

Abstract
Although a few hundred single nucleotide polymorphisms (SNPs) suffice to infer close familial relationships, high density genome-wide SNP data make possible the inference of more distant relationships such as 2nd to 9th cousinships. In order to characterize the relationship between genetic similarity and degree of kinship given a timeframe of 100–300 years, we analyzed the sharing of DNA inferred to be identical by descent (IBD) in a subset of individuals from the 23andMe customer database (n = 22,757) and from the Human Genome Diversity Panel (HGDP-CEPH, n = 952). With data from 121 populations, we show that the average amount of DNA shared IBD in most ethnolinguistically-defined populations, for example Native American groups, Finns and Ashkenazi Jews, differs from continentally-defined populations by several orders of magnitude. Via extensive pedigree-based simulations, we determined bounds for predicted degrees of relationship given the amount of genomic IBD sharing in both endogamous and ‘unrelated’ population samples. Using these bounds as a guide, we detected tens of thousands of 2nd to 9th degree cousin pairs within a heterogenous set of 5,000 Europeans. The ubiquity of distant relatives, detected via IBD segments, in both ethnolinguistic populations and in large ‘unrelated’ populations samples has important implications for genetic genealogy, forensics and genotype/phenotype mapping studies.
Introduction
The emergence of genotyping platforms that assay hundreds of thousands of sites across the genome has facilitated the rapid growth of databases with genome-wide data for human populations [1]–[3]. These large databases may include data for pairs of individuals who are cryptically related over the last 10 generations, e.g., on the order of 2nd to 9th cousins. Such cousins often share extended segments of DNA that are identical except for recent mutations. Genetic similarity metrics, such as the length of DNA segments that are consistent with identity by descent (IBD) from a common ancestor, can be used to detect relatively distantly related individuals [1], [4]. Because recombination breaks down these shared segments rapidly, pairs of relatives vary in the number and length of shared segments that they inherit from a common ancestor. Fourth cousins, for example, may or may not share any long segments that are identical by descent [5].
For a given pair of individuals, the inferred IBD pattern depends on a broad set of factors, including not only the rate and pattern of recombination, but also the accuracy of the IBD detection algorithm and the type of genetic data (e.g., autosomal SNPs) under consideration. The pattern of IBD also depends on the number of ancestors that the two individuals have in common and the number of generations since each of these common ancestors. These factors, in turn, depend on the structure of the relevant population. In a finite, panmictic population, for a random pair of individuals from the same generation, the expected degree of cousinship, that is, the n+1 number of generations back to the most recent common ancestor, is a function of the effective population size, Ne [6], [7]. For some human populations, the average pair of individuals is related as closely as 2nd–4th cousins, and in fact, may be related through multiple shared, recent ancestors. [8], [9]. However, for many large, panmictic human populations a pair of individuals chosen at random is unlikely to be closer than 10th–20th cousins [10].
Detecting these IBD patterns and predicting relationships on the basis of these patterns can be complicated by endogamy, the tendency to choose a mate from the same ethnic group or geographic location, that is associated with reduced effective population size. In smaller endogamous populations, any two individuals are likely to be genetically similar because they share either one or multiple recent common ancestors. The level of homozygosity within endogamous populations is therefore higher than in large populations and those receiving gene flow from neighboring groups [11]. Malecot [12] introduced the concept of an inbreeding coefficient, the probability of inheriting identical alleles from both parents. The inbreeding coefficient of an individual is equivalent to the coancestry, or kinship, coefficient of that individual's parents. The higher the kinship coefficient of a pair of individuals, the more likely they are to share DNA that is IBD. Inferring that DNA segments from two individuals are IBD is conceptually similar to another widely used metric of genetic similarity, runs of homozygosity. Runs of homozygosity within an individual indicate that the homologous segments trace back to a recent common ancestor.
Many genomic studies of population history have focused on analyses of genetic diversity that reflect ancient demographic events [13], [14]. However, endogamy in human populations likely reflects demographic processes occurring only on the order of hundreds, rather than thousands of years [15]–[17]. Patterns of IBD within populations provide the opportunity to examine the relationship between genetic similarity and kinship based on common ancestry between 100 and 300 years ago. In turn, the number and sizes of segments shared IBD provide a basis for estimating the kinship relationship between any two individuals.
Mountain and Ramakrishnan [18] explored the difference between ethnolinguistically- and continentally-defined populations in their distributions of genomic similarity between individuals using short tandem repeats (STRs) analyzed in the Human Genome Diversity Panel populations (HGDP-CEPH). In this study we characterize the levels of kinship within ethnolinguistically and more broadly defined populations using extensive, genome-wide single nucleotide polymorphism (SNP) data. Specifically, we infer identity by descent from unphased autosomal SNP data in order to explore the distribution of IBD segments in 121 populations from the HGDP-CEPH (n = 952) and a 23andMe dataset (n = 22,757), a subset of genomic data from the 23andMe customer database. The latter set includes data from Ashkenazi Jewish and broadly-defined “European” and “Asian” populations. In order to interpret the patterns of population-level sharing, we simulate extended pedigrees for different ethnic groups and estimated the expected extent of sharing for 1st through 9th cousins. Based on these simulations, we infer the likely relationship between each pair individuals within each population sample. This approach is an alternative to methods that use allele frequencies and identity by state (IBS) to infer relationships from low-density genetic data [19]. We show that it is possible to assign kinship with reasonable accuracy for relationships dating back 100–300 years even for a population that exhibits high levels of homozygosity.
Results
Population Structure among Ethnolinguistically- and Continentally-defined Groups
To infer identity by descent, we scanned each pair of genomes for long runs of genotype pairs that lack “opposite homozygotes” (Figure 1). We define inferred “IBDhalf” as the sum of the lengths of genomic segments where two individuals share DNA identical by state for at least one of the homologous chromosomes (see also [20]). This method is computationally feasible in large sample sets of thousands of individuals because we do not need to phase individuals; phasing is necessary for other methods of accurately inferring short IBD segments (e.g. BEAGLE, GERMLINE [1], [4], [5], [21]). We find our approach is accurate down to 7 cM per pair when we simulate the number of false positives seen in only one member of a family a trio and an un-related individual (see Methods ).
Figure 1. Schematic of IBDhalf inference method.

IBDhalf segments were inferred from unphased genotype data where a series of alleles were identical by state for at least one of the homologous chromosomes in a given pair of individuals. IBD segments are indicated in purple. The boundaries of the IBD segments are defined by “opposite homozygotes”. Additionally, an IBD region had to be minimally 5 cM in length and contains >400 genotyped SNPs that were homozygous in at least one of the two individuals being compared (see Methods ).
For each population we characterize the distribution of inferred IBD, after excluding individuals inferred to be first cousin or closer [22]. The values of mean IBDhalf, that is the average extent of inferred IBDhalf across all pairs of individuals within a population, vary widely across a number of different ethnolinguistic and broadly-defined populations (Table 1). Tables 1 and S1 summarize genomic identity by descent for pairs of individuals sampled from 121 populations from across the globe. The large 23andMe population samples of individuals with European and Asian ancestry exhibit very low levels of mean IBDhalf, ranging from about 0.05 to 0.15 cM. In contrast, estimates of mean IBDhalf for Native American population samples from the HGDP-CEPH set range from about 400 to 1900 cM. Apart from extremely high estimates of mean IBDhalf in the Native American samples, there is no clear geographic pattern to the rankings of IBDhalf for ethnic groups across different world regions. This lack of a geographic pattern suggests that mean IBDhalf is largely independent of general levels of heterozgosity in the population [14]. The correlation between haplotype heterozygosity [23] and mean IBDhalf is modest (r2 = 0.45), and that correlation is driven primarily by the inclusion of Native American samples (r2 = 0.14 without Native Americans, Figure S2A).
