

Ligand-gated ion channels (LGICs) are integral membrane proteins that contain a pore which allows the regulated flow of selected ions across the plasma membrane. Ion flux is passive and driven by the electrochemical gradient for the permeant ions. The channels are opened, or gated, by the binding of a neurotransmitter to an orthosteric site(s) that triggers a conformational change that results in the conducting state. Modulation of gating can occur by the binding of endogenous, or exogenous, modulators to allosteric sites. LGICs mediate fast synaptic transmission, on a millisecond time scale, in the nervous system and at the somatic neuromuscular junction. Such transmission involves the release of a neurotransmitter from a pre-synaptic neurone and the subsequent activation of post-synaptically located receptors that mediate a rapid, phasic, electrical signal (the excitatory, or inhibitory, post-synaptic potential). However, In addition to their traditional role in phasic neurotransmission, it is now established that some LGICs mediate a tonic form of neuronal regulation that results from the activation of extra-synaptic receptors by ambient levels of neurotransmitter. The expression of some LGICs by non-excitable cells is suggestive of additional functions.
By convention, the LGICs comprise the excitatory, cation-selective, nicotinic acetylcholine (Millar and Gotti, 2009; Changeux, 2010), 5-HT3 (Barnes et al., 2009; Walstab et al., 2010), ionotropic glutamate (Lodge, 2009; Traynelis et al., 2010) and P2X receptors (Jarvis and Khakh, 2009; Surprenant and North, 2009) and the inhibitory, anion-selective, GABAA (Olsen and Sieghart, 2008; Belelli et al., 2009) and glycine receptors (Lynch, 2009; Yevenes and Zeilhofer, 2011). The nicotinic acetylcholine, 5-HT3, GABAA and glycine receptors (and an additional zinc-activated channel) are pentameric structures and are frequently referred to as the Cys-loop receptors due to the presence of a defining loop of residues formed by a disulphide bond in the extracellular domain of their constituent subunits (Miller and Smart, 2010; Thompson et al., 2010). However, the prokaryotic ancestors of these receptors contain no such loop and the term pentameric ligand-gated ion channel (pLGIC) is gaining acceptance in the literature (Hilf and Dutzler, 2009). The ionotropic glutamate and P2X receptors are tetrameric and trimeric structures, respectively. Multiple genes encode the subunits of LGICs and the majority of these receptors are heteromultimers. Such combinational diversity results, within each class of LGIC, in a wide range of receptors with differing pharmacological and biophysical properties and varying patterns of expression within the nervous system and other tissues. The LGICs thus present attractive targets for new therapeutic agents with improved discrimination between receptor isoforms and a reduced propensity for off-target effects. The development of novel, faster screening techniques for compounds acting on LGICs (Dunlop et al., 2008) will greatly aid in the development of such agents.
Further Reading
Barnes NM, Hales TG, Lummis SCR, Peters JA (2009). The 5-HT3 receptor – the relationship between structure and function. Neuropharmacology56: 273–284.
Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW (2009). Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci29: 12757–12763.
Changeux J-P (2010). Allosteric receptors: from electric organ to cognition. Annu Rev Pharmacol Toxicol50: 1–38.
Dunlop J, Bowlby M, Peri R, Vasilyev D, Arias R (2008). High-throughput electrophysiology: an emerging paradigm for ion channel screening and physiology. Nat Rev Drug Discov7: 358–368.
Hilf RJ, Dutzler R (2009). A prokaryotic perspective on pentameric ligand-gated ion channel structure. Curr Opin Struct Biol19: 418–424.
Jarvis MF, Khakh BS (2009). ATP-gated P2X cation-channels. Neuropharmacology56: 230–236.
Lodge D (2009). The history of the pharmacology and cloning of ionotropic glutamate receptors and the development of idiosyncratic nomenclure. Neuropharmacology56: 6–21.
Lynch JW (2009). Native glycine receptors and their physiological roles. Neurpharmacology56: 303–309.
Millar NS, Gotti C (2009). Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology56: 237–246.
Miller PS, Smart TG (2010). Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci31: 161–174.
Olsen RW, Sieghart W (2009). International Union of Pharmacology. LXX. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev60: 243–260.
Surprenant A, North RA (2009). Signaling at purinergic P2X receptors. Annu Rev Physiol71: 333–359.
Thompson AJ, Lester HA, Lummis SCR (2010). The structural basis of function in Cys-loop receptors. Q Rev Biophys43: 449–499.
Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev62: 405–496.
Walstab J, Rappold G, Niesler B (2010). 5-HT3 receptors: role in disease and target of drugs. Pharmacol Ther128: 146–169.
Yevenes GE, Zeilhofer HU (2011). Allosteric modulation of glycine receptors. Br J Pharmacol164: 224–236.
5-HT3 (5-Hydroxytryptamine3)
Overview: The 5-HT3 receptor [nomenclature as agreed by the NC-IUPHAR Subcommittee on 5-hydroxytryptamine (serotonin) receptors (Hoyer et al., 1994; see also Peters et al., 2010)] is a ligand-gated ion channel of the Cys-loop family that includes the nicotinic acetylcholine, GABAA and strychnine-sensitive glycine receptors. The receptor exists as a pentamer of 4TM subunits that form an intrinsic cation selective channel (Barnes et al., 2009). Five human 5-HT3 receptor subunits have been cloned and homo-oligomeric assemblies of 5-HT3A and hetero-oligomeric assemblies of 5-HT3A and 5-HT3B subunits have been characterised in detail. The 5-HT3C (ENSG00000178084), 5-HT3D (ENSG00000186090) and 5-HT3E (ENSG00000186038) subunits (Karnovsky et al., 2003; Niesler et al., 2003), like the 5-HT3B subunit, do not form functional homomers, but are reported to assemble with the 5-HT3A subunit to influence its functional expression rather than pharmacological profile (Niesler et al., 2007; Holbrook et al., 2009; Walstab et al., 2010a). 5-HT3A, -C, -D, and -E subunits also interact with the chaperone RIC-3 which predominantly enhances the surface expression of homomeric 5-HT3A receptor (Walstab et al., 2010a). The co-expression of 5-HT3A and 5-HT3C-E subunits has been demonstrated in human colon (Kapeller et al., 2011). A recombinant hetero-oligomeric 5-HT3AB receptor has been reported to contain two copies of the 5-HT3A subunit and three copies of the 5-HT3B subunit in the order B-B-A-B-A (Barrera et al., 2005), but this is inconsistent with recent reports which show at least one A-A interface (Lochner and Lummis, 2010; Thompson et al., 2011b). The 5-HT3B subunit imparts distinctive biophysical properties upon hetero-oligomeric 5-HT3AB versus homo-oligomeric 5-HT3A recombinant receptors (Davies et al., 1999; Dubin et al., 1999; Hanna et al., 2000; Kelley et al., 2003; Stewart et al., 2003; Peters et al., 2005; Jensen et al., 2008), influences the potency of channel blockers, but generally has only a modest effect upon the apparent affinity of agonists, or the affinity of antagonists (Brady et al., 2001; but see Dubin et al., 1999; Das and Dillon, 2003; Deeb et al., 2009) which may be explained by the orthosteric binding site residing at an interface formed between 5-HT3A subunits (Lochner and Lummis, 2010; Thompson et al., 2011b). However, 5-HT3A and 5-HT3AB receptors differ in their allosteric regulation by some general anaesthetic agents, small alcohols and indoles (Solt et al., 2005; Rüsch et al., 2007; Hu and Peoples, 2008). The potential diversity of 5-HT3 receptors is increased by alternative splicing of the genes HTR3A and E (Hope et al., 1993; Bruss et al., 2000; Niesler et al., 2007, 2008; Niesler 2011). In addition, the use of tissue-specific promoters driving expression from different transcriptional start sites has been reported for the HTR3A, HTR3B, HTR3D and HTR3E genes, which could result in 5-HT3 subunits harbouring different N-termini (Tzvetkov et al., 2007; Jensen et al., 2008; Niesler, 2011). To date, inclusion of the 5-HT3A subunit appears imperative for 5-HT3 receptor function.
| Nomenclature | 5-HT3 |
|---|---|
| Former names | M |
| Ensembl ID | 5-HT3A ENSG00000166736, 5-HT3B ENSG00000149305 |
| Selective agonists (pEC50) | SR57227A (6.4), 3-chlorophenyl-biguanide (5.4–5.8), 2-methyl-5-HT (5.5–5.6), 1-phenylbiguanide (4.1) |
| Selective antagonists (pKi) | (S)-Zacopride (9.0), granisetron (8.6–8.8), tropisetron (8.5–8.8), ondansetron (7.8–8.3) |
| Channel blockers (pIC50) | TMB-8 (5.4), diltiazem (4.1–4.8), picrotoxinin (4.9 + 5-HT3B = 4.2), bilobalide (3.3 + 5-HT3B = 2.5); ginkgolide B (3.1 + 5-HT3B > 3.0) |
| Radioligands (KD) | [3H]Ramosetron (0.15 nM), [3H]granisetron (1.2 nM), [3H](S)-zacopride (2.0 nM), [3H]GR65630 (2.6 nM), [3H]LY278584 (3 nM) |
| Functional characteristics | γ = 0.4-0.8 pS [+ 5-HT3B, γ = 16 pS]; inwardly rectifying current [+ 5-HT3B, rectification reduced]; nH 2-3 [+ 5-HT3B 1-2]; relative permeability to divalent cations reduced by co-expression of the 5-HT3B subunit |
Quantitative data in the table refer to homo-oligomeric assemblies of the human 5-HT3A subunit, or the receptor native to human tissues. Significant changes introduced by co-expression of the 5-HT3B subunit are indicated in parenthesis. Methadone, although not a selective antagonist, displays multimodal and subunit-dependent antagonism of 5-HT3 receptors (Deeb et al., 2009). Similarly, TMB-8, diltiazem, picrotoxin, bilobalide and ginkgolide B are not selective for 5-HT3 receptors (e.g. Thompson et al., 2011a). The anti-malarial drugs mefloquine and quinine exert a modestly more potent block of 5-HT3A versus 5-HT3AB receptor-mediated responses (Thompson and Lummis, 2008). Varenicline, know better as a partial agonist of nicotinic acetylcholine α4β2 receptors, is also an agonist of the 5-HT3A receptor (Lummis et al. 2011). Human (Belelli et al., 1995; Miyake et al., 1995), rat (Isenberg et al., 1993), mouse (Maricq et al., 1991), guinea-pig (Lankiewicz et al., 1998) ferret (Mochizuki et al., 2000) and canine (Jensen et al., 2006) orthologues of the 5-HT3A receptor subunit have been cloned that exhibit intraspecies variations in receptor pharmacology. Notably, most ligands display significantly reduced affinities at the guinea-pig 5-HT3 receptor in comparison with other species. In addition to the agents listed in the table, native and recombinant 5-HT3 receptors are subject to allosteric modulation by extracellular divalent cations, alcohols, several general anaesthetics and 5-hydroxy- and halide-substituted indoles (see reviews by Parker et al., 1996; Thompson and Lummis, 2006, 2007; Walstab et al., 2010b).
Abbreviations: GR65630, 3-(5-methyl-1H-imidazol-4-yl)-1-(1-methyl-1H-indol-3-yl)-1-propanone; LY278584, 1-methyl-N-(8-methyl-8-azabicyclo[3.2.1]oct-3-yl)-1H-indazole- 3-carboxamide; SR57227A, 4-amino-(6-chloro-2-pyridyl)-1 piperidine hydrochloride, TMB-8, 8-(diethylamine)octyl-3,4,5-trimethoxybenzoate
Further Reading
Barnes NM, Hales TG, Lummis SCR, Peters JA (2009). The 5-HT3 receptor – the relationship between structure and function. Neuropharmacology56: 273–284.
Chameau P, Van Hooft JA (2006). Serotonin 5-HT3 receptors in the central nervous system. Cell Tissue Res326: 573–581.
Costall B, Naylor RJ (2004). 5-HT3 receptors. Curr Drug Targets CNS Neurol Disord3: 27–37.
Engleman EA, Rodd ZA, Bell RL, Murphy JM (2008). The role of 5-HT3 receptors in drug abuse and as a target for pharmacotherapy. CNS Neurol Disord Drug Targets7: 454–467.
Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ et al. (1994). International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin). Pharmacol Rev: 46, 157–203.
Jensen AA, Davies PA, Bräuner-Osborne H, Krzywkowski K (2008). 3B but which 3B? And that's just one of the questions: the heterogeneity of human 5-HT3 receptors. Trends Pharmacol Sci29: 437–444.
Machu TK (2011). Therapeutics of 5-HT3 receptor antagonists: current uses and future directions. Pharmacol Ther130: 338–347.
Modica MN, Pittalà V, Romeo G, Salerno L, Siracusa MA (2010). Serotonin 5-HT3 and 5-HT4 ligands: an update of medicinal chemistry research in the last few years. Curr Med Chem17: 334–362.
Niesler B (2011). 5-HT3 receptors: potential of individual isoforms for personalised therapy.Curr Opin Pharmacol11: 81–86.
Niesler B, Kapeller J, Hammer C, Rappold G (2008). Serotonin type 3 receptor genes: HTR3A, B, C, D, E. Pharmacogenomics9: 501–514.
Parker RM, Bentley KR, Barnes NM (1996). Allosteric modulation of 5-HT3 receptors: focus on alcohols and anaesthetic agents. Trends Pharmacol Sci17: 95–99.
Peters JA, Hales TG, Lambert JJ (2005). Molecular determinants of single channel conductance and ion selectivity in the Cys-loop transmitter-gated ion channels: insights from the 5-HT3 receptor. Trends Pharmacol Sci26: 587–594.
Peters JA, Barnes NM, Hales TG, Lummis SCR (2010). 5-HT3 receptors, introductory chapter. IUPHAR database (IUPHAR-DB), http://www.iuphar-db.org/IC/FamilyIntroductionForward?familyId = 2
Thompson AJ, Lummis SCR (2006). 5-HT3 receptors. Curr Pharm Des12: 3615–3630.
Thompson AJ, Lummis SCR (2007). The 5-HT3 receptor as a therapeutic target. Expert Opin Ther Targets11: 527–540.
Thompson AJ, Lester HA, Lummis SCR (2010). The structural basis of function in Cys-loop receptors. Q Rev Biophys43: 449–499.
Walstab J, Rappold G, Niesler B (2010b). 5-HT3 receptors: role in disease and target of drugs. Pharmacol Ther128: 146–169.
Yaakob N, Malone DT, Exintaris B, Irving HR (2011). Heterogeneity amongst 5-HT3 receptor subunits: is this significant? Curr Mol Med11: 57–68.
