

Abstract
Signal transducer and activator of transcription 3 (Stat3), a target for anticancer drug design, is activated by recruitment to phosphotyrosine residues on growth factor and cytokine receptors via its SH2 domain. We report here structure-activity relationship studies on phosphopeptide mimics targeted to the SH2 domain of Stat3. Inclusion of a methyl group on the β-position of the pTyr mimic, 4-phosphocinfnamide, enhanced affinity 2–3 fold. Bis-pivaloyloxymethyl prodrugs containing β-methyl cinnamide, dipeptide scaffolds Haic and Nle-cis-3,4-methanoproline, and glutamine surrogates were highly potent, completely inhibiting phosphorylation of Stat3 Tyr705 at 0.5–1 µM in a variety of cancer cell lines. The inhibitors were selective for Stat3 over Stat1, Stat5, Src, and p85 of PI3K, indicating ability to discriminate individual SH2 domains in intact cells. At concentrations that completely inhibited Stat3 phosphorylation, the prodrugs were not cytotoxic to a panel of tumor cells, thereby showing clear distinction between cytotoxicity and effects downstream of activated Stat3.
Introduction
Signal transducer and activator of transcription 3 (Stat3) is a member of the STAT family of transcription factors that transmits extracellular signals from receptors on the plasma membrane directly to the nucleus where it binds to various promoters and initiates gene transcription.1 In the canonical mechanism, when cytokines such as interleukin-6 (IL-6) or growth factors such as vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), or platelet-derived growth factor (PDGF) bind to their receptors, Stat3, via its Src homology 2 (SH2) domain, is recruited to phosphotyrosine residues on the receptor and becomes phosphorylated on Tyr705, either by JAK kinases, Src kinase or the kinase activity of the receptor. Phosphorylated Stat3 (pStat3) dimerizes via reciprocal pTyr705-SH2 domain interactions and is then translocated to the nucleus, where it initiates transcription of downstream genes. Introduction of antisense, dominant negative, and decoy oligonucleotides against Stat3 into tumor cells lines has been shown to reduce transcription of anti-apoptotic genes such as Bcl-2, Bcl-xL, Mcl-1, and survivin, cell cycle progression genes such as cyclin D1 and c-Myc, metastasis supporting genes including MMP-2,2, 3 and VEGF3, 4 and to result in apoptosis. Stat3 is constitutively activated (i.e. phosphorylated on Tyr705) in several cancer types, such as breast, lung, prostate, ovarian, leukemia, multiple myeloma, and others.5 Taken together, these findings support the hypothesis that phosphorylation of Tyr705 of Stat3 is a key event that contributes to increased survival and proliferation of cancer cells. Small molecule inhibitors targeted to the SH2 domain of Stat3 would be potential chemotherapeutic agents for the treatment of cancer by inhibiting receptor binding, Tyr705 phosphorylation, nuclear translocation, and transcriptional activity, resulting in decreased cell cycling and survival, and increased tumor cell death by apoptosis.5
Contrary to this hypothesis, two recent reports showed that JAK kinase inhibitors, P6 (1)6 and AZD1480 (2), at concentrations that completely eliminated Tyr705 phosphorylation, were not cytotoxic to a variety of cultured melanoma,7 breast, prostate, and pancreatic tumor cell lines.8 These results suggest that tumor cells grown in culture do not require pStat3 for survival and call into question the above hypotheses. Morevover, these studies suggest that if a compound were cytotoxic to cells grown in 2D cultures, it likely has off-target activities with respect to Stat3.8
Caveats must also be acknowledged concerning the biological activities of Stat3. Unphosphorylated Stat3 (U-Stat3) complexes with unphosphorylated NF-κB resulting in the transcription of κB-dependent genes.9 In non-transcriptional roles, Ser727-phosphorylated Stat3 has been found in electron transport complexes in mitochondria10 and in this capacity supports the growth of Ras transformed cells by sustaining glycolytic and oxidative phosphorylation.11 Thus the reported cytotoxicity and alterations in gene transcription ensuing from Stat3 knockdown and dominant-negative overexpression may, in part, be due to mechanisms not related to pTyr705-driven transcription. Therefore, highly potent and selective inhibitors of Stat3 phosphorylation are needed to understand the requirements of Tyr705 phosphorylation in cancer cell growth.
