

Abstract
Vaccines have been at the forefront of global research efforts to combat malaria, yet despite several vaccine candidates, this goal has yet to be realized. A potentially effective approach to disrupting the spread of malaria is the use of transmission-blocking vaccines (TBV), which prevent the development of malarial parasites within their mosquito vector, thereby abrogating the cascade of secondary infections in humans. Since malaria is transmitted to human hosts by the bite of an obligate insect vector, mosquito species in the genus Anopheles, targeting mosquito midgut antigens that serve as ligands for Plasmodium parasites represents a promising approach to breaking the transmission cycle. The midgut-specific anopheline alanyl aminopeptidase N (AnAPN1) is highly conserved across Anopheles vectors and is a putative ligand for Plasmodium ookinete invasion. We have developed a scalable, high-yield Escherichia coli expression and purification platform for the recombinant AnAPN1 TBV antigen and report on its marked vaccine potency and immunogenicity, its capacity for eliciting transmission-blocking antibodies, and its apparent lack of immunization-associated histopathologies in a small-animal model.
INTRODUCTION
Malaria continues to exact a devastating toll on the health of human populations worldwide, mostly among children under the age of five. In the era of malaria elimination and eradication, the need for novel interventions that specifically block transmission of the parasite is paramount. Transmission-blocking vaccines (TBV), which prevent the development of malarial parasites within the mosquito vector, fulfill such a requirement and arguably represent a critical weapon in this effort (3, 10, 11, 19, 22, 23). TBVs can target either the sexual/mosquito/sporogonic stages of the parasite or, among other things, critical molecules on the mosquito midgut lumenal surface that mediate parasite invasion (14, 15, 20, 21). The midgut-specific glycosylphosphotidyl inositol-anchored alanyl aminopeptidase N (AnAPN1) is a highly conserved molecule among divergent Anopheles vector species and is a putative ligand for both Plasmodium falciparum and Plasmodium vivax ookinetes (14). AnAPN1 was first characterized for Anopheles gambiae, the major malaria vector in sub-Saharan Africa, and rabbit antibodies against the N-terminal (135-amino-acid) domain of AnAPN1 are capable of blocking P. berghei and P. falciparum in the laboratory (15). These data suggest that AnAPN1 can form the basis of a mosquito-based malaria TBV (10, 14). In this study, we report on the expression of a recombinant AnAPN1 (rAnAPN1) antigen (in Escherichia coli), its formulation with Alhydrogel (Brenntag Biosector), its vaccine potency, immunogenicity, and histopathology in a small-animal model, and the transmission-blocking activity of antibodies isolated from rAnAPN1-immunized mice.
(Part of the data presented here was disclosed at the Gordon Research Conference on “the Science behind Malaria Control and Eradication,” 31 July to 5 August 2011, Lucca, Italy.)
MATERIALS AND METHODS
Expression, purification, and analysis of AnAPN1.
The codon-harmonized gene encoding the N-terminal 135-amino-acid fragment of AnAPN1 (AGAP004809) (4, 12, 14) was cloned into pET41a containing a C-terminal 6-His tag and expressed in E. coli BL21(DE3) by using 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). The clone with the highest expression level was selected for making a research cell bank (RCB). We used 100 μl of the RCB to inoculate 200 ml of LB containing 30 μg/ml of kanamycin. Two 1-liter cultures were grown in the same media at 30°C and 225 rpm to an optical density at 600 nm (OD600) of 0.6 and induced with IPTG (1 mM) for 20 h. The final cell pellet was resuspended (50 mM Tris-HCl, 25% sucrose, 1 mM EDTA, 0.1% sodium azide, pH 8.0) and treated with lysozyme and DNase. The postsonication inclusion body cell pellet (approximately 2 g) was dried and frozen at −20°C until further processing. Dried inclusion body material was solubilized at 10 mg/ml in 50 mM Tris-HCl, 7 M urea, and 1% Sarkosyl, pH 8.5, and the 0.22-μm filtered material was loaded onto a 5 ml Hi-Trap Ni–immobilized-metal affinity chromatography (IMAC) FastFlow (FF) column (GE Healthcare). After baseline stabilization, the column was eluted with 15% sucrose, 0.5 M imidazole, 10 mM Tris, and 0.2% Tween 80. The eluent peak was collected and analyzed via SDS-PAGE prior to further processing. The pooled fractions were loaded onto a 5-ml DEAE FF column, and fractions containing pure AnAPN1 were again pooled and dialyzed into a final buffer of 15% sucrose, 10 mM Tris, and 0.2% Tween 80 by using a 3.5-kDa Slide-a-lyzer cassette (Thermo Fisher) and sterile filtered using a 0.22-μm filter. The final protein concentration using absorbance at 280 nm (A280) and the theoretical extinction coefficient were determined. Final purified protein was stored at −80°C. SDS-PAGE analysis was performed using 14% Tris-glycine gels (Invitrogen) under both reducing and nonreducing conditions. Following electrophoresis, gels were either stained with Coomassie brilliant blue R (CBB; GE Healthcare) or silver stained (Sigma) according to the manufacturer's instructions. For immunoblots, primary antibody (rabbit polyclonal anti-AnAPN1, 1:3,000 in phosphate-buffered saline [PBS]-Tween 20 [0.02%]) was detected with anti-rabbit alkaline phosphatase and 5-bromo-4-chloro-3-indolylphosphate–nitroblue tetrazolium (BCIP-NBT) substrate solution (KPL). A Bio-Rad GS-800 instrument with Quantity One software was used to scan CBB gels and dried Western blots to assess sample purity.
Reverse-phase ultrahigh-performance liquid chromatography (RP-UPLC).