Table 1. IBD statistics for a subset of HGDP-CEPH and 23andMe population samplesa.
| Population | Region of Ancestry | Mean IBDhalf b | Sample size | FIBD c | Total Non-zero Pairs | Source |
| Surui | S. America | 1870.5 | 8 | 1.00 | 28 | CEPH/HGDP |
| Karitiana | S. America | 1229.5 | 14 | 0.88 | 80 | CEPH/HGDP |
| Kalash | C. Asia | 260.0 | 23 | 1.00 | 253 | CEPH/HGDP |
| Yakut | E. Asia | 85.4 | 25 | 0.92 | 276 | CEPH/HGDP |
| Biaka Pygmies | C. Africa | 73.2 | 21 | 0.96 | 202 | CEPH/HGDP |
| Maya | N. America | 47.3 | 21 | 0.47 | 99 | CEPH/HGDP |
| Sardinian | Europe | 12.4 | 28 | 0.38 | 143 | CEPH/HGDP |
| Tuscan | Europe | 9.3 | 8 | 0.43 | 12 | CEPH/HGDP |
| Ashkenazi | Europe/Near East | 23.0 | 847 | 0.85 | 304,539 | 23andMe |
| Finland | Europe | 10.0 | 149 | 0.53 | 5844 | 23andMe |
| Yoruba | W. Africa | 1.0 | 21 | 0.06 | 12 | CEPH/HGDP |
| Canada | Mixed | 0.6 | 373 | 0.04 | 2775 | 23andMe |
| Han Chinese | E. Asia | 0.3 | 44 | 0.01 | 13 | CEPH/HGDP |
| Italy | Europe | 0.1 | 386 | 0.01 | 743 | 23andMe |
See also Supporting Information, Table S1, for IBD statistics in all 121 populations.
“IBDhalf” is defined as the sum of the lengths of genomic segments where two individuals are inferred to share DNA identical by state for at least one of the homologous chromosomes.
“FIBD” is defined as the fraction of pairs that share at least one IBDhalf segment greater than or equal to 7 cM.
Figures 2A–2C and S1 present distributions of IBDhalf for all pairs of individuals within our sampled population. We define “FIBD” as the fraction of pairs within a population that share non-zero IBD, standardized by the total number of pairwise comparisons. Population samples where only a few pairs show evidence of IBD, that is where FIBD is less than 25%, have pairwise distributions that come close to being exponentially distributed (Figure S1). However, some population samples, including several Native American groups (Figure 2A), the Yakut (Figure 2B), and the Kalash, are largely composed of pairs with IBDhalf greater than 7 cM. Almost all pairwise values of IBDhalf within the Native American Karitiana, Pima and Surui samples fall between 500 and 2500 cM (Figure 2A, Figure S1). The Mayan IBDhalf values, in contrast, appear to be exponentially distributed (Figure S1). These differences across the mean IBDhalf distributions of the Native American groups suggest that the IBD patterns reflect either recent population endogamy or non-random population sampling rather than the ancient population founder event that occurred during initial migration into the Americas, which would have influenced all populations similarly [24], [25].
Figure 2. Distributions of IBDhalf for pairs of individuals within three human populations.

The average amount of DNA that is identical by descent (mean IBDhalf) varies widely across HGDP-CEPH, European, Asian and Ashkenazi populations. We present distributions of pairwise comparisons with IBDhalf segments ≥7 cM for the (a) Karitiana Native Americans, (b) Yakut of Siberia, (c) Ashkenazi Jews primarily from the United States. Prior to the analysis, individuals were eliminated in order to remove close relationships (sibling, parent-child, avuncular, grandparent-grandchild, and 1st cousin pairs) (see Methods ). Pairs with less than 7 cM IBDhalf are not displayed. Distributions of IBDhalf for additional HGDP-CEPH samples are presented in Supplementary Material (Figure S1).
Assigning Degrees of Kinship using IBD
We simulated genetic data within extended family pedigrees in order to characterize the correspondence between the number of IBDhalf segments or IBDhalf and the degree of cousinship for a pair of individuals from the same population. Drawing from pools of computationally-phased haplotypes from HGDP-CEPH and 23andMe population samples, we simulated extended family pedigrees of 11 generations assuming random mating of individuals from the sample (see Methods ). Using the specified family pedigree and the data simulated on the basis of that pedigree, we estimated the number of shared segments and IBDhalf for cousinship degrees of n (where n = 1–10) (Figure 3A). Our simulations suggest that IBDhalf(n) decreases as the nth cousinship increases from 1st to 5th cousins (Figure 3B). The decay in mean IBDhalf(n) asymptotes at different levels across a set of ethnolinguistically-defined populations, starting at n = 5 (e.g., Kalash) (Figure 3B). Therefore, in populations with moderate to high mean IBDhalf (Table 1), IBDhalf alone cannot be used to distinguish accurately between 5th or greater cousinships.
Figure 3. Relationship between degree of cousinship and IBDhalf metrics.

We used pedigree-based simulations to characterize the relationship between IBDhalf metrics and degrees of cousinship for multiple population samples. a) Genomic data from a European sample were used to simulate an 11-generation pedigree. The joint distribution of IBDhalf and number of IBDhalf segments is shown for each pairwise comparison from the pedigree simulations. GP/GC indicates grandparent/grandchild pairs. b) For each of eight populations, we summarize the distribution of IBDhalf by plotting IBDhalf(n) for the population by degree of cousinship. The degrees of cousinship distinguished by IBDhalf(n) asymptotes at different levels of IBD in ethnolinguistically-defined populations. Simulations were run on phased samples from several HGDP-CEPH population samples and European, Asian and Ashkenazi samples from a 23andMe customer dataset. Simulations were conducted by specifying an extended pedigree structure and simulating genomes for the pedigree by mating individuals drawn from a pool of empirical genomes (see Methods ).
We explored in greater detail the data simulated for 2nd–9th cousins based on the Ashkenazi (Figure 4A–C) and European (Figure 4D–F) samples. These figures reveal that there is a relatively clear relationship between IBDhalf plus number of IBD segments and degree of cousinship for 2nd–5th cousinships. For example, the median relationship for a pair of Ashkenazi individuals with IBDhalf of 100 cM and >9 segments is 2nd cousinship, with 1st–3rd cousin bounds (Figure 4B, 95% and 5% bounds presented in Figure 4A,C). For pairs from both populations, 3rd cousinships encompass a wide range of segmental variation, generally greater than 25 cM IBDhalf and fewer than 10 distinct IBD segments (Figure 4B,E). For the European sample, just one or two shared segments totaling less than 15 cM in length yield a median relationship of 5th cousin (Figure 4E). However, low amounts of IBDhalf (e.g., less than 4 segments totaling less than 40 cM in length) in simulated Ashkenazi pairs were not associated with an identifiable cousinship within our simulated pedigree (Figure 4A–C).
Figure 4. Distributions of IBDhalf by degree of cousinship, assessed with simulated pedigrees for Ashkenazim and Europeans.

Plotted, for each combination of IBDhalf and number of IBD segments, are the 95th percentile, 50th percentile and 5th percentile degrees of cousinship based on 1 million simulated pedigrees. A–C) Ashkenazi pairs, D–F) European pairs, G–I) The differences between Ashkenazi and European results, presented in the prior panels, are represented in grey. Darker grey indicates higher number of differences. Each nth cousinship category was scaled by the expected number of nth degree cousins given a model of population growth (Table 2, Methods ). Simulations were conducted by specifying an extended pedigree and creating simulated genomes for the pedigree by mating individuals drawn from a pool of empirical genomes. Pairs of individuals who appear to share IBDhalf that was not inherited through the specified simulated pedigree are marked in grey in the A–F panels.
