

Abstract
NMR observables, such as NOE-based distance measurements, are increasingly being used to characterize membrane protein structures. However, challenges in membrane protein NMR studies often yield a relatively small number of such restraints that can create ambiguities in defining critical side chain-side chain interactions. In the recent solution NMR structure of the DAP12-NKG2C immunoreceptor transmembrane helix complex, five functionally required interfacial residues (two Asps and two Thrs in the DAP12 dimer and one Lys in NKG2C) display a surprising arrangement in which one Asp side chain faces the membrane hydrophobic core. To explore whether these side-chain interactions are energetically optimal, we used the published distance restraints for molecular dynamics simulations in explicit micelles and bilayers. The structures refined by this protocol are globally similar to the published structure, but the side chains of those five residues form a stable network of salt bridges and hydrogen bonds, leaving the Asp side chain shielded from the hydrophobic core, which is also consistent with available experimental observations. Moreover, the simulations show similar short-range interactions between the transmembrane complex and lipid/detergent molecules in micelles and bilayers, respectively. This study illustrates the efficacy of NMR membrane protein structure refinements in explicit membrane systems.
NMR observables, such as nuclear Overhauser effect (NOE)-based distance, chemical shift, and various dipolar coupling measurements, are increasingly being used to characterize membrane protein structures (1–3). However, membrane proteins are challenging subjects for NMR and result in a relatively small number of such measurements, which can create ambiguities in determining critical side chain-side chain interactions. Additionally, most membrane protein structure calculations do not consider several unique features of the membrane environment that may affect the determined structures, such as the low degree of hydration and associated electrostatic interactions, and the spatial constraints enforced by bilayer geometry. Therefore, the resulting structures, even with few violations of NOE-based distances and/or other observables, can present side-chain conformations that may not reflect the most energetically favorable arrangements.
Recently, Call et al. (4) determined the NOE-based solution NMR structure (PDB:2L35) of a micelle-embedded transmembrane (TM) hetero-trimeric complex DAP12-NKG2C, representing the membrane-embedded portions of the natural killer cell-activating receptor complex DAP12-NKG2C/CD94. DNAX-activation protein 12 (DAP12) is a homodimer containing an immunoreceptor tyrosine-based activation motif in its cytoplasmic domain and noncovalently associated with natural killer group 2C (NKG2C). NKG2C forms a heterodimer with the C-type lectin CD94 and recognizes the human nonclassical MHC class I molecule HLA-E, delivering activating signals via the DAP12 immunoreceptor tyrosine-based activation motifs (5). Mutagenesis studies demonstrated that five polar residues, including one Asp and one Thr in each DAP12 TM helix and one Lys in the NKG2C TM helix, mediate the key TM contacts between DAP12 and NKG2C (4,6). The NMR structure provided the first structural insight into the TM contacts within an assembled immunoreceptor complex. Nonetheless, this complex structure shows a puzzling aspect in that one of Asp residues faces the hydrophobic core, which may not be energetically favorable. To explore whether these side-chain conformations are optimal in membrane environments, we have performed a refinement of the DAP12-NKG2C structure using NOE-based restrained molecular dynamics (MD) simulations in both explicit micelles and bilayers.
The representative DAP12-NKG2C-micelle and DAP12-NKG2C-bilayer systems are shown in Fig. 1 (see Table S1 in the Supporting Material for detailed system information). The average NMR structure of PDB:2L35 was used as a starting structure. For simplicity, the DAP12 with a short linker to NKG2C is named DAP12-1 in this work; the linker was introduced in the NMR study to produce a covalently stabilized three-TM complex. For the micelle simulations, 13 sodium dodecyl sulfate and 130 FOS-Choline 14 molecules (1:10 ratio) were radially distributed around the protein surface to mimic the NMR experimental conditions (4). For the bilayer simulations, DAP12-NKG2C was inserted into a bilayer of 129 dimyristoylphosphatidylcholine molecules using CHARMM-GUI Membrane Builder (7,8). Each system was replicated and assigned with different initial velocities to generate three independent simulation systems. CHARMM (9) was used to run a total of 10 ns for each micelle system and 40 ns for each bilayer system under available NOE-based distance restraint potentials.
Figure 1.
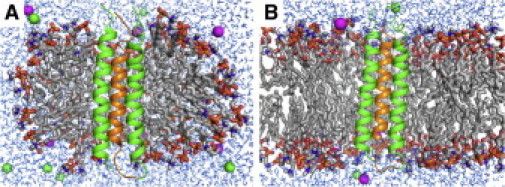
Simulation systems of DAP12 (green) and NKG2C (orange) in (A) a sodium dodecyl sulfate/ FOS-Choline 14 micelle and (B) a dimyristoylphosphatidylcholine bilayer. Detergent and lipid molecules are shown in sticks, ions in spheres, and water molecules in lines.