An Acquity-H (Waters) system with an in-line photodiode array detector and Empower 2 software was used with a BEH300 C4 1.7-μm-particle-size, 2.1- by 100-mm (Waters) column heated to 65°C. Samples of AnAPN1 were mixed 1:1 into mobile phase A (0.05% heptafluorobutyric acid [HFBA] in water). Following equilibration, 5 μl of sample was introduced in 100% mobile phase A set to 0.6 ml/min. A gradient from 0 to 4.73 min to 80% mobile phase B (0.043% HFBA in acetonitrile) was performed followed by a hold at 80% mobile phase B from 4.73 to 5 min. The spectrum from 210 to 400 nm was recorded, and UV absorbance of the resultant peaks was monitored at 215 and 280 nm. The retention time of AnAPN1 was determined, and the percent purity of the main peak was ascertained through integration using Empower 2 software. A single main peak, with the apex eluting at 2.8 ± 0.1 min, was expected for each lot.
Formulation and dose preparation.
For formulations, the recombinant AnAPN1 was diluted to a final concentration of 0.1 mg/ml in the final buffer of 15% sucrose, 10 mM Tris, and 0.2% Tween 80 and left unformulated or formulated with Alhydrogel (at a final concentration of 0.8 mg/ml) under aseptic conditions and stored at 2 to 8°C. A placebo control was prepared by formulating 0.8 mg/ml Alhydrogel (∼0.4 mg/ml aluminum) in 15% sucrose, 10 mM Tris, and 0.2% Tween 80 under aseptic conditions.
Immunogenicity study.
A total of 10 BALB/c female mice (15 g to 20 g) were divided into 2 groups of 5 mice. Mice in the control group received incomplete Freund's adjuvant (IFA), whereas mice in the treatment group received 2 μg of AnAPN1 without IFA. At day 0, mice received a subcutaneous (s.c.) injection of the appropriate inoculum in a volume of 100 μl per mouse. At 2, 4, and 6 weeks postpriming, mice were boosted i.p. with the same dose of the inoculum per group except for the controls, which received PBS. At these time points, each mouse was bled to collect sera for antibody titer determination via indirect enzyme-linked immunosorbent assay (ELISA). Briefly, Maxisorp 96-well ELISA plates (Nunc, Fisher Scientific) were incubated overnight at 4°C with antigen at 1 μg/ml in 0.1 M PBS (pH 7.2). After two washes with PBS-Tween 20 (0.05%) (PBST20), the plates were blocked for 1 h at room temperature with 1% bovine serum albumin (BSA) in distilled water. Serum samples were diluted 10-fold from 1:101 to 1:106 in 0.67% BSA in distilled water. One hundred microliters of each of six sample dilutions was added to each well in triplicate and incubated 1 h at room temperature. Plates were washed with PBST20, and 100 μl of a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG(H+L) (KPL) diluted 1:500 in 0.67% BSA was added to each well and incubated for 1 h at room temperature. Plates were washed with PBST20 and developed by adding 100 μl of TMB (3,3′,5,5′-tetramethylbenzidine) microwell peroxidase substrate (KPL) to each well. Development was stopped after 5 min by the addition of 100 μl of TMB Stop solution (KPL), and plates were read at 450 nm using a SpectraMax 384 Plus plate reader (Molecular Devices) and analyzed by SOFTmax Pro 5.3 software.
A mixed mode of immunizations was performed since (i) initial s.c. priming immunizations in rabbits and mice led to a strong priming antibody response, (ii) s.c. delivery is the route most relevant for human systemic immunizations in clinical trials, (iii) i.p. and s.c. boosts had no discernible difference in resulting serum endpoint antibody titers (data not shown), and (iv) i.p. delivery had the advantage of facilitating the process of titer monitoring with boosting as well as lessening skin irritation because of the use of IFA as the adjuvant.
Transmission-blocking assays.
For direct feeding assays (DFA) with mosquitoes, Swiss Webster “donor” mice were infected via the i.p. route with P. berghei ANKA 2.34. A week later, infected red blood cells from these infected donor mice were transferred to immunized mice by i.p. injection. These infected mice were used for DFA at 3 to 4 days postchallenge. For each DFA, mice in the control and treatment groups meeting the exflagellation threshold of ≥1 per field at 40× magnification were anesthetized and individually placed on top of a 1-pint cup containing ∼40 5- to 6-day-old female Anopheles stephensi mosquitoes. Mosquitoes were allowed to feed on mice for 15 min and were then maintained at 19°C with water and sucrose until dissection. Ten days after the DFA, midguts were dissected and stained with 0.1% merbromin (Mercurochrome) to score oocyst numbers. Passive immunization feeding assays (PIFA), which also test IgG functionality through passive transfer to infected mice prior to mosquito feeding, were performed as previously described (14). The final concentration of passively transferred IgG/mouse was calculated based on a standard 2-ml blood volume for a 24-g mouse.
Immunohistochemical analysis.
Slides of frozen, optimal cutting temperature (OCT; Tissue Tek) embedded sections of normal human liver, small intestine (jejunum), and kidney (Zyagen Life Sciences, San Diego, CA) were fixed with acetone (liver and small intestine) or a 1:1 ratio of acetone-methanol (kidney) and then blocked overnight at 4°C with PBS-BSA (3%). Anti-AnAPN1 IgG (rabbit, 10 μg/ml) and anti-human aminopeptidase N-CD13 (monoclonal antibody [MAb] WM15, 10 μg/ml; BD Pharmingen) primary antibodies diluted in blocking buffer were incubated with replicate sections of each tissue for 2 h at 37°C in a humidifying chamber, washed with PBS, and detected by the corresponding goat anti-mouse Texas Red (Invitrogen) or goat anti-rabbit Alexa Fluor 488 (Invitrogen) conjugated secondary antibodies. Images were acquired at 200× and 400× magnifications by using a Nikon E800 epifluorescence microscope and SPOT imaging software (version 4.9).
Histopathology study.