Expected and Predicted Distant Cousins
The relationship between IBD metrics and degree of relationship indicated in Figures 3 and 4 can be leveraged to predict degree of relationship on the basis of IBD metrics. After quantifying the bounds of IBD sharing for 1st–9th cousins for European and Ashkanazi population samples, we determined how common it was to find a distant relative in a heterogenous database of a specified size. In order to estimate the number of individuals who have a distant relative in the 23andMe dataset, we first removed individuals in pairs with close kinship (i.e., parent/child, sibling, grandparent/grandchild, avuncular, 1st cousin). Then, for all pairs of individuals in each of the European and Ashkenazi samples, we counted the fraction of pairs with IBDhalf ≥7 cM. Sample size was initially restricted to n = 300 for each population.
The fraction of 23andMe individuals with at least one predicted 2nd–9th degree cousin varied across the European and Ashkenazi populations. Virtually all (99%) individuals of Ashkenazi ancestry had a least one relative detectable in the 23andMe dataset. Interestingly, 70% of individuals in our 23andMe European sample were also predicted to have at least one 2nd–9th cousin (or similarly distant relationship) even with a dataset size as small as 300.
In order to examine the relationship between database size and number of detected cousins per individual, we assembled 5,000 individuals with European ancestry and randomly drew subsets of this dataset (Figure 5A). For each subset we calculated the fraction of individuals with at least one relative (2nd–9th degree cousin) given an IBDhalf threshold of ≥7 cM. As the dataset size increased, the fraction of individuals with at least one distant relative increased logarithmically (Figure 5A). With a sample size of 1,000 heterogenous Europeans, more than 90% of individuals had a predicted cousin in the 2nd–9th degree range. With a sample size of 5,000 individuals, virtually every individual had at least one predicted relative. We also catalogued the number of individuals for each nth degree of cousinship for datasets of increasing sizes in our database (Figure 5B). The 4th–6th cousinships made up the bulk of predicted relationships; there were approximately 30,000 predicted 4th cousin pairs in a dataset of 5,000 Europeans (Figure 5B). The ranking of the number of detectable cousins (Table 2) differs from our ranking of observed cousinships (Figure 5b) [please note that Ndc refers to the number of expected cousins for a single individual, while Figure 5b shows the observed number of cousin pairs.] This difference in the ranking of the expected number of detectable cousins and the observed number of cousin pairs in our dataset may be due to: deviation from the assumed 2.5 mean number of historical offspring in our model, higher than expected IBD due to sharing multiple ancestors rather than a single common ancestor, and variability in the recombination map. In sum, there are an extensive number of cousins for any given individual sampled from a large population (Table 2).
Figure 5. Fraction of 23andMe individuals with detectable distant relatives within subsamples inferred using IBDhalf.

A) The fraction of individuals with at least one predicted relative (2nd–9th cousin) given datasets of varying size. All datasets were drawn from a dataset of 5000 individuals with European ancestry. All closely related individuals (i.e., 1st or 2nd generation family) were removed before performing the analysis. B) The number of predicted cousins of each degree of cousinship given the dataset size. Predictions based on parameters obtained from simulations (Figure 4e).
Table 2. Expected extent of IBD and number of cousins for 1st–10th degrees of cousinship.
| Degree of cousinship | Expected amount of IBD (cM)a | Chance of detecting nth cousin (%) with IBDhalf b | Expected number of cousinsc | Expected number of detectable cousins (Ndc)d |
| 1 | 900 | 100 | 7.5 | 7.5 |
| 2 | 225 | 100 | 38 | 38 |
| 3 | 56 | 89.7 | 190 | 170.4 |
| 4 | 14 | 45.9 | 940 | 431.5 |
| 5 | 3.5 | 14.9 | 4,700 | 700.3 |
| 6 | 0.88 | 4.1 | 23,000 | 943 |
| 7 | 0.22 | 1.1 | 120,000 | 1,320 |
| 8 | 0.055 | 0.24 | 590,000 | 1,416 |
| 9 | 0.014 | 0.06 | >106 | NAe |
| 10 | 0.0034 | 0.002 | >106 | NAe |
Theoretical expectation of the amount of IBD across the genome shared between nth cousins, assuming 3600 cM across the entire genome. It should be emphasized this description assumes a single common ancestor for a pair of cousins; multiple shared common ancestors will increase the predicted IBD sharing.
The fraction of nth degree cousins detected using our IBD algorithm and based on simulated pedigrees of up to 10th degree cousins (see Methods ).
Assuming a specific model of pedigree and population growth over the past 11 generations (see Methods ).
The expected number of cousins detectable with our IBD algorithm (Ndc) was calculated by multiplying the probability of detecting an nth cousin by the number of nth cousins obtained from our pedigree model of population growth (see Methods ).
Given the variation in population growth at >9 generations ago, combined with a low power of detection for 9th or 10th cousins, we have indicated the number of detectable cousins for those categories as not applicable, “NA”.
We then compared the distributions of predicted kinship with expectations based on population history and database sampling. Specifically, we modeled the number of cousins (2nd–9th degree) expected for any given individual ( Methods ). The model predicted that 99% of Ashkenazim and 91% of individuals with European ancestry would have at least one 2nd–9th degree cousin in the datasets representing 300 and 5000 individuals, respectively. These predictions closely matched our observed values: 99% for Ashkenazim and 99% for Europeans. The predictions are robust to several choices of parameters (i.e., number of children per generation, shared ancestors [not shown]), but the choice of Npop parameter (i.e., the current size of the pool of potential relatives) significantly influences the theoretical predictions (Table S2).
Discussion
For any given set of genome-wide genotype data for a population sample, one can observe the number and length of DNA segments shared IBS across the entire genome for a pair of individuals and, from these observations, infer IBD. Our simulations indicate that one can then predict the degree of cousinship for each pair of individuals, at least when our IBDhalf statistic is sufficiently large (Figure 4). The total length of homologous identical by descent DNA segments that two individuals share is similar to a classical metric, runs of homozygosity (ROHs), which are inferred by comparing allele states extending along a locus within an individual. ROHs are readily calculated without phasing of genotype data, and when averaged over all individuals in a sample, have been used successfully to track population processes, such as consanguinity [8], [13], [26], [27]. An advantage of using a pairwise IBD approach rather than ROHs is that pairwise IBD comparison yields discovery of many relatives within a population, whereas ROHs only allow inference of relatedness for an individual's parents.
