

Abstract
Genetic studies have implicated Notch signaling in the maintenance of pancreatic progenitors. However, how Notch signaling regulates the quiescent, proliferative or differentiation behaviors of pancreatic progenitors at the single-cell level remains unclear. Here, using single-cell genetic analyses and a new transgenic system that allows dynamic assessment of Notch signaling, we address how discrete levels of Notch signaling regulate the behavior of endocrine progenitors in the zebrafish intrapancreatic duct. We find that these progenitors experience different levels of Notch signaling, which in turn regulate distinct cellular outcomes. High levels of Notch signaling induce quiescence, whereas lower levels promote progenitor amplification. The sustained downregulation of Notch signaling triggers a multistep process that includes cell cycle entry and progenitor amplification prior to endocrine differentiation. Importantly, progenitor amplification and differentiation can be uncoupled by modulating the duration and/or extent of Notch signaling downregulation, indicating that these processes are triggered by distinct levels of Notch signaling. These data show that different levels of Notch signaling drive distinct behaviors in a progenitor population.
Keywords: Notch, Endocrine, Pancreas, Progenitor, Transgenic, Zebrafish
INTRODUCTION
The pancreas is composed of acinar cells, which produce digestive enzymes, ductal cells and hormone-producing endocrine cells, including the insulin-secreting β-cells. Understanding β-cell development is central to our ability to devise more effective therapies for individuals with diabetes. Such therapies could use the in vitro differentiation of β-cells from stem cells or the in vivo stimulation of β-cell formation. Thus, identifying the mechanisms that can recruit and activate endocrine progenitors, as well as those that can promote renewal of the progenitor population, constitute important goals.
The Notch signaling pathway is a crucial regulator of progenitor dynamics in the mammalian pancreas. Genetic deletion of Notch signaling components in the pancreatic domain results in the excessive differentiation of presumptive Notch responsive progenitors into endocrine cells, including β-cells (Apelqvist et al., 1999; Jensen et al., 2000). Genetic lineage-tracing studies further corroborated these findings, showing that during normal development, Notch-responsive cells (NRCs) contribute to endocrine neogenesis (Kopinke et al., 2011).
Recently, an analogous population of NRCs, which contribute to duct cells and endocrine cells, has been identified in the zebrafish pancreas (Parsons et al., 2009; Wang et al., 2011). Upon γ-secretase inhibition, NRCs give rise to endocrine cells, including β-cells (Parsons et al., 2009; Wang et al., 2011). NRCs first appear in the ventral pancreatic bud and, after mixing with the dorsal pancreatic bud-derived cells, expand posteriorly to form a network of interconnected cells bounded by acinar cells (Parsons et al., 2009; Wang et al., 2011). During larval development, and as assessed by genetic lineage tracing, NRCs give rise to endocrine cells, thereby leading to the formation of secondary islets along the intrapancreatic duct (IPD) (Wang et al., 2011).
Here, we address at the single-cell level several critical questions regarding these Notch responsive progenitors in the pancreas including: (1) what are the levels of Notch signaling during cell fate transitions; (2) what proportion of Notch responsive progenitors is competent to give rise to endocrine cells; (3) how is the decision to proliferate or remain quiescent regulated; and (4) are different levels of Notch signaling linked to distinct cellular responses?
We used new as well as established reporters for Notch signaling, endocrine neogenesis and proliferation, complemented by live imaging and loss- and gain-of-function approaches to show that these progenitors experience different levels of Notch signaling that regulate distinct cellular outcomes. Our analyses uncover a novel role for Notch signaling levels in regulating quiescence, renewal and differentiation.
MATERIALS AND METHODS
Zebrafish strains and lines
Zebrafish were raised under standard conditions at 28°C. Phenylthiourea (PTU) was added at 12 hpf to prevent pigmentation. Heat shock regimens were: 14 hpf, 39.5°C for 20 minutes; 3.5 dpf, 39.5°C for 30 minutes; 4.5 dpf, 39.5°C for 25 minutes. Embryos and larvae were collected using a net and transferred using pre-warmed egg water to a plate with minimal volume of egg water at the desired temperature. We used the following lines: Tg(Tp1bglob:eGFP)um14 (Parsons et al., 2009), abbreviated Tg(Tp1:eGFP); TgBAC(neurod:EGFP)nl1 (Obholzer et al., 2008), abbreviated Tg (neurod:EGFP); Tg(–3.5nkx2.2a:GFP)ia3 (Pauls et al., 2007), abbreviated Tg(nkx2.2a:GFP); Tg(hsp70l:Gal4)1.5kca4 (Scheer et al., 2001), abbreviated Tg(hsp:Gal4); Tg(UAS:myc-Notch1a-intra)kca3 (Scheer et al., 2001) and mibta52b (Itoh et al., 2003).
DNA constructs and transgenic lines
Tg(Tp1bglob:H2BmCherry)S939, abbreviated Tg(Tp1:H2BmCherry), and Tg(TP1bglob:VenusPEST)S940, abbreviated Tg(TP1:VenusPEST), were generated using the pT2KXIGΔin backbone (Urasaki et al., 2006). Tg(hsp:ZdnSu(H)-myc; hsp:GFP)S941 was generated by placing ZdnSu(H)-myc downstream of the vector containing bidirectional heat shock elements (Bajoghli et al., 2004; Row and Kimelman, 2009) to drive GFP and ZdnSu(H)-myc on each side. Transgenics were generated in AB or TL using the tol-2 system (Kawakami et al., 2004). Multiple lines were established for each construct; a single representative line was used for all experiments.
Overexpression of myc-Notch1a-intra in LY411575- and DAPT-treated larvae
Larvae from a cross of Tg(hsp:Gal4); Tg(UAS:myc-Notch1a-intra) hemizygous and Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) fish were heat shocked at 3.5 dpf and incubated in DMSO, DAPT or 10 μM LY411575 until 6.5 dpf. Overexpression of myc-Notch1a-intra at 3.5 dpf induced Notch activation throughout the larvae, as evidenced by the upregulation of Tg(Tp1:H2BmCherry) expression (Fig. 2G) and near ubiquitous expression of myc-Notch1a-intra (data not shown), as detected by anti-Myc staining 24 hours after heat shock, consistent with previous studies (Scheer et al., 2002). Larvae not exhibiting upregulated Tg(Tp1:H2BmCherry) expression were negative for Myc immunoreactivity (data not shown).
Fig. 2.

