152/japplphysiol.00986.2004. [DOI] [PubMed] [Google Scholar]
- 244.Zhang L. Yu L. Yu CA. Generation of superoxide anion by succinate cytochrome c reductase from bovine heart mitochondria. J Biol Chem. 1998;273:33972–33976. doi: 10.1074/jbc.273.51.33972. [DOI] [PubMed] [Google Scholar]
- 245.Zhao J. Yuan X. Frodin M. Grummt I. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Molecular Cell. 2003;11:405–413. doi: 10.1016/s1097-2765(03)00036-4. [DOI] [PubMed] [Google Scholar]
- 246.Zhou BP. Liao Y. Xia W. Spohn B. Lee MH. Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 247.Zorzano A. Regulation of mitofusin-2 expression in skeletal muscle. Appl Physiol Nutr Metab. 2009;34:433–439. doi: 10.1139/H09-049. [DOI] [PubMed] [Google Scholar]
- 248.Zorzano A. Liesa M. Palacín M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol. 2009;41:1846–1854. doi: 10.1016/j.biocel.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 249.Zorzano A. Liesa M. Sebastián D. Segalés J. Palacín M. Mitochondrial fusion proteins: dual regulators of morphology and metabolism. Semin Cell Dev Biol. 2010;21:566–574. doi: 10.1016/j.semcdb.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 250.Züchner S. Mersiyanova IV. Muglia M. Bissar-Tadmouri N. Rochelle J. Dadali EL. Zappia M. Nelis E. Patitucci A. Senderek J. Parman Y. Evgrafov O. Jonghe PD. Takahashi Y. Tsuji S. Pericak-Vance MA. Quattrone A. Battaloglu E. Polyakov AV. Timmerman V. Schröder JM. Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]