

Abstract
Combination of innocuous dietary components with anticancer drugs is an emerging new strategy for cancer chemotherapy to increase anti-tumor responses. Tangeretin (TG) is a citrus flavonoid known to inhibit cancer cell proliferation. Here, we show an enhanced response of A2780/CP70 and 2008/C13 cisplatin-resistant human ovarian cancer cells to various combination treatments of cisplatin (Cis) and tangeretin. Pretreatment of cells with tangeretin prior to cisplatin treatment synergistically inhibited cancer cell proliferation. This combination was effective in activating apoptosis via caspase cascade as well as arresting cell cycle at G2/M-phase. Moreover, phospho-Akt and its downstream substrates, e.g., NF-κB, phospho-GSK-3β and phospho-BAD were down-regulated upon tangeretin-cisplatin treatment. The tangeretin-cisplatin induced apoptosis in A2780/CP70 cells was increased by phosphoinositide-3 kinase (PI3K) inhibition and siRNA-mediated Akt silencing, but reduced by over-expression of constitutively activated-Akt and GSK-3β inhibition. The overall results indicated that tangeretin exposure preconditions cisplatin-resistant human ovarian cancer cells for a conventional response to low-dose cisplatin-induced cell death occurring through down-regulation of PI3K/Akt signaling pathway. Thus, effectiveness of tangeretin combinations, as a promising modality in the treatment of resistant cancers, warrants systematic clinical studies.
Introduction
Majority of patients with ovarian cancer are not effectively treated with standard cisplatin [cisdiamminedichloroplatinum (II)] regimens primarily due to the obstacle posed by development of drug resistance (1). Like most malignancies, intrinsic drug resistance of ovarian cancers could be attributed to multiple genetic and/or epigenetic alterations resulting in the loss of tumor suppressor functions or dysregulation of survival signals (2). Identification and reversal of these traits offer valuable targets for modulating the response of cancer cells. Development of new effective strategies based on transient targeted response modulation is now in the forefront of cancer research to overcome drug resistance in cancer chemotherapy.
Amongst the current chemotherapy drug regimens, cisplatin represents one of the clinically most important antineoplastic agent with anticancer activity against a wide variety of solid tumors (3). The anti-neoplastic effect of cisplatin is mediated by the formation of functionally lethal intra-strand DNA cross-links. These lesions activate damage response pathways that result in diverse effects including DNA synthesis inhibition, RNA transcription suppression, cell cycle arrest and apoptosis. Several mechanisms are known to be responsible for cisplatin resistance. These include reduced platinum accumulation, enhanced platinum detoxification and metabolism, altered DNA damage repair, and more importantly the activation of phospholipid kinase, phosphatidyl inositol 3-kinase (PI3K)/Akt, and other cellular survival signaling pathways ultimately causing dysregulation of apoptotic pathway (4).
PI3K/Akt signal transduction plays a critical role in the control of cell growth and proliferation (5). The increased Akt activation or dysregulation due to elevated Akt expression and indirect changes in Akt regulators results in stronger cell survival signaling, which is a common feature in various forms of human cancers, including human ovarian carcinoma (6, 7). Dozens of downstream substrates of Akt kinase have been identified including those related to chemotherapeutic resistance in cancer cells, e.g., GSK-3β, BAD and transcription factor NF-κB, etc. These substrates directly or indirectly regulate apoptosis. GSK-3β, for example, is phosphorylated by Akt and, GSK-3 itself is involved in the regulation of cell proliferation, anti-apoptotic pathways and cell cycle progression (8-10). BAD, a pro-apoptotic Bcl-2 family member (11), is an Akt target directly implicated in regulating cell survival (12). Phosphorylation of BAD changes its affinity to Bcl-2 molecules and phospho-BAD (p-BAD) is unable to inhibit Bcl-2 function (13, 14). Akt also regulates NF-κB pathway via phosphorylation and activation of inhibitory kappa-B kinase (IKK) and RelA (15, 16). The NF-κB transcription factors themselves regulate several important physiological processes, e.g., inflammation and immune responses, cell growth and apoptosis. Thus, inhibition of NF-κB activation offers a potential strategy for treatment of different malignancies (17, 18).
Newer approaches using dietary flavonoids in combination therapies are being increasingly explored to achieve greater efficacy for drug resistant cancer cells. For instance, soy isoflavone genistein has been shown to increase apoptosis induced by chemotherapeutic agents in human cancer cells through inactivation of NF-κB via down-regulating Akt pathway (19). Flavonoids alone have also been shown to induce apoptosis in some cancer cells, while sparing normal cells (20). Several mechanisms have been suggested for flavonoid-induced apoptosis, including inhibition of topoisomerase I/II activity (21-24), regulation of expression of heat shock (25) and Bcl-2 family proteins (26-29) , activation of caspase-9 and 3 (22) and modulation of Akt signaling and NF-κB activation (30).
Tangeretin (Figure S1), a polymethoxylated citrus flavonoid, exhibits anti-proliferative, anti-invasive, anti-metastatic, and anti-oxidant activities (20, 31). The molecular mechanisms and potential applications of tangeretin and other citrus flavonoids in therapy have been reviewed (20, 31, 32). In this study, we tested the potential of tangeretin to sensitize cisplatin-resistant human ovarian cancer A2780/CP70 and 2008/C13 cells to cisplatin-induced cell death using different combination schedules and identified the underlying mechanism of its action. The results demonstrated that tangeretin pretreatment synergizes the low-dose cisplatin-induced cancer cell death. The synergistic effect is mediated through the down-regulation of PI3K/Akt pathway. Based on these observations, tangeretincisplatin combination may offer a promising new approach in effective treatment of human ovarian cancers.