Population Structure among Ethnolinguistically- and Continentally-defined Groups
The characterization of genomic similarity across samples of the HGDP-CEPH collection has revealed about 100 cryptically related or duplicate samples [18], [22]. Prior analyses focused on identifying familial relationships such as sibships, parent-offspring pairs, etc. (i.e., 1st–3rd degree relatives). We extended this analysis through pairwise IBD comparisons and characterized pairs of individuals who are related as 1st through 9th cousins within each HGDP population (Figure 2, Figure S1). When datasets include samples from populations where many of the individuals are closely related, analyses of population structure tend to cluster these populations more discretely than populations with greater genetic variation. For example, previous analyses of genome-wide microsatellite data identified the Kalash of Pakistan as a distinct global population in a STRUCTURE analysis of HGDP, k = 6 [28]. Our pairwise IBD metrics suggest an explanation for this result; the Kalash share on average 260 cM IBDhalf (Table 1) and thus any given pair of individuals is the genomic equivalent of second cousins. In both microsatellite and SNP genotype-based STRUCTURE plots, Native American HGDP populations emerged as a population subset at k = 4 [23], [28]. Our IBDhalf estimates for the Native Americans (Figure 2A, S1, Table 1) are consistent with an interpretation that the majority of individuals within these populations are related as the genomic equivalent of 2nd cousins or closer. We note that although putative close relatives were removed with the HGDP952 dataset, our 700 cM cutoff employed for the 23andMe dataset would have removed most pairs in the Native Americans populations (though not from most HGDP groups, see Figure S1). Populations such as the Karitiana or Pima display levels of mean IBDhalf that are 3–4 orders of magnitude higher than many HGDP Asian populations such as the Han, Japanese, Mongolians. As such, population genetic analyses of the HGDP-CEPH Native Americans will need to account for the IBD derived from their elevated levels of historic endogamy or uneven population sampling.
The high levels of mean IBDhalf and FIBD of many of the 121 globally distributed populations we analyzed are consistent with substantial structure among populations within continents. (Table 1, Table S1). On the other hand, populations that were sampled randomly across a wide ethnolinguistic and geographic space (i.e., 23andMe European and Asian continental samples, Table 1) have very low estimates of mean IBDhalf. The elevated amount of IBDhalf in the majority of ethnolinguistic populations from HGDP-CEPH, and in some 23andMe sub-continental samples, compared to that of samples of general European or Asian ancestry is indicative of small effective population size, often reflecting endogamy. Our IBD metrics complement and extend other genomic analyses, such as principal components analysis or clustering algorithms, that have recently indicated fine-scale population structure within continents [16], [29], [30]. We show that IBD distributions can be used to characterize patterns of kinship within groups over the past 100–300 years, and these genetic-based indications of endogamy can be helpful for explaining the processes generating population structure. Our results also suggest that exploring within-population structure, occurring during the last few hundred years, may be fruitful avenue for future research [31].
Differences in IBD Metrics Across Ethnolinguistically-defined Populations
Although there is no clear geographic patterning, populations practicing hunting-gathering or pastoralist subsistence strategies tend to have higher mean IBDhalf. African hunter-gatherers, former hunter-gatherers from South America, Yakut, Mozabite Berber, and Bedouin pastoralists are ranked in the top 50% of our sample set in terms of mean IBDhalf estimates (Table 1). A high estimate of mean IBDhalf suggests that the population has a relatively small effective size or has been sampled in such a manner as to over represent related individuals. Small effective size for human populations often reflects a high level of endogamy, a population bottleneck, or both.
In order to illustrate the effect of recent demographic fluctuations on IBD distributions, we highlight two populations: the Yakut and Ashkenazi Jews. The Yakut, a population of nomadic pastoralists in Siberia, have an IBDhalf distribution that is approximately normally distributed, with a mean of 85 cM (Figure 2b). Recent demographic inference from mitochondrial DNA simulations suggests that a small group of individuals (∼150 females) in southern Siberia founded the ancestral Yakut population only about 1,000 years ago (ya) [32]. Their IBDhalf distribution includes a few low identity pairwise comparisons (8%) and the remaining pairwise comparisons are more normally distributed (FIBD = 92%, Figure 2b). The high proportion of pairs of individuals who share some fraction of their genome IBD is consistent with the results from mtDNA mismatch simulations indicating that the Yakut experienced a founder event occurring within the past 1,000 years, followed by population isolation.
Historically Ashkenazi Jews have had endogamous mating patterns and are thought to have experienced two major founder events approximately 650ya and 2,000ya [33], [34]. We studied a set of over 150 individuals in the 23andMe database who reported having four Ashkenazi grandparents. Mean IBDhalf is in the bottom quartile of our population samples, 8 cM, similar to that of other European populations such as the Tuscans or Sardinians. The FIBD statistic (FIBD = 37%) for the Ashkenazi is similar to that of many other single ethnolinguistically-defined European or Asian populations (Table S1). Although the Ashkenazi and the Yakut have similar purported population histories, their patterns of genome-wide IBD differ substantially. The differences may reflect different initial population sizes or recent gene flow into the Ashkenazim.
Assigning Degrees of Kinship using IBD
Through simulation we demonstrated that it is possible use IBDhalf to accurately predict a probable level of kinship for 2nd–5th cousins. Cousinships between 6th–9th degrees are assigned less accurately due to varying levels of background relatedness in different populations and stochasticity in IBD segment lengths over many generations (Figure 3 and Figure 4). We focused on the metrics IBDhalf and number of IBDhalf segments shared by a pair of individuals in order to identify recent common ancestry. We verified the accuracy of the IBDhalf segments by checking whether the segment was found in either of the simulated parents of the pedigree. The paucity of predictions at high nth degrees suggests that our IBDhalf metrics and IBD algorithm is accurate only within the bounds of 1st–9th cousins.
A large fraction of simulated Ashkenazi pairs shared IBDhalf segments that showed no descent through the common ancestor specified in our simulated extended pedigree (Figure 4a–c, i.e., indicated by “unknown”). We initially assumed that all pairs of Ashkenazi used to seed the simulations were unrelated (after pruning close relatives, Methods ). However, the high fraction of Ashkenazi pairs with short IBDhalf segments (5–20 cM, Figure 2C) indicates that many individuals in our dataset are already related on the order of 2nd–9th cousins. Thus, a random sample of Ashkenazim, such as that incorporated into our simulations, includes pairs of individuals who share multiple, long segments that are identical by descent and whose inferred relationship, in some cases, is more recent than the cousinship specified in the simulation. Ancestry on the order of 2nd–9th cousins, or about 200 years, is considerably more recent than a common ancestor dating back to the proposed founder event for the Ashkenazim 1,500 years ago. This finding illustrates the impact of endogamy and possibly non-random mating (i.e., sub-structure) within the Ashkenazi population over the past few hundred years on patterns of genetic variation.
Alternatively, the pattern of “unknown” matches may reflect the high homozygosity among the Ashkenazim. Short, distinct but overlapping IBD segments that would otherwise not be detectable by an IBD algorithm can appear together as a longer, detectable tract of IBD with another “unrelated” individual. Such matches reflect identity by state rather than identity by descent. Preliminary simulations suggest that this is a potential explanation for the excess “unknown” matches in the Ashkenazi population sample [35] (Figure 4).
Expected and Predicted Distant Cousins
We determined how many individuals in a population sample have a predicted 2nd to 9th degree cousin in the same sample. We observed that 99% of Ashkenazim and 99% of Europeans have at least one distant relative (e.g., 2nd–9th cousin) in the 23andMe datasets (of 300 and 5,000 individuals respectively). It is possible that our dataset is enriched for second cousins, as 23andMe customers may have suggested that extended family members join the 23andMe service. However, most IBDhalf matches occurred with very low amounts of IBDhalf (e.g., 10–20 cM) (Figure 5a) indicative of likely 3rd–9th cousinship (Figure 3, Figure 4). As most Americans are unlikely to know their 3rd cousins or more distant relatives (i.e., 7 or more generational degrees of separation), the general finding is relevant to random sampling of individuals in these populations. Using identity by descent, Kong et al. [36] examined a 10 cM segment in the MHC region within a sample of Icelandic individuals. In a database of 6,300 samples, they found that 78% of Icelandic individuals share a long IBD segment with at least one other person. Both our results and those of Kong et al. [36] suggest that a large sample of individuals with European ancestry is likely to include many pairs of 2nd through 9th cousins.