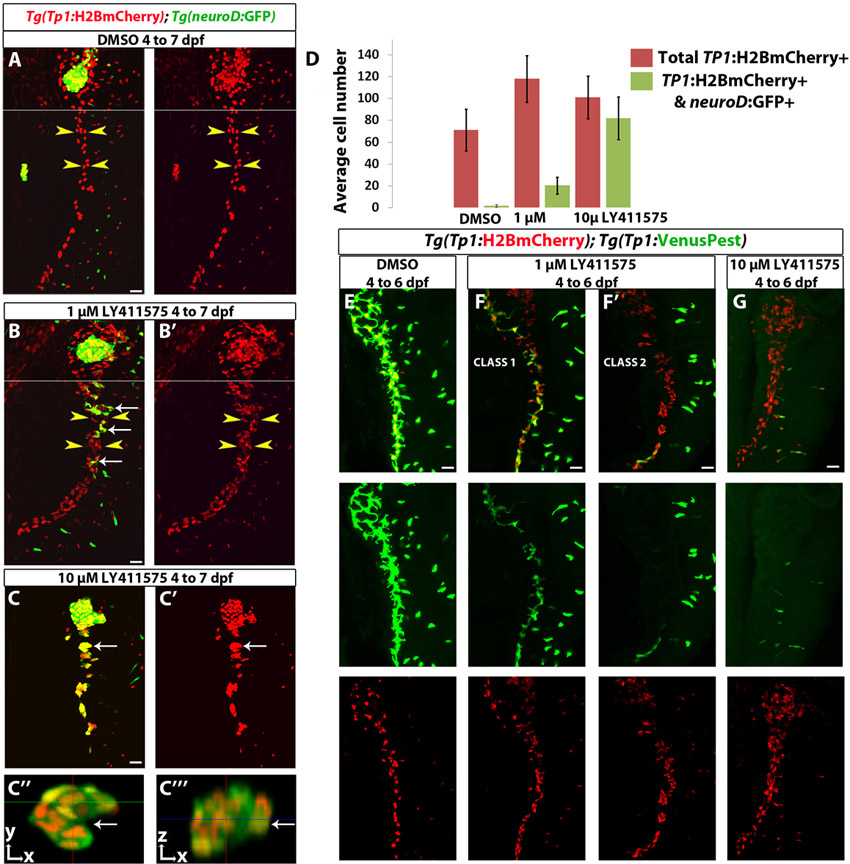
Increasing the level of Notch signaling downregulation leads to an increase in endocrine differentiation of NRCs. (A-D′) Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) larvae were used to label NRCs (red) and follow endocrine differentiation (green). Larvae were treated with (A) DMSO, (B) 100 μM DAPT, (C) 10 μM LY411575 and (D) 50 μM LY411575 from 3.5 to 6.5 dpf and analyzed at 7.5 dpf. 2F11 staining marks the IPD. (A,A′) In control larvae, the pancreatic NRCs form the IPD, as assessed by 2F11 immunoreactivity (arrowhead). White arrows indicate endocrine cells along the IPD. (B,B′) In the DAPT-treated larvae, some NRCs posterior to the PI differentiate into the endocrine lineage, as assessed by Tg(neurod:EGFP) expression (arrows). The majority of NRCs retained IPD cell identity, as assessed by 2F11 immunoreactivity (arrowheads), and did not undergo endocrine differentiation (arrowheads). (C,C′) In the larvae treated with 10 μM LY411575, a smaller number of NRCs retained IPD cell identity (compared with B), as assessed by 2F11 immunoreactivity (arrowheads). Note that the majority of Tg(Tp1:H2BmCherry)+ cells differentiated into endocrine cells, as assessed by Tg(neurod:EGFP) expression. Arrows indicate endocrine cells. (D,D′) In the larvae treated with 50 μM LY411575, the vast majority of NRCs converted into endocrine cells, turned on Tg(neurod:EGFP) expression and aggregated into secondary islets (arrows). The IPD network was absent, as assessed by 2F11 immunoreactivity. (E-G′) Larvae obtained from a cross of Tg(hsp:Gal4); Tg(UAS:myc-Notch1a-intra) hemizygous and Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) fish were heat shocked at 3.5 dpf to induce myc-Notch1a-intra expression and incubated in DMSO or 10 μM LY411575 until 6.5 dpf. (E,E′) A DMSO-treated (myc-Notch1a-intra)– larva. Upper arrowhead, PI; lower arrowhead, IPD. (F,F′) A LY411575-treated (myc-Notch1a-intra)– larva with ectopic secondary islets (lower arrowheads). Note the increased size of the PI (upper arrowhead; compare with E). [Tg(neurod:EGFP) expression is also increased in the brain (yellow arrowhead).] (G,G′) A (myc-Notch1a-intra)+ larva treated with LY411575. No secondary islets were present along the IPD (lower arrowhead) and no increase in the size of the PI was observed (upper arrowhead). Note the dramatic increase in Tg(Tp1:H2BmCherry) expression. (H-I″′) Tg(Tp1:H2BmCherry) larvae treated with (H,H′) DMSO and (I-I″′) 50 μM LY411575 from 3.5 to 6.5 dpf, and stained for glucagon and insulin at 7.5 dpf. In the LY411575-treated larvae, NRCs have formed five islets posterior to the PI (arrows). These islets contain NRC-derived α- and β-cells. (I″,I″′) High-magnification projections of several planes for two of the islets (for the full stack, see supplementary material Movie 4). Yellow and white arrowheads in I″ point to α- and β-cells, respectively. All images are lateral views, anterior towards the top, ventral towards the right. 2F11+, Tg(Tp1:H2BmCherry)+ and Tg(neurod:EGFP)+ cells ventral to the IPD are gut cells. Scale bars: 20 μm in A-D′,H-I′; 100 μm in E-G.
Immunofluorescence and imaging
Antibodies used were: GFP (1:1000, Aves Labs), c-Myc (1:100, 9E10 Santa Cruz), mouse monoclonal 2F11 (1:100, Abcam, Cambridge, UK), Pdx1 (1:200, a gift from Chris Wright, Vanderbilt University, TN, USA), Insulin (1:100, Sigma), Glucagon (1:300, Sigma) and Alexa secondary antibodies (1:300, Invitrogen). Larvae were fixed in 4% PFA overnight at 4°C. After removing the skin and yolk, larvae were permeabilized in 0.4% PBT (TritonX-100) and blocked in 4% PBTB (BSA). Primary antibody staining was performed at 4°C overnight and secondary antibody staining was at 4°C overnight or at room temperature for 3 hours. Samples were mounted in a layer of 1.2% low melt agarose, covered with Vectashield, and imaged using a Zeiss LSM5 confocal microscope. Images are projections of stacks unless otherwise indicated. For Fig. 2E-G and supplementary material Fig. S1A-F, images were acquired live using a Zeiss Lumar stereo microscope. For confocal live imaging, larvae were anesthetized with a low dose of tricaine, placed in a glass bottom Petri dish (MatTek) with a layer of 1.2% low melt agarose and imaged using a 40× water dipping objective. Movies were processed using ImageJ and Fiji. Movies are projections of the stack, except supplementary material Movie 2, which experienced time-points with severe sample movements owing to gut contractions. To correct for z-distortion, several planes spanning the endocrine cells were projected. Some time-points were removed owing to excessive movements; their position is indicated in the time-scale.
In all images, brightness and contrast were adjusted linearly and equally using the LSM software and ImageJ. Photoshop was used to add labels, arrows, scale bars and to outline cells.
Quantification of Tg(TP1:VenusPEST) fluorescence intensity
Fluorescence intensities were analyzed using Fiji in individual planes of confocal stacks. Tg(Tp1:H2BmCherry)+ EdU+ and EdU– NRCs were outlined and the mean Tg(TP1:VenusPEST) intensity for each cell was calculated by drawing a circle of defined area through the brightest plane containing the nucleus.
Chemical treatments
Stock solutions for DAPT (565784, EMD Chemicals) and LY411575 were made in DMSO. DAPT was used at 100 μM. DAPT and LY411575 were diluted in PTU egg water plus DMSO to a final concentration of 1% DMSO and mixed by vortexing. Ten larvae were placed in 1 ml of PTU-egg water in 12-well plates.
EdU analysis
For EdU cell cycle experiments starting at 4 dpf, only larvae that displayed inflation of the swimbladder were selected in order to reduce experimental variability. EdU (E10187, Invitrogen) stock in DMSO was dissolved in PTU-egg water. EdU was used at 7 mM at 1.7% final DMSO concentration for better permeability. EdU staining was revealed for 25 minutes as described previously (Hesselson et al., 2009) after antibody staining.