References
- Barrera NP, et al. Proc Natl Acad Sci U S A. 2005;102:12595–12600. doi: 10.1073/pnas.0503253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, et al. Mol Pharmacol. 1995;48:1054–1062. [PubMed] [Google Scholar]
- Brady CA, et al. Neuropharmacology. 2001;41:282–284. doi: 10.1016/s0028-3908(01)00074-0. [DOI] [PubMed] [Google Scholar]
- Bruss M, et al. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:392–401. doi: 10.1007/s002100000342. [DOI] [PubMed] [Google Scholar]
- Das P, Dillon GH. Brain Res Mol Brain Res. 2003;119:207–212. doi: 10.1016/j.molbrainres.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Davies PA, et al. Nature. 1999;397:359–363. doi: 10.1038/16941. [DOI] [PubMed] [Google Scholar]
- Deeb TZ, et al. Mol Pharmacol. 2009;75:908–917. doi: 10.1124/mol.108.053322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin A, et al. J Biol Chem. 1999;274:30799–30810. doi: 10.1074/jbc.274.43.30799. [DOI] [PubMed] [Google Scholar]
- Hanna MC, et al. J Neurochem. 2000;75:240–247. doi: 10.1046/j.1471-4159.2000.0750240.x. [DOI] [PubMed] [Google Scholar]
- Hope AG, et al. Eur J Pharmacol. 1993;245:187–192. doi: 10.1016/0922-4106(93)90128-v. [DOI] [PubMed] [Google Scholar]
- Holbrook JD, et al. J Neurochem. 2009;108:384–396. doi: 10.1111/j.1471-4159.2008.05775.x. [DOI] [PubMed] [Google Scholar]
- Hu XQ, Peoples RW. J Biol Chem. 2008;283:6826–6831. doi: 10.1074/jbc.M707571200. [DOI] [PubMed] [Google Scholar]
- Isenberg KE, et al. Neuroreport. 1993;18:121–124. doi: 10.1097/00001756-199311180-00006. [DOI] [PubMed] [Google Scholar]
- Jensen TN, et al. Eur J Pharmacol. 2006;538:23–31. doi: 10.1016/j.ejphar.2006.03.050. [DOI] [PubMed] [Google Scholar]
- Kapeller J, et al. J Comp Neurol. 2011;519:420–432. doi: 10.1002/cne.22525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnovsky AM, et al. Gene. 2003;319:137–148. doi: 10.1016/s0378-1119(03)00803-5. [DOI] [PubMed] [Google Scholar]
- Kelley SP, et al. Nature. 2003;424:321–324. doi: 10.1038/nature01788. [DOI] [PubMed] [Google Scholar]
- Lankiewicz S, et al. Mol Pharmacol. 1998;53:202–212. doi: 10.1124/mol.53.2.202. [DOI] [PubMed] [Google Scholar]
- Lochner M, Lummis SCR. Biophys J. 2010;98:1494–1502. doi: 10.1016/j.bpj.2009.12.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lummis SCR, et al. J Pharmacol Exp Ther. 2011;339:125–131. doi: 10.1124/jpet.111.185306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricq AV, et al. Science. 1991;254:432–437. doi: 10.1126/science.1718042. [DOI] [PubMed] [Google Scholar]
- Miyake A, et al. Mol Pharmacol. 1995;48:407–416. [PubMed] [Google Scholar]
- Mochizuki S, et al. Eur J Pharmacol. 2000;399:97–106. doi: 10.1016/s0014-2999(00)00371-x. [DOI] [PubMed] [Google Scholar]
- Niesler B, et al. Gene. 2003;310:101–111. doi: 10.1016/s0378-1119(03)00503-1. [DOI] [PubMed] [Google Scholar]
- Niesler B, et al. Mol Pharmacol. 2007;72:8–17. doi: 10.1124/mol.106.032144. [DOI] [PubMed] [Google Scholar]
- Rüsch D, et al. J Pharmacol Exp Ther. 2007;321:1069–1074. doi: 10.1124/jpet.106.118752. [DOI] [PubMed] [Google Scholar]
- Solt K, et al. J Pharmacol Exp Ther. 2005;315:771–776. doi: 10.1124/jpet.105.090621. [DOI] [PubMed] [Google Scholar]
- Stewart A, et al. Neuropharmacology. 2003;44:214–223. doi: 10.1016/s0028-3908(02)00376-3. [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Lummis SCR. Br J Pharmacol. 2008;153:1686–1696. doi: 10.1038/bjp.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, et al. Neuropharmacology. 2011a;60:488–495. doi: 10.1016/j.neuropharm.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, et al. J Physiol. 2011b;589:4243–4257. doi: 10.1113/jphysiol.2011.208439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzvetkov MV, et al. Gene. 2007;386:52–62. doi: 10.1016/j.gene.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Walstab J, et al. J Biol Chem. 2010a;285:26956–26965. doi: 10.1074/jbc.M110.122838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Acetylcholine (nicotinic)
Overview: Nicotinic acetylcholine receptors are members of the Cys-loop family of transmitter-gated ion channels that includes the GABAA, strychnine-sensitive glycine and 5-HT3 receptors (Sine and Engel, 2006; Albuquerque et al., 2009; Millar and Gotti, 2009; Taly et al., 2009, Wu and Lukas, 2011). All nicotinic receptors are pentamers in which each of the five subunits contains four α-helical transmembrane domains. Genes (Ensembl family ID ENSF00000000049) encoding a total of 17 subunits (α1-10, β1-4, γ, δ and ε) have been identified (Kalamida et al., 2007). All subunits with the exception of α8 (present in avian species) have been identified in mammals. All α subunits possess two tandem cysteine residues near to the site involved in acetylcholine binding, and subunits not named α lack these residues (Millar and Gotti, 2009). The orthosteric ligand binding site is formed by residues within at least three peptide domains on the α subunit (principal component), and three on the adjacent subunit (complementary component). nAChRs contain several allosteric modulatory sites. One such site, for positive allosteric modulators (PAMs) and allosteric agonists, has been proposed to reside within an intrasubunit cavity between the four transmembrane domains (Young et al., 2008; Gill et al., 2011; see also Hibbs and Gouaux, 2011). The high resolution crystal structure of the molluscan acetylcholine binding protein, a structural homologue of the extracellular binding domain of a nicotinic receptor pentamer, in complex with several nicotinic receptor ligands (e.g., Celie et al., 2004) and the crystal structure of the extracellular domain of the α1 subunit bound to α-bungarotoxin at 1.94 Å resolution (Dellisanti et al., 2007), has revealed the orthosteric binding site in detail (reviewed Sine and Engel, 2006; Kalamida et al., 2007; Changeux and Taly, 2008; Rucktooa et al., 2009). Nicotinic receptors at the somatic neuromuscular junction of adult animals have the stoichiometry (α1)2β1δε, whereas an extrajunctional (α1)2β1γδ receptor predominates in embryonic and denervated skeletal muscle and other pathological states. Other nicotinic receptors are assembled as combinations of α(2-6) and β(2-4) subunits. For α2, α3, α4 and β2 and β4 subunits, pairwise combinations of α and β (e.g., α3β4, α4β2) are sufficient to form a functional receptor in vitro, but far more complex isoforms may exist in vivo (reviewed by Gotti et al., 2006, 2009, Millar and Gotti, 2009). There is strong evidence that the pairwise assembly of some α and β subunits can occur with variable stoichiometry [e.g., (α4)2(β2)2, or (α4)3(β2)2] which influences the biophysical and pharmacological properties of the receptor (Millar and Gotti, 2009). α5 and β3 subunits lack function when expressed alone, or pairwise, but participate in the formation of functional hetero-oligomeric receptors when expressed as a third subunit with another α and β pair [e.g., α4α5αβ2, α4αβ2β3, α5α6β2, see Millar and Gotti (2009) for further examples]. The α6 subunit can form a functional receptor when co-expressed with β4 in vitro, but more efficient expression ensues from incorporation of a third partner, such as β3 (Yang et al., 2009). The α7, α8, and α9 subunits form functional homo-oligomers, but can also combine with a second subunit to constitute a hetero-oligomeric assembly (e.g., α7β2 and α9α10). For functional expression of the α10 subunit, co-assembly with α9 is necessary. The latter, along with the α10 subunit, appears to be largely confined to cochlear and vestibular hair cells. Comprehensive listings of nicotinic receptor subunit combinations identified from recombinant expression systems, or in vivo, are given in Millar and Gotti (2009). In addition, numerous proteins interact with nicotinic ACh receptors modifying their assembly, trafficking to and from the cell surface, and activation by ACh (reviewed by Millar, 2008; Araud et al., 2010; Jones et al., 2010).
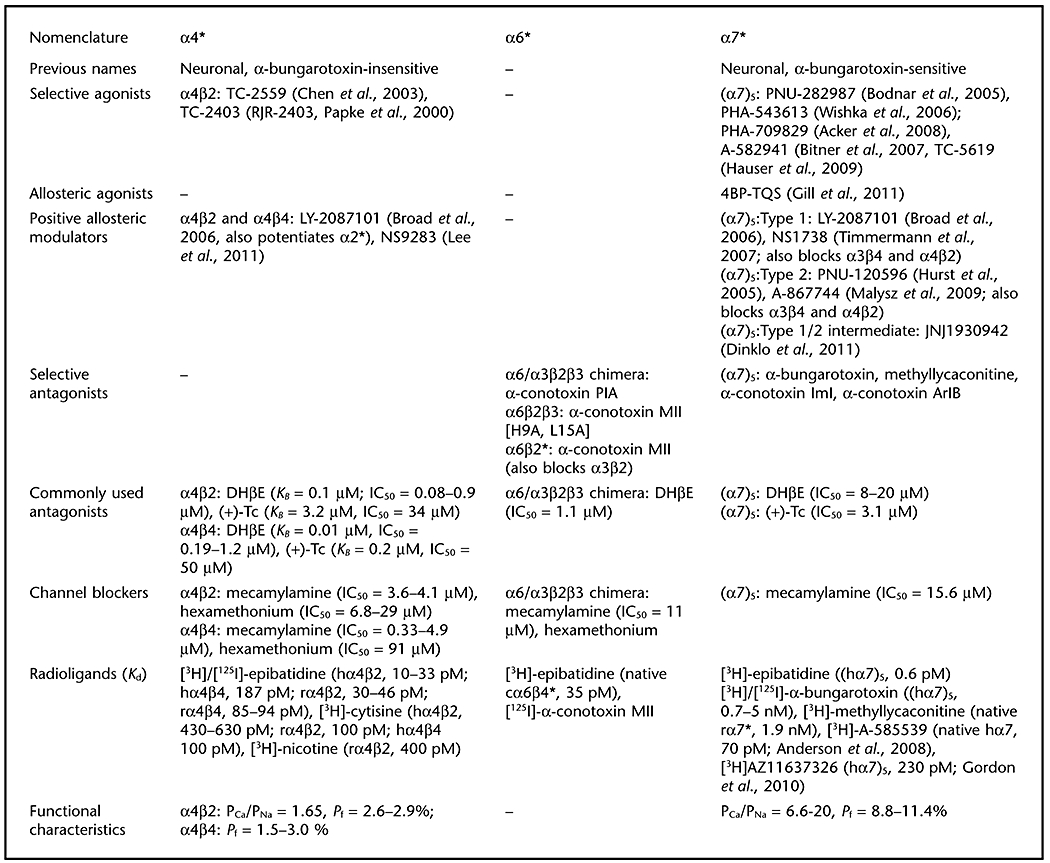
The nicotinic receptor subcommittee of NC-IUPHAR has recommended a nomenclature and classification scheme for nicotinic acetylcholine (nACh) receptors based on the subunit composition of known, naturally- and/or heterologously-expressed nACh receptor subtypes (Lukas et al., 1999). Headings for this table reflect abbreviations designating nACh receptor subtypes based on the predominant α subunit contained in that receptor subtype. An asterisk following the indicated α subunit denotes that other subunits are known to, or may, assemble with the indicated α subunit to form the designated nACh receptor subtype(s). Where subunit stoichiometries within a specific nACh receptor subtype are known, numbers of a particular subunit larger than 1 are indicated by a subscript following the subunit (enclosed in parentheses – see also Collingridge et al., 2009).
| Nomenclature | α1* | α2* | α3* |
|---|---|---|---|
| Previous names | Muscle-type, muscle | – | Autonomic, ganglionic |
| Selective agonists | Succinylcholine (selective for (α1)2β1γδ) | – | – |
| Positive allosteric modulators | – | LY-2087101 (Broad et al., 2006, also potentiates α4β2 and α4β4) | – |
| Selective antagonists | Waglerin-1 (selective for α(1)2β1δε), α-bungarotoxin, α-conotoxin GI, α-conotoxin MI, pancuronium | – | α3β2: α-conotoxin MII (also blocks α6-containing), α-conotoxin-GIC, α-conotoxin PnIA, α-conotoxin TxIA |
| α3β4: α-conotoxin AuIB | |||
| Commonly used antagonists | (α1)2β1γδ and (α1)2β1δε: α-bungarotoxin, > pancuronium > vecuronium > rocuronium > (+)-Tc (IC50 = 43–82 nM) | α2β2: DHβE (KB = 0.9 µM), (+)-Tc (KB = 1.4 µM) | α3β2: DHβE (KB = 1.6 µM, IC50 = 2.0 µM), (+)-Tc (KB = 2.4 µM) |
| α2β4: DHβE (KB = 3.6 µM), (+)-Tc (KB = 4.2 µM) | α3β4: DHβE (KB = 19 µM, IC50 = 26 µM), (+)-Tc (KB = 2.2 µM) | ||
| Channel blockers | α(1)2β1δε and α(1)2β1yδ: gallamine (IC50∼ 1 µM) | mecamylamine, hexamethonium | α3β2: mecamylamine (IC50 = 7.6 µM), hexamethonium |
| α(1)2β1δε: mecamylamine (IC50∼ 1.5 µM) | α3β4: mecamylamine (IC50 = 0.39 µM), hexamethonium | ||
| Radioligands (Kd) | [3H]/[125I]-α-bungarotoxin | [3H]/[125I]-epibatidine (hα2β4, 42 pM; rα2β2, 10–21 pM; rα2β4, 84–87 pM), [3H]-cytisine | [3H]/[125I]-epibatidine (hα3β2, 7 pM; hα3β4, 230 pM; rα3β2, 14–34 pM, rα3β4, 290–304 pM), [3H]-cytisine |
| Functional characteristics | α(1)2βγδ: PCa/PNa = 0.16–0.2, Pf = 2.1–2.9%; α(1)2βδε: PCa/PNa = 0.65–1.38, Pf = 4.1–7.2% | α2β2: PCa/PNa∼ 1.5 | α3β2: PCa/PNa = 1.5; α3β4: PCa/PNa = 0.78–1.1, Pf = 2.7–4.6% |
 |
| Nomenclature | α8* (avian) | α9* |
|---|---|---|
| Previous names | Neuronal, α-bungarotoxin-sensitive | – |
| Selective agonists | – | – |
| Selective antagonists | – | (α9)5: α-bungarotoxin, strychnine, nicotine, muscarine |
| α9α10: α-contoxin RgIA, α-bungarotoxin, strychnine, nicotine, muscarine | ||
| Commonly used antagonists | (α8)5: α-bungarotoxin > atropine ≥ (+)-Tc ≥ strychnine | (α9)5: α-bungarotoxin > methyllycaconitine > strychnine ∼ tropisetron > (+)-Tc |
| α9α10: α-bungarotoxin > tropisetron = strychnine > (+)-Tc | ||
| Channel blockers | – | – |
| Radioligands (Kd) | [3H]-epibatidine ((α8)5, 0.2 nM) | [3H]-methyllycaconitine (hα9α10, 7.5 nM) |
| [3H]/[125I]-α-bungarotoxin (native α8*, 5.5 nM) | [3H]/[125I]-α-bungarotoxin | |
| Functional characteristics | – | (α9)5: PCa/PNa = 9; α9α10: PCa/PNa = 9, Pf = 22% |
Commonly used agonists of nicotinic acetylcholine receptors that display limited discrimination in functional assays between receptor subtypes include A-85380, cytisine, DMPP, epibatidine, nicotine and the natural transmitter, ACh. A summary of their profile across differing receptors is provided in Gotti et al. (2006) and quantitative data across numerous assay systems are summarised in Jensen et al. (2005). Quantitative data presented in the table for commonly used antagonists and channel blockers for human receptors studied under voltage-clamp are from Buisson et al., 1996, Chavez-Noriaga et al., (1997), Papke et al. (2001, 2008), Paul et al. (2002) and Wu et al. (2006). Type I PAMs increase peak agonist-evoked responses but have little, or no, effect on the rate of desensitization of α7 nicotinic ACh receptors whereas type II PAMs also cause a large reduction in desensitization (reviewed by Williams et al., 2011).
Abbreviations: 4BP-TQS, 4-(4-bromophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide; A-582941, 2-methyl-5-(6-phenyl-pyridazin-3-yl)-octahydro-pyrrolo[3,4-c]pyrrole; A-585539, (1S,4S)-2,2-dimethyl-5-(6-phenylpyridazin-3-yl)-5-aza-2-azaniabicyclo[2.2.1]heptane; A-867744, 4-(5-(4-chlorophenyl)-2-methyl-3-propionyl-1H-pyrrol-1-yl)benzenesulfonamide; ABT-594, (R)-5-(2-azetidinylmethoxy)-2-chloropyridine; ACh, acetylcholine; AZ11637326, (5′-(2-fluoro[3,4,5(-3)H3]phenyl)-spiro[1-azabicyclo [2.2.2]octane-3,2′(3′H)-furo[2,3-b]pyridine, DHβE, dihydro-β-erythroidine; DMPP, 1,1-dimethyl-4-phenylpiperazinium; JNJ-1930942, 2-[[4-fluoro-3-(trifluoromethyl)phenyl]amino]-4-(4-pyridinyl)-5-thiazolemethanol; LY-2087101, see Broad et al. (2006) for structure; NS1738, 1-(5-chloro-2-hydroxy-phenyl)-3-(2-chloro-5-trifluoromethyl-phenyl)-urea; NS9283, 3-(3-(pyridine-3-yl)-1,2,4-oxadiazol-5-yl)benzonitrile; PHA-543613, N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide; PHA-709829, N-[(3R,5R)-1-azabicyclo[3.2.1]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide; PNU-120596, 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea; PNU-282987N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride; PSAB-OFP, (R)-(-)-5′phenylspiro[1-azabicyclo[2.2.2] octane-3,2′-(3′H)furo[2,3-b]pyridine; TC-2403 (RJR-2403), (E)-N-methyl-4-(3-pyridinyl)-3-butene-1-amine;TC-2559, (E)-N-methyl-4-[3-(5-ethoxypyridin)yl]-3-buten-1-amine; TC-5619, N-[2-(pyridin-3-ylmethyl)-1-azabicyclo[2.2.2]oct-3-yl]-1-benzofuran-2-carboxamide; (+)-Tc, (+)-tubocurarine
Further Reading
Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev89: 73–120.