BALB/c female mice that were immunized (i.p.) with 5 μg/dose of the E. coli rAnAPN1-Alhydrogel formulation (treatment) or Alhydrogel alone (control) were sacrificed at day 14 and day 90 to assess acute and chronic histopathologies resulting from vaccination, respectively. At each time point, slides from the heart, brain, sera, small intestines, large intestines, uterus, lungs, liver, spleen, kidneys, inguinal node, and mesenteric nodes were prepared for a complete gross histopathological analysis. To provide additional functional analysis to corroborate the results from the histopathology data, a satellite study was performed to evaluate the efficacy of anti-rAnAPN1 antibodies generated in these mice. To maintain consistency between the histopathology and subsequent potency experiments, i.p. immunizations were used in these studies.
Potency assay.
Mice were immunized with AnAPN1 with and without Alhydrogel to determine the doses of AnAPN1-Alhydrogel to estimate the median 50% effective dose (ED50) as a measure of vaccine potency. For biologics, the ED50 is commonly used to describe the lowest dose of a product that induces a response in 50% of the animals in a dose group (9). Twelve groups of 10 female BALB/c mice (Taconic) 6 to 8 weeks old were administered (i.p.) a range of AnAPN1-Alhydrogel doses in a prime (day 0) and boost (day 28) regimen and bled on days −1, 14, and 28 (boost), with a terminal bleed on day 42 (Table 1). To maintain consistency between the histopathology and potency experiments, i.p. immunizations were also used in these studies. The i.p. route of delivery is advantageous in this context, because it allows for larger volumes and doses of antigens. Doses were prepared by “fractionating” AnAPN1-Alhydrogel by volume prior to immunization. Two control groups were included in the study following the same prime-boost and bleed schedule: 10 mice each receiving either 400 μg of Alhydrogel alone (adjuvant control) or 10 mice receiving 50 μg of AnAPN1 alone (antigen control).
Table 1.
The numbers of responders for potency study days 14, 28, and 42 by dose group for AnAPN1-Alhydrogel in BALB/c micea
| Group | Amt (μg) |
No. of responders/group |
|||
|---|---|---|---|---|---|
| AnAPN1 | Alhydrogel | Day 14 | Day 28 | Day 42 | |
| 1 | 400 | 0 | 0 | 0 | |
| 2 | 50.00 | 1 | 7 | 10 | |
| 3 | 50.00 | 400.00 | 2 | 10 | 10 |
| 4 | 28.57 | 228.57 | 2 | 7 | 8 |
| 5 | 16.33 | 130.61 | 2 | 2 | 9 |
| 6 | 9.33 | 74.64 | 1 | 1 | 9 |
| 7 | 5.33 | 42.65 | 0 | 0 | 8 |
| 8 | 3.05 | 24.37 | 0 | 0 | 2 |
| 9 | 1.74 | 13.93 | 0 | 0 | 0 |
| 10 | 0.99 | 7.96 | 0 | 0 | 0 |
| 11 | 0.57 | 4.55 | 0 | 0 | 0 |
| 12 | 0.32 | 2.60 | 0 | 0 | 0 |
| 13 | 0.19 | 1.48 | 0 | 0 | 0 |
| 14 | 0.11 | 0.85 | 0 | 0 | 0 |
| ED50 (μg) | |||||
| Value | 127.61 | 21.25 | 5.35 | ||
| 95% fiducial limits | |||||
| Lower | 16.37 | 3.00 | |||
| Upper | 28.28 | 9.05 | |||
Mice were considered positive if the anti-AnAPN1 IgG level was above the response threshold (RT; 2 AU) established by the standard calibration curve. There were 10 mice per group.
Polysorp 96-well ELISA plates (Nunc F96; Fisher Scientific) were incubated overnight at 4°C with antigen in 0.15 M PBS (pH 7.2). After being washed with PBST20 (0.05%), the plates were blocked for 2 h at room temperature with 3% BSA in PBST20. One hundred microliters of sample serum was added to each well and incubated overnight at 4°C. Plates were washed with PBST20, and 0.1 ml of an HRP-conjugated anti-mouse IgG (gamma) (Sigma) diluted in 3% BSA in PBST20 was added to each well and incubated for 2 h at room temperature. Plates were developed by adding o-phenylenediamine dihydrochloride for 30 min at room temperature and read at 492 nm by using a SpectraMax 340PC reader (Molecular Devices) and analyzed by GXP Validated SOFTmax Pro software. The indirect ELISA was qualified for precision (reproducibility), linearity, and ruggedness. The reactivity of IgG against AnAPN1 was quantified into arbitrary units of antibody (AU) as determined from OD values of test serum dilutions interpolated onto a standard calibration curve derived from standard reference sera (see Fig. S2 in the supplemental material for the development of the standard calibration curve and the assignment of AU). The standard reference serum was developed by pooling the sera from 10 female BALB/c mice (Taconic) 6 to 8 weeks old that were administered 50 μg of AnAPN1-Alhydrogel (i.p.) in a prime (day 0) and boost (day 28) regimen and bled on days −1, 14, and 28 (boost), with a terminal bleed on day 42 (Table 1). Sera were pooled from the terminal bleed and then tested for the presence of anti-AnAPN1 IgG (see Fig. S2).
Lymphoproliferation assay.
Splenocytes were harvested from BALB/c mice at day 42 following two immunizations with various doses of AnAPN1 formulated with or without Alhydrogel. Splenocytes were added in duplicate (1 × 105 cells/well) and incubated with 35 μg/ml of AnAPN1 for 5 days. Lymphoproliferation is expressed as the stimulation index (SI) with levels of bromodeoxyuridine (BrdU) in cells cultured with AnAPN1 over the levels in cells in control wells (no antigen) after 5 days. All wells contained RPMI medium supplemented with 10% fetal bovine serum and 1% of 100× antibiotic/antimycotic. The addition of BrdU and its measurement on a microplate reader were done following the manufacturer's instructions (Roche).
Statistical methods.