Statistical analysis
P-values were calculated using an unpaired two-tailed t-test with unequal variance using Excel (Microsoft).
RESULTS
Dynamic regulation of Notch signaling in pancreatic endocrine progenitors
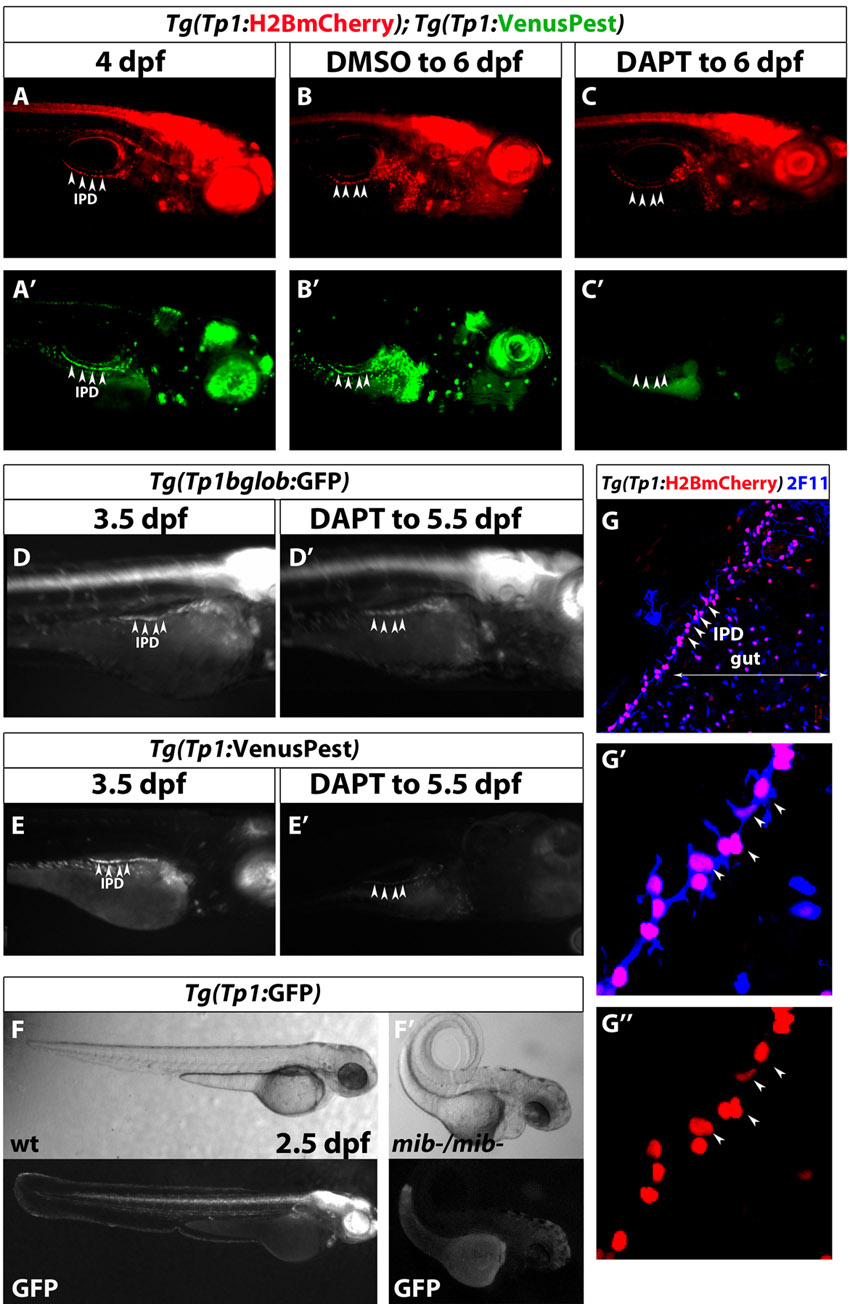
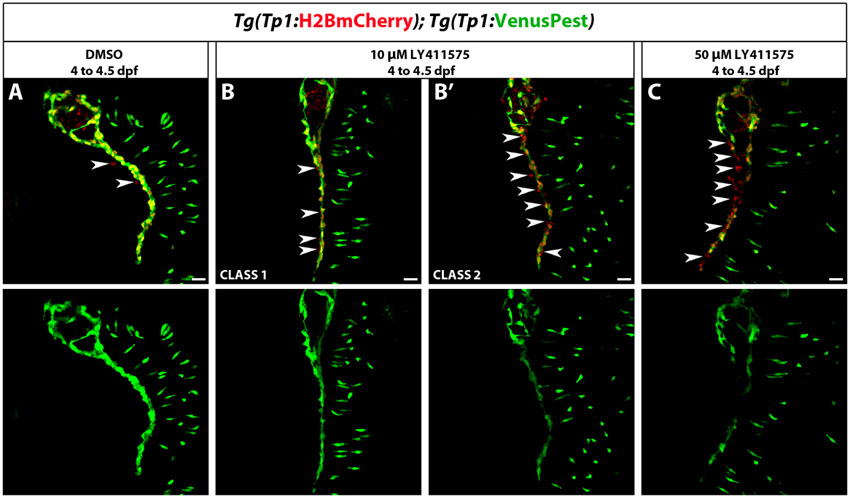
Cells experiencing Notch signaling, including the pancreatic NRCs, can be visualized using Notch reporter lines based on the TP1 module (multiple RBP-Jk-binding sites in front of a minimal promoter) (Kato et al., 1997; Kohyama et al., 2005; Parsons et al., 2009). The currently available Notch reporter transgenic lines, however, use fluorescent proteins with relatively long perdurance (Parsons et al., 2009). In order to follow Notch signaling dynamics in pancreatic NRCs, we generated double transgenic lines expressing a destabilized fluorescent protein (VenusPEST) (Aulehla et al., 2008) and a fluorescent protein with enhanced stability (Histone2BmCherry) under the TP1 element. PEST degradation dramatically reduces the half-life of fusion proteins (from 23 to 2 hours for GFP-PEST in mammalian cells) (Li et al., 1998), whereas fluorescent proteins fused to histones are conferred a long half-life (Brennand et al., 2007). To assess whether the Tg(TP1:H2BmCherry); Tg(TP1:VenusPEST) combination could be used to distinguish NRCs that have experienced, but have switched off, Notch signaling from those that are currently experiencing Notch signaling (Fig. 1A), we treated Tg(TP1:H2BmCherry); Tg(TP1:VenusPEST) larvae with the γ-secretase inhibitor (GSI) DAPT, in order to block Notch signaling. After 2 days of DAPT treatment, pancreatic NRCs lost Tg(TP1:VenusPEST) fluorescence but maintained Tg(TP1:H2BmCherry) labeling at high levels (supplementary material. Fig. S1A-C). Furthermore, direct comparison with the published Tg(TP1:eGFP) line (Parsons et al., 2009) showed that Tg(TP1:VenusPEST) provided a much more sensitive readout of Notch signaling dynamics (supplementary material Fig. S1D,E). As a further test of the sensitivity of the VenusPEST reporter, we induced a brief but strong downregulation of Notch signaling using a second generation DAPT derivative (LY411575) (Fauq et al., 2007), which in zebrafish has been shown to be as potent as DAPT at one-tenth of the concentration (Rothenaigner et al., 2011). Larvae treated with 10 μM LY411575 for 12 hours (4-4.5 dpf) showed variable downregulation of Tg(TP1:VenusPEST) expression in pancreatic NRCs; however, larvae treated with 50 μM LY411575 displayed consistent downregulation of Tg(TP1:VenusPEST) expression, as multiple NRCs were Tg(Tp1:H2BmCherry)+ but Tg(Tp1:VenusPest)– (supplementary material Fig. S2). These data indicate that Tg(TP1:VenusPEST) provides a sensitive readout of Notch signaling in pancreatic NRCs. Finally, to assess the specificity of the TP1 element to Notch signaling, we examined Tg(TP1:eGFP) expression in 60 hpf embryos homozygous for the mind bomb (mib) mutation, which abrogates Notch signaling (Itoh et al., 2003). Homozygosity for the mib mutation inhibited the activity of the TP1 module (supplementary material Fig. S1F).
Fig. 1.

Distinct levels of Notch signaling determine differentiated or quiescent states of NRCs. (A) Tg(Tp1:H2BmCherry) drives expression of H2BmCherry, which has a very long half-life, in NRCs. Tg(Tp1:VenusPest) drives expression of a destabilized VenusPEST fluorescent protein, which has a short half-life, in NRCs. Cells with active Notch signaling are Tg(Tp1:H2BmCherry)+ and Tg(Tp1:VenusPest)+. Cells that had active Notch signaling but lost Notch signaling are Tg(Tp1:H2BmCherry)+ but Tg(Tp1:VenusPest)– (supplementary material Fig. S1A-C, Fig. S2). (B-D″) Tg(Tp1:H2BmCherry); Tg(Tp1:VenusPest) larvae were fixed at progressive developmental stages and stained for Insulin to visualize β-cells. NRCs undergo cell shape changes, migration and proliferation to build the IPD. (D-D”) At 4.5 dpf, a pair of β-cells (white arrowhead) have formed a secondary islet along the IPD. To better visualize these cells, a high magnification single plane is shown in D′,D″ with separate channels. The two β-cells (arrowheads) are Tg(Tp1:H2BmCherry)+ but Tg(Tp1:VenusPest)–, indicating that they originated from NRCs that turned off Notch signaling. (E,E’) Tg(Tp1:H2BmCherry); Tg(Tp1:VenusPest) larvae were fixed and examined at 5.5 dpf. A single plane through the IPD is shown. Based on the fluorescence intensity of Tg(Tp1:VenusPest) expression, NRCs are Tg(Tp1:VenusPest)hi (yellow arrowheads), Tg(Tp1:VenusPest)low (white arrowheads) or Tg(Tp1:VenusPest)– (arrow), indicating that they experience different levels of Notch signaling. (F,F’) Live imaging of Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) larva at 3.5 dpf. Tg(Tp1:H2BmCherry) expression labels NRCs (red) and Tg(neurod:EGFP) expression labels endocrine cells and their direct progenitors (green). White arrowheads indicate a differentiating endocrine cell. Yellow arrowheads indicate a single NRC that, over time, upregulates Tg(neurod:EGFP) expression, suggesting onset of endocrine lineage differentiation. The cells shift position over time owing to the morphogenesis of the pancreas. (G-G″) Tg(TP1:H2BmCherry); Tg(TP1:VenusPEST) larvae were incubated with EdU from 4 to 5 dpf for cell cycle analysis. Yellow arrowheads indicate Tg(TP1:VenusPEST)hi NRCs; the majority are EdU–, whereas neighboring NRCs with lower levels of Tg(TP1:VenusPEST) expression are EdU+ (white arrowheads). (H) Mean Tg(TP1:VenusPEST) fluorescence intensity was calculated for 163 EdU– and 109 EdU+ NRCs (from confocal stacks of four larvae). The average mean fluorescence intensity value (±s.d.) between larvae for EdU– NRCs was 148 (s.d.=17.4) and for EdU+ NRCs was 63.9 (s.d.=34.2) (P<0.01). All images are lateral views, anterior towards the top, ventral towards the right. The NRCs ventral to the IPD in C, D and F are gut cells. Scale bars: 20 μm.