Factors that contribute to the large number of relative pairs within moderately sized datasets include the large numbers of actual cousinships, especially beyond 4th cousins (Table 2), and the small effective sizes of some populations. Given a model of 2.5 children per generation for every mating event, the number of relatives in the 5th through 10th degree cousinship categories runs from the thousands into the millions (Table 2). While we made a number of simplifying assumptions (i.e., perfect survivorship, non-overlapping generations), the expected number of detectable cousins between 1st and 8th, (Ndc) for a given individual is very high (Table 2). The expected number of individuals with at least one predicted relative in a dataset of a given size using our Ndc estimates, was similar to the observed values.
Additionally, the exponentially growing number of ancestors for any individual, constrained by finite population size means that the most recent common ancestor for any two individuals can be quite recent, especially in small populations. Indeed, Rohde et al. [37] used a spatially structured model to calculate that every pair of living humans has a most recent common ancestor as few as 200 generations ago. The number of individuals with at least one detected relative in our dataset varies between populations (Figure 5a). Ashkenazim are more likely than other Europeans or Asians to have at least one cousin in a dataset of 300 (99%) because the effective size of the Ashkenazi population is small. This small effective size most likely reflects population bottlenecks and low rates of migration into the population.
We predicted the number of distant cousins we could detect for a given individual with a moderately sized database given simulations and a model that takes into account average sibship size and effective population size (see below). We found that our expectations matched empirical results when we assumed that we are drawing individuals from population that is 10% of the actual US census sizes. Two confounding factors are relevant in the comparison of the number of observed and expected cousins: population size and population structure (i.e. random mating). We assumed panmictic mating in our calculation of the exponential number of ancestors for each individual. However, we did not assume panmictic mating for the pool of individuals who have opted in to the 23andMe customer dataset presented here; we reduced the population size to account for population stratification.
If we were to incorporate non-random mating (i.e. population structure) into our calculation for the number of ancestors for a given individual, then in small populations a pair of individuals would share multiple ancestors thus reducing the ancestral population size and consequently the number of nth degree cousins for that population. For timescales on the order of 10 generation and populations larger 100,000, a random pair of individuals will rarely have overlapping ancestors [10].
Broad Significance
By identifying genomic segments that are inherited identically by descent we can characterize kinship among individuals within the last three hundred years. We observed tens of thousands of 2nd to 9th degree cousin pairs within a heterogenous set of 5,000 Europeans. The high frequency of such relative pairs in some populations is likely due to population bottlenecks followed by recent population growth over the past 10 generations and/or to endogamous mating within ethnolinguistic populations. Patterns of inferred IBD suggest, for example, that the Ashkenazi Jewish population has remained relatively small and endogamous over the last few centuries.
We emphasize that the high prevalence of IBD in many populations can advance disease association studies conducted within those populations. Similarly, disease phenotype mapping research has demonstrated the feasibility of homozygosity haplotype analysis in autism, Parkinson's disease and breast cancer studies, among others [38]. Homozygosity haplotype mapping identifies a disease locus with extended runs of homozygosity within an affected individual, indicative of a recent common ancestor for the segment. Homozygosity haplotype mapping studies have typically focused on autosomal recessive diseases and/or family pedigrees with known consanguinity. However, this approach can be extended to incorporate IBD between affected individuals, i.e., pairwise homozygosity mapping or relatedness mapping [39], [40]. Recent pairwise IBD analysis of individuals with breast cancer suggests that there are significant differences in local IBD between cases and controls [39]. We have demonstrated that, even among individuals from broadly-defined population samples, IBD segmental detection and inference of recent shared ancestry on the order of 10 generations is quite common (Figure 5). Thus, an IBD-based approach can facilitate disease association even in large genomic databases where the family pedigrees are unknown or individuals are considered unrelated.
Materials and Methods
Datasets
Our data consist of genotypes for 650,000 SNPs (publicly available) from the Illumina 650K platform typed in 52 worldwide human populations from the HGDP-CEPH collection [23]. Additionally, genotypes for about 580,000 SNPs typed on the 23andMe customized Illumina 550K+v1 or +v2 platforms were obtained from populations in the 23andMe customer database. All individuals provided informed consent and answered surveys online according to our human subjects protocol, which was reviewed and approved by Independent Review Consulting, now part of Ethical & Independent Review Services, a private institutional review board (http://www.eandireview.com). Consent was obtained electronically; in a subset of cases a waiver of consent was obtained. All consent processes were approved by the above-named institutional review board. In order to protect the privacy of 23andMe participants, the 580K data are not publicly available. We used the overlapping 550,000 SNPs from HGDP and 23andMe for analyses presented herein. We selected 5,000 unrelated individuals inferred to be of European ancestry, 300 inferred to be unrelated Ashkenazim, and 300 inferred to be unrelated East Asians. Most of these individuals were likely born in the United States. An additional set of # individuals is included in Table S1.
Ancestry was assigned using two methods. We used self-reported ancestry as obtained from customer surveys, where membership in a given ancestry was defined by an individual reporting all four grandparents born in the same country. For Ashkenazi Jewish, we identified a grandparent to be of Ashkenazi descent if this was stated in a field denoting ethnic identity. In order to increase the numbers of some subpopulations we also used principal components analysis to partition individuals into clusters. Individuals falling into predefined northern or southern European, Asian or Ashkenazi clusters were grouped for subsequent analyses. In order to obtain “unrelated” individuals, we removed close relationships (i.e., sibling, parent, grandparent, avuncular, 1st cousin) among 23andMe customer groups by employing a greedy search that chooses individuals who have less than 700 cM IBD shared between each person already in the set of conforming individuals. Similarly, we used the HGDP952 subset identified by Rosenberg [22] to eliminate closely related individuals (between siblings, parent-offspring, grandparents) among HGDP-CEPH individuals.
Identification and Calculation of IBD Segments
We inferred that two individuals share DNA IBD from unphased data. We inferred boundaries of IBD by comparing two individuals' genotypes at a locus and identifying SNPs where one individual's genotype is homozygous for one allele and the other individual's genotype is homozygous for a second allele. By characterizing stretches that lacked these “opposite homozygotes”, we defined regions that contain at least half IBD between two individuals (Figure 1, see also similar method in [36]). That is, an IBDhalf segment was characterized by a series of alleles that were identical by state for at least one of the homologous chromosomes in a given pair of individuals. We define “IBDhalf” as the sum of the lengths of genomic segments where two individuals are inferred to share DNA identical by descent for at least one of the homologous chromosomes. We additionally enforced two criteria to increase our confidence that a region represents DNA that is IBD: first, the region is minimally 5 cM in length (Figure 6) and second, it contains >400 genotyped SNPs that are homozygous in at least one of the two individuals being compared, ensuring that there is both sufficient genotype coverage and genetic distance defining the IBD region. Finally, we accepted a comparison as IBD if the longest segment in the comparison was at least 7 cM (Figure 6, see below). Pairs of individuals sharing less than the above IBD were considered to have zero IBDhalf. A recently published method that accounts for LD between SNPs has accuracy for IBD segments as low as 2 cM, however this also requires phasing large datasets [4]. We used the June 2006 version of the HapMap genetic map for estimates of genetic distance [41]. This genetic map was used for all populations in our dataset.
Figure 6. Precision and accuracy of implemented IBD algorithm.