The SH2 domain of Stat3 has been targeted in several laboratories by a variety of phosphopeptides,12–16 peptidomimetics,17–12 and small molecules.23–25 We are targeting the SH2 domain of Stat3 with inhibitors based on our lead peptide, Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2.26–31 We recently reported the conversion of a conformationally constrained version of the lead peptide29 to a cell-permeable, phosphatase-stable peptidomimetic, BP-PM6 (3, Chart 1), that completely inhibited constitutive phosphorylation of Stat3 Tyr705 (pStat3) in MDA-MB-468 breast cancer cells at a concentration of 10 µM.32 The X-ray structure33 and molecular models of peptides bound to the SH2 domain29, 34 suggest that a methyl group on the β-carbon of phosphotyrosine or a suitable mimic might increase affinity due to increased hydrophobic interaction. In this communication we demonstrate that a β-methyl group on the phosphocinnamate pTyr mimic enhances affinity for Stat3. This modification as well as recently described glutamine analogues30 were incorporated into a series of peptidomimetic prodrugs that displayed >10-fold enhanced potency over 3, inhibiting pStat3 at concentrations of 0.1–0.5 µM. We show that these prodrugs are selective for the SH2 domain of Stat3 over those of Stat1, Stat5, Src, and the p85 regulatory domain of the phosphatidylinositol-3-kinase in intact cells. There was no effect on p38MAPK or S473Akt phosphorylation. However, as reported for the JAK inhibitors,7, 8 they are not cytotoxic to a panel of tumor cells in 2D culture on plastic plates at concentrations that inhibit Stat3 phosphorylation.
Chart 1.

Names for some of the prodrugs are given after their compound number
Results
Chemistry
Phosphopeptide inhibitors were synthesized using a convergent strategy. Amino acid sequences were assembled by manual solid phase synthesis on Rink amide resin by first coupling Fmoc-Glu-NHBn27 or modified Fmoc glutamic acids30 via the side chain. After addition of the remaining amino acids, peptidomimetics 4a, 5a, 6a, and 7a (Figure 1) were prepared by capping with 4-(di-tert-butoxyphosphoryloxy)-cinnamic acid.29 Inhibitors 4b, 5b, 6b, 7b, and 8–19 (Table 1) were capped with pentachlorophenyl (2E)-4-phosphoryloxyphenylbutenoic acid (25, Scheme 1). Peptides and mimetics were cleaved and purified by reverse phase HPLC.
Figure 1.

Left. Model of pYLPQ-NHBn34 docked to the SH2 domain of Stat3 showing space between inhibitor and Glu638. Surface electrostatic potential was calculated with APBS tools in PyMol.65 Right. Incorporation of methyl group on the β-position of 4-phosphoryloxycinnamide enhances affinity for Stat3, as judged by a fluorescence polarization assay. 4a and 7a were from reference 29. 5a was from reference 30.
Table 1.
Effect of substitution of Gln-NHBn with glutamine mimics on peptidomimetics 3b, 4b, 5b, and 6b.a
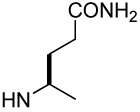
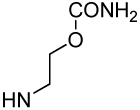
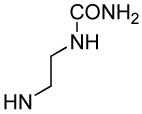
| Xxx |
||||
|---|---|---|---|---|
 |
 |
 |
||
| Compound | βMpCinn-Leu-Pro-Xxx | 8 | 9 | 10 |
| KI (nM) | 144 ± 9 | 203 ± 32 | 94 ± 7 | |
| Compound | βMpCinn-Leu-mPro- Xxx |
11 | 12 | 13 |
| KI (nM) | 83 ± 10 | 193 ± 38 | 39 ± 10 | |
| Compound | βMpCinn-Nle-mPro-Xxx | 14 | 15 | 16 |
| KI (nM) | 66 ± 13 | 114±14 | 46 ± 6 | |
| Compound | PMpCinn-Haic-Xxx | 17 | 18 | 19 |
| KI (nM) | 105 ± 22 | 188 ± 13 | 386 ± 41 | |
KI values were determined by fluorescence polarization.27
Scheme 1.

Reagents and condition: (i) Diethylchlorophosphate, TEA, CH2Cl2, 0°C to r.t. overnight; (ii) LitOBu, tBuOH, (EtO)2POCH2CO2tBu, r.t. overnight ; (iii) TFA/CH2Cl2 (95:5) 1h; (iv) C6Cl5OH, DCC, DMAP (cat.) EtOAc, r.t. 24 h; (v) TMSI, BSTFA, CH2Cl2, 0°C 1h then r.t. 1 h.