Next, we followed Notch signaling dynamics in the pancreatic NRCs (Fig. 1B-E). These cells undergo cell shape changes, migration and proliferation to build the IPD (Fig. 1B-D), as assessed by co-staining for the IPD marker 2F11 (Dong et al., 2007) (supplementary material Fig. S1G). During these stages, pancreatic NRCs clearly exhibited different levels of Tg(TP1:VenusPEST) expression (Fig. 1E). At 3.5 dpf, an average of 6% (s.d.=2%) of the NRCs (29 NRCs per larva, s.d.=5 cells, n=4 larvae) in the pancreatic tail were Tg(TP1:VenusPEST)–, whereas by 5.5 dpf, their numbers increased to an average of 10% (s.d.=3%) (55 NRCs per larva, s.d.=3 cells, n=4 larvae). In this interval, we also observed insulin+ β-cells seeding the secondary islets along the IPD (Fig. 1D). Importantly, β-cells were Tg(Tp1:H2BmCherry)+ but Tg(Tp1:VenusPest)–, indicating that they originated from NRCs that turned off Notch signaling (Fig. 1D′) (eight cells in four larvae).
To gain further insight into endocrine differentiation of NRCs, we conducted live imaging of transgenic larvae. To follow endocrine differentiation, we used the Tg(neurod:EGFP) line, which labels all pancreatic endocrine cells as well as their direct progenitors (Soyer et al., 2010; Dalgin et al., 2011; Kimmel et al., 2011). At 3.5 dpf, we observed the upregulation of Tg(neurod:EGFP) expression in individual NRCs (Fig. 1F; supplementary material Movie 1), indicating the onset of endocrine differentiation. At 4.5 dpf, endocrine cells differentiating in proximity to each other coalesced to form small secondary islets (supplementary material Movie 2). Interestingly, out of eight endocrine cells imaged for up to 12 hours (starting at 3.5 and 4.5 dpf) none were observed to divide, suggesting that differentiation in the endocrine lineage is associated with cell cycle exit or very slow proliferation. In addition, we observed via live imaging that the NRCs not undergoing endocrine differentiation divided actively between 3.5 and 4 dpf (supplementary material Movie 3).
To determine whether the levels of Notch signaling correlated with cell cycle progression of the NRCs, we performed cell cycle analysis by incubating Tg(TP1:H2BmCherry); Tg(TP1:VenusPEST) larvae with the replication marker 5-ethynyl-2′-deoxyuridine (EdU) from 4 to 5 dpf (Fig. 1G). We observed that on average, EdU– NRCs exhibited higher mean fluorescence intensity of Tg(TP1:VenusPEST) expression (148, s.d.=17.4) than EdU+ NRCs (63.9, s.d.=34.2) (Fig. 1H). These data indicate that between 4 and 5 dpf, NRCs with higher levels of Tg(TP1:VenusPEST) expression were less likely to progress through the cell cycle than NRCs with lower levels, suggesting that high levels of Notch signaling correlate with quiescence/slow cycling.
Endocrine differentiation of a majority of NRCs requires strong downregulation of Notch signaling
As noted above, very few NRCs enter the endocrine lineage during larval development (Fig. 1D,F); this low level of endocrine differentiation could be because only a few NRCs are competent to become endocrine cells or because Notch signaling downregulation is so tightly regulated that very few NRCs lose Notch signaling to an extent compatible with their differentiation into the endocrine lineage.
To distinguish between these possibilities, we induced strong downregulation of Notch signaling and assessed the proportion of NRCs that differentiated into endocrine cells. We treated Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) larvae with DAPT or LY411575 from 3.5 to 6.5 dpf and examined them at 7.5 dpf (Fig. 2). DMSO-treated larvae, used as controls, showed few endocrine cells along the IPD at 7.5 dpf (Fig. 2A), as assessed by Tg(neurod:EGFP) expression. Larvae treated with DAPT showed enlarged Tg(neurod:EGFP)+ endocrine islets along the IPD (Fig. 2B). These islets formed via the clustering of differentiating endocrine cells (data not shown). Importantly, we found that out of 275 Tg(neurod:EGFP)+ cells (n=8 larvae) in the pancreatic tail, 99% were Tg(Tp1:H2BmCherry)+, indicating that upon Notch signaling downregulation, all endocrine cells derive from NRCs. However, we detected on average 35 NRCs in the pancreatic tail (s.d.=18 cells, n=12 larvae) that did not differentiate into endocrine cells even upon DAPT treatment. These cells maintained high 2F11 immunoreactivity and exhibited duct morphology (Fig. 2B). Treatment with 10 μM LY411575 from 3.5 to 6.5 dpf significantly reduced the number of NRCs in the pancreatic tail that did not undergo endocrine differentiation to an average of 13 cells (s.d.=8 cells, n=11 larvae) (Fig. 2C). Strikingly, in two out of 20 larvae treated with 50 μM LY411575, the vast majority of NRCs in the pancreatic tail converted into endocrine cells (Fig. 2D). These observations strongly suggest that a majority of NRCs are competent to give rise to endocrine cells upon efficient Notch signaling downregulation.
To further test whether the endocrine differentiation induced by the GSIs, which block Notch intracellular domain (NICD) cleavage, was specific to the Notch pathway, we overexpressed NICD (myc-Notch1a-intra) using the Tg(hsp:Gal4); Tg(UAS:myc-Notch1a-intra) system by heat shocking larvae at 3.5 dpf, followed by treatment with LY411575 (10 μM) and DAPT until 6.5 dpf. LY411575-treated (myc-Notch1a-intra)– larvae formed more than 50 endocrine cells posterior to the principal islet (PI), whereas (myc-Notch1a-intra)+ larvae treated with LY411575 formed on average four endocrine cells posterior to the PI (s.d.=3.25 cells, n=9 larvae) (Fig. 2E-G).
We also examined the expression of specific pancreatic hormones, including insulin and glucagon after treatment with 50 μM LY411575, and observed abundant differentiation of NRC-derived α- and β-cells (Fig. 2H,I; supplementary material Movie 4). Altogether, these data indicate that upon Notch signaling downregulation, NRCs differentiate into various endocrine cell types.
The downregulation of Notch signaling induces cell cycle entry prior to endocrine differentiation
High levels of Notch signaling correlate with quiescence/slow cycling, whereas lower levels are associated with progenitor amplification (Fig. 1G,H); in addition, endocrine differentiation requires strong Notch signaling downregulation (Fig. 2). To further characterize how cell cycle progression and endocrine differentiation are interlinked during the transition from an NRC to an endocrine cell, we analyzed cell cycle progression and differentiation upon Notch signaling downregulation. We incubated Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) larvae with both 10 μM LY411575 and EdU for 52 hours (4 to 6 dpf), such that we could identify all cells that went through S-phase during this period (Fig. 3A-C). We found that 65% (s.d.=16%, 129 cells counted, n=7 larvae) of the newly formed endocrine cells were EdU+ (Fig. 3B,C). Importantly, when examining the NRCs that had not undergone endocrine differentiation, we found that twice as many were EdU+ in LY411575-treated larvae (85% EdU+ cells, s.d.=16%, 140 cells counted, n=7 larvae) than in controls (43%, s.d.=7%, 140 cells counted, n=7 larvae) (Fig. 3C), indicating that the downregulation of Notch signaling leads a majority of NRCs to enter the cell cycle.
Fig. 3.