Materials and Methods
Cell culture and reagents
Human ovarian cancer cells A2780 and its cognate cisplatin-resistant A2780/CP70 were provided by Dr. Paul Modrich (Duke University, NC). Another pair of cisplatin-sensitive and -resistant human ovarian cancer cell lines 2008 and 2008/C13 was provided by Drs. Francois Claret and Qingxiu Zhang (M.D. Anderson Cancer Center, TX). Cells were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum at 37°C in a humidified 5% CO2 atmosphere. Tangeretin, cisplatin, GSK-3β inhibitor SB216763, PI3K inhibitor LY294002 and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Stock solutions of SB216763 (25 mM) and LY294002 (10 mM) in DMSO, were used at final concentrations of 50 and 10 μM, respectively, according to the manufacture's recommendations. Antibodies against Akt, phospho-Akt [p-Akt (Ser473)], BAD, phospho-BAD [p-BAD (Ser136)], GSK-3ß, phospho-GSK-3ß [(p-GSK-3ß (Ser9)], NF-kB p65, PARP and SignalSilence Akt siRNA kit were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against β-actin and lamin-B were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against CyclinB1 and Cdc25c were purchased from Abcam Inc. (Cambridge, MA). Antibody against p53 was purchased from Neomarkers (Fremont, CA). Horseradish peroxidaseconjugated secondary antibodies, protease inhibitor and the Cell Death ELISA Plus kit were from Roche (Indianapolis, IN). Chemiluminescence substrate was obtained from Pierce (Rockford, IL). The DC Bio-Rad protein quantitation reagents were from Bio-Rad (Hercules, CA). Plasmid 14751, harboring a constitutively active mutant Akt gene was purchased from Addgene (Cambridge, MA).
Cell growth and MTT cytotoxicity assay
Cells were seeded in 96-well plates, treated with different concentration of cisplatin or tangeretin and maintained in culture for 72 hours. For the combination experiments, A2780/CP70 and 2008/C13 cells were treated with tangeretin (150 μM) either 24 hours prior to, or after cisplatin (3 or 6 μM) treatment. Drug-treated cells were incubated for 72 hours and then MTT was added to the cultures for an additional 2 hours. The medium was replaced with acidified isopropanol for 1 hour and the absorbance was determined by Spectramax M5 microplate reader (Molecular Devices, California, CA). Drug interactions and isobologram were analyzed using CalcuSyn software (Biosoft, Ferguson, MO).
DNA fragmentation analysis for detecting apoptosis
Cells were treated with 150 μM tangeretin or vehicle for 24 hours and then exposed to 3 or 6 μM cisplatin or vehicle for another 72 hours. Adherent and floating cells were recovered and DNA was isolated and evaluated for fragmentation as described earlier (33).
Quantitative measurement of apoptosis by Histone/DNA ELISA
Cell death ELISA plus kit was used for detection of apoptosis according to manufacturer's protocol. Briefly, cells were treated with tangeretin and cisplatin as described above. The cytoplasmic histone/DNA fragments in cell lysates were bound to the immobilized biotin-conjugated histone antibodies in a 96-well streptavidin-coated plate. Wells were exposed to peroxidase-conjugated DNA antibody followed by peroxidase substrate and the absorbance at 405 nm was measured colorimetrically using Spectramax M5 microplate reader.
Western blot analysis
Cells were treated with tangeretin, cisplatin or tangeretin-cisplatin combination. The cells were harvested and boiled in lysis buffer containing protease inhibitors. Samples were subjected to SDS-PAGE and electrophoretically transferred to a PVDF membrane. Membranes were incubated with the primary antibodies at 4°C overnight, washed with TBST buffer, and incubated again with an appropriate HRP-conjugated secondary antibody at 37°C for 1 hour. The membranes were washed and examined by chemiluminescence detection.
Flow cytometric analysis of cell cycle and apoptosis
Tangeretin and/or cisplatin treated cells were collected and fixed with 70% ice-cold ethanol overnight at -20°C. Cells were centrifuged, resuspended in a mix of propidium iodide and RNase A and incubated at 37°C for 40 minutes. Cells were pelleted, washed and resuspended in PBS to a final concentration of 1×106/ml and analyzed using BD FACS Calibur (BD Biosciences, San Jose, CA).
Transfection of constitutively active Akt plasmid
Cells were seeded in an antibiotic-free medium at 37°C for 24 hours and transfected with constitutively active Akt plasmid 14751 or empty vector DNA using Lipofectamine-2000 transfection reagent according to the manufacturer's instructions. After 24 hours transfection, cells were treated as described above and analyzed for cellular proteins and apoptosis.
Akt small interfering RNA (siRNA) transfection
The Akt siRNA kit was used to silence Akt expression according to the manufacturer's protocol. Briefly, cells were grown for 24 hours to ~50% confluency and medium was replaced with 500 μl of an antibiotic-free medium. siRNA-transfection reagent mixture was applied to cultured cells for 48 hours. Cells were then treated as described above and analyzed for cellular proteins and apoptosis.