Synthesis of the phosphotyrosine mimic, (2E)-4-phosphoryloxyphenylbutenoic acid
The phenolic hydroxyl group of 4-hydroxyacetophenone (20) was phosphorylated with diethylchlorophosphate at the beginning of the synthesis to install the phosphate. The modified acetophenone (21) was elaborated by Horner-Emmons vinylogation with tert-butyl (diethylphosphono)acetate. The use of EtOH as a solvent resulted in 100% stereoselectivity for the trans isomer. Unfortunately, transesterification of the carboxyl group to an ethyl ester occurred and selective cleavage of the carboxy ester could not be achieved as cleavage of one or more ethyl groups on the phosphate was observed. However, the use of tert-butanol as the solvent avoided the side reaction. The stereoselectivity was not as high as with ethanol and resulted in approximately 25% of the cis isomer, which could readily be separated using silica gel chromatography. The resulting tert-butyl ester (22) was cleaved with TFA to give 23 which was esterified with pentachlorophenol (24). Removal of the ethyl groups with trimethylsilyl iodide (TMSI) gave the phosphate 25 ready for coupling to amino acid sequences.
Synthesis of prodrugs
To inhibit Stat3 in intact cells, we employed the same prodrug strategy as with 3 (Chart 1).32 The phosphate group of β-methyl cinnamate was substituted with the isosteric difluoromethylphosphonate (F2Pm) group to render inhibitors stable to phosphatases.32, 35 The negatively charged oxygen atoms on the F2Pm group were capped with carboxyesterase-labile pivaloyloxymethyl (POM)36 groups to facilitate cell penetration. The active ester bis-POM building block approach32 was used to assemble the prodrugs. Starting from iodoacetophenone (26), Horner-Emmons coupling with tert-butyl (diethylphosphono)acetate gave the iodocinnamate, 27 (Scheme II). As in the case of 22, t-BuOH was used as the solvent and the cis and trans isomers were separated by silica gel chromatography. Copper-cadmium cross coupling with diethyl bromodifluoromethylphosphonate37 provided phosphonate 28. Acidolytic removal of the tert-butyl ester followed by esterification with pentachlorophenol gave intermediate 29a. Trimethylsilyl iodide treatment removed the phosphonate ethyl groups resulting in phosphonic acid 30a. The phosphonate was neutralized with two equivalents of NaOH and the sodium counterions were exchanged with silver. The silver salt was alkylated with two equivalents of pivaloyloxymethyl iodide in toluene to give prodrug building block 31a. 4-Nitrophenyl esters were also synthesized using identical reaction schemes (29b, 30b, 31b).
Scheme 2.

Synthesis of bis-POM-protected difluoromethylphosphonocinnamates for incorporation into prodrugs.
Reagents and conditions (i) (EtO)2POCH2CO2tBu; (ii) BrCdCF2PO3Et2, CuCl; (iii) (a) TFA, (b) C6Cl5OH, DCC; (iv) TMS-I; (v) (a) NaOH, (b) AgNO3, (c) POM-I.
Prodrugs were formed by solution phase coupling of 31a or 31b to Haic-XXX, or Nle-mPro-XXX intermediates, which were synthesized on Rink resin and were purified by reverse phase HPLC before use. Acylation of dipeptides was accomplished with catalytic amounts of dimethylaminopyridine under anhydrous conditions. Prodrugs were purified by RP-HPLC using gradients of MeCN in water with no TFA or other additives. All prodrugs were >98% pure by reverse phase HPLC and gave the correct mass by high resolution mass spectrometry.
Unprotected difluoromethylphosphonates were prepared by acylation of amino acid sequences by intermediate 30a on solid supports, followed by cleavage with TFA and HPLC purification. To prepare mono-POM-protected prodrugs, 31a was coupled to presynthesized peptides in solution using DMF and HOBt hydrate, conditions that result in premature hydrolysis of one of the POM groups.32 HPLC purification yielded both mono and bis-POM prodrugs. To prepare 40 (Chart 1), the diethyl ester analogue of 34, 29a was coupled to resin-bound Haic-Apa (Apa = (R)-4-aminopentamide) followed by TFA cleavage and HPLC purification.