A) We considered how accurately we detect IBD segments that were transmitted between parent and child. We compared distant cousins with trios; we expect to observe sharing between a distant cousin and the child to also be observed in one of the parents for the same (or a longer) segment. Using this approach, we calculated the precision of our algorithm at different IBD segment lengths in a large sample of European-Americans. IBD segment lengths greater than 7 cM were observed 90% of the time in at least one parent. Preliminary data suggest that 7 cM segments shared between a distant cousin and child that were not observed in the parents were due to false negatives in the parents. B) We also examined our ability to detect IBD segments in simulated genotypes. After simulating large pedigrees, we examined 30,000 segments shorter than 200 cM resulting from 1st to 10th cousin relationships. We calculated the percentage of true IBD segments were detected by the IBDhalf algorithm at different cM lengths. IBD segment lengths greater than 7 cM were detected over 90% of the time. C) This schematic illustrates the pedigree simulations, where actual genotypes reflect individuals randomly sampled from a given population and simulated children with known degree relationships were tracked. The simulated genotypes were then analyzed using the IBD algorithm (see Methods for additional details).
We minimized the effects of genotyping error in several ways: 1) we removed genotyped SNPs from consideration (for all individuals) if there were a large number of Mendelian errors when comparing data from known parent/offspring trios (more than 1 in 1000), 2) we removed SNPs that had a high “no call” rate (>40% no calls at a site) or otherwise failed quality control measures, and 3) in a putative IBD segment, we allowed for one opposite homozygotes if there was a region of at least 3 cM, containing 300 SNPs, surrounding the opposite homozygote that were free of opposite homozygotes. Segments identified as sharing IBD via our IBD algorithm and criteria described above are referred to as “IBDhalf” segments in this paper; note however, the lack of opposite homozygotes in a segment is evidence for IBD across either one or both chromosomes.
Accuracy of IBD detection algorithm
We assessed the accuracy of the IBD algorithm in two ways. First, we considered how accurately IBD segments were transmitted between parent and child. Given real IBD sharing, we expect that when comparing a distant cousin with a trio, sharing observed in the child should also be observed in one of the parents for the same (or longer) segment. We calculated the accuracy of the algorithm for a broad range of IBD segment lengths. IBD segment lengths greater than 7 cM showed 90% accuracy (Figure S6A).
We also compared the inferred IBD segments with IBD segments generated via simulation. By looking at a series of 30,000 simulated IBD segments shorter than 200 cM, we determined the percentage of IBD segments that were detected by our IBD algorithm for different simulated IBD lengths. IBD segment lengths greater than 7 cM were detected over 90% of the time (Figure S6B). Given the similar results of the two analyses, we concluded that a minimum longest segment length of 7 cM provided sufficient evidence for IBD and therefore removed pairwise relationships that did not meet this threshold in subsequent analyses.
IBD metrics by population and pairwise distributions
We define mean IBDhalf as average IBDhalf across all pairs of individuals within a population sample. Considering 7 cM of IBDhalf segment length identified between individuals to be a minimum, we calculated the mean IBDhalf for all HGDP-CEPH and 23andMe customer populations. Many pairwise comparisons within a population were characterized by less than 7 cM IBDhalf; these pairs were considered to have zero DNA IBD. We define a FIBD as the fraction of pairs within a population that share non-zero IBD, standardized by the total number of pairwise comparisons.
For each HGDP-CEPH and 23andMe customer populations we characterized the pairwise IBD distribution within each sample via mean IBDhalf, distributions of IBDhalf, and FIBD. We removed closely related individuals as described above, ensuring that the majority of individuals in each population sample should be only cryptically related.
Simulating IBD patterns from different genomic datasets
We expected that for different populations in our dataset the amount of DNA shared IBD by nth cousins would depend upon the amount of inbreeding the population has experienced in its recent past. To calculate population-specific expectations for the amount of DNA shared IBD between distant cousins, we simulated inheritance along extended pedigrees for each population of interest, starting from reference genotype data of known ancestry, and resulting in simulated genotypes of individuals with known relationships (Figure 6C). We generated a pedigree for pairs of nth cousins in the following manner: we began with two parents, at the top of the pedigree, who had two children. The two children then mated with two randomly-chosen individuals, sampled without replacement, from the population sample and produced only one child per union, resulting in a pair of individuals who are first cousins. By mating each of the two first cousins with a randomly-chosen individual from the population, producing exactly one child in each union, we obtained second cousins. Further repetitions resulted in more distant simulated cousins of known degree. Our method assumes that the composition of currently available genomes is representative of genomic diversity throughout the past 200 years, because we paired contemporary individuals with simulated ones throughout the different generations of the pedigree.
To simulate data for a pair of nth cousins, we required 2n+2 phased autosomal genomes. We used SNP genotypes from the 23andMe database that were of inferred Ashkenazi ancestry and European ancestry and from a subset of the HGDP-CEPH populations (see Datasets above). Genotype data were phased using version 3.0 of BEAGLE [42]. Data were phased in groups of 1000 subjects, grouped by ethnicity. Within each group, chromosomes were split into regions of 10,000 SNPs (with a 1000 SNP overlap between adjacent regions). All regions were phased using the default settings of BEAGLE. The resulting overlapping haplotypes were resolved into entire chromosomes by selecting the orientation that minimized differences between the overlapping haplotypes. Each population sample was screened for parent-offspring pairs, 1st degree cousins, grandparent and avuncular relationships. Populations shown in Figure 2,3 were simulated separately. It is expected that phasing error would result in an underestimate of shared IBD since errors would introduce false breakpoints in a shared IBD segment.
With each mating specified by the pedigree, crossovers were introduced between the two haplotypes in each parent, and each parent then contributed one of their haplotypes to the offspring. Crossover breakpoints were distributed according to the HapMap fine-scale recombination map [41], where the number of crossovers per chromosome per generation was modeled as a Poisson random variable with mean equal to the total genetic distance spanned by that chromosome. The same genetic map was used for all populations in our dataset.
The simulated genotypes were then analyzed using the IBD algorithm. The detected IBD segments were then compared to the “theoretical” IBD segments, since we kept track of the breakpoint locations.
We ran the simulations across both the HGDP-CEPH and 23andMe population samples. For samples that had fewer than 22 genomes (required for generating a 10th cousin), we generated as deep of a cousinship as possible. For the 23andMe datasets, the simulation was repeated one million times for each population sample; for the HGDP-CEPH datasets, the simulation was repeated 1,000 times.
Calculation of expected bounds of relationships using simulated data
We estimated the expected relationship range for a pair of distant relatives based on the simulation results. We first counted the number of times each relationship type led to different combinations of IBDhalf (rounded to the nearest 10 cM) and number of IBD segments. Pairs of individuals with IBD segments that did not match the IBD segments in their simulated ancestors were categorized as “unknown”, reflecting the possibility of having a common ancestor not due to the simulated pedigree. Because the simulations generated an equal number of cousins per cousinship type, we additionally weighted each cousinship type by the expected number of cousins for each cousinship type in order to account for the fact that there are many more cousins at distant degrees of relationship than at close degrees (Table 2); we estimated the number of each type of cousins using a model of pedigree. For each amount of IBDhalf and number of segments, we calculated a distribution of 1st through 10th cousins (and unknown cousins) using the simulation results and the pedigree model. From this distribution, we extracted relationships found at the 95th and 5th percentile, which represent the expected bounds of the relationship, as well as at the 50th percentile, which represents the median expected relationship.