The average distance violation is a direct measurement to check whether the MD-refined structures satisfy the NMR observables. The total number of restraints is 238. Given a cutoff value of 0.5 Å, 15 structures with the least violations were selected for each system, as in the conventional NMR structure determination. The average numbers of violated restraints are <1 for all these structures in the different systems, which are comparable to the 15 structures in PDB:2L35 (see Fig. S1 in the Supporting Material). The root mean-squared deviations of the TM helix backbone atoms from the average NMR structure are 1.0 ± 0.1 Å in both micelle and bilayer systems. There are no significant differences between PDB:2L35 and MD-refined structures in terms of helix-helix distance and crossing angles (see Table S2). This result indicates that the MD-refined complex structures well satisfy the NMR observables and their overall structures are similar to the PDB:2L35 structures.
Compared to PDB:2L35, however, a different side-chain conformation of DAP12-1 Asp16 (numbering based on PDB:2L35) is observed in the MD-refined structures (Fig. 2). This Asp16 orients toward the complex interface in the MD-refined structures, but faces the membrane hydrophobic core in PDB:2L35 (Fig. 2 and see Fig. S2). Because there is no distance restraint for the Asp16 side chain, its orientation can be varied, depending on how interactions and environments are treated. Interestingly, the side chain of DAP12-2 Leu19 is also reoriented to shield this Asp16 from the hydrophobic core (Fig. 2, B2 and B3).
Figure 2.
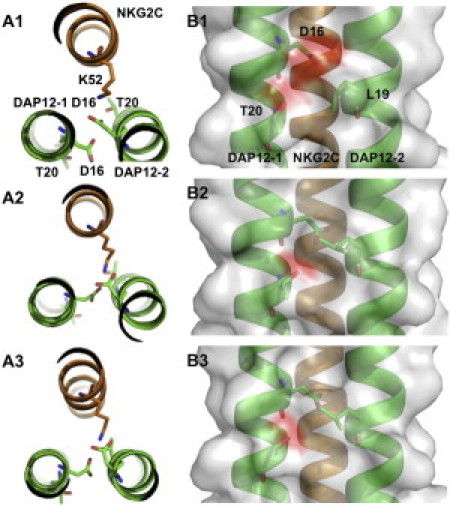
Top and side views of the DAP12-NKG2C complex showing the interactions between the key interfacial residues in (A1, B1) PDB:2L35, (A2, B2) micelle systems, and (A3, B3) bilayer systems, respectively. All other side chains are omitted for clarity. (Red and gray) Negatively charged and neutral protein surfaces, respectively.
The Asp16 rotation is related to Asp16s' electrostatic interactions with NKG2C Lys52 (Fig. 2, A2 and A3). The distances between DAP12 Asp16s and NKG2C Lys52 indicate that both Asp16s can form stable salt bridges with Lys52 in the MD-refined structures (see Fig. S3), which provides a plausible explanation for the strict requirement for two DAP12 and only one NKG2C TM domains in the complex (4). The opposite face of DAP12 in the refined structures is essentially devoid of an appropriate NKG2C TM association site due to the rotation (see Fig. S4), which attractively explains the experimental observation that a second NKG2C TM domain cannot join in the assembly (4).
In addition, DAP12-2 Thr20 in the MD-refined structures shows higher probabilities of hydrogen bond (H-bond) formations with DAP12-1 Asp16 and with NKG2C Lys52 than in PDB:2L35 (see Fig. S5). DAP12-1 Thr20 also has a chance to form intrahelical H-bond with DAP12-1 Asp16. These Thr20-associated intra- and interhelical H-bonds can keep the DAP12 dimer compact and further stabilizes the TM physical contact (Fig. 2). Therefore, Thr20s in the refined structures show direct contributions to the complex stability, which is supported by the mutagenesis study in which the substitution of DAP12 Thr20 to Ala leads to serious defects in the complex assembly (4).
The Asp ionization states and the presence of water near the polar residues influence their electrostatic interactions and can yield different side-chain conformations. In the refinement, both Asp16s were unprotonated. However, two additional normal MD simulations with protonated DAP12-1 Asp16 (atom types OD1 or OD2) showed that the Asp rotated from the hydrophobic core and formed stable interactions with NKG2C Lys52 (data not shown). Although this result does not provide an answer to the Asp16 ionization state in the DAP12-NKG2C complex, it indicates that the critical polar interactions in this refinement do not depend on the Asp16 ionization state. Although a small number (<5) of water molecules were observed near the key polar residues in some micelle or bilayer systems (see Fig. S6), the interaction pattern of the interfacial residues remained identical in those systems, illustrating that the presence of such water does not interfere with the key residue interactions (see Fig. S7).