The ED50 and its 95% fiducial limits were estimated using the Proc Probit procedure of SAS 9.2. The dose versus percent response curves generated for each bleed were tested for linearity as determined by the Pearson chi-square test for goodness of fit using the Proc Probit procedure of SAS 9.2. Statistical significance for all PIFA data was assessed using a Kruskal-Wallis test (GraphPad Prism). A Poisson generalized estimating equations (GEE) model (30) was used to compare oocyst count data for treatment and control mice in the DFA. The GEE model allows for overdispersion and accounts for the correlation between mosquitoes from the same mouse. GEE models were fitted using R 2.13 and the geepack package (18, 26).
RESULTS
Expression of recombinant AnAPN1.
Given the lack of cysteines in the sequence and a relatively simple secondary structure (>59% β sheet), we used E. coli to express the predicted 16.4-kDa N-terminal 135-amino-acid protein fragment of AnAPN1. We produced inclusion bodies containing the target protein and achieved total yields of ∼1 g/liter and a relative purity of solubilized inclusion body material of >80% (see Fig. S1a, lane 1, in the supplemental material). Immobilized-metal affinity chromatography (IMAC) as a capture step eliminated the majority of host cell proteins (Fig. S1a, lanes 2 and 3) and also served as a buffer exchange for the target molecule. While various ion-exchange chromatography steps were initially explored, a weak anion-exchange column (DEAE) was selected for an additional purification step. The target molecule was bound in its entirety and was successfully separated from minor contaminating bands at ∼0.25 M NaCl. From approximately 1 g of inclusion bodies, one-tenth of the material was purified to yield approximately 60 mg with an overall recovery of >80% of the target (data not shown). The final buffer, comprised of 15% sucrose, 10 mM Tris, and 0.2% Tween 80, was selected for stabilization of the recombinant protein and prevention of aggregation (data not shown).
SDS-PAGE with CBB staining and densitometry reported an overall purity of 95% (Fig. S1a, lane 4). Given the relatively small molecular weight, we used RP-UPLC to further analyze the target and any product-derived degradation forms (Fig. S1b, panel I). In-process samples were used to demonstrate the resolution of the full-length target (at approximately 2.8 min) and an observed clipped form at ∼3.2 min (Fig. S1b, panel II). This clipped form was used in assay development to demonstrate separation capabilities of full-length AnAPN1 from other product-derived species and represents a cleavage at amino acid 132, creating a 14.5-kDa form of AnAPN1 (data not shown). This separation had a resolution of >2 between peaks. In addition, the main target peak (2.8 min) had a peak asymmetric factor of 1.18 and a peak tailing factor of 1.22. Upon further analysis using half height measurement, the plate count was determined to be 134,075. Utilizing the photodiode array, we observed a minor contaminating peak in the final purified material seen in Fig. S1b in both panel I (∼2.3 min) and panel II. The contaminating peak is thought to represent minor contaminants, such as residual dimers of the molecule. The main target peak of AnAPN1 (2.8 min) was also analyzed for peak purity and demonstrated peak purity over 3 wavelength passes (Figure S1c). The 0.1 mg/ml AnAPN1 and 0.8 mg/ml Alhydrogel formulation is the expected clinical formulation and is within the FDA and WHO guidelines of maximum aluminum content (∼0.4 mg/ml aluminum). Ongoing stability analysis has shown that AnAPN1 remains completely adsorbed to Alhydrogel for >9 months and the AnAPN1 molecule remains intact (data not shown).
Immunogenicity of AnAPN1.
We immunized mice with E. coli-expressed recombinant AnAPN1 (2 μg/dose/mouse) following prime (s.c.)–three-boost (i.p.) (Fig. 1a and b) and prime (s.c.)–single-boost (i.p.) regimens (Fig. 1c and d), both in the absence of adjuvant to understand the baseline immunogenicity of this antigen. We found the antigen to be highly immunogenic, and although we noted differences in the kinetics of the antibody response, the overall antibody titers from the two regimens did not greatly differ. Individual mice immunized with AnAPN1 formulated with Alhydrogel (2 μg/dose/mouse) using the prime (s.c.)–three-boost (i.p.) regimen also elicited a strong humoral response (Fig. 2a and b).
Fig 1.

Recombinant AnAPN1, produced in E. coli, exhibits intrinsic antigenicity in mice and is capable of eliciting functional malaria transmission-blocking antibodies in the absence of adjuvant. (a and b) A single-prime and three-boost immunization dose regimen (2 μg/dose at 2-week intervals) produces a robust antigen-specific antibody response in mice compared to that seen in controls (mice receiving only incomplete Freund's adjuvant). Sera have been pooled from 4 or 5 mice/cohort. (c and d) A single prime and -boost dose at day 28 (2 μg/dose) produce an antibody response for week 8 postimmunization sera comparable to that seen in panel b. Sera were pooled from 4 or 5 mice/cohort. (e and f) Direct feeding assay (DFA) results demonstrated that mice receiving AnAPN1 (2 μg/dose) in the absence of adjuvant elicited transmission-blocking antibodies against Plasmodium berghei in Anopheles stephensi mosquitoes. Control group values are shown as open circles (corresponding to panel c). Treatment group values are shown as filled circles (corresponding to panel d). Horizontal bars indicate median oocyst numbers per mosquito midgut. The lower table highlights the statistical significance of the Pr value of 0.031430 as determined by the GEE model. Data are representative of two independent biological replicate cohort immunization studies.
Fig 2.