Inclusion of a methyl group on the β-position of a pTyr mimic increases affinity
Examination of the original crystal structure of Stat333 and molecular models developed by us29, 34 showed that there was space between the β-carbon of phosphotyrosine or pCinn and the side chain methylene groups of Glu638 that could be filled to increase hydrophobic interaction between the inhibitor and protein (Figure 1). Addition of a methyl group to the β-position of phosphocinnamate resulted in 1.5–3 fold increases in affinity in a series of phosphopeptide mimetics, as judged by a fluorescence polarization assay (Figure 1).27 Note that commercially available 3,4-cis-methanoproline is sold as a mixture of enantiomers and peptides incorporating them can be separated into the individual diastereomers, one of which exhibits higher affinity than the other.27 The results presented for 5a, 5b, 6a, and 6b in Figure 1 are from the more active stereoisomers.
Unfortunately, we have not been able to obtain a crystal structure of Stat3 complexed with any of the β-methylcinnamide-containing inhibitors to determine the nature of the increase in affinity. To gain an understanding of the effect of β-methyl substitution on the conformation of the cinnamate, we determined the crystal structure of a model compound, 4-iodo-β-methylcinnamoyl-leucine tert-butyl ester (manuscript in preparation). In this structure the aromatic ring deviates 27–30 degrees from the plane of the α-β double bond to avoid steric clash with the β-methyl group (Figure S1). The cinnamide carbonyl oxygen is on the same side of the C-Cα bond as the double bond, which was observed in the crystal structure of several cinnamides.38, 39 The double bond is slightly distorted because of collision between the carbonyl oxygen and the β-methyl group. This conformation was supported by 1H ROESY NMR spectroscopy of the dipeptide mimic in DMSO-d6 in which there was a strong cross peak between the Leu NH and the α-proton of the cinnamate. The corresponding cross peak was observed in ROESY spectra of the non-POM version of prodrug 33 (Chart 1), βMF2PmCinn-Haic-Gln-NHBn, as well as 3, F2PmCinn-Haic-Gln-NHBn,32 possessing no methyl group on the β-position of the cinnamate. It is uncertain if the increases in affinity of the β-methylcinnamide-possessing inhibitors are the result of the extra hydrophobic interaction between the β-methyl group and Glu638, a more favorable conformation of the aromatic ring, or both.
Modifications of Glutamine
Glutamine at pY+3 is an essential part of the recognition determinant for Stat3.31, 40, 41 To further reduce the peptide nature of our inhibitors, we replaced the C-terminal Gln-NHBn groups of compounds 4b, 5b, 6b, and 7b with glutamine mimics Apa, 2-aminoethyl carbamate (Aec), and 2-aminoethyl urea (Aeu), recently reported by our laboratory (Table 1).30 Overall, the Gln mimics were 1.2–4 fold less avid than the Gln-NHBn leads. In cases of proline or methanoproline, the urea analogues (10, 13, and 16) were the highest affinity inhibitors, displaying KI values of 39–94 nM. The (R)-4-aminopentamides (8, 11, and 13) were approximately 2 fold less potent than the corresponding Gln-NHBn-containing inhibitors and the carbamates (9, 11, and 15) were the least tolerated. The pattern with the Haic-containing compounds was slightly different. The urea, 19, showed the least affinity and the amino pentanamide, 17, the greatest.
Effect of the β-methyl group and C-terminal substitution on the inhibition of constitutive Stat3 phosphorylation in intact breast tumor cells
To determine the effect of the β-methyl group on the inhibition of Stat3 phosphorylation in intact cells prodrug 3 was compared with 32, possessing a methyl group on the β-position of the cinnamate (Chart 1 and Figure 2). The MDA-MB-468 breast cancer cell line was utilized as these cells possess constitutively active Stat3.42 Cells were treated with compounds for two hours and total and Tyr705-phosphorylated Stat3 levels were estimated by Western blotting of cell lysates (Figure 2, column A). Both compounds reduced the level of pStat3 in a dose-dependent manner, suggesting that the prodrugs enter cells, are stripped of the POM groups, and the resulting phosphonates bind to the SH2 domain of Stat3 resulting in (1) breakup of preformed dimers followed by de-phosphorylation and/or (2) prevention of binding to growth factor or cytokine receptors and the subsequent phosphorylation of Tyr705. Addition of the β-methyl group provided a slight but detectable enhancement in the inhibition of the phosphorylation of Stat3.
Figure 2.