Araud T, Wonnacott S, Bertrand D (2010). Associated proteins: the universal toolbox controlling ligand gated ion channel function. Biochem Pharmacol80: 160–169.
Arneric SP, Holladay M, Williams M (2007). Neuronal nicotinic receptors: a perspective on two decades of drug discovery research. Biochem Pharmacol74: 1092–1101.
Arias HR (2010). Positive and negative modulation of nicotinic receptors. Adv Protein Chem Struct Biol80: 153–203.
Balfour DJK (2009). The neuronal pathways mediating the behavioral and addictive properties of nicotine. Handb Exp Pharmacol192: 209–233.
Benowitz NL (2009). Pharmacology of nicotine: addiction, smoking-induced disease, and therapeutics. Annu Rev Pharmacol Toxicol49: 57–71.
Champtiaux N, Changeux J-P (2004). Knockout and knockin mice to investigate the role of nicotinic receptors in the central nervous system. Prog Brain Res145: 235–251.
Changeux JP, Taly A (2008). Nicotinic receptors, allosteric proteins and medicine. Trends Mol Med14: 93–102.
Collingridge GL, Olsen RW, Peters J, Spedding M (2009). A nomenclature for ligand-gated ion channels. Neuropharmacology56: 2–5.
Dajas-Bailador F, Wonnacott S (2004). Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci25: 317–324.
D'hoedt D, Bertrand D (2009). Nicotinic acetylcholine receptors: an overview on drug discovery. Expert Opin Ther Targets13: 395–411.
Faghih R, Gopalakrishnan M, Briggs CA (2008). Allosteric modulators of the α7 nicotinic acetylcholine receptor. J Med Chem51: 701–712.
Fucile S (2004). Ca2+-permeability of nicotinic acetylcholine receptors. Cell Calcium35: 1–8.
Gotti C, Zoli M, Clementi F (2006). Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci27: 482–491.
Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I et al. (2009). Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol78: 703–711.
Hogg RC, Bertrand D (2004). Nicotinic acetylcholine receptors as drug targets. Curr Drug Targets CNS Neurol Disord3: 123–130.
Hogg RC, Raggenbass M, Bertrand D (2003). Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol147: 1–46.
Jensen AA, Frøland B, Liljefors T, Krogagaard-Larsen P. (2005). Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem48: 4705–4745.
Jones AK, Buckingham SD, Sattelle DB (2010). Proteins interacting with nicotinic acetylcholine receptors: expanding functional and therapeutic horizons. Trends Pharmacol Sci31: 455–462.
Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K et al. (2007). Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J274: 3799–3845.
Letchworth SR, Whiteaker P (2011). Progress and challenges in the study of α6-containing nicotinic acetylcholine receptors. Biochem Pharmacol82: 862–872.
Lukas RJ, Changeux J-P, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK et al. (1999). International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev51: 397–401.
Millar NS (2008). RIC-3: a nicotinic acetylcholine receptor chaperone. Br J Pharmacol153 (Suppl. 1): S177–S183.
Millar NS, Gotti C (2009). Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology56: 237–246.
Millar NS, Harkness PC (2008). Assembly and trafficking of nicotinic acetylcholine receptors. Mol Membr Biol25: 279–292.
Miwa JM, Freedman R, Lester HA (2011). Neural systems governed by nicotinic acetylcholine receptors: emerging hypotheses. Neuron70: 20–33.
Olivera BM, Quik M, Vincler M, MacIntosh JM (2008). Subtype-selective conopeptides targeted to nicotinic receptors. Channels (Austin)2: 143–152.
Pandya A, Yakel JL (2011). Allosteric modulators of the α4β2 subtype of neuronal nicotinic acetylcholine receptors. Biochem Pharmacol82: 862–872.
Romanelli MN, Gratteri P, Guandalini L, Martini E, Bonaccini C, Gualtieri F (2007). Central nicotinic receptors: structure, function, ligands, and therapeutic potential. ChemMedChem2: 746–767.
Rucktooa P, Smit AB, Sixma TK (2009). Insight in nAChR subtype selectivity from AChBP crystal structures. Biochem Pharmacol78: 777–787.
Sharma G, Vijayaraghavan S (2008). Nicotinic receptors containing the α7 subunit: a model for rational drug design. Curr Med Chem15: 2921–2932.
Steinlein OK, Bertrand D (2008). Neuronal nicotinic acetylcholine receptors: from the genetic analysis to neurological diseases. Biochem Pharmacol76: 1175–1183.
Sine SM, Engel AG (2006). Recent advances in Cys-loop receptor structure and function. Nature440: 448–455.
Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP (2009). Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov8: 733–750.
Tsetlin V, Hucho F (2009). Nicotinic acetylcholine receptors at atomic resolution. Curr Opin Pharmacol9: 306–310.
Tsetlin V, Utkin Y, Kasheverov I (2009). Polypeptide and peptide toxins, magnifying lenses for binding sites in nicotinic acetylcholine receptors. Biochem Pharmacol78: 720–731.
Tsetlin V, Kuzmin D, Kasheverov I (2011). Assembly of nicotinic and other Cys-loop receptors. J Neurochem116: 734–741.
Unwin N (2005). Refined structure of the nicotinic acetylcholine receptor at 4Å resolution. J Mol Biol346: 967–989.
Williams DK, Wang J, Papke RL (2011). Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem Pharmacol82: 915–930.
Yang KC, Jin GZ, Wu J (2009). Mysterious α6-containing nAChRs: function, pharmacology, and pathophysiology. Acta Pharmacol Sin30: 740–751.
Wu J, Lukas RJ (2011). Naturally expressed nicotinic acetylcholine receptor subtypes. Biochem Pharmacol82: 800–807.
Zouridakis M, Zisimopoulou P, Poulas K, Tzartos SJ (2009). Recent advances in understanding the structure of nicotinic acetylcholine receptors. IUBMB Life61: 407–423.
References
- Acker BA, et al. Bioorg Med Chem Lett. 2008;18:3611–3625. doi: 10.1016/j.bmcl.2008.04.070. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, et al. J Pharmacol Exp Ther. 2008;324:179–187. doi: 10.1124/jpet.107.130062. [DOI] [PubMed] [Google Scholar]
- Bitner RS, et al. J Neurosci. 2007;27:10578–10587. doi: 10.1523/JNEUROSCI.2444-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AL, et al. J Med Chem. 2005;48:905–908. doi: 10.1021/jm049363q. [DOI] [PubMed] [Google Scholar]
- Broad LM, et al. J Pharmacol Exp Ther. 2006;318:1108–1117. doi: 10.1124/jpet.106.104505. [DOI] [PubMed] [Google Scholar]
- Buisson B, et al. J Neurosci. 1996;16:7880–7891. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celie PH, et al. Neuron. 2004;25:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriaga LE, et al. J Pharmacol Exp Ther. 1997;280:346–356. [PubMed] [Google Scholar]
- Chen Y, et al. Neuropharmacology. 2003;45:334–344. doi: 10.1016/s0028-3908(03)00189-8. [DOI] [PubMed] [Google Scholar]
- Dellisanti CD, et al. Nat Neurosci. 2007;10:953–962. doi: 10.1038/nn1942. [DOI] [PubMed] [Google Scholar]
- Dinklo T, et al. J Pharmacol Exp Ther. 2011;336:560–574. doi: 10.1124/jpet.110.173245. [DOI] [PubMed] [Google Scholar]
- Gill JK, et al. Proc Natl Acad Sci U S A. 2011;108:5867–5872. doi: 10.1073/pnas.1017975108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JC, et al. Eur J Pharmacol. 2010;645:63–69. doi: 10.1016/j.ejphar.2010.07.035. [DOI] [PubMed] [Google Scholar]
- Hauser TA, et al. Biochem Pharmacol. 2009;78:803–812. doi: 10.1016/j.bcp.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst RS, et al. J Neurosci. 2005;25:4396–4405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, et al. Biochem Pharmacol. 2011;82:959–966. doi: 10.1016/j.bcp.2011.06.044. [DOI] [PubMed] [Google Scholar]
- Malysz J, et al. J Pharmacol Exp Ther. 2009;330:257–267. doi: 10.1124/jpet.109.151886. [DOI] [PubMed] [Google Scholar]
- Papke RL, et al. J Neurochem. 2000;75:204–216. doi: 10.1046/j.1471-4159.2000.0750204.x. [DOI] [PubMed] [Google Scholar]
- Papke RL, et al. J Pharmacol Exp Ther. 2001;297:646–656. [PubMed] [Google Scholar]
- Papke RL, et al. Neuropharmacology. 2008;54:1189–1200. doi: 10.1016/j.neuropharm.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M, et al. Anesth Analg. 2002;94:597–603. doi: 10.1097/00000539-200203000-00022. [DOI] [PubMed] [Google Scholar]
- Timmermann DB, et al. J Pharmacol Exp Ther. 2007;323:294–307. doi: 10.1124/jpet.107.120436. [DOI] [PubMed] [Google Scholar]
- Wishka DG, et al. J Med Chem. 2006;49:4425–4436. doi: 10.1021/jm0602413. [DOI] [PubMed] [Google Scholar]
- Wu J, et al. J Physiol. 2006;576:103–118. doi: 10.1113/jphysiol.2006.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GT, et al. Proc Natl Acad Sci U S A. 2008;105:14686–14691. doi: 10.1073/pnas.0804372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
GABAA (γ-aminobutyric acid)
Overview: The GABAA receptor is a ligand-gated ion channel of the Cys-loop family that includes the nicotinic acetylcholine, 5-HT3 and strychnine-sensitive glycine receptors. GABAA receptor-mediated inhibition within the CNS occurs by fast synaptic transmission, sustained tonic inhibition and temporally intermediate events that have been termed ‘GABAA, slow’ (Campogna and Pearce, 2011). GABAA receptors exist as pentamers of 4TM subunits that form an intrinsic anion selective channel. Sequences of six α, three β, three γ, one δ, three ρ, one ε, one π and one θ GABAA receptor subunits (Ensembl gene family ID ENSF00000000053) have been reported in mammals (Korpi et al., 2002; Whiting, 2003; Sieghart, 2006; Olsen and Sieghart, 2008, 2009). The π-subunit is restricted to reproductive tissue. Alternatively spliced versions of many subunits exist (e.g.α4- and α6- (both not functional) α5-, β2-, β3- and γ2), along with RNA editing of the α3 subunit (Daniel and Ohman, 2009). The three ρ-subunits, (ρ1-3) function as either homo- or hetero-oligomeric assemblies (Zhang et al., 2001; Chebib, 2004). Receptors formed from ρ-subunits, because of their distinctive pharmacology that includes insensitivity to bicuculline, benzodiazepines and barbiturates, have sometimes been termed GABAC receptors (Zhang et al., 2001), but they are classified as GABAA receptors by NC-IUPHAR on the basis of structural and functional criteria (Barnard et al., 1998; Olsen and Sieghart, 2008, 2009).
Many GABAA receptor subtypes contain α-, β- and γ-subunits with the likely stoichiometry 2α.2β.1γ (Korpi et al., 2002, Olsen and Sieghart, 2008). It is thought that the majority of GABAA receptors harbour a single type of α- and β-subunit variant. The α1β2γ2 hetero-oligomer constitutes the largest population of GABAA receptors in the CNS, followed by the α2β3γ2 and α3β3γ2 isoforms. Receptors that incorporate the α4- α5-or α6-subunit, or the β1-, γ1-, γ3-, δ-, ε- and θ-subunits, are less numerous, but they may nonetheless serve important functions. For example, extrasynaptically located receptors that contain α6- and δ-subunits in cerebellar granule cells, or an α4- and δ-subunit in dentate gyrus granule cells and thalamic neurones, mediate a tonic current that is important for neuronal excitability in response to ambient concentrations of GABA (see Mody and Pearce, 2004; Semyanov et al., 2004; Farrant and Nusser, 2005; Belelli et al., 2009). GABA binding occurs at the β+/α- subunit interface and the homologous γ+/α- subunits interface creates the benzodiazepine site. A second site for benzodiazepine binding has recently been postulated to occur at the α+/β- interface (Ramerstorfer et al., 2011; reviewed by Sigel and Lüscher, 2011). The particular α-and γ-subunit isoforms exhibit marked effects on recognition and/or efficacy at the benzodiazepine site. Thus, receptors incorporating either α4- or α6-subunits are not recognised by ‘classical’ benzodiazepines, such as flunitrazepam (but see You et al., 2010). The trafficking, cell surface expression, internalisation and function of GABAA receptors and their subunits are discussed in detail in several recent reviews (Chen and Olsen, 2007; Jacob et al., 2008; Lüscher et al., 2011; Vithlani et al., 2011) but one point worthy of note is that receptors incorporating the γ2 subunit (except when associated with α5) cluster at the postsynaptic membrane (but may distribute dynamically between synaptic and extrasynaptic locations), whereas as those incorporating the δ subunit appear to be exclusively extrasynaptic.
NC-IUPHAR (Barnard et al. 1998; Olsen and Sieghart, 2008) class GABAA receptors according to their subunit structure, pharmacology and receptor function. Currently, eleven native GABAA receptors are classed as conclusively identified (i.e., α1β2γ2, α1βγ2, α3βγ2, α4βγ2, α4β2δ, α4β3δ, α5βγ2, α6βγ2, α6β2δ, α6β3δ and ρ) with further receptor isoforms occurring with high probability, or only tentatively (Olsen and Sieghart, 2008, 2009). It is beyond the scope of this Guide to discuss the pharmacology of individual GABAA receptor isoforms in detail; such information can be gleaned in the reviews by Barnard et al. (1998), Frolund et al. (2002), Korpi et al. (2002), Krogsgaard-Larsen et al. (2002), Johnston (2005), Sieghart (2006), Möhler (2007), Olsen and Sieghart (2008, 2009) and Atack (2008, 2010). Agents that discriminate between α-subunit isoforms are noted in the table and additional agents that demonstrate selectivity between receptor isoforms, for example viaβ-subunit selectivity, are indicated in the text below. The distinctive agonist and antagonist pharmacology of ρ receptors is summarised in the table and additional aspects are reviewed by Zhang et al. (2001), Chebib (2004), Johnston et al. (2010) and Ng et al. (2011).