Results
Tangeretin potentiates cisplatin-induced growth inhibition in a drug sequence-dependent manner
First, the anti-proliferative effect of cisplatin and tangeretin was examined in cisplatin-resistant A2780/CP70 and 2008/C13 human ovarian cancer cell lines (Figure S2). Next, we tested whether tangeretin can sensitize cells to low-dose cisplatin-induced death. Cells were exposed to varying concentrations of tangeretin (25-150 μM) and cisplatin (1.5-6 μM) under two different drug administration scenarios: an initial 24 hours tangeretin exposure followed by 72 hours cisplatin treatment and vice versa. The cell growth was determined and the values of CI, a quantitative measure of drug interaction, were calculated (Figure 1, Table S1). As shown in Figure 1, pretreatment of cells with tangeretin for 24 hours synergized the cytotoxic effect of cisplatin. The CI ranged from ~0.5-0.7 for every tangeretin-cisplatin combination tested. On the other hand, when cisplatin was administered prior to tangeretin, CI was between 1 and 1.1, indicating additive rather than synergistic effects. We selected two cisplatin doses (3 and 6 μM) that kill about 20-30 percent of resistant cells and combined them with 150 μM tangeretin to further test whether this combination can enhance the growth inhibition of cisplatin-resistant cells. Figure 1C shows that pretreatment of cells with tangeretin increased the potency of cisplatin over 2-fold. For instance, single treatment of cells with 3 and 6 μM cisplatin caused about 19% and 31.4% growth inhibition in A2780, respectively. However, the growth inhibition increased to 60% and 72.9% when the cells were pretreated with tangeretin. For the drug administration sequences, using cisplatin prior to tangeretin, the growth inhibition was only 42.1 and 57.9% for 3 and 6 μM cisplatin, respectively. A similar response was seen with 2008/C13 cells. Treatment of 2008/C13 cells with 3 and 6 μM cisplatin caused about 21% and 33% inhibition of cell survival. The inhibition was increased further from 41 & 49% to 58% and 66%, respectively upon tangeretin pretreatment compared to when tangeretin was added 24 hours after cisplatin administration. These effects were confirmed by colony forming assay (Figure S2). Taken together, the results indicated that the combination of tangeretin with low dose of cisplatin elicits significantly higher cytotoxic response in cisplatin-resistant human ovarian cancer cells.
Figure 1.

Synergism of tangeretin-cisplatin combination is dependent on the drug administration sequence. A2780/CP70 (A) and 2008/C13 (B) were treated with different concentration of cisplatin and tangeretin in the indicated drug administration sequences. Cells were allowed to grow for 72 hours and cell growth was determined by MTT assay. CI and normalized isobologram were calculated and processed using CalcuSyn software. Numbered symbols 1, 2, 3, 4 and 5 represent CI of drug combination where cisplatin to tangeretin ratio were 1:16.67, 1:25.00, 1:33.33, 1:50.00 and 1:25.00, respectively. C, Cells were treated with tangeretin 24 hours before cisplatin (columns 5&6) or with cisplatin 24 hours prior to tangeretin (columns 7&8) as indicated by arrow. Symbol** represents significant difference between the corresponding single treatment groups at p<0.001 by Tukey-Kramer Multiple Comparisons Test; n=3.
Tangeretin enhances cisplatin-induced apoptosis via a caspase-dependent mechanism
Cellular apoptosis was assessed first by gel electrophoretic analysis of inter-nucleosomal DNA fragmentation as well as by a quantitative ELISA assay measuring cytoplasmic histone/DNA fragments. Treatment of A2780/CP70 and 2008/C13 cells with single exposures of tangeretin or cisplatin showed the presence of mostly high molecular weight DNA as seen with untreated control. However, a DNA ladder pattern, the typical feature of apoptosis, was distinctly observed upon combination of tangeretin with different concentrations of cisplatin. This apoptotic response was reversed upon pretreatment with the general caspase inhibitor, z-VAD-FMK (Figure 2A, lanes 5, 6, 13, and 14 vs. 7, 8, 15, and 16). The quantitative measurement of apoptosis showed that tangeretin alone caused negligible increase in cell death over the background. Also, treatment with cisplatin up to 6 μM revealed a minimal extent of cell death. On the other hand, tangeretin prior to cisplatin treatment caused a robust increase in apoptosis index of treated cells (Figure 2B, column 5, 6, 13, and 14) that was reversed upon caspase inhibition (Figure 2B, column 7, 8, 15 & 16). Moreover, the cleavage of PARP, a hallmark of caspase-dependent apoptosis, and activation of the caspase-7 and -3 were also seen in parallel for the same combination treatments. Consistent with cell growth inhibition, the cleavage of caspase-7, caspase-3 and PARP were abolished by z-VAD-FMK, indicating that the apoptosis was specifically mediated through activation of the caspase pathway. Notably, the overall apoptotic response was significantly higher in combined tangeretin and 3 μM cisplatin dually exposed than in 6 μM cisplatin singly treated cells. Thus, these results also suggested a synergized apoptotic effect of tangeretin-cisplatin combination on cisplatin-resistant A2780/CP70 and 2008/C13 cells.
Figure 2.

Tangeretin enhances cisplatin-induced apoptosis. A, Cells were treated with either 150 μM tangeretin or vehicle for 24 hours then treated for another 72 hours with cisplatin or vehicle. Caspase inhibitor Z-VAD was applied to cells 1 hour prior to tangeretin treatment. Cells were processed for DNA fragmentation through agarose gel electrophoresis. B, Quantitative assessment of apoptosis was performed using Cell Death ELISA kit. Symbol** represents significant difference between the corresponding single treatment groups at p<0.001; n=3. C, PARP, caspase-7 and caspase-3 cleavage were determined by Western blot analysis.