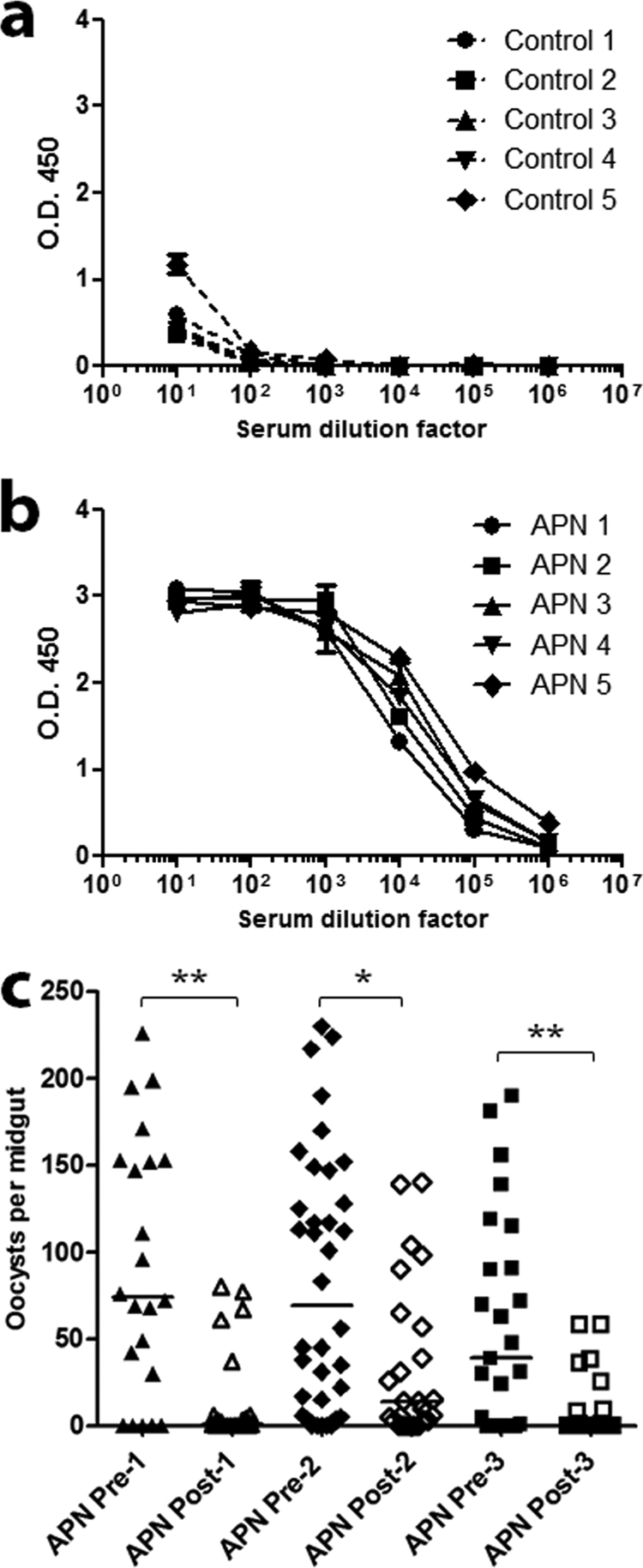
Immunization of mice with AnAPN1-Alhydrogel elicited a potent antigen-specific humoral response and functional, malaria parasite transmission-blocking antibodies. Sera were collected from the satellite mouse control cohort C (a) and APN cohort D (b). Only sera from cohort D demonstrated an AnAPN1-specific antibody response. Mice were immunized following the prime–three-boost schedule (5 μg/dose) used for mouse cohorts A and B, which were subsequently analyzed for gross and subpatent histopathologies. (c) Representative results from passive immunization feeding assays (PIFA) that demonstrate the potent transmission-blocking activity of AnAPN1-specific IgG (100 μg/ml) collected from mice in cohorts B and D against Plasmodium berghei in Anopheles stephensi mosquitoes. We observed reductions in median oocyst infection intensity between 80 to 100%. Each set of pre- and postexperiment values represents a single biological replicate. *, P < 0.05; **, P < 0.01.
Antibodies demonstrate functional transmission-blocking efficacy.
We also sought to evaluate whether AnAPN1 can generate functional transmission-blocking antibodies in these mice. Antibodies raised against AnAPN1 (2 μg) without adjuvant were found to have transmission-blocking activity (Fig. 1e and f), as indicated by an increased absence of oocysts (i.e., reduction in infected mosquito prevalence) or a reduction in oocyst intensity (i.e., the number of oocysts counted) in the midguts. These results highlighted the intrinsic immunogenicity of AnAPN1 and suggested that a day 0 and 28 prime and boost schedule was appropriate for a subsequent potency study. Moreover, we demonstrated through PIFA studies that inoculation of 100 μg/ml of AnAPN1-specific IgG from a satellite histopathology cohort of “donor” mice immunized with AnAPN1-Alhydrogel into nonimmunized P. berghei-infected “acceptor” mice also exhibited potent transmission-blocking efficacy (Fig. 2c).
Immunohistochemistry.
Although AnAPN1 and its putative human ortholog, alanyl (membrane) aminopeptidase (CD13; EC 3.4.11.2), share less than 30% identity at the amino acid level for both the full-length and recombinant N-terminal 135-amino-acid proteins (see Fig. S3 in the supplemental material), immunofluorescence analyses were performed to assess anti-AnAPN1 IgG cross-specific recognition of human APN homologs that localize to the kidney, the liver, and the jejunum of the small intestine (a few of the primary tissues expressing human APN). Anti-CD13/human APN staining of the normal human tissue sections was consistent with published results (13, 25, 27, 28). The data suggested that rabbit anti-AnAPN1 antibody does not stain human homologs of APN in frozen tissues, and the signal observed corresponds to the high autofluorescence background of tissue sections (Fig. 3a). While we interpret these results to suggest the lack of antibody cross-reactivity, such an assay does not include the complete repertoire of tissues that express CD13, including distinct macrophage populations and stromal cells, and thus falls short of assessing the myriad of effects that would result following immunization with AnAPN1 in vivo.
Fig 3.