| Nomenclature | GABAA |
|---|---|
| Ensembl Gene family ID | ENSF00000000053 |
| Selective agonists (GABA site) | Muscimol (partial agonist at ρ subunits), isoguvacine (partial agonist at ρ subunits), THIP (gaboxadol; δ subunit preferring, antagonist at ρ subunits), piperidine-4-sulphonic acid (low efficacy at α4 and α6 subunits, antagonist at ρ subunits), isonipecotic acid (α4 and α6 subunit selective via relatively high efficacy, antagonist at ρ subunits), (±)-cis-2-CAMP (ρ subunit selective), 5-MeIAA (ρ subunit selective) |
| Selective antagonists (GABA site) | Bicuculline (not active at ρ subunits), gabazine (SR95531; weakly active on ρ subunits), TPMPA (ρ subunit selective), cis- and trans-3-ACPBPA (ρ subunit selective), Aza-THIP (ρ subunit selective) |
| Selective agonists (positive allosteric modulators) (benzodiazepine site) | Diazepam (not α4- or α6-subunits), flunitrazepam (not α4- or α6-subunits), bretazenil (including α4- and α6-subunits, zolpidem, zaleplon and indiplon (α1 subunit selective via high affinity), ocinaplon (α1 subunit selective as essentially a full agonist versus partial agonist at α2, α3 and α5 subunit-containing receptors), L838417 (α2, α3 and α5 subunit selective as a partial agonist versus antagonist at α1-subunit-containing receptors), Ro154513 (selective for α4- and α6-subunit-containing receptors as an agonist versus inverse agonist at α1-, α2-, α3- and α5-subunit-containing receptors), TP003 (selective for α3-subunit-containing receptors as a high efficacy partial agonist versus essentially antagonist activity at α1- α2- and α5-subunit-containing receptors), TPA023 (selective for α2- and α3-subunit-containing receptors as a low efficacy partial agonist versus essentially antagonist activity at α1- and α5-subunit-containing receptors) |
| Selective antagonists (neutral allosteric modulators) (benzodiazepine site) | Flumazenil (low affinity for α4- or α6-subunits and partial agonist), ZK93426, L838417 (α1 subunit selective via antagonist activity versus partial agonist at α2-, α3- and α5-subunit subunit containing receptors) |
| Inverse agonists (negative allosteric modulators) (benzodiazepine site) | DMCM, Ro194603, α3IA (α3 selective via higher affinity and greater inverse agonist activity versusα1, α2 and α5-subunit containing receptors), L655708, RY024 (α5 selective via high affinity), α5IA, MRK016 (α5 selective versusα1, α2 and α3-subunit containing receptors via greater inverse agonist efficacy), Ro4938581 (α5 selective versusα1, α2 and α3-subunit containing receptors via higher affinity and greater inverse agonist activity) |
| Endogenous allosteric modulators | 5α-pregnan-3α-ol-20-one (potentiation), tetrahydrodeoxycorticosterone (potentiation), Zn2+ (potent inhibition of receptors formed from binary combinations of α and β subunits, incorporation of a δ- or γ-subunit causes a modest, or pronounced, reduction in inhibitory potency, respectively, Krishek et al., 1998), extracellular protons (subunit dependent activity, Krishek et al., 1996) |
| Channel blockers | Picrotoxin, TBPS |
| Probes | |
| GABA site | [3H]Muscimol, [3H]gabazine (SR95531) |
| benzodiazepine site | [3H]Flunitrazepam (not α4- or α6-subunit), [3H]zolpidem (α1-subunit selective), [3H]L655708 (α5-subunit selective), [3H]RY80 (α5-subunit selective), [3H]Ro154513 [selectively labels α4- and α6-subunit-containing receptors in the presence of a saturating concentration of a ‘classical’ benzodiazepine (e.g., diazepam)], [3H]CGS8216, [11C]flumazenil (PET ligand with low affinity for α4- or α6-subunits), [18F]fluoroethylflumazenil (PET ligand) |
| Anion channel | [35S]TBPS |
The potency and efficacy of many GABA agonists varies between receptor GABAA receptor isoforms (Frolund et al., 2002; Krogsgaard-Larsen et al., 2002). For example, THIP (gaboxadol) is a partial agonist at receptors with the subunit composition α4β3γ2, but elicits currents in excess of those evoked by GABA at the α4β3δ receptor where GABA itself is a low efficacy agonist (Brown et al., 2002; Bianchi and MacDonald, 2003). The antagonists bicuculline and gabazine differ in their ability to suppress spontaneous openings of the GABAA receptor, the former being more effective (Thompson et al. 1999). The presence of the γ subunit within the heterotrimeric complex reduces the potency and efficacy of agonists (Stórustovu and Ebert, 2006). The GABAA receptor contains distinct allosteric sites that bind barbiturates and endogenous (e.g., 5α-pregnan-3α-ol-20-one) and synthetic (e.g., alphaxalone) neuroactive steroids in a diastereo- or enantio-selective manner (see Belelli and Lambert 2005; Herd et al., 2007; Hosie et al., 2007; Veleiro and Burton, 2009). Picrotoxinin and TBPS act at an allosteric site within the chloride channel pore to negatively regulate channel activity; negative allosteric regulation by γ-butyrolactone derivatives also involves the picrotoxinin site, whereas positive allosteric regulation by such compounds is proposed to occur at a distinct locus. Many intravenous (e.g., etomidate, propofol) and inhalational (e.g., halothane, isoflurane) anaesthetics and alcohols also exert a regulatory influence upon GABAA receptor activity (Bonin and Orser, 2008; Olsen and Li, 2011). Specific amino acid residues within GABAA receptor α- and β-subunits that influence allosteric regulation by anaesthetic and non-anaesthetic compounds have been identified (Hemmings et al., 2005; Hosie et al., 2007). Photoaffinity labelling of distinct amino acid residues within purified GABAA receptors by the etomidate derivative, [3H]-azietomidate, has also been demonstrated (Li et al., 2006) and this binding subject to positive allosteric regulation by anaesthetic steroids (Li et al., 2009). An array of natural products including flavonoid and terpenoid compounds exert varied actions at GABAA receptors (reviewed in detail by Johnston, 2005).
In addition to the agents listed in the table, modulators of GABAA receptor activity that exhibit subunit dependent activity include: salicylidene salicylhydrazide [negative allosteric modulator selective for β1- versusβ2-, or β3-subunit-containing receptors (Thompson et al., 2004)]; fragrent dioxane derivatives [positive allosteric modulators selective for β1- versusβ2-, or β3-subunit-containing receptors (Sergeeva et al., 2010)]; loreclezole, etomidate, tracazolate mefenamic acid, etifoxine, stiripentol, valerinic acid amide [positive allosteric modulators with selectivity for β2/β3- over β1-subunit-containing receptors, see Korpi et al. (2002), Fisher (2009), Khom et al., (2010)]; tracazolate [intrinsic efficacy, i.e., potentiation, or inhibition, is dependent upon the identity of the γ1-3-, δ-, or ε-subunit co-assembed with α1- and β1-subunits (Thompson et al., 2002)]; amiloride [selective blockade of receptors containing an α6-subunit (Fisher, 2002)]; furosemide [selective blockade of receptors containing an α6-subunit co-assembled with β2/β3-, but not β1-subunit (see Korpi et al. (2002)]; La3+[potentiates responses mediated by α1β3γ2L receptors, weakly inhibits α6β3γ2L receptors, and strongly blocks α6β3δ and α4β3δ receptors (Saxena et al., 1997, Brown et al., 2002)]; ethanol [selectively potentiates responses mediated by α4β3δ and α6β3δ receptors versus receptors in which β2 replaces β3, or γ replaces δ (Wallner et al., 2006, but see also Korpi et al., 2007)]; DS1 and DS2 [selectively potentiate responses mediated by δ-subunit-containing receptors (Wafford et al., 2009)]. It should be noted that the apparent selectivity of some positive allosteric modulators (e.g., neurosteroids such as 5α-pregnan-3α-ol-20-one for δ-subunit-containing receptors (e.g., α1β3δ) may be a consequence of the unusually low efficacy of GABA at this receptor isoform (Bianchi and MacDonald, 2003; Belelli et al., 2009).
Abbreviations: 3-ACPBPA, 3-amino-cyclopentenylbutylphosphonic acid; 5-Me-IAA, 5-methy-1H-imidazole-4-acetic acid; (±)-cis-2-CAMP, (±)-cis-2-aminomethylcyclopropane carboxylic acid; α3IA, 6-(4-pyridyl)-5-(4-methoxyphenyl)-3-carbomethoxy-1-methyl-1H-pyridin-2-one; α5IA, 3-(5-methylisoxazol-3-yl)-6-[(1-methyl-1,2,3-triazol-4-yl)methyloxy]-1,2,4-triazolo[3,4-a]phthalazine; CACA, cis-aminocrotonic acid; CGS8216, 2-phenylpyrazolo[4,3-c]quinolin-3(5)-one; DMCM, methy-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate; DS1, 4-chloro-N-[6,8-dibromo-2-(2-thienyl)imidazo[1,2-a]pyridine-3-yl benzamide; DS2, 4-chloro-N-[2-(2-thienyl)imidazo[1,2-a]pyridine-3-yl benzamide; L655708, ethyl(s)-(11,12,13,13a-tetrahydro-7-methoxy-9-oxo)-imidazo[1,5-a]pyrrolo[2,1-c][1,4]benzodiazepine-1-carboxylate; L838417, 7-tert-butyl-3-(2,5-difluoro-phenyl)-6-(2-methyl-2H-[1,2,4]triazol-3-ylmethoxy)-[1,2,4]triazolo[4,3-b]pyridazine; MRK016, 3-tert-butyl-7-(5-methylisoxazol-3-yl)-2-(1-methyl-1H-1,2,4-triazol-5-ylmethoxy)-pyrazolo[1,5-d]-[1,2,4]triazine; Ro154513, ethyl-8-azido-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[1,5-a][1,4] benzodiazepine-3-carboxylate; Ro194603, imidazo[1,5-a]1,4-thienodiazepinone; Ro4938581, 3-bromo-10-difluoromethyl-9H-imidazo[1,5-a][1,2,4]triazolo[1,5-d][1,4]benzodiazepine; SR95531, 2-(3′-carboxy-2′-propyl)-3-amino-6-p-methoxyphenylpyridazinium bromide; TBPS, tert-butylbicyclophosphorothionate; TP003, 4,2′-difluro-5′-[8-fluro-7-(1-hydroxy-1-methylethyl)imidazo[1,2-á]pyridine-3-yl]biphenyl-2-carbonitrile; TPA023, 7-(1,1-dimethylethyl)-6-(2-ethyl-2H-1,2,4-triazol-3-ylmethoxy)-3-(2-fluorphenyl)-1,2,4-triazolo[4,3-b]pyridazine; TPMPA, (1,2,5,6-tetrahydropyridine-4-yl)methylphosphinic acid; RY024, tert-butyl-8-ethynyl-5,6-dihydro-5-methyl-6-oxo-4H-imidazol[1,5-α][1,4]benzodiazepine-3-carboxylate; RY80, ethyl-8-acetylene-5, 6-dihydro-5-methyl-6-oxo-4H-imidazo[1,5a][1, 4]benzodiazepine-3-carboxylate; ZK93423, 6-benzyloxy-4-methoxymethy-β-carboline-3-carboxylate ethyl ester; ZK93426, 5-isopropyl-4-methyl-β-carboline-3-carboxylate ethyl ester
Further Reading
Atack JR (2008). GABAA receptor subtype-selective efficacy: TPA023, an α2/α3 selective non-sedating anxiolytic and α5IA, an α5 selective cognition enhancer. CNS Neurosci Ther14: 25–35.
Atack JR (2010). GABAA receptor α2/α3 subtype-selective modulators as potential nonsedating anxiolytics. Curr Top Behav Neurosci2: 331–360.
Barnard EA, Skolnick P, Olsen RW, Mohler H, Sieghart W, Biggio G et al. (1998). International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function Pharmacol Rev50: 291–313.
Belelli D, Lambert JJ (2005). Neurosteroids: endogenous regulators of the GABAA receptor. Nat Rev Neurosci6: 565–575.
Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW (2009). Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci29: 12757–12763.
Bonin RP, Orser BA (2008). GABAA receptor subtypes underlying general anesthesia. Pharmacol Biochem Behav90: 105–112.
Bowery NG, Smart TG (2006). GABA and glycine as neurotransmitters: a brief history. Br J Pharmacol147 (Suppl. 1): S109–S119.
Capogna M, Pearce RA (2011). GABA A,slow: causes and consequences. Trends Neurosci34: 101–112.
Chebib M (2004). GABAC receptor ion channels. Clin Exp Pharmacol Physiol31: 800–804.
Daniel C, Ohman M (2009). RNA editing and its impact on GABAA receptor function. Biochem Soc Trans37: 1399–1403.
Chen ZW, Olsen RW (2007). GABAA receptor associated proteins: a key factor regulating GABAA receptor function. J Neurochem100: 279–294.
D'Hulst C, Atack JR, Kooy RF (2009). The complexity of the GABAA receptor shapes unique pharmacological properties. Drug Discov Today14: 866–875.
Farrant M, Nusser Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci6: 215–229.
Farrant M, Kaila K (2007). The cellular, molecular and ionic basis of GABAA receptor signalling. Prog Brain Res160: 59–87.
Frolund B, Ebert B, Kristiansen U, Liljefors T, Krogsgaard-Larsen P (2002). GABAA receptor ligands and their therapeutic potentials. Curr Top Med Chem2: 817–832.
Galanopoulou AS (2010). Mutations affecting GABAergic signaling in seizures and epilepsy. Pflugers Arch460: 505–523.
Hanchar HJ, Wallner M, Olsen RW (2004). Alcohol effects on γ-aminobutyric acid type A receptors: are extrasynaptic receptors the answer? Life Sci76: 1–8.
Hemmings HC, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL (2005). Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci26: 503–510.
Herd MB, Belelli D, Lambert JJ (2007). Neurosteroid modulation of synaptic and extrasynaptic GABAA receptors. Pharmacol Ther116: 20–34.
Hosie AM, Wilkins ME, Smart TG (2007). Neurosteroid binding sites on GABAA receptors. Pharmacol Ther116: 7–19.
Jacob TC, Moss SJ, Jurd R (2008). GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci9: 331–343.
Johnston GA (2005). GABAA receptor channel pharmacology. Curr Pharm Des11: 1867–1885.
Johnston GA, Chebib M, Hanrahan JR, Mewett KN (2010). Neurochemicals for the investigation of GABAC receptors. Neurochem Res35: 1970–1977.
Korpi ER, Grunder G, Luddens H (2002). Drug interactions at GABAA receptors. Prog Neurobiol67: 113–159.
Korpi ER, Debus F, Linden AM, Malecot C, Leppa E, Vekovischeva O et al. (2007). Does ethanol act preferentially via selected brain GABAA receptor subtypes? The current evidence is ambiguous. Alcohol41: 163–176.
Krogsgaard-Larsen P, Frolund B, Liljefors T (2002). Specific GABAA agonists and partial agonists. Chem Rec2: 419–430.
Luscher B, Fuchs T, Kilpatrick CL (2011). GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron70: 385–409.
Mody I, Pearce RA (2004). Diversity of inhibitory neurotransmission through GABAA receptors. Trends Neurosci27: 569–575.
Möhler H (2006). GABAA receptors in central nervous system disease: anxiety, epilepsy, and insomnia. J Recept Signal Transduct Res26: 731–740.
Möhler H (2007). Molecular regulation of cognitive functions and developmental plasticity: impact of GABAA receptors. J Neurochem102: 1–12.
Möhler H, Fritschy JM, Vogt K, Crestani F, Rudolph U (2005). Pathophysiology and pharmacology of GABAA receptors. Handb Exp Pharmacol169: 225–247.
Munro G, Ahring PK, Mirza NR (2009). Developing analgesics by enhancing spinal inhibition after injury: GABAA receptor subtypes as novel targets. Trends Pharmacol Sci30: 453–459.
Ng CK, Kim HL, Gavande N, Yamamoto I, Kumar RJ, Mewett KN et al. (2011). Medicinal chemistry of ρ GABAC receptors. Future Med Chem3: 197–209.
Nutt DJ, Stahl SM (2010). Searching for perfect sleep: the continuing evolution of GABAA receptor modulators as hypnotics. J Psychopharmacol24: 1601–1612.
Olsen RW, Li GD (2011). GABAA receptors as molecular targets of general anesthetics: identification of binding sites provides clues to allosteric modulation. Can J Anaesth58: 206–215.
Olsen RW, Sieghart W (2008). IUPHAR, LXX. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev60: 243–260.
Olsen RW, Sieghart, W (2009). GABAA receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology56: 141–148.
Olsen RW, Chang CS, Li G, Hanchar HJ, Wallner M (2004). Fishing for allosteric sites on GABAA receptors. Biochem Pharmacol68: 1675–1684.
Rudolph U, Möhler H (2004). Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol44: 475–498.
Rudolph U, Möhler H (2006). GABA-based therapeutic approaches: GABAA receptor subtype functions. Curr Opin Pharmacol6: 18–23.
Semyanov A, Walker MC, Kullmann DM, Silver RA (2004). Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends Neurosci27: 263–269.
Sieghart W (2006). Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharmacol54: 231–263.
Sigel E, Lüscher BP (2011). A closer look at the high affinity benzodiazepine binding site on GABAA receptors. Curr Top Med Chem11: 241–246.
Tan KR, Rudolph U, Lüscher C (2011). Hooked on benzodiazepines: GABAA receptor subtypes and addiction. Trends Neurosci34: 188–197.
Veleiro AS, Burton G (2009). Structure-activity relationships of neuroactive steroids acting on the GABAA receptor. Curr Med Chem16: 455–472.