Tangeretin-cisplatin combination arrests resistant cells in G2/M-phase
Asynchronously growing A2780/CP70 cells, treated with 150 μM tangeretin followed by 3 μM cisplatin, were examined for their cycle progression by flow cytometry (Figure 3A&B). In untreated control, the percentage of cells in G1-, S- and G2/M-phases were 63.47%, 21.22% and 10.51 %, respectively. Single exposure with cisplatin had no significant effect on the cell cycle while tangeretin caused a small redistribution to G2/M-phase. The tangeretin-cisplatin treatment, however, resulted in a pronounced G2/M arrest. The percentage of G1-, S- and G2/M-phase cells following tangeretin-cisplatin treatment was 15.95%, 14.82% and 51.98%, respectively. Thus, the accumulation of G2/M-phase cells was largely at the expense of cells failing to cycle into G1-phase. As expected, flow cytometry revealed the presence of a high proportion of sub-G1-phase population (apoptotic cell) in cultures treated with tangeretin-cisplatin combination. Consistent with the flow cytometric analysis, tangeretin-cisplatin treatment reduced the levels of Cdc25C and cyclin B1, while the p53 level was constitutively elevated in these cells (Figure 3C).
Figure 3.

Tangeretin-cisplatin combination induces G2/M arrest in A2780/CP70 cells. Cells were treated with either 150 μM tangeretin or its vehicle for 24 hours, followed by treatment with cisplatin or its vehicle for another 72 hours. The cell cycle distribution and apoptosis of drug treated cells were analyzed using flow cytometry (A&B). Symbol** represent significant difference at p<0.001 the corresponding single treatment groups by Tukey-Kramer Multiple Comparisons Test; n=3. C, Cyclin B1, Cdc25C and p53 were determined by Western blot analysis.
p-Akt and its substrates are down-regulated by tangeretin-cisplatin combination
Since cisplatin resistance of human ovarian cancer is related to activation of PI3K/Akt signaling pathway (4), we wanted to determine the potential attenuation of this pathway by tangeretin-cisplatin treatment. Western blot analysis (Figure 4) showed a specific reduction in the p-Akt protein in A2780/CP70 and 2008/C13 cells treated with tangeretin-cisplatin, as compared with that of untreated as well as singly treated controls. The total Akt level, however, remained unaffected by all treatment conditions. Relatively insignificant decrease of p-Akt by tangeretin was consistent with the high IC50 observed for this treatment. The expression levels of Akt downstream substrates NF-κB, BAD and GSK-3β were also assessed. The level of NF-κB p65 showed the expected decrease by tangeretin-cisplatin combination. The combination treatment also decreased p-BAD and p-GSK-3β without affecting their non-phosphorylated forms. Taken together, this data suggested that regulation of Akt pathway is intimately associated with tangeretin-cisplatin induced growth inhibition, apoptosis and G2/M arrest.
Figure 4.
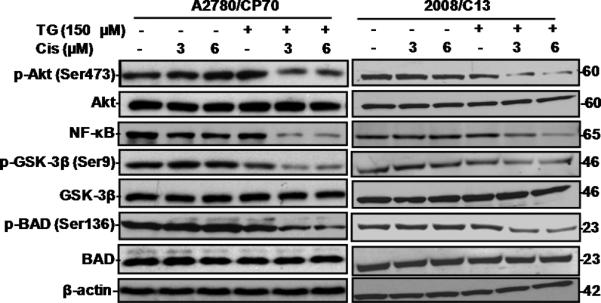
Tangeretin-cisplatin combination down-regulates p-Akt and its downstream substrates. The A2780/CP70 and 2008/C13 cells were treated with either 150 μM tangeretin or its vehicle for 24 hours and then treated for another 72 hours with cisplatin or its vehicle. The cellular proteins p-Akt, Akt, p-Bad, Bad, NFκB, p-GSK-3β and GSK-3β were determined by Western blot analysis.
Tangeretin-cisplatin combination induces apoptosis in resistant cells by down-regulating Akt signaling
To understand whether modulation of PI3K/Akt signaling pathway is involved in tangeretin-cisplatin induced apoptosis in resistant cells, we performed a series of experiments based on PI3K inhibition, siRNA-mediated Akt silencing and ectopic Akt over-expression in A2780/CP70 cells. We pretreated the cells with PI3K inhibitor LY294002 before drug administration. The results showed that PI3K inhibition induced a dose-dependent PARP cleavage and DNA fragmentation in tangeretin-treated cells (Figure 5A, lanes and columns 7&8), indicating that PI3K acts as an upstream factor in tangeretin induced cellular pro-apoptotic signaling. Moreover, pretreatment of the cells with PI3K inhibitor alongside tangeretin-cisplatin combination treatment caused a dramatic increase of PARP cleavage and DNA fragmentation (Figure 5A, lanes and columns 10 vs. 2, 5, 7&9). This data clearly identified PI3K as the upstream regulatory factor for invoking the tangeretin's effect in augmenting the cisplatin-induced cytotoxicity and apoptosis.
Figure 5.

Tangeretin-cisplatin combination induces apoptosis in A2780/CP70 cells by modulating Akt signaling pathway. A, PI3K inhibitor LY294002 enhances tangeretin-cisplatin combination induced apoptosis. One hour prior to drug treatment, 50 μM LY294002 was applied to A2780/CP70 cells. The cells were then treated with either 100 or 150 μM tangeretin or vehicle for 24 hours and followed by cisplatin or vehicle treatments for another 48 hours. B, silencing of Akt by siRNA potentiates tangeretincisplatin combination to induce apoptosis in A2780/CP70 cells. The cells were transfected with 50 nM Akt siRNA for 24 hours and then treated with tangeretin-cisplatin combination. C, overexpression of constitutively activated Akt hinders the ability of tangeretin-cisplatin combination to induce apoptosis. Cells were transfected with Akt cDNA for 24 hours, prior to treatment of tangeretin-cisplatin combination. For each of the above treatments, cellular PARP was determined by Western blotting (Top panels) and apoptosis was quantitatively assessed using Cell Death ELISA (bottom panels). Symbols * and ** represent significant difference at p<0.01 and p<0.001 between the groups with and without LY294002 or Akt siRNA, or Akt cDNA treatment by Tukey-Kramer Multiple Comparisons Test; n=3.