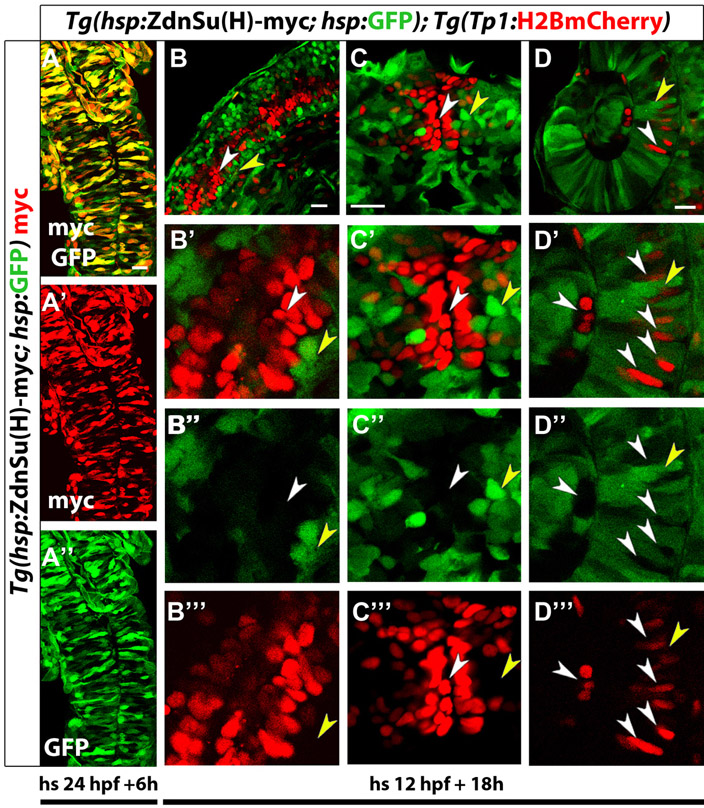
Notch signaling downregulation induces cell cycle progression of NRCs, whereas Notch signaling upregulation triggers their quiescence. (A-C) Larvae were treated with DMSO or 10 μM LY411575 from 4 to 6 dpf (52 hours) in the presence of EdU. (A) DMSO-treated control larva. Yellow arrowheads indicate EdU+ NRCs; white arrowheads indicate two endocrine cells along the IPD (projection of several planes). (B) The LY411575-treated larva showed an increase in the number of endocrine cells. Note that the majority of the new endocrine cells are EdU+ (arrows) and only two endocrine cells are EdU– (arrowheads) (projection of several planes). Upon endocrine differentiation, however, NRCs exit the cell cycle (see supplementary material Fig. S3B,C). (C) Quantification of average percentage of EdU+ cells for A and B. Blue bar: on average, 43% of the NRCs in DMSO controls were EdU+ (140 cells counted in 7 larvae). Red bar: on average, 85% of the NRCs not undergoing endocrine differentiation in LY411575-treated larvae were EdU+ (140 cells counted in 7 larvae) (P<0.001). Green bar: the majority (65%) of new endocrine cells were EdU+ (129 cells counted in 7 larvae). Error bars indicate s.d. (D,E) Larvae were heat shocked at 4.5 dpf to induce mosaic expression of (D) ZdnSu(H)-myc or (E) myc-Notch1a-intra, and incubated in EdU for 24 hours (4.5 to 5.5 dpf). EdU incorporation was scored in cells positive or negative for the transgenes. (D) (ZdnSu(H)-myc)+ cells are identified by GFP expression (supplementary material Fig. S4A). Yellow arrowheads indicate five GFP+ NRCs, all of which are EdU+. Neighboring GFP– NRCs (white arrowheads) incorporate EdU stochastically (projection of several planes). Eighty-one percent of GFP+ NRCs were EdU+ (104 cells counted in 52 larvae), whereas 42% of GFP– NRCs were EDU+ (351 cells counted in nine larvae). (E) Myc staining marks myc-Notch1a-intra overexpressing cells (red) and Tg(Nkx2.2a:GFP) expression marks IPD cells (green). Myc+ IPD cells are EdU– (yellow arrowheads). EdU incorporation is restricted to neighboring Myc- IPD cells (white arrowheads) (projection of several planes). All images are lateral views, anterior towards the top, ventral towards the right. Scale bars: 20 μm.
The observation that most endocrine cells were EdU+ could be because they derived from NRCs that proliferated before endocrine differentiation, or because they proliferated after differentiation. To distinguish between these possibilities, we analyzed cell cycle progression 36 hours after initiating downregulation of Notch signaling. We incubated Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) larvae with 10 μM LY411575 for 60 hours (4 to 6.5 dpf) and added EdU after a delay of 36 hours (5.5 to 6.5 dpf). Strikingly, only 8% (s.d.=3.3%, 182 cells counted, n=4 larvae) of NRC-derived endocrine cells were EdU+ (supplementary material Fig. S3B,C). In contrast to the endocrine cells, a significant proportion of NRCs that did not undergo endocrine differentiation were cycling, as assessed by EdU incorporation (60% EdU+ cells, s.d.=7%, 140 cells counted, n=7 larvae). This percentage is much higher than the percentage of EdU+ NRCs in DMSO controls (11%, s.d.=6.5%, 100 cells counted, n=5 larvae) (supplementary material Fig. S3A-C). These results indicate (1) that the downregulation of Notch signaling induces many pancreatic NRCs to progress through the cell cycle prior to endocrine differentiation and (2) that upon endocrine differentiation NRCs exit the cell cycle.
Notch signaling levels regulate the decision to proliferate or remain quiescent
To test whether downregulation of Notch signaling induces cell cycle progression in NRCs cell-autonomously, we generated transgenic lines expressing a dominant-negative version of the NICD transcriptional co-factor Suppressor of Hairless [ZdnSu(H)-myc] under a synthetic bidirectional heat shock promoter (Bajoghli et al., 2004). This construct allows the expression of GFP and ZdnSu(H)-myc in the same cells such that their behavior can be traced (supplementary material Fig. S4A). Overexpression of Tg(hsp:ZdnSu(H)-myc; hsp:GFP) in the embryo impaired the activation of Notch signaling in the Tg(hsp:ZdnSu(H)-myc; hsp:GFP)-expressing cells in several organs (supplementary material Fig. S4B-D), validating this line as a tool to downregulate Notch signaling cell-autonomously. We then triggered expression of Tg(hsp:ZdnSu(H)-myc; hsp:GFP) by heat shocking larvae at 4.5 dpf and analyzed cell cycle progression in pancreatic NRCs (Fig. 3D). We detected between 0 and 8 GFP+ NRCs per larva (Fig. 3D), owing to the mosaic activity of the heat-inducible promoter at this developmental stage. Analysis of 104 GFP+ NRCs (n=52 larvae) showed that 81% were EdU+, a two fold increase compared with the GFP– NRCs (42% EdU+ out of 351 cells counted, n=9 larvae). Thus, cell-autonomous downregulation of Notch activity through the canonical branch of the pathway leads to increased proliferation of pancreatic NRCs.
These data indicate that the decision of NRCs to remain quiescent or proliferate is regulated by the levels of Notch signaling. To further test this hypothesis, we used Tg(hsp:Gal4); Tg(UAS:myc-Notch1a-intra) larvae to overexpress myc-Notch1a-intra (hyperactivating Notch signaling) by heat shock and scored EdU incorporation in NRCs. Expression of myc-Notch1a-intra was induced at 4.5 dpf and EdU was added after 1.5 hours and maintained until 5.5 dpf. We observed that with this heat shock regimen, and at this developmental stage, the expression of Tg(UAS:myc-Notch1a-intra) was mosaic, allowing us to compare directly cell cycle progression of (myc-Notch1a-intra)+ cells with (myc-Notch1a-intra)– cells (Fig. 3E; supplementary material Fig. S3E). To label and count the NRCs, we stained for Pdx1, which marks the nuclei of the NRCs in the pancreatic tail (supplementary material Fig. S3D). We found that 2.5% (3 out of 118, n=9 larvae) of (myc-Notch1a-intra)+ NRCs were EdU+ whereas, on average, 49% (115 out of 235, n=9 larvae) of (myc-Notch1a-intra)– NRCs were EdU+. Similar observations were made using Tg(nkx2.2a:GFP), which marks IPD cells (Pauls et al., 2007) (Fig. 3E). In summary, downregulation of Notch signaling induces Su(H)-dependent cell cycle entry in pancreatic NRCs, whereas hyperactivation of the Notch pathway induces their quiescence.
Progenitor amplification and differentiation can be uncoupled by modulating the duration and/or extent of Notch signaling downregulation
The downregulation of Notch signaling in pancreatic NRCs elicits a dual response: transient amplification of the progenitors prior to endocrine differentiation. However, it remained unclear whether these events could be uncoupled. First, we explored whether cell cycle progression and endocrine differentiation of NRCs could be uncoupled based on the duration of the downregulation of Notch signaling. We induced a short-term downregulation of Notch signaling and scored cell cycle progression in NRCs. Larvae were treated with 10 μM LY411575 for 12 hours (4 dpf to 4.5 dpf), after which the inhibitor was washed away and EdU was added for 24 hours (4.5 to 5.5). In DMSO controls, 23% of NRCs were EdU+ (s.d.=13.6%, 480 cells counted, n=12 larvae), whereas in LY411575-treated fish, on average, 69% of NRCs were EdU+ (s.d.=21%, 480 cells counted, n=12 larvae) (Fig. 4A-C), indicating that a short period of downregulation of Notch signaling is sufficient to trigger proliferation in a majority of NRCs. We then compared the extent of endocrine differentiation after short- or long-term downregulation of Notch signaling (Fig. 4D-F). Treatment with 10 μM LY411575 for 12 hours at 4 dpf induced a small fraction of NRCs to differentiate into endocrine cells 3 days later. On average, 12 endocrine cells (Fig. 4E) were present posterior to the PI (s.d.=4 cells, n=8 larvae), which translates into ∼14% of the NRCs in the pancreatic tail differentiating into the endocrine lineage. DMSO controls showed on average one cell (s.d.=1 cell, n=7 larvae). As a comparison, treatment with 10 μM LY411575 for 3 days induced abundant endocrine differentiation (Fig. 4F).
Fig. 4.