Vithlani M, Terunuma M, Moss SJ (2011). The dynamic modulation of GABAA receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol Rev91: 1009–1022.
Wallner M, Hanchar HJ, Olsen RW (2006). Low dose acute alcohol effects on GABAA receptor subtypes. Pharmacol Ther112: 513–528.
Whiting PJ (2003). The GABAA receptor gene family: new opportunities for drug development. Curr Opin Drug Discov Devel6: 648–657.
Zeller A, Jurd R, Lambert S, Arras M, Drexler B, Grashoff C et al. (2008). Inhibitory ligand-gated ion channels as substrates for general anesthetic actions. Handb Exp Pharmacol182: 31–51.
Zhang D, Pan ZH, Awobuluyi M, Lipton SA (2001). Structure and function of GABAC receptors: a comparison of native versus recombinant receptors. Trends Pharmacol Sci22: 121–132.
References
- Bianchi MT, MacDonald RL. J Neurosci. 2003;23:10934–10943. doi: 10.1523/JNEUROSCI.23-34-10934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N, et al. Br J Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JL. Mol Pharmacol. 2002;61:1322–1328. doi: 10.1124/mol.61.6.1322. [DOI] [PubMed] [Google Scholar]
- Fisher JL. Neuropharmacology. 2009;56:190–197. doi: 10.1016/j.neuropharm.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khom S, et al. Br J Pharmacol. 2010;161:65–78. doi: 10.1111/j.1476-5381.2010.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishek BJ, et al. J Physiol. 1996;492:431–443. doi: 10.1113/jphysiol.1996.sp021319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishek BJ, et al. J Physiol. 1998;507:639–652. doi: 10.1111/j.1469-7793.1998.639bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, et al. J Neurosci. 2006;26:11599–11605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, et al. J Biol Chem. 2009;284:11771–11775. doi: 10.1074/jbc.C900016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramerstorfer J, et al. J Neurosci. 2011;31:870–877. doi: 10.1523/JNEUROSCI.5012-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NC, et al. Mol Pharmacol. 1997;51:328–335. doi: 10.1124/mol.51.2.328. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, et al. J Biol Chem. 2010;285:23985–23993. doi: 10.1074/jbc.M110.103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stórustovu SI, Ebert B. J Pharmacol Exp Ther. 2006;316:3151–3159. doi: 10.1124/jpet.105.092403. [DOI] [PubMed] [Google Scholar]
- Thompson SA, et al. Br J Pharmacol. 1999;127:1349–1358. doi: 10.1038/sj.bjp.0702687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SA, et al. Mol Pharmacol. 2002;61:861–869. doi: 10.1124/mol.61.4.861. [DOI] [PubMed] [Google Scholar]
- Thompson SA, et al. Br J Pharmacol. 2004;142:97–106. doi: 10.1038/sj.bjp.0705689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wafford KA, et al. Neuropharmacology. 2009;56:182–189. doi: 10.1016/j.neuropharm.2008.08.004. [DOI] [PubMed] [Google Scholar]
- You H, et al. Neuropharmacology. 2010;59:527–533. doi: 10.1016/j.neuropharm.2010.07.011. [DOI] [PubMed] [Google Scholar]
Glutamate (ionotropic)
Overview: The ionotropic glutamate receptors comprise members of the NMDA (N-methyl-d-aspartate), AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid) and kainate receptor classes, named originally according to their preferred, synthetic, agonist (Dingledine et al., 1999; Lodge, 2009; Traynelis et al., 2010). Receptor heterogeneity within each class arises from the homo-oligomeric, or hetero-oligomeric, assembly of distinct subunits into cation-selective tetramers. Each subunit of the tetrameric complex comprises an extracellular amino terminal domain (ATD), an extracellular ligand binding domain (LBD), a transmembrane domain (TMD) composed of three membrane spans (M1, M3 and M4) with a channel lining re-entrant ‘p-loop’ (M2) located between M1 and M3 and an intracellular carboxy- terminal domain (CTD) (see Mayer, 2006; Kaczor and Matosiuk, 2010; Nakagawa, 2010; Traynelis et al., 2010). The X-ray structure of a homomeric ionotropic glutamate receptor (GluA2 – see below) has recently been solved at 3.6Å resolution (Sobolevsky et al., 2009) and although providing the most complete structural information current available may not representative of the subunit arrangement of, for example, the heteromeric NMDA receptors (Karakas et al., 2011). It is beyond the scope of this supplement to discuss the pharmacology of individual ionotropic glutamate receptor isoforms in detail; such information can be gleaned from Dingledine et al. (1999), Jane et al. (2000), Cull-Candy and Leszkiewicz (2004), Kew and Kemp (2005), Erreger et al. (2007), Paoletti and Neyton (2007), Chen et al. (2008), Jane et al. (2009) and Traynelis et al. (2010). Agents that discriminate between subunit isoforms are, where appropriate, noted in the tables and additional compounds that distinguish between receptor isoforms are indicated in the text below.
The classification of glutamate receptor subunits has been recently been re-addressed by NC-IUPHAR (Collingridge et al., 2009). The scheme developed recommends a revised nomenclature for ionotropic glutamate receptor subunits that is adopted here.
NMDA receptors: NMDA receptors assemble as obligate heteromers that may be drawn from GluN1, GluN2A, GluN2B, GluN2C, GluN2D, GluN3A and GluN3B subunits. Alternative splicing can generate eight isoforms of GluN1 with differing pharmacological properties. Various splice variants of GluN2B, 2C, 2D and GluN3A have also been reported. Activation of NMDA receptors containing GluN1 and GluN2 subunits requires the binding of two agonists, glutamate to the S1 and S2 regions of the GluN2 subunit and glycine to S1 and S2 regions of the GluN1 subunit (Erreger et al. 2004; Chen and Wyllie, 2006). The minimal requirement for efficient functional expression of NMDA receptors in vitro is a di-heteromeric assembly of GluN1 and at least one GluN2 subunit variant, as a dimer of heterodimers arrangement in the extracellular domain (Furukawa et al, 2005; Mayer, 2006; Karakas et al., 2011). However, more complex tri-heteromeric assemblies, incorporating multiple subtypes of GluN2 subunit, or GluN3 subunits, can be generated in vitro and occur in vivo. The NMDA receptor channel commonly has a high relative permeability to Ca2+ and is blocked, in a voltage-dependent manner, by Mg2+ such that at resting potentials the response is substantially inhibited.
| Nomenclature | NMDA |
|---|---|
| Ensembl Gene family ID | ENSF00000000436 |
| Selective agonists (glutamate site) | NMDA (GluN2D > GluN2C > GluN2B > GluN2A), L-aspartate (GluN2D = GluN2B > GluN2C = GluN2A), D-aspartate (GluN2D > GluN2C = GluN2B > GluN2A), (RS)-(tetrazol-5-yl)glycine (GluN2D > GluN2C = GluN2B > GluN2A), homoquinolinic acid (GluN2B ≥ GluN2A ≥ GluN2D > GluN2C; partial agonist at GluN2A and GluN2C) |
| Selective antagonists (glutamate site) | d-AP5, CGS19755 (selfotel), CGP37849, LY233053, d-CCPene (GluN2A = GluN2B > GluN2C = GluN2D), UBP141 (GluN2D > GluN2D > GluN2A > GluN2A, Morley et al., 2005), NVP-AAM077 (GluN2A > GluN2B (human), Auberson et al. 2002; but weakly selective for rat GluN2A versus GluN2B Feng et al., 2004; Frizelle et al., 2006; Neyton and Paoletti, 2006), conantokin-G (GluN2B > GluN2D = GluN2C = GluN2A) |
| Selective agonists (glycine site) | Glycine (GluN2D > GluN2C > GluN2B > GluN2A), D-serine (GluN2D > GluN2C > GluN2B > GluN2A), (+)-HA966 (partial agonist, GluN2B > GluN2A) |
| Selective antagonists (glycine site) | 5,7-Dichlorokynurenate, L689560, L701324, GV196771A |
| Channel blockers | Mg2+ (GluN2A = GluN2B > GluN2C = GluN2D), (+)-MK801, ketamine, phencyclidine, memantine (GluN2C ≥ GluN2D ≥ GluN2B > GluN2A), amantidine (GluN2C = GluN2D ≥ GluN2B ≥ GluN2A), N1-dansyl-spermine (GluN2A = GluN2B >> GluN2C = GluN2D) |
| Probes | |
| Glutamate site | [3H]CPP, [3H]CGS19755, [3H]CGP39653 |
| Glycine site | [3H]Glycine, [3H]L689560, [3H]MDL105519, [3H]CGP61594 (photoaffinity ligand) |
| Cation channel | [3H]-MK801 (dizocilpine) |
Potency orders unreferenced in the table are from Kuner and Schoepfer (1996), Dravid et al. (2007), Erreger et al. (2007), Paoletti and Neyton (2007), Chen et al. (2008) and Traynelis et al. (2010). In addition to the glutamate and glycine binding sites documented in the table, physiologically important inhibitory modulatory sites exist for Mg2+, Zn2+, and protons (see Dingledine et al., 1999; Cull-Candy and Leszkiewicz, 2004; Traynelis et al., 2010). Voltage-independent inhibition by Zn2+ binding with high affinity within the ATD is highly subunit selective (GluN2A >> GluN2B > GluN2C ≥ GluN2D; Paoletti and Neyton, 2007, Traynelis et al., 2010). The receptor is also allosterically modulated, in both positive and negative directions, by endogenous neuroactive steroids in a subunit dependent manner (Malayev et al., 2002, Horak et al., 2006). Tonic proton blockade of NMDA receptor function is alleviated by polyamines and the inclusion of exon 5 within GluN1 subunit splice variants, whereas the non-competitive antagonists ifenprodil and CP101606 (traxoprodil) increase the fraction of receptors blocked by protons at ambient concentration. Inclusion of exon 5 also abolishes potentiation by polyamines and inhibition by Zn2+ that occurs through binding in the ATD (Traynelis et al., 1998). Ifenprodil, CP101606, haloperidol, felbamate and Ro84304 discriminate between recombinant NMDA receptors assembled from GluN1 and either GluN2A, or GluN2B, subunits by acting as selective, non-competitive, antagonists of hetero-oligomers incorporating GluN2B through a binding site at the ATD GluN1/GluN2B subunit interface (Karakas et al., 2011). LY233536 is a competitive antagonist that also displays selectivity for GluN2B over GluN2A subunit-containing receptors. Similarly, CGP61594 is a photoaffinity label that interacts selectively with receptors incorporating GluN2B versus GluN2A, GluN2D and, to a lesser extent, GluN2C subunits. In addition to influencing the pharmacological profile of the NMDA receptor, the identity of the GluN2 subunit co-assembled with GluN1 is an important determinant of biophysical properties that include sensitivity to block by Mg2+, single-channel conductance and maximal open probablity and channel deactivation time (Cull-Candy and Leszkiewicz, 2004; Erreger et al., 2004; Gielen et al., 2009). Incorporation of the GluN3A subunit into tri-heteromers containing GluN1 and GluN2 subunits is associated with decreased single-channel conductance, reduced permeability to Ca2+ and decreased susceptibility to block by Mg2+ (Cavara and Hollmann, 2008; Henson et al., 2010). Reduced permeability to Ca2+ has also been observed following the inclusion of GluN3B in tri-heteromers. The expression of GluN3A, or GluN3B, with GluN1 alone forms, in Xenopus laevis oocytes, a cation channel with unique properties that include activation by glycine (but not NMDA), lack of permeation by Ca2+ and resistance to blockade by Mg2+ and NMDA receptor antagonists (Chatterton et al., 2002). The function of heteromers composed of GluN1 and GluN3A is enhanced by Zn2+, or glycine site antagonists, binding to the GluN1 subunit (Madry et al., 2008). Zn2+ also directly activates such complexes. The co-expression of GluN1, GluN3A and GluN3B appears to be required to form glycine-activated receptors in mammalian cell hosts (Smothers and Woodward, 2007).
AMPA and Kainate receptors: AMPA receptors assemble as homomers, or heteromers, that may be drawn from GluA1, GluA2, GluA3 and GluA4 subunits. Transmembrane AMPA receptor regulatory proteins (TARPs) of class I (i.e.γ2, γ3, γ4 and γ8) act, with variable stoichiometry, as auxiliary subunits to AMPA receptors and influence their trafficking, single channel conductance gating and pharmacology (reviewed by Esteban, 2008; Milstein and Nicoll, 2008; Tomita, 2010; Jackson and Nicoll, 2011). Functional kainate receptors can be expressed as homomers of GluK1, GluK2 or GluK3 subunits. GluK1-3 subunits are also capable of assembling into heterotetramers (e.g. GluK1/K2; see Lerma, 2006; Pinheiro and Mulle, 2006; Perrais et al., 2010). Two additional kainate receptor subunits, GluK4 and GluK5, when expressed individually, form high affinity binding sites for kainate, but lack function, but can form heteromers when expressed with GluK1-3 subunits (e.g. GluK2/K5; reviewed by Pinheiro and Mulle, 2006; Jane et al., 2009; Perrais et al., 2010). Kainate receptors may also exhibit ‘metabotropic’ functions (Lerma, 2006; Rodriguez-Morino and Sihra, 2007). As found for AMPA receptors, kainate receptors are modulated by auxiliary subunits (Neto proteins, see Perrais et al., 2010; Lerma, 2011). An important function difference between AMPA and kainate receptors is that the latter require extracellular Na+ and Cl- for their activation (Bowie, 2010; Plested, 2011). RNA encoding the GluA2 subunit undergoes extensive RNA editing in which the codon encoding a p-loop glutamine residue (Q) is converted to one encoding arginine (R). This Q/R site strongly influences the biophysical properties of the receptor. Recombinant AMPA receptors lacking RNA edited GluA2 subunits are: (1) permeable to Ca2+; (2) blocked by intracellular polyamines at depolarized potentials causing inward rectification (the latter being reduced by TARPs); (3) blocked by extracellular argiotoxin and Joro spider toxins and (4) demonstrate higher channel conductances than receptors containing the edited form of GluA2 (Seeburg and Hartner, 2003; Isaac et al., 2007). GluK1 and GluK2, but not other kainate receptor subunits, are similarly edited and broadly similar functional characteristics apply to kainate receptors lacking either an RNA edited GluK1, or GluK2, subunit (Lerma, 2006; Perrais et al., 2010). Native AMPA and kainate receptors displaying differential channel conductances, Ca2+ permeabilites and sensitivity to block by intracellular polyamines have been identified (Cull-Candy et al., 2006; Isaac et al., 2007, Liu and Zukin, 2007). GluA1-4 can exist as two variants generated by alternative splicing (termed ‘flip’ and ‘flop’) that differ in their desensitization kinetics and their desensitization in the presence of cyclothiazide which stabilises the non-desensitized state. TARPs also stabilise the non-desensitized conformation of AMPA receptors and facilitate the action of cyclothiazide (Milstein and Nicoll, 2008). Splice variants of GluK1-3 also exist which affects their trafficking (Lerma, 2006; Perrais et al., 2010).
| Nomenclature | AMPA | Kainate |
|---|---|---|
| Ensembl Gene family ID | ENSF00000000118 | ENSF00000000118 |
| Selective agonists | AMPA, (S)-5-fluorowillardiine | ATPA, (S)-4-AHCP, 8-deoxy-neodysiherbaine, (S)-5-iodowillardiine, LY339434 (all selective for receptors containing a GluK1 subunit), (2S,4R)-4-methylglutamate (SYM2081), dysiherbaine, domoic acid (inactive at GluK3), kainate (low potency at GluK3) |
| Selective antagonists | NBQX, ATPO, LY293558, GYKI53655/LY300168 (active isomer GYKI53784/LY303070) (noncompetitive) | UBP302, UBP310, ACET, LY382884, LY466195 (all selective for receptors containing a GluK1 subunit), NS3763 (non-competitive, GluK1 selective), MSVIII-19 (GluK1 selective), 2,4-epi-neodysiherbaine (GluK1 and GluK2 selective) |
| Positive modulators | Pyrrolidinones (piracetam, aniracetam), benzothiadiazides (cyclothiazide, S18986, IDRA-21), benzylpiperidines (CX-516 (BDP-12), CX-546), biarylpropylsulfonamides (LY392098, LY404187 and LY503430) | Concanavalin A (GluK1 and GluK2, not GluK3) |
| Channel blockers | Intracellular polyamines, extracellular argiotoxin, extracellular Joro toxin, (selective for channels lacking GluA2) | Intracellular polyamines (subtype selective; GluK3 >> GluK2) |
| Probes (Kd) | [3H]AMPA, [3H]CNQX | [3H]Kainate, [3H](2S,4R)-4-methylglutamate, [3H]UBP310 (GluK1, 21 nM, GluK3, 0.56 µM, Atlason et al., 2010) |
All AMPA receptors are additionally activated by kainate (and domoate) with relatively low potency, (EC50∼ 100 µm). Inclusion of TARPs within the receptor complex increases the potency and maximal effect of kainate (Milstein and Nicoll, 2008; Jackson and Nicoll, 2011). AMPA is weak partial agonist at GluK1 and at heteromeric assemblies of GluK1/GluK2, GluK1/GluK5 and GluK2/GluK5 (Jane et al., 2009). Quinoxalinediones such as CNQX and NBQX show limited selectivity between AMPA and kainate receptors. LY293558 also has kainate (GluK1) receptor activity as has GYKI53655 (GluK3 and GluK2/GluK3) (Jane et al., 2009). ATPO is a potent competitive antagonist of AMPA receptors, has a weaker antagonist action at kainate receptors comprising GluK1 subunits, but is devoid of activity at kainate receptors formed from GluK2 or GluK2/GluK5 subunits. The pharmacological activity of ATPO resides with the (S)-enantiomer. ACET and UBP310 may block GluK3, in addition to GluK1 (Perrais et al., 2009; Atlason et al., 2010). (2S,4R)-4-methylglutamate (SYM2081) is equipotent in activating (and desensitising) GluK1 and GluK2 receptor isoforms and, via the induction of desensitisation at low concentrations, has been used as a functional antagonist of kainate receptors. Both (2S,4R)-4-methylglutamate and LY339434 have agonist activity at NMDA receptors. (2S,4R)-4-methylglutamate is also an inhibitor of the glutamate transporters EAAT1 and EAAT2.