An immunohistochemical study of normal human tissue sections with rabbit anti-AnAPN1 IgG and a complete histopathological analysis of mice following AnAPN1-Alhydrogel immunizations demonstrated the complete absence of cross-specific staining of human APN homologs and gross histopathologies in mice. (a) Frozen sections were stained with anti-CD13, MAb (WM15 clone, positive control), and polyclonal rabbit anti-AnAPN1 IgG and detected with anti-mouse Texas Red and anti-rabbit Alexa Fluor 488-conjugated antibody. Images were acquired at 200× and 400× magnifications. Differential interference contrast (DIC) images are provided for orientation at 200×, and the inset corresponds to the anti-CD13 and anti-AnAPN1 staining and the merged image, which were all acquired at 400×. DAPI (4′,6-diamidino-2-phenylindole) was used to stain nuclei and appear blue. The signal observed in the 460- to 490-nm band pass filter range across all AnAPN1 images corresponds to the marked autofluorescence observed for these tissues. (b) A summary of the acute (days 1 to 14) and chronic (90 days) histopathology results in control (Alhydrogel) and treatment (AnAPN1-Alhydrogel) mouse cohorts A and B (see the Fig. 2 legend), respectively. SHL, small hepatic lesions; LNE, lymph node enlargements; UE, urinary tract edema; ME, mild enteritis.
Histopathology.
Given our immunohistochemistry results, we performed a complete histopathological study of the heart, brain, blood (sera), small intestines, large intestines, uterus, lungs, liver, spleen, kidneys, and inguinal and mesenteric nodes taken from vaccinated BALB/c mice to ascertain both acute (first 14 days post-priming immunization) and chronic (90 days postimmunization) subpatent histopathologies, which include but are not limited to general inflammation, excessive tissue infiltration by lymphocytes admixed with plasma cells, interstitial edema, interstitial fibrosis, tubular degeneration, and other tissue-differentiated characteristics frequently associated with autoimmune disorders of the respective tissues analyzed in this study.
Acute-phase histopathology results (days 1 to 14).
For the Alhydrogel-immunized control mice, we observed lesions that were distributed across different time points (days 1, 3, 7, and 14) but were limited to enlargement/hyperplasia of mesenteric and other gastrointestinal (GI) lymph nodes (LN) and small, inconsequential hepatic inflammatory infiltrates including extramedullary hematopoiesis. Similarly, for the AnAPN1-Alhydrogel-immunized mice, we also observed, distributed across different time points, lesions that were limited to hyperplasia of mesenteric and other GI lymph nodes (Fig. 3b). One animal had evidence of very mild focal enteritis, which was interpreted as inconsequential for the study. Overall, there was no evidence of histopathology, and what is described above is likely a result of immune stimulation following immunization.
Chronic-phase histopathology results (day 90).
For the Alhydrogel-immunized control mice, we also observed lesions that were limited to hyperplasia of mesenteric and other GI lymph nodes, small inconsequential hepatic inflammatory infiltrates, and the occasional hepatocyte apoptosis. Some of these lesions can be consistent with the Helicobacter-positive status in the colony. Lymph nodes were hyperplastic although there was no gross evidence of hyperplasia. In contrast, for the AnAPN1-Alhydrogel-immunized mice, we observed fewer lesions in general (Fig. 3b). Moreover, no gross abnormalities in any of the mice were noted, and all mice across both groups had gained weight and thus had more fat stores at necropsy.
Taken together, in both phases of the study, we found no evidence across all mice of LN enlargement in the thoracic cavity or of increased bronchiole-associated lymphoid tissue and noted that both thymic and splenic tissue architectures appeared normal across all animals.
Dose-ranging potency study.
We conducted an exploratory dose-ranging study in female BALB/c mice immunized with AnAPN1 with and without Alhydrogel. The objective was to test a wide range of doses (from 50 μg to 0.11 μg) of AnAPN1-Alhydrogel to optimize the bioassay and immunoassay (indirect ELISA) conditions (doses, vaccination schedule, and route of immunization, etc.) (Table 1) required to generate the linear dose-response curve necessary to estimate the ED50. We observed that using sera collected on day 28 post-priming immunization provides the most linear range of responses needed to generate the ED50 (data not shown). The dose-response curve on this day did not significantly depart from linearity (P = 0.9984, with 5 degrees of freedom). We selected 2 arbitrary units of anti-AnAPN1 IgG as the response threshold (RT) (Fig. 4a and b; see Fig. S2 in the supplemental material). The RT was used to determine a threshold of murine IgG against AnAPN1, above which we designated an individual test serum as “positive.” Table 1 shows the ED50 and 95% fiducial limits from days 14, 28, and 42. We also observed IgG1, IgG2a, and IgG2b Abs following immunization to AnAPN1 alone or AnAPN1-Alhydrogel, suggesting a mixed Th1/Th2 humoral response (Fig. 4c). The Probit dose-response curves for days 14 and 42 showed a significant departure from linearity (P values of 0.0201 and <0.0001, respectively, with 10 degrees of freedom). However, the Probit dose-response curve from day 28 did not show a significant departure from linearity (P value of 0.9984, with 5 degrees of freedom) (Fig. 5a). We observed that a wide range of doses, from 50 μg to 0.11 μg, in eight groups with a 1.75-fold difference between dilutions generated a reproducible and comparable linear dose-response curve using sera collected at 4 weeks after prime immunization (day 28). The 50-μg dose of AnAPN1 alone (without Alhydrogel) was immunogenic as measured on day 42 (14 days post-single-boost vaccination) and seroconverted 100% of the animals (Fig. 4b). On day 28, we estimated the ED50 in the presence of adjuvant to be 21.25 μg (95% fiducial limits, 16.37 μg and 28.28 μg) (Fig. 5a).
Fig 4.