Next, we investigated the functional relevance of Akt to sensitization of A2780/CP70 cells by tangeretin-cisplatin treatment. A2780/CP70 cells were transfected with Akt siRNA prior to drug treatment and quantitative Akt knockdown was confirmed by Western blot analysis. Akt knockdown induced a dose-dependent PARP cleavage and DNA fragmentation in tangeretin-treated cells (Figure 5B, lanes and columns 7&8), indicating that Akt is also an upstream factor of tangeretin induced cellular pro-apoptotic signaling. Akt silencing resulted in partial PARP cleavage by 3 μM cisplatin treatment which was prominently elevated upon tangeretin-cisplatin combination treatment (Figure 5B, lanes and columns 9&10 vs. 4&5). The results of PARP cleavage were corroborated by parallel measurements of apoptosis. These results firmly emphasize the central role of Akt in regulation of apoptotic response in resistant A2780/CP70 cells to tangeretin-cisplatin treatment.
Finally, we transfected Akt expression construct into A2780/CP70 cells to test if ectopically over-expressed, constitutively active form of Akt can block tangeretin-cisplatin-induced apoptosis. Western blotting confirmed that Akt was significantly over-expressed in A2780/CP70 cells as compared to vector DNA transfected controls (Figure 5C). Here, the combination treatment of the controls exhibited the signature PARP cleavage and apoptosis responses. However, in cells over-expressing Akt, both PARP cleavage and DNA fragmentation were dramatically blocked in tangeretin-cisplatin treated cells (Figure 5C, lanes 5&6). These results demonstrated that Akt expression overrode the effect of tangeretin treatment and prevented the drug treated cells from undergoing apoptosis. The results further suggested that the p-Akt and its downstream substrates were vital signaling pathway components responsive for tangeretin-cisplatin treatment. We surmised that PI3K/Akt signaling is a key mediator of cisplatin resistance of A2780/CP70 cells. Down-regulation of PI3K/Akt pathway by tangeretin-cisplatin treatment eliminates the downstream apoptosis inhibitory signals, e.g., NF-κB and activated BAD, and consequently renders the resistant A2780/CP70 cells susceptible to the cytotoxic effects of cisplatin.
GSK-3β is PI3K/Akt downstream effector of tangeretin-cisplatin induced apoptosis
Since PI3K/Akt influences apoptosis through multiple auxiliary pathways, we wanted to assess the extent of contribution of GSK-3β in tangeretin-cisplatin induced apoptosis. A2780/CP70 cells were pretreated with a specific GSK-3β inhibitor, SB216763, prior to the tangeretin-cisplatin treatment for measuring the effects on apoptosis parameters. The inhibition of GSK-3β resulted in complete blockage of tangeretincisplatin induced PARP cleavage and apoptosis (Figure 6A&B, lanes 5&6 vs. 2&3). Interestingly, quantitative measurement of DNA fragmentation showed that GSK-3β inhibitor blocked ~60% of apoptosis resulting from the combination treatment. This data indicated that GSK-3β is another major target of acquired cisplatin resistance through PI3K/Akt signaling pathway and its down-regulation, through inactivating PI3K/Akt signaling, contributes significantly to tangeretin-cisplatin induced apoptosis.
Figure 6.
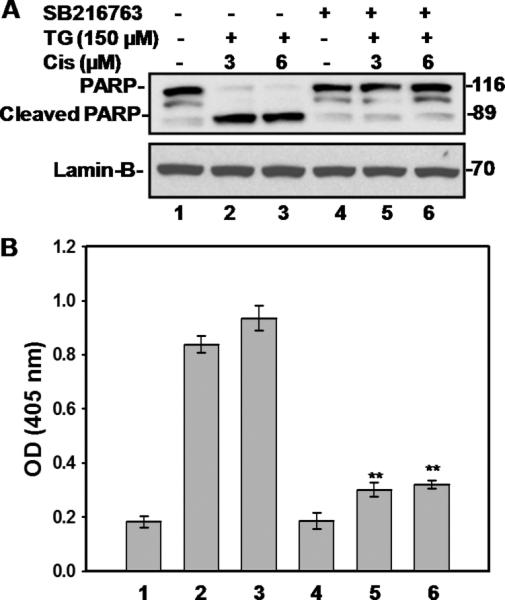
GSK-3β inhibitor SB216763 compromises the ability of tangeretin-cisplatin combination to induce apoptosis. SB216763 was applied 1 hour prior to treatment combination. A, PARP cleavage was determined by Western blot analysis. B, Apoptosis of drug treated cells was quantitatively examined using Cell Death ELISA. Symbol ** shows significant difference (p<0.001) of the corresponding combination group without SB216763 by Tukey-Kramer Multiple Comparisons Test; n=3.
Discussion
Although cisplatin has been widely used for decades to treat a wide range of cancers, de novo and acquired resistance, restricting the successful use of this potent chemotherapeutic agent, continues to pose a major clinical challenge. The dose escalation necessary to overcome even a small increase in cellular resistance can cause severe cytotoxicity to dose-limiting normal tissue. Thus, strategies using dual agents that act through distinct molecular mechanisms, rather than using single agents, represent the most useful alternatives for achieving higher curability with least toxicity during cancer chemotherapy. Recently, there has been an increasing interest in evaluating synergistic cancer cell cytotoxicity from combining chemotherapeutic agents with highly promising and relatively innocuous dietary flavonoids (34, 35). In this study, we investigated the potential of a dietary flavonoid, tangeretin, to sensitize the cisplatin-resistant ovarian cancer cells to cisplatin-induced cytotoxicity. We have demonstrated that tangeretin reduces the recalcitrance of human ovarian cancer cells to low dose cisplatin-induced cell death through down-regulation of PI3K/Akt signaling pathway.