Delta subunits: GluD1 and GluD2 comprise, on the basis of sequence homology, an ‘orphan’ class of ionotropic glutamate receptor subunit. They do not form a functional receptor when expressed solely, or in combination with other ionotropic glutamate receptor subunits, in transfected cells (Yuzaki, 2003). However, GluD2 subunits bind D-serine and glycine and GluD2 subunits carrying the mutation A654T form a spontaneously open channel that is closed by D-serine (Naur et al., 2007).
Abbreviations: (S)-4-AHCP, (S)-2-amino-3-(3-hydroxy-7,8-dihydro-6H-cyclohepta[d]isoxazol-4-yl)propionic acid; ACET, (S)-1-(2-amino-2-carboxyethyl)-3-(2-carboxy-5-phenylthiophene-3-yl-methyl)-5-methylpyrimidine-2,4-dione; AMPA, (RS)-α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; APTA, (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl)propionic acid; ATPO, (RS)-2-amino-3-(3-[5-tert-butyl-3-(phosphonomethoxy)-4-isoxazolyl]propionic acid; CGP37849, (RS)-(E)-2-amino-4-methyl-5-phosphono-3-pentenoic acid; CGP39653, (RS)-(E)-2-amino-4-propyl-5-phosphono-3-pentenoic acid; CGS19755, (±)-cis-4-phosphonomethylpiperidine-2-carboxylic acid; CGP 61594, (±)-trans-4-[2-(4-azidophenyl)-acetylamino]-5,7-dichloro-1,2,3,4-tetrahydro-quinoline-2-carboxylic acid; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione; CP101606, (1S,2S)-1-4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol; CPP, (R)-3-(2-carboxypiperazine-4-yl)propyl-1-phosphonic acid; CX-516; 1-(quinoxalin-6-yl-carbonyl)piperidine; CX-546, 1-(1,4-benzodioxan-6-ylcarbonyl)piperidine; d-AP5, (R)-2-amino-5-phosphonopentanoate; d-CCPene, (R)-(E)-3-(2-carboxypiperazine-4-yl)propenyl-1-phosphonic acid; GV196771A, E-4,6-dichloro-3-(2-oxo-1-phenylpyrrolidin-3-ylidenemethyl)-1H-indole-2-carboxylic acid; GYKI53655, (±)-1-(4-aminophenyl)-3-methylcarbamoyl-4-methyl-3,4-dihydro-7,8-(methylenedioxy)-5H-2,3-benzodiazepine; also known as LY300168; GYKI53784, (-)-1-(4-aminophenyl)-3-methylcarbamoyl-4-methyl-3,4-dihydro-7,8-(methylenedioxy)-5H-2,3-benzodiazepine, also known as LY303070; HA966, 3-amino-1-hydroxypyrrolidin-2-one; IDRA-21, 7-chloro-3-methyl-3,4-dihydro-2H-1,2,4-benzothiadiazine-S,S-dioxide; L689560, trans-2-carboxy-5,7-dichloro-4-phenylaminocarbonylamino-1,2,3,4-tetrahydroquinoline; L701324, 7-chloro-4-hydroxy-3-(3-phenoxy)phenyl-2(H)quinolone; LY233053, (±)-cis-4-[(2H-tetrazol-5-yl)methyl]piperidine-2-carboxylic acid; LY233536, (±)-6-(1H-tetrazol-5-ylmethyl)decahydraisoquinoline-3-carboxylic acid; LY293558, (3S,4aR,6R,8aR)-6-[2-(1H)-tetrazol-5-yl)ethyl]decahydroisoquinoline-3-carboxylic acid; LY339434, (2S,4R,6E)-2-amino-4-carboxy-7-(2-naphthyl)hept-6-enoic acid; LY382884, (3S,4aR,6S,8aR)-6-((4-carboxyphenyl)methyl-1,2,3,4,4a,5,6,7,8,8a-decahydroisoquinoline-3-carboxylic acid; LY392098, propane-2-sulfonic acid [2-(4-thiophen-3-yl-phenyl)propyl]amide; LY404187, propane-2-sulfonic acid [2-(4′-cyanobiphenyl-4-yl)propyl]amide; LY466195, (3S,4aR,6S,8aR)-6-[[(2S)-2-carboxy-4,4-difluoro-1-pyrrolidinyl]-methyl]decahydro-3-isoquinolinecarboxylic acid; LY503430, (R)-4′-[1-fluoro-1-methyl-2-(propane-2-sulfonylamino)ethyl]biphenyl-4-carboxylic acid methylamide; MDL105519, (E)-3-(2-phenyl-2-carboxyethenyl)-4,6-dichloro-1H-indole-2-carboxylic acid; MSVIII-19, (2R,3aR,7aR)-2-[(2S)-2-amino-2-carboxyethyl]-hexahydro-furo-[3,2-b]pyran-2-carboxylic acid; NBQX, 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo(f)quinoxaline; NS3763, 5-carboxy-2,4-di-benzamidobenzoic acid; NVP-AAM077, (R)-[(S)-1-(4-bromophenyl)ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)methyl]phosphonic acid; Ro8-4304, 4-3-[4-(4-fluorophenyl)-3,6-dihydro-2H-pyridin-1-yl]-2-hydroxypropoxybenzamide; S18986, (S)-2,3-dihydro-[3,4]cyclopentano-1,2,4-benzothiadiazine-1,1-dioxide; UBP141, (2R*,3S*)-1-(phenanthrenyl-3-carbonyl)piperazine-2,3-dicarboxylic acid; UBP302, (S)-1-(2-amino-2-carboxyethyl)-3-(2-carboxybenzyl)pyrimidine-2,4-dione, UBP310, (S)-1-(2-amino-2-carboxyethyl)-3-(2-carboxythiophene-3-yl-methyl)-5-methylpyrimidine-2,4-dione
Further Reading
Bowie D (2008). Ionotropic glutamate receptors & CNS disorders. CNS Neurol Disord Drug Targets7: 129–143.
Bowie D (2010). Ion dependent gating of kainate receptors. J Physiol588: 67–81.
Cavara NA, Hollmann M (2008). Shuffling the deck anew: how NR3 tweaks NMDA receptor function. Mol Neurobiol38: 16–26.
Chen PD, Wyllie DJ (2006). Pharmacological insights obtained from structure-function studies of ionotropic glutamate receptors. Br J Pharmacol147: 839–853.
Collingridge GL, Olsen RW, Peters JA, Spedding M (2009). A nomenclature for ligand-gated ion channels. Neuropharmacology56: 2–5.
Contractor A, Mulle C, Swanson GT (2010). Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci34: 154–163.
Cull-Candy SG, Leszkiewicz DN (2004). Role of distinct NMDA receptor subtypes at central synapses. Sci STKE255: re16.
Cull-Candy SG, Kelly L, Farrant M (2006). Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol16: 288–297.
Dingledine R, Borges K, Bowie D, Traynelis SF (1999). The glutamate receptor ion channels. Pharmacol Rev51: 7–61.
Esteban JA (2008). Intracellular machinery for the transport of AMPA receptors. Br J Pharmacol153 (Suppl. 1): S35–S43.
Erreger K, Chen PE, Wyllie DJ, Traynelis SF (2004).Glutamate receptor gating. Crit Rev Neurobiol16: 187–224.
Hall BJ, Ghosh A (2008). http://www.ncbi.nlm.nih.gov/pubmed/18201773?ordinalpos = 25&itool = EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSumRegulation of AMPA receptor recruitment at developing synapses. Trends Neurosci31: 82–89.
Hardingham GE, Bading H (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci11: 682–696.
Isaac JTR, Ashby MC, McBain CJ (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron54: 859–871.
Hansen KB, Yuan H, Traynelis SF (2007). Structural aspects of AMPA receptor activation, desensitization and deactivation. Curr Opin Neurobiol17: 281–288.
Henson MA, Roberts AC, Pérez-Otaño I, Philpot BD (2010). Influence of the NR3A subunit on NMDA receptor functions. Prog Neurobiol91: 23–37.
Jackson AC, Nicoll RA (2011). The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron70: 178–199.
Jane DE, Tse H-W, Skifter DA, Christie JM, Monaghan DT (2000). Glutamate receptor ion channels: activators and inhibitors In: Endo M, Kurachi Y, Mishina M (eds). Handbook of Experimental Pharmacology, Pharmacology of Ionic Channel Function: Activators and Inhibitors, Vol. 147. Springer: Berlin. pp. 415–478.
Jane DE, Lodge D, Collingridge GL (2009). Kainate receptors: pharmacology, function and therapeutic potential. Neuropharmacology56: 90–113.
Kaczor AA, Matosiuk D (2010). Molecular structure of ionotropic glutamate receptors. Curr Med Chem17: 2608–2635.
Kessels HW, Malinow R (2009). Synaptic AMPA receptor plasticity and behavior. Neuron61: 340–350.
Kew JN, Kemp JA (2005). Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology179: 4–29.
Kloda A, Martinac B, Adams DJ (2007). Polymodal regulation of NMDA receptor channels. Channels (Austin)1: 334–343.
Lerma J (2006). Kainate receptor physiology. Curr Opin Pharmacol6: 89–97.
Lerma J (2011). Net(o) excitement for kainate receptors. Nat Neurosci14: 808–810.
Liu SJ, Zukin RS (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci30: 126–134.
Lodge D (2009). The history of the pharmacology and cloning of ionotropic glutamate receptors and the development of idiosyncratic nomenclature. Neuropharmacology56: 6–21.
Low CM, Wee KS (2010). New insights into the not-so-new NR3 subunits of N-methyl-D-aspartate receptor: localization, structure, and function. Mol Pharmacol78: 1–11.
Mayer ML (2006). Glutamate receptors at atomic resolution. Nature440: 456–462.
Mellor IR (2010). The AMPA receptor as a therapeutic target: current perspectives and emerging possibilities. Future Med Chem2: 877–891.
Milstein AD, Nicoll RA (2008). http://www.ncbi.nlm.nih.gov/pubmed/18514334?ordinalpos = 2&itool = EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSumRegulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci29: 333–339.
Monaghan DT, Jane DE (2009). Pharmacology of NMDA Receptors. In: Van Dongen AM (ed.). Biology of the NMDA Receptor. Chapter 12. CRC Press: Boca Raton.
Mony L, Kew JN, Gunthorpe MJ, Paoletti P (2009). Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol157: 1301–1317.
Nakagawa T (2010). The biochemistry, ultrastructure, and subunit assembly mechanism of AMPA receptors. Mol Neurobiol42: 161–184.
O'Neill MJ, Witkin JM (2007). AMPA receptor potentiators: application for depression and Parkinson's disease. Curr Drug Targets8: 603–620.
O'Neill MJ, Bleakman D, Zimmerman DM, Nisenbaum ES (2004). AMPA receptor potentiators for the treatment of CNS disorders. Curr Drug Targets CNS Neurol Disord3: 181–194.
Paoletti P, Neyton J (2007). NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol7: 39–47.
Paoletti P, Vergnano AM, Barbour B, Casado M (2009). Zinc at glutamatergic synapses. Neuroscience158: 126–136.
Perrais D, Veran J, Mulle C (2010). Gating and permeation of kainate receptors: differences unveiled. Trends Pharmacol Sci31: 516–522.
Pinheiro P, Mulle C (2006). Kainate receptors. Cell Tissue Res326: 457–482.
Planells-Cases R, Lerma J, Ferrer-Montiel A (2006). Pharmacological intervention at ionotropic glutamate receptor complexes. Curr Pharm Des12: 3583–3596.
Plested AJ (2011). Kainate receptor modulation by sodium and chloride. Adv Exp Med Biol717: 93–113.
Prorok M, Castellino FJ (2007). The molecular basis of conantokin antagonism of NMDA receptor function. Curr Drug Targets8: 633–642.
Rodriguez-Morino A, Sihra TS (2007). Kainate receptors with a metabotropic modus operandi. Trends Neurosci30: 630–637.
Seeburg PH, Hartner J (2003). Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr opin Neurobiol13: 279–283.
Stawski P, Janovjak H, Trauner D (2010). Pharmacology of ionotropic glutamate receptors: a structural perspective. Bioorg Med Chem18: 7759–7772.
Stephenson FA, Cousins SL, Kenny AV (2008). Assembly and forward trafficking of NMDA receptors. Mol Membr Biol25: 311–320.
Swanson GT, Sakai R (2009). Ligands for ionotropic glutamate receptors. Prog Mol Subcell Biol46: 123–157.
Tomita, S. (2010). Regulation of ionotropic glutamate receptors by their auxiliary subunits. Physiology (Bethesda)25: 41–49.
Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev62: 405–496.
Ward SE, Bax BD, Harries M (2010). Challenges for and current status of research into positive modulators of AMPA receptors. Br J Pharmacol160: 181–190.
Waxman EA, Lynch DR (2005). N-methyl-D-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist11: 37–49.
Yuzaki M (2003). The delta2 glutamate receptor: 10 years later. Neurosci Res46: 11–22.