Calculation of expected number of individuals sharing DNA IBD
We developed theoretical expectations for the number of nth cousins for any given individual assuming an average sibship size, and, given these expectations plus simulation results, calculated the probability of detecting a cousin via inference of sharing of DNA IBD. We began by calculating the number of ith degree cousins “Nc(i)” for one individual (equation 1). For each degree, “i” there are 2i possible pairs of ancestors, each of which can generate multiple children. For instance, for i = 1 (1st cousin), the number of couples in the relevant ancestral generation is 2 (two sets of grandparents) and a 1st cousin could descend from either the individual's mother's or father's siblings. If each set of parents produces some number of children “z”, then the number of cousins for one side of the family is equal to the product of the number of non-ancestral offspring of the ancestral pair (z−1) and the total number of ith degree cousins generated by those non-ancestral offspring (zi): 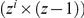 . Therefore, Nc(i) can be calculated as follows:
. Therefore, Nc(i) can be calculated as follows:
| (1) |
We assumed, for z, bounds of 2–3 children per parent couple, where 2 children per couple is consistent with constant population size and 3 children per couple is equivalent to a population growth rate of 40% per generation, as discussed by Slatkin [20] for recent population history among the Ashkenazim. Estimates of population growth for Europeans and East Asians calculated using coalescent models tend to be closer to 2.01–2.15 children per couple, that is 0.7–7% growth per generation assuming 30 years per generation [43], [44]. We present results for the median estimate of 2.5 children per generation per couple (Table S2).
We then asked how many relatives should be detectable in a large heterogenous database using our IBD algorithm and data on 580,000 SNPs per individual. The fraction of detectable ith cousins “f(i)” was calculated based on results of simulations (described above). Those simulations yielded the fraction of ith cousins who share IBD greater than 5 cM and are therefore detectable using our IBD algorithm (Table 2). Ndc(i), the number of detectable ith degree cousins, is defined as the product of the total number of ith cousins (Nc(i)) and the fraction of those cousins that is detectable (f(i)):
| (2) |
We then calculated the probability of detecting at least one ith cousin for a given individual in a database of size Ndb. If the relevant population is of size Npop, the chance that a given member of the population is a detectable, ith cousin is Ndc/Npop. Therefore the chance that a given member of the population is not a detectable ith cousin is 1−(Ndc(i)/Npop). The chance that none of the individuals in a database of size Ndb is a detectable ith cousin is (1−(Ndc(i)/Npop))∧Ndb. The chance of detecting a cousin is, therefore:
| (3) |
We assumed that the number of ancestors for a given individual increases at a constant rate going back in time. At some point in the past, the number of ancestors for two individuals overlaps and the number of ancestors for an individual ceases to grow exponentially. However, Derrida et al. [10] showed that the proportion of ancestors that overlap over 10 generations, given a large present-day population size (>100,000) and panmixia, is quite minimal. Thus, an exponential model is appropriate for our calculation of the number of ancestors for a pair of individuals when we assume random mating within populations throughout the past 10 generations.
In order to determine the relevant values of Npop, we first approximated the size of the Ashkenazi population of the United States as 6 million, and the size of the European-American population as 200 million [45]. Individuals who self-select for 23andMe may tend to have ancestry from particular subsets of the United States population. Ethnic ancestry is not homogeneously distributed throughout the United States [45]. For example, individuals from Maine are more likely to have multiple pairs of ancestors tracing back to England, while individuals from Wisconsin are more likely to have ancestry from Germany. Since our dataset is not a random subset of individuals from the European-American or Ashkenazi population, we then calculated reductions in population size from which potential relatives are drawn by 5th percentiles of 200 million (see Table S2 for percentile calculations given our Ashkenazi data). Since the US population is unlikely to be characterized by truly random mating, we chose our Npop to be 10% of US census sizes.
Supporting Information
Distributions of IBDhalf for pairs of individuals within HGDP-CEPH populations. The average amount of DNA that is identical by descent varies widely among HGDP-CEPH, European, Asian and Ashkenazi populations. We present distributions of pairwise comparisons with IBDhalf segments ≥5 cM for all HGDP populations. Prior to the analysis, individuals were eliminated in order to remove close relationships (sibling, parent-child, avuncular, grandparent-grandchild, and 1st cousin pairs) (see Methods ). Segments of less than 5 cM are not displayed.
(PDF)
Comparison and correlation of HGDP-CEPH population statistics. a) We compare the mean haplotype heterozygosity statistic obtained from Li et al. [23] with the mean IBDhalf for each of 46 populations from HGDP-CEPH. Native American populations (Karitiana, Surui, Pima) were removed from the analysis due to their extreme levels of IBDhalf, indicating many close relatives within each sample. b) The length of runs of homozygosity (ROHs) for each individual were calculated following the same procedure as Nalls et al. [26]. ROHs were averaged for each population and presented as percent of the genome. We contrast the percent of the genome with ROHs to mean IBDhalf for each population. The correlation between is driven primarily by populations with very high IBDhalf, such as Native Americans.
(TIFF)
IBD and ROH statistics for HGDP-CEPH and 23andMe population samples.
(DOC)
Detectability of Ashkenazi Relatives.
(DOC)
Acknowledgments
We gratefully acknowledge Stan Nelson for consultation on methods of calculating IBD and Howard Cann for discussion of HGDP-CEPH. We especially thank the 23andMe customers who contributed the genetic data and ancestry information that made this study possible. We are grateful to all the employees of 23andMe, Inc., for creating and supporting the resources necessary for this research.
Footnotes
Competing Interests: BMH, LH, JMM, NE, SS, and JLM are or were previously employed by 23andMe and own stock options in the company. The research described in this paper is related to a web site feature developed by 23andMe. This does not alter the authors' adherence to all the PLoS ONE policies on sharing data and materials. However, the authors' obligations to protect their customers' privacy (as outlined in our Terms of Service and Privacy Statement) prevent them from making their customers' individual-level data publicly available. Aggregate-level data (for example, in the form of tables that were used for the authors' statistics) can be made available upon request. This research was designed by the 23andMe, Inc. research team and funded by 23andMe, Inc. (www.23andme.com/research). There are no current external funding sources for this study. 23andMe, Inc. non-research staff had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding: The authors have no support or funding to report.
References
- 1.Gusev A, Lowe JK, Stoffel M, Daly MJ, Altshuler D, et al. Whole population, genome-wide mapping of hidden relatedness. Genom Res. 2009;19:318–326. doi: 10.1101/gr.081398.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott L, Mohlke K, Bonnycastle L, Willer C, Li Y, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WTCCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Browning SR, Browning BL. High-resolution detection of identity by descent in unrelated individuals. Am J Hum Genet. 2010;86:526–539. doi: 10.1016/j.ajhg.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huff CD, Witherspoon DJ, Simonson TS, Xing J, Watkins WS, et al. Maximum-likelihood estimation of recent shared ancestry (ERSA). Genom Res. 2011;21:768–774. doi: 10.1101/gr.115972.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg N, Nordborg M. Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nature Reviews Genetics. 2002;3:380–390. doi: 10.1038/nrg795. [DOI] [PubMed] [Google Scholar]
- 7.Hudson RR. Gene genealogies and the coalescent process. Oxford surveys in evolutionary biology. 1990;7:44. [Google Scholar]
- 8.McQuillan R, Leutenegger A, Abdel-Rahman R, Franklin C, Pericic M, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss L, Pan L, Abney M, Ober C. The sex-specific genetic architecture of quantitative traits in humans. Nature genetics. 2006;38:218–222. doi: 10.1038/ng1726. [DOI] [PubMed] [Google Scholar]
- 10.Derrida B, Manrubia S, Zanette D. Distribution of repetitions of ancestors in genealogical trees. Physica A: Statistical Mechanics and its Applications. 2000;281:1–16. [Google Scholar]
- 11.Bodmer W, Cavalli-Sforza L. The genetics of human populations. San Francisco: Dover Publications; 1971. [Google Scholar]
- 12.Malecot G. 1948. Les mathematiques de l'Heredite: Masson.