Progenitor amplification and differentiation can be uncoupled by modulating the duration and/or extent of Notch signaling downregulation. (A-B′) Tg(Tp1:H2BmCherry) larvae were used to label NRCs (red). Larvae were treated with DMSO or 10 μM LY411575 from 4 to 4.5 dpf. At 4.5 dpf, the chemicals were washed away and EdU was added from 4.5 to 5.5 dpf for cell cycle analysis. The positions of the EdU+ NRCs are indicated by white outlines. This brief period of LY411575 treatment was sufficient to induce cell cycle entry in the majority of NRCs compared with controls. (C) Average percentage of EdU+ NRCs in A and B (±s.d.). Forty Tg(Tp1:H2BmCherry)+ cells per larva were counted, starting from the posterior part of the pancreas (n=12 larvae for DMSO and n=12 larvae for LY411575). In DMSO controls, on average 23% of the NRCs were EdU+, whereas in LY411575-treated larvae an average of 69% were EdU+ (P<0.001). (D-F) Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) larvae were treated with (D) DMSO or (E,F) 10 μM LY411575. (E) Larvae treated with LY411575 from 4 to 4.5 dpf. At 4.5 dpf, LY411575 was washed away and the animals were allowed to develop until 7 dpf. On average, 12 endocrine cells were present posterior to the PI (s.d.=4 cells, n=8 larvae), which translates into ∼14% of the NRCs in the pancreatic tail differentiating into the endocrine lineage. DMSO controls showed on average 1 endocrine cell (s.d.=1 cell, n=7 larvae). Arrowheads indicate NRC-derived endocrine cells. Arrows indicate Tg(neurod:EGFP)+ cells in the gut. (F) Larvae treated with LY411575 from 4 to 7 dpf. White arrowheads indicate three new islets composed of numerous endocrine cells. Only two NRCs did not undergo endocrine differentiation (yellow arrowheads). (G-H′) To induce moderate Notch signaling downregulation, Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) larvae were treated with (G) DMSO or (H,H′) 1 μM LY411575 from 4 to 7 dpf. In each case, the chemicals were replaced every 24 hours. Larvae treated with 1 μM LY411575 (H,H′) exhibited a significant increase in the total number of Tg(Tp1:H2BmCherry)+ cells posterior to the PI (yellow arrowheads) (118 cells, s.d.=21 cells, n=20 larvae) compared with DMSO controls (71 cells, s.d.=19 cells, n=18 larvae) (P<0.0001). NRC-derived endocrine cells represented, on average, 17% (s.d.=7.26%, n=19 larvae) of the total number of Tg(Tp1:H2BmCherry)+ cells posterior to the PI (white arrowheads) (see also supplementary material Fig. S5A,B,D for low-magnification views and data quantitation). All images are lateral views, anterior towards the top, ventral towards the right. Scale bars: 20 μm. (I) NRCs experience different levels of Notch signaling regulating their quiescent, proliferative or differentiation states. (J) Upon downregulation of Notch signaling, NRCs (red) enter the cell cycle. Under sustained downregulation of Notch signaling, they differentiate into endocrine cells and exit from the cell cycle. Some NRCs can differentiate directly without entering the cell cycle; some NRCs differentiate into endocrine cells even after a brief and/or a moderate level of Notch signaling downregulation.
We reasoned that a brief pulse of GSI treatment was insufficient to bring Notch signaling levels down to a crucial threshold necessary for endocrine differentiation of a majority of NRCs, yet it provided a drop in Notch activity sufficient to trigger cell cycle progression. To further test whether the extent of downregulation of Notch signaling could regulate proliferative or differentiation outcomes, we induced graded Notch signaling downregulation using various concentrations of LY411575 by treating Tg(Tp1:H2BmCherry); Tg(neurod:EGFP) larvae with 0.1, 0.5, 1, 2, 5 and 10 μM LY411575 from 4 to 7 dpf. In each case, the inhibitor was replaced every 24 hours. We observed a progressive decline in endocrine differentiation with decreasing concentrations of the inhibitor such that, at 0.1 μM LY411575, it was comparable with DMSO controls (data not shown). For a further detailed analysis, we elected to compare larvae treated with 1 μM versus 10 μM LY411575, and counted the number of pancreatic Tg(Tp1:H2BmCherry)+ cells per larva and the proportion of Tg(Tp1:H2BmCherry)+ cells that had differentiated into endocrine cells. In larvae treated with 1 μM LY411575, NRC-derived endocrine cells represented on average 17% (s.d.=7.26%, n=19 larvae) of the total number of Tg(Tp1:H2BmCherry)+ cells posterior to the PI. Importantly, these larvae exhibited a significant increase in the total number of Tg(Tp1:H2BmCherry)+ cells posterior to the PI (118 cells, s.d.=21 cells, n=20 larvae) compared with DMSO controls (71 cells, s.d.=19 cells, n=18 larvae) (Fig. 4G,H; supplementary material Fig. S5A,B,D). In larvae treated with 10 μM LY411575, NRC-derived endocrine cells represented, on average, 79% (s.d.=12%, n=14 larvae) of the total number of Tg(Tp1:H2BmCherry)+ cells posterior to the PI (supplementary material Fig. S5C,D). Furthermore, they displayed a more limited increase in the number of Tg(Tp1:H2BmCherry)+ cells posterior to the PI (101 cells, s.d.=19.5 cells, n=14 larvae) over DMSO controls (supplementary material Fig. S5D), consistent with the transient amplification of a majority of the progenitors, followed by cell cycle exit upon endocrine differentiation. To further test that the treatment with 1 μM LY411575 resulted in a more modest downregulation of Notch signaling compared with 10 μM LY411575, we used Tg(TP1:H2BmCherry); Tg(TP1:VenusPEST) larvae. The majority of larvae treated with 1 μM LY415575 from 4 to 6 dpf retained Tg(TP1:VenusPEST) expression in pancreatic NRCs, albeit at lower levels than DMSO controls, whereas larvae treated with 10 μM of the inhibitor showed complete loss of Tg(TP1:VenusPEST) expression (supplementary material Fig. S5E-G). Altogether, these data indicate that moderate Notch signaling downregulation favors proliferation of NRCs while avoiding endocrine differentiation, whereas strong Notch signaling downregulation leads to transient amplification, followed by endocrine differentiation and cell cycle exit, and, as a result, progenitor depletion.
DISCUSSION
During the stages we examined, very few NRCs lose Notch signaling and differentiate into endocrine cells, whereas the rest of the NRCs exhibit different levels of Notch signaling that regulate their proliferation. NRCs with low levels of Notch signaling are more likely to proliferate, whereas NRCs with high levels are usually slow cycling. Moreover, hyperactivation of Notch signaling triggers proliferative NRCs to enter quiescence, whereas Notch signaling downregulation leads a majority of the NRCs to enter the cell cycle prior to endocrine differentiation. By modulating the dose and/or duration of Notch signaling downregulation, we further show that proliferation and differentiation are regulated by different levels of Notch signaling. In addition, NRCs differ in their propensity for endocrine differentiation, and some can differentiate even under a brief or moderate level of Notch signaling downregulation, whereas the majority requires strong and sustained downregulation (Fig. 4I,J). In summary, and consistent with the models proposed in neural stem and progenitor cells (Shimojo et al., 2008; Chapouton et al., 2010), in a population of cells with active Notch signaling, individual cells can exhibit different behaviors that result from different doses of Notch activity. An important message from these analyses is that when addressing the functions of the Notch pathway, it is necessary to consider not only the on and off states but also the differences in Notch signaling levels at the single-cell level. This approach should be applied to better understand the dichotomous roles of Notch signaling (discussed below) across different cell populations, especially in regulating cell cycle progression and differentiation during normal development as well as in pathological conditions.
Quiescent versus proliferative roles for Notch signaling
The mechanisms that link high Notch signaling with slow cycling and quiescence remain to be elucidated; however, sustained expression of Hes1, a well-characterized Notch target, has been shown to downregulate the expression of G1 cyclins (Shimojo et al., 2008). In addition, high levels of Notch signaling induce the expression of the CDK inhibitor p21, and this induction is necessary for Notch-mediated growth arrest in epithelial cells (Niimi et al., 2007), whereas downregulation of Notch signaling prevents the accumulation of the cell cycle inhibitor Ink4b in zebrafish adult neural stem cells (Chapouton et al., 2010). Furthermore, in the Drosophila bristle lineage, Notch signaling counteracts cyclin E and maintains cell cycle arrest (Simon et al., 2009), while in the developing eye, a deletion of the Notch locus suppresses the phenotypes induced by overexpression of the G2 inhibitors Myt1 and Wee1 (Price et al., 2002).
However, in many other instances, Notch signaling is linked to cell cycle progression, and exerts a mitogenic function, often maintaining progenitors in a proliferative state (for a review, see Kageyama et al., 2008; Pierfelice et al., 2011). How can such dichotomous functions be explained? An interesting insight comes from the analysis of the effects of Notch signaling on the proliferation of mammary epithelial cells. In this system, overexpression of NICD triggered a binary response – hyperproliferation or cell cycle arrest. Interestingly, the hyperproliferative response was linked to lower levels of NICD expression, whereas high levels of overexpression triggered cell cycle block, and stimulated expression of the cell cycle inhibitor p21. Furthermore, at lower levels of activity, Notch signaling interacted with JAK/STAT3 and EGF, and these two signaling pathways were required for Notch to function as a mitogen (Mazzone et al., 2010). It would therefore be important to compare Notch signaling levels in the various tissues where Notch has been shown to regulate quiescence as well as proliferation, in order to assess whether these differences are linked to different endogenous levels of Notch signaling.