References
- Atlason PT, et al. Mol Pharmacol. 2010;78:1036–1045. doi: 10.1124/mol.110.067934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auberson YP, et al. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/s0960-894x(02)00074-4. [DOI] [PubMed] [Google Scholar]
- Chatterton JE, et al. Nature. 2002;415:793–798. doi: 10.1038/nature715. [DOI] [PubMed] [Google Scholar]
- Chen PE, et al. J Physiol. 2008;586:227–245. doi: 10.1113/jphysiol.2007.143172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravid SM, et al. J Physiol. 2007;581:107–128. doi: 10.1113/jphysiol.2006.124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K, et al. Mol Pharmacol. 2007;72:907–920. doi: 10.1124/mol.107.037333. [DOI] [PubMed] [Google Scholar]
- Feng B, et al. Br J Pharmacol. 2004;141:508–516. doi: 10.1038/sj.bjp.0705644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizelle PA, et al. Mol Pharmacol. 2006;70:1022–1032. doi: 10.1124/mol.106.024042. [DOI] [PubMed] [Google Scholar]
- Furukawa H, et al. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- Gielen M, et al. Nature. 2009;459:703–707. doi: 10.1038/nature07993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak M, et al. Neuroscience. 2006;137:93–102. doi: 10.1016/j.neuroscience.2005.08.058. [DOI] [PubMed] [Google Scholar]
- Karakas E, et al. Nature. 2011;475:249–253. doi: 10.1038/nature10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner T, Schoepfer R. J Neurosci. 1996;16:3549–3558. doi: 10.1523/JNEUROSCI.16-11-03549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madry C, et al. Proc Natl Acad Sci U S A. 2008;105:12563–12568. doi: 10.1073/pnas.0805624105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malayev A, et al. Br J Pharmacol. 2002;135:901–909. doi: 10.1038/sj.bjp.0704543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley RM, et al. J Med Chem. 2005;48:2627–2637. doi: 10.1021/jm0492498. [DOI] [PubMed] [Google Scholar]
- Naur P, et al. Proc Natl Acad Sci U S A. 2007;104:14116–11421. doi: 10.1073/pnas.0703718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyton J, Paoletti P. J Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais D, et al. Neuropharmacology. 2009;56:131–140. doi: 10.1016/j.neuropharm.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Smothers CT, Woodward JJ. J Pharmacol Exp Ther. 2007;322:739–748. doi: 10.1124/jpet.107.123836. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, et al. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, et al. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Glycine
Overview: The inhibitory glycine receptor [nomenclature as agreed by the NC-IUPHAR sub-committee on glycine receptors (Lynch, 2009a)] is a member of the Cys-loop superfamily of transmitter-gated ion channels that includes the GABAA, nicotinic acetylcholine and 5-HT3 receptors (Lynch 2009b). The receptor is expressed either as a homo-pentamer of α subunits, or a complex now thought to harbour 2α and 3β subunits (Grudzinska et al., 2005; Betz and Laube, 2006), that contain an intrinsic anion channel. Four differentially expressed isoforms of the α-subunit (α1-α4) and one variant of the β-subunit (β1, ENSG00000109738) have been identified by genomic and cDNA cloning. Further diversity originates from alternative splicing of the primary gene transcripts for α1 (α1INS and α1del), α2 (α2A and α2B), α3 (α3S and α3L) and β (βΔ7) subunits and by mRNA editing of the α2 and α3 subunit (Meier et al., 2005; Oertel et al., 2007; Eichler et al., 2008). Both α2 splicing and α3 mRNA editing can produce subunits (i.e., α2B and α3P185L) with enhanced agonist sensitivity. Predominantly, the mature form of the receptor contains α1 (or α3) and β subunits while the immature form is mostly composed of only α2 subunits. RNA transcripts encoding the α4-subunit have not been detected in adult humans. The N-terminal domain of the α-subunit contains both the agonist and strychnine binding sites that consist of several discontinuous regions of amino acids. Inclusion of the β-subunit in the pentameric glycine receptor contributes to agonist binding, reduces single channel conductance and alters pharmacology. The β-subunit also anchors the receptor, via an amphipathic sequence within the large intracellular loop region, to gephyrin. The latter is a cytoskeletal attachment protein that binds to a number of subsynaptic proteins involved in cytoskeletal structure and thus clusters and anchors hetero-oligomeric receptors to the synapse (see Moss and Smart, 2001; Kirsch, 2006; Kneussel and Loebrich, 2007). G-protein βγ subunits enhance the open state probability of native and recombinant glycine receptors by association with domains within the large intracellular loop (Yevenes et al., 2003; 2006;). Intracellular chloride concentration modulates the kinetics of native and recombinant glycine receptors (Pitt et al., 2008). Intracellular Ca2+ appears to increase native and recombinant glycine receptor affinity, prolonging channel open events, by a mechanism that does not involve phosphorylation (Fucile et al., 2000).
| Nomenclature | α1 | α2 | α3 |
|---|---|---|---|
| Ensembl ID | ENSG00000145888 | ENSG00000101958 | ENSG00000145451 |
| Selective agonists (potency order) | Glycine > β-alanine > taurine | Glycine > β-alanine > taurine | Glycine > β-alanine > taurine |
| Selective antagonists and modulators with subunit selectivity | Strychnine, PMBA, bilobalide (IC50 = 20 µM +β = 204 µM), pregnenolone sulphate (Ki = 1.9 µM; +β = 2.7 µM), tropisetron (Ki = 84 µM), colchicine (IC50 = 324 µM), nifedepine (IC50 = 3.3 µM +β = 1.2 µM), ginkgolide X (IC50 = 0.76 µM +β > 300 µM), HU308 (weak inhibition) | Strychnine, PMBA, bilobalide (8 µM +β = 50 µM), pregnenolone sulphate (Ki = 5.5 µM; +β = 10.1 µM), tropisetron (Ki = 13 µM +β = 5.4 µM), colchicine (IC50 = 64 µM), DCKA (IC50 = 188 µM), ginkgolide X (IC50 = 2.8 µM +β > 300 µM), HU210 (90 nM), HU308 (1.1 µM), WIN55,212-2 (220 nM) | Strychnine, nifedepine (IC50 = 29.2µM +β = 11.4 µM), HU210 (50 nM), HU 308 (97 nM), WIN55212-2 (97 nM), (12E,20Z,18S)-8-hydroxyvariabilin (IC50 = 7.0 µM) |
| Selective potentiators (EC50) | HU210 (270 nM), anandamide (38 nM), Δ9-tetrahydrocannabinol (∼3µM, ∼1500% potentiation) | Δ9-tetrahydrocannabinol (∼1µM, ∼230% potentiation) | Δ9-tetrahydrocannabinol (∼5µM, ∼1500% potentiation) |
| Endogenous potentiators (EC50) | Zn2+ (37nM) (not affected by β) | Zn2+ (540 nM) (not affected by β) | |
| Endogenous inhibitors (IC50) | Zn2+ (15 µM; +β = 13 µM), Cu2+ (4-15 µM) (not affected by β), H+ | Zn2+ (360 µM; +β = 180 µM), Cu2+ (17 µM) | Zn2+ (150 µM), Cu2+ (9µM) |
| Channel blockers (IC50) | cyanotriphenylborate (1.3 µM +β = 2.8 µM), picrotoxin (6.3 µM +β = 219 µM), picrotoxinin (5.1 µM +β = 27µM), picrotin (5.2 µM +β = 27 µM), ginkgolide B (0.6–8.0 µM +β = 0.18–2.5 µM), | cyanotriphenylborate (>>20 µM; +β = 7.5 µM), picrotoxin (2.3 µM +β = 29.7 µM), picrotoxinin (0.41 µM), picrotin (13.1 µM), ginkgolide B (3.7–11.4 µM +β = 0.14–0.8 µM) | picrotoxin (+β weakens block), picrotoxinin (0.43 µM +β = 8.9 µM), picrotin (6.0 µM +β = 24 µM), ginkgolide B (1.8 µM +β = 0.55 µM) |
| Probes | [3H]strychnine | [3H]strychnine | [3H]strychnine |
| Functional characteristics | γ = 86 pS (main state) (+β = 44 pS) | γ = 111 pS (main state) (+β = 54 pS) | γ = 105 pS (main state) (+β = 48) |
Data in the table refer to homo-oligomeric assemblies of the α-subunit, significant changes introduced by co-expression of the β1 subunit are indicated in parenthesis. Not all glycine receptor ligands are listed within the table, but some that may be useful in distinguishing between glycine receptor isoforms are indicated (see Lynch (2009a) for a more comprehensive listing). Pregnenolone sulphate, tropisetron and colchicine, for example, although not selective antagonists of glycine receptors, are included for this purpose. Strychnine is a potent and selective competitive glycine receptor antagonist with affinities in the range 5–15 nM. RU5135 demonstrates comparable potency, but additionally blocks GABAA receptors. There are conflicting reports concerning the ability of cannabinoids to inhibit (Lozovaya et al., 2005), or potentiate and at high concentrations activate (Hejazi et al., 2006; Yang et al., 2008; Ahrens et al., 2009; Demir et al., 2009; Xiong et al., 2011) glycine receptors. Nonetheless, cannabinoid analogues may hold promise in distinguishing between glycine receptor subtypes (Yang et al., 2008). In addition, potentiation of glycine receptor activity by cannabinoids has been claimed to contribute to cannabis-induced analgesia relying on Ser296/307 (α1/α3) in M3 (Xiong et al., 2011). Several analogues of muscimol and piperidine act as agonists and antagonists of both glycine and GABAA receptors. Picrotoxin acts as an allosteric inhibitor that appears to bind within the pore, and shows strong selectivity towards homomeric receptors. While its components, picrotoxinin and picrotin, have equal potencies at α1 receptors, their potencies at α2 and α3 receptors differ modestly and may allow some distinction between different receptor types (Yang et al., 2007). Binding of picrotoxin within the pore has recently been demonstrated in the crystal structure of the related C. elegans GluCl Cys-loop receptor (Hibbs and Gouaux, 2011) In addition to the compounds listed in the table, numerous agents act as allosteric regulators of glycine receptors (comprehensively reviewed by Laube et al., 2002; Lynch, 2004; Webb and Lynch, 2007; Yevenes and Zeilhofer, 2011). Zn2+ acts through distinct binding sites of high- and low-affinity to allosterically enhance channel function at low (<10 µM) concentrations and inhibits responses at higher concentrations in a subunit selective manner (Miller et al., 2005). The effect of Zn2+ is somewhat mimicked by Ni2+. Endogenous Zn2+ is essential for normal glycinergic neurotransmission mediated by α1 subunit-containing receptors (Hirzel et al., 2006). Elevation of intracellular Ca2+ produces fast potentiation of glycine receptor-mediated responses. Dideoxyforskolin (4 µM) and tamoxifen (0.2–5 µM) both potentiate responses to low glycine concentrations (15 µM), but act as inhibitors at higher glycine concentrations (100 µM). Additional modulatory agents that enhance glycine receptor function include inhalational, and several intravenous general anaesthetics (e.g. minaxolone, propofol and pentobarbitone) and certain neurosteroids. Ethanol and higher order n-alcohols also enhance glycine receptor function although whether this occurs by a direct allosteric action at the receptor (Mascia et al., 2000), or through G-protein βγ subunits (Yevenes et al., 2010) is debated. Recent crystal structures of the bacterial homologue, GLIC, have identified transmembrane binding pockets for both anaesthetics (Nury et al., 2011) and alcohols (Howard et al., 2011). Solvents inhaled as drugs of abuse (e.g. toluene, 1-1-1-trichloroethane) may act at sites that overlap with those recognising alcohols and volatile anaesthetics to produce potentiation of glycine receptor function. The function of glycine receptors formed as homomeric complexes of α1 or α2 subunits, or hetero-oligomers of α1/β or α2/β subunits, is differentially affected by the 5-HT3 receptor antagonist tropisetron (ICS 205-930) which may evoke potentiation (which may occur within the femtomolar range at the homomeric glycine α1 receptor), or inhibition, depending upon the subunit composition of the receptor and the concentrations of the modulator and glycine employed. Potentiation and inhibition by tropeines involves different binding modes (Maksay et al., 2009). Additional tropeines, including atropine, modulate glycine receptor activity.
Abbreviations: DCKA, dichlorokynurenic acid, HU210, (6aR,10aR)-9-(hydroxymethyl)-6,6-dimethyl-3-(2-methyloctan-2-yl)-6a,7,10,10a-tetrahydrobenzo[c]chromen-1-ol; HU308, [(1R,2R,5R)-2-[2,6-dimethoxy-4-(2-methyloctan-2-yl)phenyl]-6,6-dimethyl-4-bicyclo[3.1.1]hept-3-enyl]methanol; PMBA, 3-[2′-phosphonomethyl[1,1′-biphenyl]-3-yl]alanine; RU5135, 3α-hydroxy-16-imino-5β-17-azaandrostan-11-one, WIN55212-2, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate
Further Reading
Betz H, Laube B (2006). Glycine receptors: recent insights into their structural organization and functional diversity. J Neurochem97: 1600–1610.
Bowery NG, Smart TG (2006). GABA and glycine as neurotransmitters: a brief history. Br J Pharmacol47 (Suppl. 1): S109–S119.
Callister RJ, Graham BA (2010). Early history of glycine receptor biology in mammalian spinal cord circuits. Front Mol Neurosci3: 13.
Cascio M (2004). Structure and function of the glycine receptor and related nicotinicoid receptors. J Biol Chem279: 19383–19386.
Colquhoun D, Sivilotti LG (2004). Function and structure in glycine receptors and some of their relatives. Trends Neurosci27: 337–344.
Dumoulin A, Triller A, Kneussel M (2010). Cellular transport and membrane dynamics of the glycine receptor. Front Mol Neurosci2: 28.
Gilbert DF, Islam R, Lynagh T, Lynch JW, Webb TI (2009). High Throughput Techniques for Discovering New Glycine Receptor Modulators and their Binding Sites. Front Mol Neurosci2: 17.
Harvey RJ, Topf M, Harvey K, Rees MI (2008). The genetics of hyperekplexia: more than startle! Trends Genet24: 439–447.
Harvey VL, Caley A, Müller UC, Harvey RJ, Dickenson AH (2009). A selective role for α3 subunit glycine receptors in inflammatory pain. Front Mol Neurosci2: 14.
Hernandes MS, Troncone LR (2009). Glycine as a neurotransmitter in the forebrain: a short review. J Neural Transm116: 1551–1560.
Kirsch J (2006). Glycinergic transmission. Cell Tissue Res326, 535–540.
Kneussel M, Loebrich S (2007). Trafficking and synaptic anchoring of ionotropic inhibitory neurotransmitter receptors. Biol Cell99: 297–309.
Laube B, Maksay G, Schemm R, Betz H (2002). Modulation of glycine receptor function: a novel approach for therapeutic intervention at inhibitory synapses. Trends Pharmacol Sci23: 519–527.
Legendre P (2001). The glycinergic inhibitory synapse Cell Mol Life Sci58: 760–793.
Lewis T M, Schofield PR (1999). Structure-function relationships for the human glycine receptor: insights from hyperekplexia mutations. Ann NY Acad Sci868: 681–684.
Lobo IA, Harris RA (2005). Sites of alcohol and volatile anesthetic action on glycine receptors. Int Rev Neurobiol65: 53–87.
Lynch JW (2004). Molecular structure and function of the glycine receptor chloride channel. Physiol Rev84: 1051–1095.
Lynch JW (2009a). Glycine receptors, introductory chapter. IUPHAR database (IUPHAR-DB), http://www.iuphar-db.org/IC/FamilyIntroductionForward?familyId = 9
Lynch JW (2009b) Native glycine receptor subtypes and their physiological roles. Neuropharmacology56: 303–309.
Lynch JW, Callister RJ.(2006). Glycine receptors: a new therapeutic target in pain pathways. Curr Opin Investig Drugs7: 48–53.
Moss SJ, Smart TG (2001). Constructing inhibitory synapses. Nature Neurosci Rev2: 240–250.
Perkins DI, Trudell JR, Crawford DK, Alkana RL, Davies DL (2010). Molecular targets and mechanisms for ethanol action in glycine receptors. Pharmacol Ther127: 53–65.
Sivilotti LG (2010). What single-channel analysis tells us of the activation mechanism of ligand-gated channels: the case of the glycine receptor. J Physiol588: 45–58.
Webb TI, Lynch JW (2007). Molecular pharmacology of the glycine receptor chloride channel. Curr Pharm Des13: 2350–2367.
Xu TL, Gong N (2010). Glycine and glycine receptor signaling in hippocampal neurons: diversity, function and regulation. Prog Neurobiol91: 349–361.
Yevenes GE, Zeilhofer HU (2011). Allosteric modulation of glycine receptors. Br J Pharmacol164: 224–236.