Inhibition of the constitutive phosphorylation of Tyr705 of Stat3 in MDA-MB-468 breast tumor cells. Column A. Effect of substitution on the cinnamide β-position and the C-terminus. Column B. Inhibition of Nle-mPro-based prodrugs. Column C. Time course of inhibition. Prodrug concentrations were 5 µM. Column D. Effect of zero, one, or two POM groups on inhibition. Prodrug concentrations were 5 µM. Cells were treated with the indicated concentrations for two h (A, B, and D) or for the indicated time intervals C. After cell lysis pStat3 and total Stat3 were determined by western blots. For each pair of gels the top is pStat3 and the lower is total Stat3.
Even though the C-terminal methyl group resulted in 2-fold lower affinity than the benzyl amide in the phosphate series (17 and 7b, respectively, Table 1 and Figure 1) the potencies of prodrugs incorporating the simpler structure were substantially enhanced (34 and 33, Figure 2A). There was noticeable inhibition of pStat3 at 0.1 µM and the signal was completely gone at 0.5 µM. It is unclear whether this is due to reduced cell penetration, increased clearance, or degradation of the benzylamide-containing compounds.
Additional prodrugs incorporating the Nle-mPro and Haic scaffolds and glutamine mimics in Table 1 were synthesized. For the first group, compounds 6b, 14, 15 and 16 were converted to their corresponding prodrugs. In a previous study from our laboratory30 it was discovered that the glutamine surrogate, (4R,5S)-4-amino-5-benzyloxyhexanamide, in addition to being isosteric to Gln-NHBn, was equipotent in the context of the pCinn-Leu-mPro (but not pCinn-Haic). Therefore this mimic was included in the Nle-mPro prodrug series. In addition to the Haic containing prodrugs 32–34, compounds incorporating the urea and carbamate groups of 18 and 19 in the Haic series were converted to their corresponding prodrugs. This series was screened for the ability to inhibit constitutive phosphorylation of Stat3 in BT20 breast tumor cells. From this series, 35 and 37 stood out as having considerable potency (Chart 1). This pair of prodrugs, possessing the Nle-cis-3,4-methanoproline dipeptide scaffold, was also highly potent and completely inhibited pStat3 at 0.5 µM (Figure 2, column B).
During the synthesis of 35 and 37, diastereomers possessing the opposite enantiomers of cis-3,4-methanoproline, 36 and 38, respectively, were isolated during the HPLC runs and were tested for their abilities to inhibit Stat3 phosphorylation in MDA-MB-468 breast cancer cells (Figure 2, column B). In each case, the first eluting isomers from HPLC purification runs were very potent inhibitors. Inhibition of pStat3 was evident at 10 nM and was nearly complete at 100 nM. As expected, the second stereoisomers were very poor inhibitors: the intensity of the band at 25 µM was only partially reduced. This corresponds to the reduced affinity for isolated Stat3 measured for the diastereomeric mPro containing peptidomimetics.27 Compound 35 has a benzyloxyethyl group at the α-position of the glutamine surrogate, whereas 37 possesses a methyl group. The high potency of the latter as well as 33 and 34 suggests that in intact cells, C-terminal benzyl appendages are not necessary for efficient inhibition of Stat3 phosphorylation.
Compound 16, possessing 2-aminomethylurea in place of glutamine, was a very high affinity inhibitor in the fluorescence polarization assay (KI = 49 nM). In the case of the corresponding prodrug, 39, we were unable to separate the diastereomers so the compound was tested as a mixture of stereoisomers. In spite of the high affinity of the parent compound, there was little inhibition of the phosphorylation of Stat3 in the breast tumor cells up to 1 µM (Figure 2, column B). At 5 µM complete inhibition was observed. The alkyl carboxamides impart greater cellular potency than the ethyl urea.
The time course of inhibition of constitutive phosphorylation of Stat3 in MDA-MB-468 cells is shown in Figure 2, column C. A single dose of 5 µM of the high affinity prodrugs 34, 35, and 37 completely inhibited pStat3 formation at 30 min and the effect was sustained for 4h. Partial recovery was evident at 8h and recovery was complete at 16h.
Inhibition of pStat3 nuclear translocation
MDA-MB-468 cells were treated with 34 for 2 h and were then stained with fluorescent antibodies for pTyr705 Stat3 (Figure 3). In vehicle-treated controls, pStat3 had a strong presence in the nucleus. Treatment with the prodrug not only greatly reduced the level of pStat3, but also abrogated nuclear localization.
Figure 3.