The bivalent role of Notch signaling as a mitogen and a cell cycle inhibitor is especially intriguing in the pancreas. For example, in the early mammalian pancreas, the global downregulation of Notch signaling results in hypoplastic pancreata, indicating that Notch signaling plays a mitogenic role (Ahnfelt-Ronne et al., 2012). Moreover, deletion of Rbpj in the Hes1-positive centroacinar cells does not increase their replication (Kopinke et al., 2012), yet downregulation of Notch signaling through temporal treatment with GSIs or deletion of presenilin 1 and 2 results in an increase in cell cycle progression (Cras-Meneur et al., 2009). These distinct phenotypes might be due to the different roles of Notch signaling depending on its activity levels, or to changes in the activities of interacting pathways (for a review, see Andersson et al., 2011). In this light, it will be important to define whether the role of high Notch signaling levels in restricting proliferation of pancreatic NRCs in zebrafish is merely a consequence of counteracting pathways that promote pancreatic proliferation.
The final outcome of downregulation of Notch signaling in pancreatic NRCs in zebrafish; however, is cell cycle exit, and this exit correlates with the transition into the endocrine lineage, a cell population we and others have shown is characterized by different cell cycle kinetics, namely slow proliferation (Desgraz and Herrera, 2009; Hesselson et al., 2009). One likely candidate for instructing this transition is NeuroD, the expression of which is induced upon inhibition of Notch signaling and is able to increase the expression of CDK inhibitors (Mutoh et al., 1998; Farah et al., 2000; Ochocinska and Hitchcock, 2009), as well as to act as a pan-endocrine differentiation factor (Naya et al., 1997). However, the activity of NeuroD must later be modulated to allow for the expansion of endocrine cells in the adult pancreas.
Dose-dependent effects of Notch signaling – from quiescence to endocrine differentiation
Importantly, a dose-dependent role for Notch signaling may be easier to uncover when examining hypomorphic conditions rather than loss-of-function mutations of crucial components of the Notch pathway. Elegant recent analyses of Notch signaling in Drosophila intestinal stem cells, using mutants for GDP-mannose 4,6-dehydratase (Gmd), a modulator of Notch signaling, as well as temperature-sensitive alleles of the Notch glycosyltransferase Rumi, uncovered a dose-dependent role for Notch activity levels in cell cycle exit and differentiation (Perdigoto et al., 2011). A modest downregulation of Notch signaling prevented cell cycle exit of enteroblasts; however, it did not affect their ability to differentiate into endocrine cells or enterocytes, indicating that higher levels of Notch signaling are required for cell cycle exit than for differentiation (Perdigoto et al., 2011). In our experiments, we aimed to induce graded downregulation of Notch signaling using different concentrations of GSIs. Recently, in vitro experiments using a dynamic complementation sensor for NICD/Rbpj interactions (Ilagan et al., 2011) showed that high concentrations of GSIs led to a gradual reduction of Notch signaling and maximum inhibition occurred 6 hours after the initiation of drug treatment. However, at lower concentrations of GSI treatment, Notch signaling was downregulated at a much slower rate, so that the cells experienced a prolonged state of intermediate Notch signaling. Analogously, we observed that, at high concentrations, LY411575 led to a more rapid downregulation of Notch signaling (supplementary material Fig. S2, Fig. S5E-G), which resulted in a transient increase in proliferation, followed by the endocrine differentiation of a majority of NRCs (Fig. 3A-C, Fig. 4A-C,F). At lower concentrations of the inhibitor, however, Notch signaling decayed at a slower rate, as indicated by Tg(TP1:VenusPest) expression (supplementary material Fig. S5F-G), such that the majority of the NRCs entered the cell cycle, but they were not able to differentiate during the time window examined (4 to 7 dpf) (Fig. 4H; supplementary material Fig. S5B). These analyses lead us to propose that the strength and duration of Notch signaling downregulation dictate different cell cycle and differentiation outcomes. A similar dose-dependent role for Notch signaling may regulate proliferative and differentiation decisions in stem cell populations where abrogating Notch signaling has been shown to trigger exit from quiescence, for instance in the adult mouse and zebrafish brain (Chapouton et al., 2010; Ehm et al., 2010; Imayoshi et al., 2010) and in adult muscle (Bjornson et al., 2012; Mourikis et al., 2012).
What are the potential mechanisms by which Notch could exert specific and distinct outcomes at different activity levels? It is possible that the cell cycle is more sensitive to changes in Notch signaling, as modulating a single cell cycle regulator could have strong effects, whereas differentiation may require stable changes in the regulation of multiple genes. For example, a requirement for sustained and strong downregulation of Notch signaling for endocrine differentiation may reflect the need for global changes in chromatin organization in NRCs. Indeed, Notch signaling is known to act together with chromatin remodeling factors, including histone deacetylases and acetyltransferases (for a review, see Bray, 2006). Whether modulating the activity of such factors could enhance or suppress endocrine differentiation remains to be explored. In this context, the high temporal and spatial resolution used in our analyses revealed that a subset of NRCs can differentiate into endocrine cells even after a brief and/or a moderate level of Notch signaling downregulation (Fig. 4E,H). Whether this enhanced capacity for differentiation is mainly due to weaker basal Notch activity in these cells compared with the rest of the NRCs, or whether additional genetic mechanisms, including epigenetic regulators, confer higher plasticity will be an interesting avenue to pursue. Endocrine differentiation from Notch-responsive progenitors in the zebrafish pancreas constitutes a powerful system with which to further investigate mechanisms regulating cellular plasticity and the competence for endocrine neogenesis. A precise understanding of these cellular behaviors constitutes an important goal towards improving regenerative strategies related to type 1 diabetes.
Supplementary Material
Acknowledgments
We thank Ana Ayala and Milagritos Alva for expert fish assistance; Daniel Hesselson, David Staudt and Alethia Villasenor for critical reading of the manuscript; and all members of the Stainier group for helpful discussions. LY411575 was a generous gift from Abdul H. Fauq (Mayo Clinic Florida, Jacksonville, FL, USA). The cDNA encoding hsp:ZdnSu(H)-myc was a generous gift from Tim Simmons and Bruce Appel (University of Colorado, Denver, CO, USA). Special thanks to Jiandong Liu for mind bomb mutants and for helpful discussions, and to Anne-Sophie de Preux Charles and David Staudt (USCF) for Tg(hsp70l:Gal4) and Tg(UAS:myc-Notch1a-intra) fish.