References
- Ahrens J, et al. Pharmacology. 2009;83:217–222. doi: 10.1159/000201556. [DOI] [PubMed] [Google Scholar]
- Demir R, et al. Pharmacology. 2009;83:270–274. doi: 10.1159/000209291. [DOI] [PubMed] [Google Scholar]
- Eichler SA, et al. J Cell Mol Biol Med. 2008;12:2848–2866. doi: 10.1111/j.1582-4934.2008.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fucile S, et al. Neuron. 2000;28:571–583. doi: 10.1016/s0896-6273(00)00134-3. [DOI] [PubMed] [Google Scholar]
- Grudzinska J, et al. Neuron. 2005;45:727–739. doi: 10.1016/j.neuron.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Hejazi N, et al. Mol Pharmacol. 2006;69:991–997. doi: 10.1124/mol.105.019174. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirzel K, et al. Neuron. 2006;52:679–690. doi: 10.1016/j.neuron.2006.09.035. [DOI] [PubMed] [Google Scholar]
- Howard RJ, et al. Proc Natl Acad Sci U S A. 2011;108:12149–12154. doi: 10.1073/pnas.1104480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozovaya N, et al. J Neurosci. 2005;25:7499–7506. doi: 10.1523/JNEUROSCI.0977-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksay G, et al. J Neurochem. 2009;109:1725–1732. doi: 10.1111/j.1471-4159.2009.06083.x. [DOI] [PubMed] [Google Scholar]
- Mascia MP, et al. Proc Natl Acad Sci U S A. 2000;97:9305–9310. doi: 10.1073/pnas.160128797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JC, et al. Nat Neurosci. 2005;8:736–744. doi: 10.1038/nn1467. [DOI] [PubMed] [Google Scholar]
- Miller PS, et al. J Physiol (Lond) 2005;566:657–670. doi: 10.1113/jphysiol.2005.088575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nury H, et al. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- Oertel J, et al. J Biol Chem. 2007;282:2798–2807. doi: 10.1074/jbc.M608941200. [DOI] [PubMed] [Google Scholar]
- Pitt SJ, et al. J Neurosci. 2008;28:11454–11467. doi: 10.1523/JNEUROSCI.3890-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, et al. Nat Chem Biol. 2011;7:296–303. doi: 10.1038/nchembio.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, et al. J Neurochem. 2007;103:580–589. doi: 10.1111/j.1471-4159.2007.04850.x. [DOI] [PubMed] [Google Scholar]
- Yang Z, et al. Biochem Pharmacol. 2008;76:1014–1023. doi: 10.1016/j.bcp.2008.07.037. [DOI] [PubMed] [Google Scholar]
- Yevenes GE, et al. Nat Neurosci. 2003;6:819–824. doi: 10.1038/nn1095. [DOI] [PubMed] [Google Scholar]
- Yevenes GE, et al. J Biol Chem. 2006;281:39300–39307. doi: 10.1074/jbc.M608272200. [DOI] [PubMed] [Google Scholar]
- Yevenes GE, et al. J Biol Chem. 2010;285:30203–30213. doi: 10.1074/jbc.M110.134676. [DOI] [PMC free article] [PubMed] [Google Scholar]
P2X
Overview: P2X receptors (nomenclature as agreed by NC-IUPHAR Subcommittee on P2X Receptors, Collingridge et al., 2009; Khakh et al., 2001) are ligand-gated ion channels with a trimeric topology (Jiang et al., 2003; Kawate et al., 2009; Nicke et al., 1998), gating primarily Na+, K+ and Ca2+, exceptionally Cl- with two putative TM domains, where the endogenous ligand is ATP. The Nomenclature Subcommittee has recommended that for P2X receptors, structural criteria should be the initial criteria for nomenclature where possible.The P2X receptor nomenclature recommended below reflects the newly accepted format for ligand-gated ion channels (see Collingridge et al., 2009). Functional P2X receptors exist as polymeric transmitter-gated channels; the native receptors may occur as either homopolymers (e.g. P2X1 in smooth muscle) or heteropolymers (e.g. P2X2:P2X3 in the nodose ganglion and P2X1:P2X5 in mouse cortical astrocytes, Lalo et al., 2008). P2X2, P2X4 and P2X7 receptors have been shown to form functional homopolymers which, in turn, activate pores permeable to low molecular weight solutes (see Surprenant and North, 2009). The hemi-channel pannexin-1 has been implicated in the pore formation induced by P2X7 (Pelegrin and Surprenant, 2009), but not P2X2 (Chaumont and Khakh, 2008), receptor activation.
| Nomenclature | P2X1 | P2X2 | P2X3 | P2X4 |
|---|---|---|---|---|
| Ensembl ID | ENSG00000108405 | ENSG00000187848 | ENSG00000109991 | ENSG00000135124 |
| Potent agonists | L-βγ-meATP, αβ-meATP, BzATP | – | αβ-meATP, BzATP | – |
| Potent antagonists | TNP-ATP (pIC50 8.9, Virginio et al., 1998), Ip5I (pIC50 8.5), NF023 (pIC50 6.7); NF449 (pIC50 6.3, Kassack et al., 2004) | – | TNP-ATP (pIC50 8.9, Virginio et al., 1998), AF353 (pIC50 8.0, Gever et al., 2010), A317491 (7.5, Jarvis et al., 2002), RO3 (pIC50 7.5, Ford et al., 2006) | – |
A317491 and RO3 also block the P2X2:P2X3 heteromultimer (Jarvis et al., 2002; Ford et al., 2006). NF449, A317491 and RO3 are more than 10-fold selective for P2X1 and P2X3 receptors, respectively.
| Nomenclature | P2X5 | P2X6 | P2X7 |
|---|---|---|---|
| Other names | – | – | P2Z |
| Ensembl ID | ENSG00000083454 | ENSG00000099957 | ENSG00000089041 |
| Potent antagonists | – | – | Brilliant Blue G (pIC50 8.0, Jiang et al., 2000), A804598 (pIC50 8.0), A839977 (pIC50 7.7, Donnelly-Roberts and Jarvis, 2007; Donnelly-Roberts et al., 2009, Honore et al., 2009), decavanadate (pA2 7.4, Michel et al., 2006a), KN62 (Gargett and Wiley, 1997), A740003 (pIC50 7.4), A438079 (pIC50 6.9, Donnelly-Roberts and Jarvis, 2007) |
Agonists listed show selectivity within recombinant P2X receptors of ca. one order of magnitude. A804598, A839977, A740003 and A438079 are at least 10-fold selective for P2X7 receptors and show similar affinity across human and rodent receptors (Donnelly-Roberts and Jarvis, 2007, Donnelly-Roberts et al., 2009; Honore et al., 2009).
Several P2X receptors (particularly P2X1 and P2X3) may be inhibited by desensitisation using stable agonists (e.g. αβ-meATP); suramin and PPADS are non-selective antagonists at r & hP2X1–3,5 and hP2X4, but not rP2X4,6,7 (Buell et al., 1996), and can also inhibit ATPase activity (Crack et al., 1994). Ip5I is inactive at rP2X2, an antagonist at rP2X3 (pIC50 5.6) and enhances agonist responses at rP2X4 (King et al., 1999). Antagonist potency of NF023 at recombinant P2X2, P2X3 and P2X5 is two orders of magnitude lower than that at P2X1 receptors (Soto et al., 1999). The P2X7 receptor may be inhibited in a non-competitive manner by the protein kinase inhibitors KN62 and chelerythrine (Shemon et al., 2004), while the p38 MAP kinase inhibitor SB202190 and the cyclic imide AZ11645373 show a species-dependent non-competitive action (Donnelly-Roberts et al., 2004; Michel et al., 2006b; Stokes et al., 2006; Michel, 2009). The pH-sensitive dye used in culture media, phenol red, is also reported to inhibit P2X1 and P2X3 containing channels (King et al., 2005). Some recombinant P2X receptors expressed to high density bind [35S]-ATPγS and [3H]-αβ-meATP, although the latter can also bind to 5′-nucleotidase (Michel et al., 1995). [3H]-A317491 and [3H]-A804598 have been used as high affinity antagonist radioligands for P2X3 (and P2X2/3) and P2X7 receptors, respectively (Donnelly-Roberts et al., 2009).
Abbreviations: αβ-meATP, αβ-methylene-adenosine 5′-triphosphate; βγ-meATP, βγ-methylene-adenosine 5′-triphosphate; A317491, 5-({[3-phenoxybenzyl][(1S)-1,2,3,4-tetrahydro-1-naphthalenyl]amino}carbonyl)-1,2,4-benzenetricarboxylic acid; A438079, 3-(5-(2,3-dichlorophenyl)-1H-tetrazol-1-yl) methyl pyridine; A740003, (N-(1-{[(cyanoimino)(5-quinolinylamino) methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide; A839977,1-(2,3-dichlorophenyl)-N-[2-(pyridin-2-yloxy)benzyl]-1H-tetrazol-5-amine; A804598, (s)-1-(1-(4-bromophenyl)ethyl)-2-cyano-3-(quinoline-5-yl)guanidine; AF353, (5-(5-iodo-2-isopropyl-4-methoxy-phenoxy)-pyrimidine-2,4-diamine; ATPγS, adenosine 5′-(3-thio)triphosphate; AZ11645373, 3-[1-[4-(3-nitrophenyl)phenoxy]-4-pyridin-4-ylbutan-2-yl]-1,3-thiazolidine-2,4-dione; Ip5I, diinosine-5′,5″-pentaphosphate; KN62, 1-(N,O-bis[5-isoquinolinesulphonyl]-N-methyl-l-tyrosyl)-4-phenylpiperazine; NF023, 8,8′-(carbonylbis[imino-3,1-phenylene carbonylimino])bis-1,3,5-naphthalenetrisulfonic acid; NF449, 4,4′,4″,4′″-(carbonylbis[imino-5,1,3-benzenetriyl-bis{carbonylimino}])tetrakisbenzene-1,3-disulfonic acid octasodium salt; PPADS, pyridoxalphosphate-6-azophenyl-2′,4′-disulphonate; RO3, 5-(methyl[2-methylethyl-4,5-dimethoxyphenyl]-2,4pyridinediamine; SB202190, 4-[4-(4-fluorophenyl)-5-pyridin-4-yl-1H-imidazol-2-yl]phenol; TNP-ATP, 2′,3′-O-(2,4,6-trinitrophenyl)-ATP
Further Reading
Burnstock G (2008). Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov7: 575–590.
Browne LE, Jiang LH, North RA (2010). New structure enlivens interest in P2X receptors. Trends Pharmacol Sci31: 229–237.
Browne LE, Cao L, Broomhead HE, Bragg L, Wilkinson WJ, North RA (2011). P2X receptor channels show threefold symmetry in ionic charge selectivity and unitary conductance. Nat Neurosci14: 17–18.
Collingridge GL, Olsen RW, Peters J, Spedding M (2009). A nomenclature for ligand-gated ion channels. Neuropharmacology, 56: 2–5.
Donnelly-Roberts D, McGaraughty S, Shieh CC, Honore P, Jarvis MF (2008). Painful purinergic receptors. J Pharmacol Exp Ther324: 409–415.
Evans RJ (2010). Structural interpretation of P2X receptor mutagenesis studies on drug action. Br J Pharmacol161: 961–971.
Guile SD, Alcaraz L, Birkinshaw TN, Bowers KC, Ebden MR, Furber M et al. (2009). Antagonists of the P2X7 receptor. From lead identification to drug development. J Med Chem52: 3123–3141.
Hu H, Hoylaerts MF (2010). The P2X1 ion channel in platelet function. Platelets21: 153–166.
Jarvis MF (2010). The neural-glial purinergic receptor ensemble in chronic pain states. Trends Neurosci33: 48–57.
Jarvis MF, Khakh BS (2009). ATP-gated P2X cation-channels. Neuropharmacology56: 208–215.
Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Séguéla P et al. (2001). International Union of Pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev53: 107–118.
Pankratov Y, Lalo U, Krishtal OA, Verkhratsky A (2009). P2X receptors and synaptic plasticity. Neuroscience158: 137–148.
Skaper SD, Debetto P, Giusti P (2010). The P2X7 purinergic receptor: from physiology to neurological disorders. FASEB J24: 337–345.
Surprenant A, North RA (2009). Signaling at Purinergic P2X receptors. Annu Rev Physiol, 71: 333–359.
Young MT (2010). P2X receptors: dawn of the post-structure era. Trends Biochem Sci35: 83–90.
Zemkova H, Balik A, Jindrichova M, Vavra V (2008). Molecular structure of purinergic P2X receptors and their expression in the hypothalamus and pituitary. Physiol Res57 (Suppl. 3): S23–S38.
References
- Buell G, et al. EMBO J. 1996;15:55–62. [PMC free article] [PubMed] [Google Scholar]
- Chaumont S, Khakh BS. Proc Natl Acad Sci U S A. 2008;105:12063–12068. doi: 10.1073/pnas.0803008105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crack BE, et al. Br J Pharmacol. 1994;113:1432–1438. doi: 10.1111/j.1476-5381.1994.tb17157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, et al. J Pharmacol Exp Ther. 2004;308:1053–1061. doi: 10.1124/jpet.103.059600. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, et al. Br J Pharmacol. 2009;157:1203–1214. doi: 10.1111/j.1476-5381.2009.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Jarvis MF. Br J Pharmacol. 2007;151:571–579. doi: 10.1038/sj.bjp.0707265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford APDW, et al. Br J Pharmacol. 2006;147:S132–S143. doi: 10.1038/sj.bjp.0706637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargett CE, Wiley JS. Br J Pharmacol. 1997;120:1483–1490. doi: 10.1038/sj.bjp.0701081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gever JR, et al. Br J Pharmacol. 2010;160:1387–1398. doi: 10.1111/j.1476-5381.2010.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, et al. Behav Brain Res. 2009;204:77–81. doi: 10.1016/j.bbr.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, et al. Proc Natl Acad Sci USA. 2002;99:17179–17184. doi: 10.1073/pnas.252537299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LH, et al. Mol Pharmacol. 2000;58:82–88. [PubMed] [Google Scholar]
- Jiang LH, et al. J Neurosci. 2003;23:8903–8910. doi: 10.1523/JNEUROSCI.23-26-08903.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassack MU, et al. Eur J Med Chem. 2004;39:345–357. doi: 10.1016/j.ejmech.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Kawate T, et al. Nature. 2009;460:592–598. doi: 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BF, et al. Br J Pharmacol. 1999;128:981–988. doi: 10.1038/sj.bjp.0702876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BF, et al. Br J Pharmacol. 2005;145:313–322. doi: 10.1038/sj.bjp.0706187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, et al. J Neurosci. 2008;28:5473–5480. doi: 10.1523/JNEUROSCI.1149-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD. Br J Pharmacol. 2009;156:1312–1325. doi: 10.1111/j.1476-5381.2009.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, et al. Br J Pharmacol. 1995;115:767–774. doi: 10.1111/j.1476-5381.1995.tb14999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, et al. Eur J Pharmacol. 2006a;534:19–29. doi: 10.1016/j.ejphar.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Michel AD, et al. Br J Pharmacol. 2006b;149:948–957. doi: 10.1038/sj.bjp.0706938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicke A, et al. EMBO J. 1998;17:3016–3028. doi: 10.1093/emboj/17.11.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Purinergic Signal. 2009;5:129–137. doi: 10.1007/s11302-009-9141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemon AN, et al. Br J Pharmacol. 2004;142:1015–1019. doi: 10.1038/sj.bjp.0705868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto F, et al. Neuropharmacology. 1999;38:141–149. doi: 10.1016/s0028-3908(98)00158-0. [DOI] [PubMed] [Google Scholar]
- Stokes L, et al. Br J Pharmacol. 2006;149:880–887. doi: 10.1038/sj.bjp.0706933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virginio C, et al. Mol Pharmacol. 1998;53:969–973. [PubMed] [Google Scholar]
ZAC (Zinc-activated channel)
Overview: The zinc-activated channel [ZAC, nomenclature as agreed by the NC-IUPHAR Subcommittee for the zinc activated channel (Hales and Peters (2010)] is a member of the Cys-loop family that includes the nicotinic acetylcholine, 5-HT3, GABAA and strychnine-sensitive glycine receptors (Davies et al., 2003; Houtani et al., 2005). The channel is likely to exist as a homopentamer of 4TM subunits that form an intrinsic cation selective channel displaying constitutive activity that can be blocked by (+)-tubocurarine (Davies et al, 2003). ZAC is present in the human, chimpanzee, dog, cow and opossum genomes, but is functionally absent from mouse, or rat, genomes (Davies et al., 2003; Houtani et al., 2005).
| Nomenclature | ZAC |
|---|---|
| Other names | L2 |
| Ensembl ID | ENSG00000186919 |
| Selective agonists (pEC50) | Zn2+ (3.3) |
| Selective antagonists (pIC50) | (+)-Tubocurarine (5.2) |
| Functional characteristics | Outwardly rectifying current (both constitutive and evoked by Zn2+) |
Although tabulated as an antagonist, it is possible that (+)-tubocurarine acts as a channel blocker.
References
- Davies PA, et al. J Biol Chem. 2003;278:712–717. doi: 10.1074/jbc.M208814200. [DOI] [PubMed] [Google Scholar]
- Hales TG, Peters JA. ZAC. 2010. IUPHAR database (IUPHAR-DB), http://www.iuphar-db.org/DATABASE/ObjectDisplayForward?objectId = 587.
- Houtani T, et al. Biochem Biophys Res Commun. 2005;335:277–285. doi: 10.1016/j.bbrc.2005.07.079. [DOI] [PubMed] [Google Scholar]