- 13.Auton A, Bryc K, Boyko A, Lohmueller K, Novembre J, et al. Global distribution of genomic diversity underscores rich complex history of continental human populations. Genom Res. 2009;19:795–803. doi: 10.1101/gr.088898.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramachandran S, Deshpande O, Roseman C, Rosenberg N, Feldman M, et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henn BM, Gravel S, Moreno-Estrada A, Acevedo-Acevedo S, Bustamante CD. Fine-scale population structure and the era of next-generation sequencing. Human molecular genetics. 2010;19:R221–R226. doi: 10.1093/hmg/ddq403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko A, et al. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Dushlaine C, McQuillan R, Weale ME, Crouch DJM, Johansson, et al. Genes predict village of origin in rural Europe. Eur J Hum Genet. 2010;18:1269–1270. doi: 10.1038/ejhg.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mountain JL, Ramakrishnan U. Impact of human population history on distributions of individual-level genetic distance. Hum Genomics. 2005;2:4–19. doi: 10.1186/1479-7364-2-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epstein M, Duren W, Boehnke M. Improved inference of relationship for pairs of individuals. Am J Hum Genet. 2000;67:1219–1231. doi: 10.1016/s0002-9297(07)62952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leibon G, Rockmore DN, Pollak MR. A SNP streak model for the identification of genetic regions identical-by-descent. Statistical Applications in Genetics and Molecular Biology. 2008;7:1–17. doi: 10.2202/1544-6115.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browning BL, Browning SR. A Fast, Powerful Method for Detecting Identity by Descent. Am J Hum Genet. 2011;88:173–182. doi: 10.1016/j.ajhg.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenberg N. Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Annal of Hum Genet. 2006;70:841–847. doi: 10.1111/j.1469-1809.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Absher D, Tang H, Southwick A, Casto A, et al. Worldwide Human Relationships Inferred from Genome-Wide Patterns of Variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 24.Kitchen A, Miyamoto M, Mulligan C, Harpending H. A three-stage colonization model for the peopling of the Americas. PLoS One. 2008;3:e1596. doi: 10.1371/journal.pone.0001596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang S, Lewis C, Jakobsson M, Ramachandran S, Ray N, et al. Genetic variation and population structure in Native Americans. PLoS Genet. 2007;3:e185. doi: 10.1371/journal.pgen.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nalls M, Simon-Sanchez J, Gibbs J, Paisan-Ruiz C, Bras J, et al. Measures of Autozygosity in Decline: Globalization, Urbanization, and Its Implications for Medical Genetics. PLoS Genet. 2009;5:e1000415. doi: 10.1371/journal.pgen.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, et al. Genomic runs of homozygosity record population history and consanguinity. PLoS One. 2010;5:e13996. doi: 10.1371/journal.pone.0013996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenberg N, Pritchard J, Weber J, Cann H, Kidd K, et al. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 29.McEvoy B, Montgomery G, McRae A, Ripatti S, Perola M, et al. Geographical structure and differential natural selection among North European populations. Genom Res. 2009;19:804. doi: 10.1101/gr.083394.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tishkoff S, Reed F, Friedlaender F, Ehret C, Ranciaro A, et al. The Genetic Structure and History of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price A, Helgason A, Palsson S, Stefansson H, Clair D, et al. The Impact of Divergence Time on the Nature of Population Structure: An Example from Iceland. PLoS Genet. 2009;5:e1000505. doi: 10.1371/journal.pgen.1000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zlojutro M, Tarskaia L, Sorensen M, Snodgrass J, Leonard W, et al. Coalescent simulations of Yakut mtDNA variation suggest small founding population. Am J Phys Anthro. 2009;139:474–482. doi: 10.1002/ajpa.21003. [DOI] [PubMed] [Google Scholar]
- 33.Behar D, Metspalu E, Kivisild T, Achilli A, Hadid Y, et al. The matrilineal ancestry of Ashkenazi Jewry: portrait of a recent founder event. Am J Hum Genet. 2006;78:487–497. doi: 10.1086/500307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slatkin M. A population-genetic test of founder effects and implications for Ashkenazi Jewish diseases. Am J Hum Genet. 2004;75:282–293. doi: 10.1086/423146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atzmon G, Hao L, Pe'er I, Velez C, Pearlman A, et al. Abraham's children in the genome era: major Jewish diaspora populations comprise distinct genetic clusters with shared Middle Eastern Ancestry. Am J Hum Genet. 2010;86:850–859. doi: 10.1016/j.ajhg.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kong A, Masson G, Frigge M, Gylfason A, Zusmanovich P, et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nature Genetics. 2008;40:1068–1075. doi: 10.1038/ng.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rohde D, Olson S, Chang J. Modelling the recent common ancestry of all living humans. Nature. 2004;431:562–566. doi: 10.1038/nature02842. [DOI] [PubMed] [Google Scholar]
- 38.Alkuraya FS. Homozygosity mapping: one more tool in the clinical geneticist's toolbox. Genetics in Medicine. 2010;12:236–239. doi: 10.1097/GIM.0b013e3181ceb95d. [DOI] [PubMed] [Google Scholar]
- 39.Albrechtsen A, Sand Korneliussen T, Moltke I, van Overseem Hansen T, Nielsen F, et al. Relatedness mapping and tracts of relatedness for genome wide data in the presence of linkage disequilibrium. Genet Epi. 2009;33:266–274. doi: 10.1002/gepi.20378. [DOI] [PubMed] [Google Scholar]
- 40.Moltke I, Albrechtsen A, Hansen TO, Nielsen FC, Nielsen R. A method for detecting IBD regions simultaneously in multiple individuals–with applications to disease genetics. Genom Res. 2011;21:1168–1180. doi: 10.1101/gr.115360.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Browning S, Browning B. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macpherson JM, Ramachandran S, Diamond L, Feldman MW. Demographic estimates from Y chromosome microsatellite polymorphisms: analysis of a worldwide sample. Hum Genomics. 2004;1:345–354. doi: 10.1186/1479-7364-1-5-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gignoux CR, Henn BM, Mountain JL. Rapid, global demographic expansions after the origins of agriculture. Proc Natl Acad Sci USA. 2011;108:6044–6049. doi: 10.1073/pnas.0914274108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.U.S. Census Bureau. 2007. Census 2000 Summary File 3.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distributions of IBDhalf for pairs of individuals within HGDP-CEPH populations. The average amount of DNA that is identical by descent varies widely among HGDP-CEPH, European, Asian and Ashkenazi populations. We present distributions of pairwise comparisons with IBDhalf segments ≥5 cM for all HGDP populations. Prior to the analysis, individuals were eliminated in order to remove close relationships (sibling, parent-child, avuncular, grandparent-grandchild, and 1st cousin pairs) (see Methods ). Segments of less than 5 cM are not displayed.
(PDF)
Comparison and correlation of HGDP-CEPH population statistics. a) We compare the mean haplotype heterozygosity statistic obtained from Li et al. [23] with the mean IBDhalf for each of 46 populations from HGDP-CEPH. Native American populations (Karitiana, Surui, Pima) were removed from the analysis due to their extreme levels of IBDhalf, indicating many close relatives within each sample. b) The length of runs of homozygosity (ROHs) for each individual were calculated following the same procedure as Nalls et al. [26]. ROHs were averaged for each population and presented as percent of the genome. We contrast the percent of the genome with ROHs to mean IBDhalf for each population. The correlation between is driven primarily by populations with very high IBDhalf, such as Native Americans.
(TIFF)
IBD and ROH statistics for HGDP-CEPH and 23andMe population samples.
(DOC)
Detectability of Ashkenazi Relatives.
(DOC)