Footnotes
Funding
This work was supported by a Post-Doctoral Fellowship Award from the Canadian Diabetes Association (N.N.) as well as by grants from the National Institutes of Health [DK075032] and the Packard Foundation to D.Y.R.S. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.076000/-/DC1
References
- Ahnfelt-Ronne J., Jorgensen M. C., Klinck R., Jensen J. N., Fuchtbauer E. M., Deering T., Macdonald R. J., Wright C. V., Madsen O. D., Serup P. (2012). Ptf1a-mediated control of Dll1 reveals an alternative to the lateral inhibition mechanism. Development 139, 33–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson E. R., Sandberg R., Lendahl U. (2011). Notch signaling: simplicity in design, versatility in function. Development 138, 3593–3612 [DOI] [PubMed] [Google Scholar]
- Apelqvist A., Li H., Sommer L., Beatus P., Anderson D. J., Honjo T., Hrabe de Angelis M., Lendahl U., Edlund H. (1999). Notch signalling controls pancreatic cell differentiation. Nature 400, 877–881 [DOI] [PubMed] [Google Scholar]
- Aulehla A., Wiegraebe W., Baubet V., Wahl M. B., Deng C., Taketo M., Lewandoski M., Pourquie O. (2008). A beta-catenin gradient links the clock and wavefront systems in mouse embryo segmentation. Nat. Cell Biol. 10, 186–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajoghli B., Aghaallaei N., Heimbucher T., Czerny T. (2004). An artificial promoter construct for heat-inducible misexpression during fish embryogenesis. Dev. Biol. 271, 416–430 [DOI] [PubMed] [Google Scholar]
- Bjornson C. R., Cheung T. H., Liu L., Tripathi P. V., Steeper K. M., Rando T. A. (2012). Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30, 232–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray S. J. (2006). Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell. Biol. 7, 678–689 [DOI] [PubMed] [Google Scholar]
- Brennand K., Huangfu D., Melton D. (2007). All beta cells contribute equally to islet growth and maintenance. PLoS Biol. 5, e163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapouton P., Skupien P., Hesl B., Coolen M., Moore J. C., Madelaine R., Kremmer E., Faus-Kessler T., Blader P., Lawson N. D., et al. (2010). Notch activity levels control the balance between quiescence and recruitment of adult neural stem cells. J. Neurosci. 30, 7961–7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cras-Meneur C., Li L., Kopan R., Permutt M. A. (2009). Presenilins, Notch dose control the fate of pancreatic endocrine progenitors during a narrow developmental window. Genes Dev. 23, 2088–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgin G., Ward A. B., Hao L. T., Beattie C. E., Nechiporuk A., Prince V. E. (2011). Zebrafish mnx1 controls cell fate choice in the developing endocrine pancreas. Development 138, 4597–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgraz R., Herrera P. L. (2009). Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development 136, 3567–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong P. D., Munson C. A., Norton W., Crosnier C., Pan X., Gong Z., Neumann C. J., Stainier D. Y. (2007). Fgf10 regulates hepatopancreatic ductal system patterning and differentiation. Nat. Genet. 39, 397–402 [DOI] [PubMed] [Google Scholar]
- Ehm O., Goritz C., Covic M., Schaffner I., Schwarz T. J., Karaca E., Kempkes B., Kremmer E., Pfrieger F. W., Espinosa L., et al. (2010). RBPJkappa-dependent signaling is essential for long-term maintenance of neural stem cells in the adult hippocampus. J. Neurosci. 30, 13794–13807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah M. H., Olson J. M., Sucic H. B., Hume R. I., Tapscott S. J., Turner D. L. (2000). Generation of neurons by transient expression of neural bHLH proteins in mammalian cells. Development 127, 693–702 [DOI] [PubMed] [Google Scholar]
- Hesselson D., Anderson R. M., Beinat M., Stainier D. Y. (2009). Distinct populations of quiescent and proliferative pancreatic beta-cells identified by HOTcre mediated labeling. Proc. Natl. Acad. Sci. USA 106, 14896–14901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan M. X., Lim S., Fulbright M., Piwnica-Worms D., Kopan R. (2011). Real-time imaging of notch activation with a luciferase complementation-based reporter. Sci. Signal. 4, rs7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I., Sakamoto M., Yamaguchi M., Mori K., Kageyama R. (2010). Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 30, 3489–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M., Kim C. H., Palardy G., Oda T., Jiang Y. J., Maust D., Yeo S. Y., Lorick K., Wright G. J., Ariza-McNaughton L., et al. (2003). Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev. Cell 4, 67–82 [DOI] [PubMed] [Google Scholar]
- Jensen J., Pedersen E. E., Galante P., Hald J., Heller R. S., Ishibashi M., Kageyama R., Guillemot F., Serup P., Madsen O. D. (2000). Control of endodermal endocrine development by Hes-1. Nat. Genet. 24, 36–44 [DOI] [PubMed] [Google Scholar]
- Kageyama R., Ohtsuka T., Shimojo H., Imayoshi I. (2008). Dynamic Notch signaling in neural progenitor cells and a revised view of lateral inhibition. Nat. Neurosci. 11, 1247–1251 [DOI] [PubMed] [Google Scholar]
- Kato H., Taniguchi Y., Kurooka H., Minoguchi S., Sakai T., Nomura-Okazaki S., Tamura K., Honjo T. (1997). Involvement of RBP-J in biological functions of mouse Notch1 and its derivatives. Development 124, 4133–4141 [DOI] [PubMed] [Google Scholar]
- Kawakami K., Takeda H., Kawakami N., Kobayashi M., Matsuda N., Mishina M. (2004). A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev. Cell 7, 133–144 [DOI] [PubMed] [Google Scholar]
- Kimmel R. A., Onder L., Wilfinger A., Ellertsdottir E., Meyer D. (2011). Requirement for Pdx1 in specification of latent endocrine progenitors in zebrafish. BMC Biol. 9, 75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohyama J., Tokunaga A., Fujita Y., Miyoshi H., Nagai T., Miyawaki A., Nakao K., Matsuzaki Y., Okano H. (2005). Visualization of spatiotemporal activation of Notch signaling: live monitoring and significance in neural development. Dev. Biol. 286, 311–325 [DOI] [PubMed] [Google Scholar]
- Kopinke D., Brailsford M., Shea J. E., Leavitt R., Scaife C. L., Murtaugh L. C. (2011). Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development 138, 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopinke D., Brailsford M., Pan F. C., Magnuson M. A., Wright C. V., Murtaugh L. C. (2012). Ongoing Notch signaling maintains phenotypic fidelity in the adult exocrine pancreas. Dev. Biol. 362, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Zhao X., Fang Y., Jiang X., Duong T., Fan C., Huang C. C., Kain S. R. (1998). Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 273, 34970–34975 [DOI] [PubMed] [Google Scholar]
- Mazzone M., Selfors L. M., Albeck J., Overholtzer M., Sale S., Carroll D. L., Pandya D., Lu Y., Mills G. B., Aster J. C., et al. (2010). Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc. Natl. Acad. Sci. USA 107, 5012–5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourikis P., Sambasivan R., Castel D., Rocheteau P., Bizzarro V., Tajbakhsh S. (2012). A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 30, 243–252 [DOI] [PubMed] [Google Scholar]
- Mutoh H., Naya F. J., Tsai M. J., Leiter A. B. (1998). The basic helix-loop-helix protein BETA2 interacts with p300 to coordinate differentiation of secretin-expressing enteroendocrine cells. Genes Dev. 12, 820–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya F. J., Huang H. P., Qiu Y., Mutoh H., DeMayo F. J., Leiter A. B., Tsai M. J. (1997). Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 11, 2323–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi H., Pardali K., Vanlandewijck M., Heldin C. H., Moustakas A. (2007). Notch signaling is necessary for epithelial growth arrest by TGF-beta. J. Cell Biol. 176, 695–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obholzer N., Wolfson S., Trapani J. G., Mo W., Nechiporuk A., Busch-Nentwich E., Seiler C., Sidi S., Sollner C., Duncan R. N., et al. (2008). Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J. Neurosci. 28, 2110–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochocinska M. J., Hitchcock P. F. (2009). NeuroD regulates proliferation of photoreceptor progenitors in the retina of the zebrafish. Mech. Dev. 126, 128–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M. J., Pisharath H., Yusuff S., Moore J. C., Siekmann A. F., Lawson N., Leach S. D. (2009). Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech. Dev. 126, 898–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls S., Zecchin E., Tiso N., Bortolussi M., Argenton F. (2007). Function and regulation of zebrafish nkx2.2a during development of pancreatic islet and ducts. Dev. Biol. 304, 875–890 [DOI] [PubMed] [Google Scholar]
- Perdigoto C. N., Schweisguth F., Bardin A. J. (2011). Distinct levels of Notch activity for commitment and terminal differentiation of stem cells in the adult fly intestine. Development 138, 4585–4595 [DOI] [PubMed] [Google Scholar]
- Pierfelice T., Alberi L., Gaiano N. (2011). Notch in the vertebrate nervous system: an old dog with new tricks. Neuron 69, 840–855 [DOI] [PubMed] [Google Scholar]
- Price D. M., Jin Z., Rabinovitch S., Campbell S. D. (2002). Ectopic expression of the Drosophila Cdk1 inhibitory kinases, Wee1 and Myt1, interferes with the second mitotic wave and disrupts pattern formation during eye development. Genetics 161, 721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenaigner I., Krecsmarik M., Hayes J. A., Bahn B., Lepier A., Fortin G., Gotz M., Jagasia R., Bally-Cuif L. (2011). Clonal analysis by distinct viral vectors identifies bona fide neural stem cells in the adult zebrafish telencephalon and characterizes their division properties and fate. Development 138, 1459–1469 [DOI] [PubMed] [Google Scholar]
- Row R. H., Kimelman D. (2009). Bmp inhibition is necessary for post-gastrulation patterning and morphogenesis of the zebrafish tailbud. Dev. Biol. 329, 55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer N., Groth A., Hans S., Campos-Ortega J. A. (2001). An instructive function for Notch in promoting gliogenesis in the zebrafish retina. Development 128, 1099–1107 [DOI] [PubMed] [Google Scholar]
- Scheer N., Riedl I., Warren J. T., Kuwada J. Y., Campos-Ortega J. A. (2002). A quantitative analysis of the kinetics of Gal4 activator and effector gene expression in the zebrafish. Mech. Dev. 112, 9–14 [DOI] [PubMed] [Google Scholar]
- Shimojo H., Ohtsuka T., Kageyama R. (2008). Oscillations in notch signaling regulate maintenance of neural progenitors. Neuron 58, 52–64 [DOI] [PubMed] [Google Scholar]
- Simon F., Fichelson P., Gho M., Audibert A. (2009). Notch and Prospero repress proliferation following cyclin E overexpression in the Drosophila bristle lineage. PLoS Genet. 5, e1000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyer J., Flasse L., Raffelsberger W., Beucher A., Orvain C., Peers B., Ravassard P., Vermot J., Voz M. L., Mellitzer G., et al. (2010). Rfx6 is an Ngn3-dependent winged helix transcription factor required for pancreatic islet cell development. Development 137, 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urasaki A., Morvan G., Kawakami K. (2006). Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics 174, 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Rovira M., Yusuff S., Parsons M. J. (2011). Genetic inducible fate mapping in larval zebrafish reveals origins of adult insulin-producing beta-cells. Development 138, 609–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}