A recent report has identified tangeretin as belonging to a superior cancer preventive flavonoid subclass that exhibits anti-proliferative activity against several human cancer cell lines (20). Its anti-proliferative effects have been attributed to alteration of activities related to apoptosis and cell cycle arrest. However, the underlying molecular events mediating growth inhibition, especially the basis of synergized anti-proliferative effects on cancer cells, are not completely understood. Tangeretin has been reported to induce G1 cell cycle arrest in human breast and colon cancer cells (36). In human colorectal carcinoma cells, tangeretin increases the cellular expression of p21 and p27 (37). We, however, observed an accumulation of G2/M cell population in tangeretin singly treated as well as the tangeretin-cisplatin treated A2780/CP70 cisplatin-resistant human ovarian cancer cells (Figure 3). Similar to our data, tangeretin has been reported to induce G2/M arrest in p53-null HL-60 promyelocytic leukemia cells (38). As G2/M arrest is typically linked to DNA damage response, we posit that pretreatment with tangeretin sensitizes these cells to respond to DNA damage especially in the context of their p53 function. The A2780/CP70 cell line initially developed their resistance to cisplatin from repeated exposures of the parental A2780 cells to challenge doses of cisplatin. Despite severe and cumulative genotoxic exposures, A2780/CP70 cells retain their wild-type p53 gene sequence. Nevertheless, these exposures render p53 protein transcriptionally inactive in A2780/CP70 cells. For instance, DNA damage fails to induce the accumulation of p21 protein in cisplatin-resistant and this induction is normal in cisplatin-sensitive parental cells (39)our unpublished data). More importantly, A2780/CP70 cisplatin-resistant cells constitutively express p53 protein in both wild-type as well as mutant conformation as detected by conformation-specific pAb240 antibody (39). The A2780 cells, on the other hand, express functional p53 protein recognizable by pAb1620 antibody. Moreover, transfection of V143A mutant p53 into A2780 cells increase resistance of the cells to DNA damaging agents but not to paclitaxel (40). It is possible that the inherently altered conformation of p53 associated with the abrogated p53 function occurred in A2780/CP70 cells, contributing to their cisplatin resistance (39). The changes in p53 functional status in A2780/CP70 cisplatin-resistant cells could also make them amenable to the observed G2/M arrest in response to tangeretin-cisplatin treatment.
Increased apoptosis induced by cisplatin is another feature of cellular response to combined tangeretin and cisplatin treatment, which manifests as the synergetic growth inhibitory effect on cisplatin-resistant cells. Here, we have shown that specific PI3K inhibition by LY294002 induced a tangeretin dose-dependent PARP cleavage and DNA fragmentation in the tangeretin-treated cells. Furthermore, pretreatment of the cells with PI3K inhibitor caused a dramatic increase of PARP cleavage and DNA fragmentation in tangeretin-cisplatin treated A2780/CP70 cells. In parallel, knockdown of Akt had a similar effect on tangeretin-cisplatin induced apoptosis. In contrast, the overexpression of constitutively active Akt expression overrides the pro-apoptotic effect of tangeretin-cisplatin treatment and prevented drug treated cells from apoptosis. These results demonstrated that PI3K/Akt signaling pathway mechanistically regulates the tangeretin-cisplatin induced apoptosis. Besides, clear down-regulation of p-Akt and its downstream substrates NF-κB, p-GSK-3β and p-BAD were only observed in tangeretin-cisplatin treated cells. Tangeretin treatment alone did not appreciably alter the cellular levels of these cell survival regulators. Given that tangeretin potentiates growth inhibition of cells caused by cisplatin in a drug administration sequence dependent manner, we reason that tangeretin pretreatment could condition the cells to respond to cisplatin induced DNA damage by muffling the constitutive activation of p-Akt. The A2780/CP70 and 2008/C13 cells express high levels of p-Akt (Figure 4), indicating that the cells have a constitutively activated cell survival signaling. Such a scenario of activated signaling in cells would inevitably institute an enhanced survival and impose a barrier for desired killing effects of DNA damaging therapeutic agents. Consistent with this notion, the A2780/CP70 cells are not only resistant to cisplatin, but also cross resistant to irradiation, melphalan and adriamycin (41). Therefore, implementing tangeretin pretreatment would condition A2780/CP70 and 2008/C13 cells, via down-regulated Akt mediated cell survival signaling, to become increasingly susceptible to cisplatin as well as other therapeutic agents. It should be noted that the tangeretin suppresses IL-1β-induced Akt activation in human lung carcinoma cells (42). Thus, it is envisaged that the down-regulation of p-Akt and its downstream substrates, NF-κB, p-GSK-3β and p-BAD, by combined treatment reflects the synergy between tangeretin and cisplatin in modulation of Akt phosphorylation (activation).
Amongst many downstream factors of PI3K/Akt signaling pathway, we have only examined the contribution of GSK-3β inhibition to tangeretin-cisplatin induced apoptosis. However, GSK-3β is not the only PI3K/Akt downstream culprit factor involved in regulating cisplatin-induced apoptosis. In fact, GSK-3β inhibitor blocked only ~60 % of tangeretin-cisplatin induced apoptosis. Other downstream factors of PI3K/Akt pathway could also participates and account for the rest of the observed apoptotic effect. We also observed down-regulation of NF-κB, p-GSK-3β and p-BAD by tangeretin-cisplatin treatment. At present, it is difficult to attribute the extent of individual contribution of these regulatory factors to tangeretin-cisplatin induced apoptosis. However, it is certain that modulation of Akt activation by the combined treatment represents a major intra-cellular switch to mechanistically control the tangeretin-cisplatin-induced tumor cells apoptosis.