AnAPN1 mouse potency study. (a) Anti-AnAPN1 IgG on day 28 post-prime vaccination. Sera were diluted at 1:6,000. Note that mice vaccinated with 50 μg of AnAPN1 alone generated an anti-AnAPN1 IgG response corroborating the data in Fig. 1. However, a statistically significant (P = 0.0237) increase in anti-AnAPN1 IgG (AU) on day 42 post-prime vaccination (day 14 postboost) was found on addition of 400 μg of Alhydrogel to 50 μg of AnAPN1. The reactivity threshold is the concentration of antibody corresponding to the interpolated intersection of the lower asymptote of the upper 95% confidence limit with the lower 95% confidence limit of the standard data. (b) Anti-AnAPN1 IgG on day 42 post-prime vaccination (14 days postboost). Sera were diluted at 1:6,000. (c) Immunoglobulin isotype subclassification of antisera from each mouse that received the 50-μg dose of AnAPN1 formulated with or without Alhydrogel.
Fig 5.

Potency of AnAPN1 as demonstrated by the median 50% effective dose (ED50) and lymphoproliferative responses. (a) Proc Probit (SAS 9.2) modeling of the dose-response curve using sera collected on day 28 (4 weeks after prime vaccination of BALB/c mice) indicates an ED50 in the presence of adjuvant of 21.25 μg (95% fiducial limits of 16.37 μg and 28.28 μg). (b) Immunization with unformulated and formulated AnAPN1 induces cellular proliferation in spleen cells of BALB/c mice. Splenic lymphocytes harvested from BALB/c mice 42 days after primary immunization and (14 days after boost) with various doses of AnAPN1 formulated with Alhydrogel (groups 1 to 3 [G1 to G3]). Lymphoproliferation is expressed as the stimulation index (SI) with levels of bromodeoxyuridine (BrdU) in cells cultured with concanavalin A (Con A) or AnAPN1 (17.5 and 35 μg/ml) over the levels in cells in control wells (no antigen) after 5 days.
We also performed lymphoproliferation assays to confirm the data we observed in the potency study. Spleens from animals for different test groups used in the potency study were isolated and splenic lymphocytes were incubated with concanavalin A (control mitogen) and AnAPN1 (Fig. 5b). As expected, we observed a significant induction in an antigen-specific proliferation of lymphocytes in spleen cells at 42 days after primary immunization and 14 days after the boost with formulated and unformulated AnAPN1.
DISCUSSION
We developed a scalable expression and purification platform for AnAPN1. The lack of disulfide bonds and a relatively simple structure of the antigen (N-terminal 135-amino-acid fragment of AnAPN1) made it an ideal candidate for high-purity, high-yield inclusion body expression that can potentially obviate an IMAC capture step, given its current purity directly from inclusion bodies and DEAE purification. Additionally, since the pilot lot discussed within this study was generated, both low-cell-density fermentation and high-cell-density fermentation have been explored, generating >400 g of inclusion bodies/10 liters of fermentation broth. The intrinsic immunogenicity of AnAPN1, which induces functional malaria parasite transmission-blocking antibodies, its complete adsorption to a highly safe adjuvant, the augmentation that Alhydrogel provides to the antigen following a single-prime-boost regimen, and the safety of AnAPN1-Alhydrogel in the rodent model underscore AnAPN1's candidacy for further TBV development and testing. Importantly, we observed a correlation between potency and the induction of functional antibodies. Our potency assay can be used as an indicator of manufacturing consistency and stability by comparing lot-to-lot variability of the good manufacturing practice (GMP)-manufactured AnAPN1 clinical product (i.e., relative potency) (16). We anticipate that parallel head-to-head testing of more potent adjuvants as well as conjugation of AnAPN1 to molecular adjuvants and its formulation with Alhydrogel is needed to further potentiate this mosquito-based TBV.
Among candidate malaria vaccines to date, only the RTS,S preerythrocytic vaccine has progressed toward phase III clinical trials; however, protection conferred by this vaccine has been largely marginal (1, 2, 5, 6, 7, 8, 17, 24, 29). A mosquito-based TBV with the potential for blocking efficacy against all species of human malaria parasites across the global spectrum of mosquito vectors holds considerable promise as a new tool to combat malaria. Our data provide evidence in favor of progressing the AnAPN1-Alhydrogel TBV toward phase I clinical testing.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sanjay Kumar for critical suggestions and insight.
This work was funded in part by the PATH-Malaria Vaccine Initiative, the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH) (grant 1K22AI077707), the Bloomberg Family Foundation and the Johns Hopkins Malaria Research Institute (JHMRI) (to R.R.D.), a JHMRI Predoctoral Fellowship (to J.S.A.), and the Calvin A. and Helen L. Lang Postdoctoral Fellowship (to D.K.M.). This publication was also made possible by the NIH National Center for Research Resources (grant number UL1 RR 025005).
We declare that there exist no competing interests such as those described under the guidelines or that might be perceived to influence the results and discussion reported in this paper.