Possibly, nonadditive effects, not captured in this study, could play a role in such electrostatic interactions (including H-bonds) in membranes, and our results need to be further confirmed by simulations when a polarizable force field becomes available in the future. Nonetheless, it is the advantage of a structure refinement in explicit membranes (with currently available additive molecular force field) to obtain optimal side-chain conformations (through side chain-side chain and side chain-lipid/detergent interactions) in a more realistic environment. Obviously, the overall distribution of detergent/lipid molecules is different in the micelle and bilayer systems (Fig. 1). Nevertheless, the local chemical environments surrounding the DAP12-NKG2C complex in both systems are surprisingly similar (Fig. 3 and see Fig. S8). This result indicates that the protein side chains show similar conformations in the similar local environments. An additional advantage of the bilayer simulation is to refine the TM helix orientation with respect to the bilayer normal. This orientation information, which is not available from micelle simulations, could be important structural information for membrane protein function. In the case of DAP12-NKG2C, the tilt angle of the complex principal axis with respect to the bilayer normal is 9.0 ± 4.8° and the tilt angle of each helix is 10.8 ± 4.6° (DAP12-1), 10.3 ± 5.2° (DAP12-2), and 9.1 ± 4.2° (NKG2C).
Figure 3.
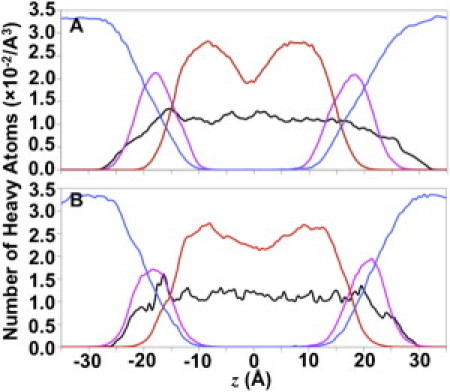
Density profiles of system components along z axis in (A) micelle systems and (B) bilayer systems. In the micelle systems, the principal axis of DAP12-NKG2C was aligned to the z axis. In the bilayer systems, the z axis corresponds to the membrane normal. The profiles involve the heavy atoms of the protein (black), detergent/lipid headgroup (magenta), carbon tail (red), and water (blue) within a radius of 20 Å around the z axis based on Fig. S8 in the Supporting Material.
In conclusion, we refined the DAP12-NKG2C structure (PDB:2L35) in explicit micelle and bilayer membranes using the NOE-based distance restraints. The refined structures are globally similar to PDB:2L35, but show different side-chain orientations/conformations of the five functionally required interfacial residues in the middle of the TM domains. Instead of being exposed to the hydrophobic core, DAP12-1 Asp16 stays in the complex interface and forms a stable salt bridge with NKG2C Lys52 in the refined structures. In addition, the refined DAP12 Thr20s form H-bonds with Asp16 and Lys52, which also enhances the complex's structural stability. These features of side-chain interactions are also consistent with the available mutagenesis data. The refined structures provide novel structural information to understand the key TM contact. In the case of the DAP12-NKG2C TM complex, the detergent molecules provide effective local environments similar to lipid bilayers. Given the considerable challenges in collecting sufficient NMR observables (NOE-based distance, chemical shift, and various dipolar coupling) to define all critical side chain-side chain interactions in membrane protein NMR studies, our study illustrates the efficacy of a structure refinement using restrained MD simulations in explicit micelles and bilayers to provide side-chain orientations in more realistic environments and the protein's orientation relative to bilayers.
Acknowledgments
The authors are grateful to Matthew E. Call for providing restraint files and for many helpful/critical discussions.
This work was supported by the National Science Foundation (grant No. MCB-0918374) and TeraGrid resources (National Science Foundation grant No. OCI-0503992).
Supporting Material
References and Footnotes
- 1.Kim H.J., Howell S.C., Sanders C.R. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2009;55:335–360. doi: 10.1016/j.pnmrs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tamm L.K., Liang B.Y. NMR of membrane proteins in solution. Prog. Nucl. Magn. Reson. Spectrosc. 2006;48:201–210. [Google Scholar]
- 3.Opella S.J., Marassi F.M. Structure determination of membrane proteins by NMR spectroscopy. Chem. Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Call M.E., Wucherpfennig K.W., Chou J.J. The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat. Immunol. 2010;11:1023–1029. doi: 10.1038/ni.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lanier L.L. Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Call M.E., Wucherpfennig K.W. Common themes in the assembly and architecture of activating immune receptors. Nat. Rev. Immunol. 2007;7:841–850. doi: 10.1038/nri2186. [DOI] [PubMed] [Google Scholar]
- 7.Jo S., Kim T., Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jo S., Lim J.B., Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.