In summary, our data experimentally showed that tangeretin pre-exposure can overcome resistance of human ovarian cancer cells to growth inhibition by cisplatin. From a mechanistic standpoint, tangeretin-cisplatin combination down-regulates PI3K/Akt pathway, leading to the sensitization of cancer cells to cisplatin induced-cell death through apoptosis. The data provide a firm molecular basis for the pharmacological effect underlying the use of tangeretin as a valuable therapeutic adjuvant. Given the broad-spectrum organ safety of tangeretin, already demonstrated in laboratory animals in vivo (43, 44), the present work should expedite the exploration and use of tangeretin in enhancing the efficacy of cisplatin in experimental animal studies as well as clinical trials, with the caveat of successfully attaining effective tangeretin doses through human consumption of citrus fruits. It would also be appealing to determine whether tangeretin interacts with additional physical and chemical therapeutic agents in killing a variety of other drug-resistant cancer cells.
Supplementary Material
Acknowledgments
We thank Drs. Paul Modrich (Duke University, NC), Francois Claret and Qingxiu Zhang (M.D. Anderson Cancer Center, TX) for providing cisplatin-sensitive and -resistant cell lines. This work is supported by Public Health Service Grants ES2388, ES12991 and CA93413 to A.A.W. El-Shaimaa A.A. is sponsored by a Scholar Exchange Program of the Egyptian Education Ministry.
References
- 1.Stewart JJ, White JT, Yan X, et al. Proteins associated with Cisplatin resistance in ovarian cancer cells identified by quantitative proteomic technology and integrated with mRNA expression levels. Mol Cell Proteomics. 2006;5:433–43. doi: 10.1074/mcp.M500140-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Balch C, Huang TH, Brown R, Nephew KP. The epigenetics of ovarian cancer drug resistance and resensitization. Am J Obstet Gynecol. 2004;191:1552–72. doi: 10.1016/j.ajog.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 3.Previati M, Lanzoni I, Corbacella E, et al. Cisplatin-induced apoptosis in human promyelocytic leukemia cells. Int J Mol Med. 2006;18:511–6. [PubMed] [Google Scholar]
- 4.Ohmichi M, Hayakawa J, Tasaka K, Kurachi H, Murata Y. Mechanisms of platinum drug resistance. Trends Pharmacol Sci. 2005;26:113–6. doi: 10.1016/j.tips.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–98. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 6.Cantley LC, Auger KR, Carpenter C, et al. Oncogenes and signal transduction. Cell. 1991;64:281–302. doi: 10.1016/0092-8674(91)90639-g. [DOI] [PubMed] [Google Scholar]
- 7.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–5. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeda S, Kishida S, Yamamoto H, et al. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17:1371–84. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–32. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 10.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gajewski TF, Thompson CB. Apoptosis meets signal transduction: elimination of a BAD influence. Cell. 1996;87:589–92. doi: 10.1016/s0092-8674(00)81377-x. [DOI] [PubMed] [Google Scholar]
- 12.Franke TF, Cantley LC. Apoptosis. A Bad kinase makes good. Nature. 1997;390:116–7. doi: 10.1038/36442. [DOI] [PubMed] [Google Scholar]
- 13.del PL, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–9. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 14.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 15.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 16.Ozes ON, Mayo LD, Gustin JA, et al. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–5. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 17.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo JL, Kamata H, Karin M. The anti-death machinery in IKK/NF-kappaB signaling. J Clin Immunol. 2005;25:541–50. doi: 10.1007/s10875-005-8217-6. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Ahmed F, Ali S, et al. Inactivation of nuclear factor kappaB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005;65:6934–42. doi: 10.1158/0008-5472.CAN-04-4604. [DOI] [PubMed] [Google Scholar]
- 20.Walle T. Methoxylated flavones, a superior cancer chemopreventive flavonoid subclass? Semin Cancer Biol. 2007;17:354–62. doi: 10.1016/j.semcancer.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bailly C. Topoisomerase I poisons and suppressors as anticancer drugs. Curr Med Chem. 2000;7:39–58. doi: 10.2174/0929867003375489. [DOI] [PubMed] [Google Scholar]
- 22.Wang IK, Lin-Shiau SY, Lin JK. Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of caspase-9 and caspase-3 in leukaemia HL-60 cells. Eur J Cancer. 1999;35:1517–25. [PubMed] [Google Scholar]
- 23.Markovits J, Linassier C, Fosse P, et al. Inhibitory effects of the tyrosine kinase inhibitor genistein on mammalian DNA topoisomerase II. Cancer Res. 1989;49:5111–7. [PubMed] [Google Scholar]
- 24.Sukardiman, Darwanto A, Tanjung M, Darmadi MO. Cytotoxic mechanism of flavonoid from Temu Kunci (Kaempferia pandurata) in cell culture of human mammary carcinoma. Clin Hemorheol Microcirc. 2000;23:185–90. [PubMed] [Google Scholar]