Footnotes
Published ahead of print 6 February 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Abdulla S, et al. 2008. Safety and immunogenicity of RTS,S/AS02D malaria vaccine in infants. N. Engl. J. Med. 359:2533–2544 doi:10.1056/NEJMoa0807773 [DOI] [PubMed] [Google Scholar]
- 2. Aide P, et al. 2011. Four year immunogenicity of the RTS,S/AS02(A) malaria vaccine in Mozambican children during a phase IIb trial. Vaccine 29:6059–6067 doi:10.1016/j.vaccine.2011.03.041 [DOI] [PubMed] [Google Scholar]
- 3. Alonso PL, et al. 2011. A research agenda to underpin malaria eradication. PLoS Med. 8:e1000406 doi:10.1371/journal.pmed.1000406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Angov E, Hillier CJ, Kincaid RL, Lyon JA. 2008. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS One 3:e2189 doi:10.1371/journal.pone.0002189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aponte JJ, et al. 2007. Safety of the RTS,S/AS02D candidate malaria vaccine in infants living in a highly endemic area of Mozambique: a double blind randomised controlled phase I/IIb trial. Lancet 370:1543–1551 doi:10.1016/S0140-6736(07)61542-6 [DOI] [PubMed] [Google Scholar]
- 6. Asante KP, et al. 2011. Safety and efficacy of the RTS,S/AS01(E) candidate malaria vaccine given with expanded-programme-on-immunisation vaccines: 19 month follow-up of a randomised, open-label, phase 2 trial. Lancet Infect. Dis. 11:741–749 doi:10.1016/S1473-3099(11)70100-1 [DOI] [PubMed] [Google Scholar]
- 7. Barbosa A, et al. 2009. Plasmodium falciparum-specific cellular immune responses after immunization with the RTS,S/AS02D candidate malaria vaccine in infants living in an area of high endemicity in Mozambique. Infect. Immun. 77:4502–4509 doi:10.1128/IAI.00442-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bojang K, et al. 2009. Five-year safety and immunogenicity of GlaxoSmithKline's candidate malaria vaccine RTS,S/AS02 following administration to semi-immune adult men living in a malaria-endemic region of The Gambia. Hum. Vaccin. 5:242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bolton S. 2003. Pharmaceutical statistics: practical and clinical applications. Marcel Dekker, Inc., New York, NY [Google Scholar]
- 10. Bousema T, Drakeley C. 2011. Epidemiology and infectivity of Plasmodium falciparum and Plasmodium vivax gametocytes in relation to malaria control and elimination. Clin. Microbiol. Rev. 24:377–410 doi:10.1128/CMR.00051-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carter R. 2001. Transmission blocking malaria vaccines. Vaccine 19:2309–2314 [DOI] [PubMed] [Google Scholar]
- 12. Chowdhury DR, Angov E, Kariuki T, Kumar N. 2009. A potent malaria transmission blocking vaccine based on codon harmonized full length Pfs48/45 expressed in Escherichia coli. PLoS One 4:e6352 doi:10.1371/journal.pone.0006352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Di Matteo P, et al. 2011. Enhanced expression of CD13 in vessels of inflammatory and neoplastic tissues. J. Histochem. Cytochem. 59:47–59 doi:10.1369/jhc.2010.956644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dinglasan RR, Jacobs-Lorena M. 2008. Flipping the paradigm on malaria transmission-blocking vaccines. Trends Parasitol. 24:364–370 doi:10.1016/j.pt.2008.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dinglasan RR, et al. 2007. Disruption of Plasmodium falciparum development by antibodies against a conserved mosquito midgut antigen. Proc. Natl. Acad. Sci. U. S. A. 104:13461–13466 doi:10.1073/pnas.0702239104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giersing BK, et al. 2006. Potency assay design for adjuvanted recombinant proteins as malaria vaccines. Vaccine 24:4264–4270 [DOI] [PubMed] [Google Scholar]
- 17. Greenwood BM, Targett GA. 2011. Malaria vaccines and the new malaria agenda. Clin. Microbiol. Infect. 17:1600–1607 doi:10.1111/j.1469-0691.2011.03612.x [DOI] [PubMed] [Google Scholar]
- 18. Halekoh U, Hojsgaard S, Yan J. 2006. The R package geepack for generalized estimating equations. J. Stat. Soft. 15(2):1–11 [Google Scholar]
- 19. Kaslow DC. 1997. Transmission-blocking vaccines: uses and current status of development. Int. J. Parasitol. 27:183–189 [DOI] [PubMed] [Google Scholar]
- 20. Lavazec C, et al. 2007. Carboxypeptidases B of Anopheles gambiae as targets for a Plasmodium falciparum transmission-blocking vaccine. Infect. Immun. 75:1635–1642 doi:10.1128/IAI.00864-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lavazec C, Bourgouin C. 2008. Mosquito-based transmission blocking vaccines for interrupting Plasmodium development. Microbes Infect. 10:845–849 doi:10.1016/j.micinf.2008.05.004 [DOI] [PubMed] [Google Scholar]
- 22. malERA Consultative Group on Basic Science and Enabling Technologies 2011. A research agenda for malaria eradication: basic science and enabling technologies. PLoS Med. 8:e1000399 doi:10.1371/journal.pmed.1000399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. malERA Consultative Group on Vaccines 2011. A research agenda for malaria eradication: vaccines. PLoS Med. 8:e1000398 doi:10.1371/journal.pmed.1000398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Owusu-Agyei S, et al. 2009. Randomized controlled trial of RTS,S/AS02D and RTS,S/AS01E malaria candidate vaccines given according to different schedules in Ghanaian children. PLoS One 4:e7302 doi:10.1371/journal.pone.0007302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Quaroni A, et al. 1992. Expression and different polarity of aminopeptidase N in normal human colonic mucosa and colonic tumors. Int. J. Cancer 51:404–411 [DOI] [PubMed] [Google Scholar]
- 26. R Development Core Team 2011. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 27. Röcken C, Licht J, Roessner A, Carl-McGrath S. 2005. Canalicular immunostaining of aminopeptidase N (CD13) as a diagnostic marker for hepatocellular carcinoma. J. Clin. Pathol. 58:1069–1075 doi:10.1136/jcp.2005.026328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stefanovic V, Vlahovic P, Ardaillou N, Ronco P, Ardaillou R. 1992. Cell surface aminopeptidase A and N activities in human glomerular epithelial cells. Kidney Int. 41:1571–1580 [DOI] [PubMed] [Google Scholar]
- 29. Vekemans J, Leach A, Cohen J. 2009. Development of the RTS,S/AS malaria candidate vaccine. Vaccine 27(Suppl 6):G67–G71 doi:10.1016/j.vaccine.2009.10.013 [DOI] [PubMed] [Google Scholar]
- 30. Zeger SL, Liang KY. 1986. Longitudinal data analysis for discrete and continuous outcomes. Biometrics 42:121–130 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.