- 25.Rong Y, Yang EB, Zhang K, Mack P. Quercetin-induced apoptosis in the monoblastoid cell line U937 in vitro and the regulation of heat shock proteins expression. Anticancer Res. 2000;20:4339–45. [PubMed] [Google Scholar]
- 26.Chen YC, Shen SC, Lee WR, et al. Wogonin and fisetin induction of apoptosis through activation of caspase 3 cascade and alternative expression of p21 protein in hepatocellular carcinoma cells SK-HEP-1. Arch Toxicol. 2002;76:351–9. doi: 10.1007/s00204-002-0346-6. [DOI] [PubMed] [Google Scholar]
- 27.Wenzel U, Kuntz S, Brendel MD, Daniel H. Dietary flavone is a potent apoptosis inducer in human colon carcinoma cells. Cancer Res. 2000;60:3823–31. [PubMed] [Google Scholar]
- 28.Iwashita K, Kobori M, Yamaki K, Tsushida T. Flavonoids inhibit cell growth and induce apoptosis in B16 melanoma 4A5 cells. Biosci Biotechnol Biochem. 2000;64:1813–20. doi: 10.1271/bbb.64.1813. [DOI] [PubMed] [Google Scholar]
- 29.Konig A, Schwartz GK, Mohammad RM, Al-Katib A, Gabrilove JL. The novel cyclin-dependent kinase inhibitor flavopiridol downregulates Bcl-2 and induces growth arrest and apoptosis in chronic B-cell leukemia lines. Blood. 1997;90:4307–12. [PubMed] [Google Scholar]
- 30.Yin F, Giuliano AE, Van Herle AJ. Signal pathways involved in apigenin inhibition of growth and induction of apoptosis of human anaplastic thyroid cancer cells (ARO). Anticancer Res. 1999;19:4297–303. [PubMed] [Google Scholar]
- 31.Kandaswami C, Perkins E, Soloniuk DS, Drzewiecki G, Middleton E., Jr Antiproliferative effects of citrus flavonoids on a human squamous cell carcinoma in vitro. Cancer Lett. 1991;56:147–52. doi: 10.1016/0304-3835(91)90089-z. [DOI] [PubMed] [Google Scholar]
- 32.Nijveldt RJ, van NE, van Hoorn DE, et al. Flavonoids: a review of probable mechanisms of action and potential applications. Am J Clin Nutr. 2001;74:418–25. doi: 10.1093/ajcn/74.4.418. [DOI] [PubMed] [Google Scholar]
- 33.El-Mahdy MA, Zhu Q, Wang QE, Wani G, Wani AA. Thymoquinone induces apoptosis through activation of caspase-8 and mitochondrial events in p53-null myeloblastic leukemia HL-60 cells. Int J Cancer. 2005;117:409–17. doi: 10.1002/ijc.21205. [DOI] [PubMed] [Google Scholar]
- 34.Sarkar FH, Li Y. Using chemopreventive agents to enhance the efficacy of cancer therapy. Cancer Res. 2006;66:3347–50. doi: 10.1158/0008-5472.CAN-05-4526. [DOI] [PubMed] [Google Scholar]
- 35.Bava SV, Puliappadamba VT, Deepti A, et al. Sensitization of taxol-induced apoptosis by curcumin involves down-regulation of nuclear factor-kappaB and the serine/threonine kinase Akt and is independent of tubulin polymerization. J Biol Chem. 2005;280:6301–8. doi: 10.1074/jbc.M410647200. [DOI] [PubMed] [Google Scholar]
- 36.Morley KL, Ferguson PJ, Koropatnick J. Tangeretin and nobiletin induce G1 cell cycle arrest but not apoptosis in human breast and colon cancer cells. Cancer Lett. 2007;251:168–78. doi: 10.1016/j.canlet.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 37.Pan MH, Chen WJ, Lin-Shiau SY, Ho CT, Lin JK. Tangeretin induces cell-cycle G1 arrest through inhibiting cyclin-dependent kinases 2 and 4 activities as well as elevating Cdk inhibitors p21 and p27 in human colorectal carcinoma cells. Carcinogenesis. 2002;23:1677–84. doi: 10.1093/carcin/23.10.1677. [DOI] [PubMed] [Google Scholar]
- 38.Hirano T, Abe K, Gotoh M, Oka K. Citrus flavone tangeretin inhibits leukaemic HL-60 cell growth partially through induction of apoptosis with less cytotoxicity on normal lymphocytes. Br J Cancer. 1995;72:1380–8. doi: 10.1038/bjc.1995.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones NA, Turner J, McIlwrath AJ, Brown R, Dive C. Cisplatin- and paclitaxel-induced apoptosis of ovarian carcinoma cells and the relationship between bax and bak up-regulation and the functional status of p53. Mol Pharmacol. 1998;53:819–26. [PubMed] [Google Scholar]
- 40.Vasey PA, Jones NA, Jenkins S, Dive C, Brown R. Cisplatin, camptothecin, and taxol sensitivities of cells with p53-associated multidrug resistance. Mol Pharmacol. 1996;50:1536–40. [PubMed] [Google Scholar]
- 41.Behrens BC, Hamilton TC, Masuda H, et al. Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 1987;47:414–8. [PubMed] [Google Scholar]
- 42.Chen KH, Weng MS, Lin JK. Tangeretin suppresses IL-1beta-induced cyclooxygenase (COX)-2 expression through inhibition of p38 MAPK, JNK, and AKT activation in human lung carcinoma cells. Biochem Pharmacol. 2007;73:215–27. doi: 10.1016/j.bcp.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 43.Datla KP, Christidou M, Widmer WW, Rooprai HK, Dexter DT. Tissue distribution and neuroprotective effects of citrus flavonoid tangeretin in a rat model of Parkinson's disease. Neuroreport. 2001;12:3871–5. doi: 10.1097/00001756-200112040-00053. [DOI] [PubMed] [Google Scholar]
- 44.Vanhoecke BW, Delporte F, Van BE, et al. A safety study of oral tangeretin and xanthohumol administration to laboratory mice. In Vivo. 2005;19:103–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.