Fluorescence immunohistochemical monitoring of the unhibition of Stat3 phosphorylation by compound 34 in MDA-MB-468 breast tumor cells. Cells were treated with 5 µM 34 for 2 h, fixed, permeabilized, stained with anti pTyr705 Stat3 antibodies, and examined with confocal microscopy. All photos were taken with the same microscope settings. (NOTE TO EDITOR- This figure can be reduced to single column size to save space)
Two POM groups are required for inhibition of Stat3
Analogs of 34, 35, and 37 possessing zero and one POM group were assayed for their ability to inhibit Stat3 phosphorylation in MDA-MB-468 cells. At concentrations of 5 µM and two hour treatment the unprotected (NP) and mono-POM (MP) esters did not inhibit pStat3 formation, whereas the bis-POM (BP) prodrugs did (Figure 2, Column D). Thus, consistent with the first generation prodrug, 3,32 two POM groups are required for efficient cell penetration and inhibition of Stat3 phosphorylation.
Selectivity for Stat3
Ladbury et al. argued that selective disruption of individual signaling pathways with phosphopeptide mimics in intact cells may be difficult to achieve because the differences in measured affinities of phosphopeptides for the SH2 domains of different proteins is relatively small.43, 44 To study the selectivity of our prodrugs, we assayed for the inhibition of the phosphorylation of Tyr701 of Stat1, Tyr694 of Stat5, Ser473 of Akt, and Tyr861 of the focal adhesion kinase (FAK), which are all mediated directly or indirectly by SH2 domains binding to pTyr residues on proteins. MDA-MB-468 cells were treated with 5 µM 34, 35, and 37 for 1.5 h and were then stimulated with EGF. After 30 min the cells were lysed and pStat3, phosphoTyr694 Stat5, and phosphoSer473 Akt levels were analyzed by Western blots (Figure 4A). The prodrugs completely inhibited the increase in Stat3 phosphorylation induced by EGF. Like Stat3, Stat5 binds to pTyr residues on receptors via its SH2 domains and becomes phosphorylated on Tyr694. Stat5 phosphorylation was not inhibited by our prodrugs. Phosphatidylinositol-3-kinase (PI3K) is recruited to EGFR via the SH2 domains of p85, the regulatory subunit, which activates the kinase domain resulting in the phosphorylation of phosphatidylinositol-2,4-diphosphate on the 3-position. Phosphatidylinositol-2,3,4-triphosphate recruits both phosphatidylinositol-dependant kinase (PDK) and Akt via their plekstrin homology domains. Akt is then phosphorylated on Ser473 by PDK. PI3K is constitutively activated in MDA-MB-468 cells due to loss of PTEN and ATP competitive inhibitors of this enzyme have been shown to reduce phosphorylation of Akt.45, 46 The fact that our prodrugs do not inhibit Akt phosphorylation suggests that they do not bind to the SH2 domains of p85 and prevent downstream signaling of PI3K. Via its SH2 domain, Src kinase binds to FAK and phosphorylates the unique substrate, Tyr861.47 MDA-MB-468 cells express constitutive phosphorylation of Tyr861 of FAK48 and levels of Tyr861 phosphorylation have been shown to decrease on treatment of tumor cells with the Src inhibitor, dasatinib.49, 50 After 2 h treatment with the prodrugs no reduction of Tyr861 phosphorylation was observed (Figure 4B). Therefore we conclude that our prodrugs do not bind to the SH2 domain of Src. To test for effects on Stat1, cells were treated with increasing concentrations of the prodrugs for 1.5 h followed by 30 min stimulation with interferon γ (IFNγ). Tyr701 phosphorylation of Stat1 was determined by Western blotting. There was a dose dependent inhibition of Stat1 phosphorylation with complete inhibition at 5 µM, ca 10-fold higher than that required for Stat3 (Figure 4C). In HCC-827 NSCLC cells, 34 had no effect on the phosphorylation of p38 MAPK and Ser473 of Akt.
Figure 4.

Selectivity of Stat3 inhibitors. A. MDA-MB-468 cells were treated with prodrugs for 1.5 h and were stimulated with EGF. After 30 min cells were lysed and levels of pStat3, pSer473 Akt, and pTyr699 Stat5 were estimated by western blots. B. Inhibiton of constitutive phosphorylation of FAK Y861. C. Inhibition of IFN-γ stimulated phsphorylation of Stat1. D and E. Effect of 34 (5 µM) on downstream genes in HCC-827 NSCLC cells.
No inhibition of the expression of the canonical downstream genes, cyclin D1 and Bcl-xL, was observed in MDA-MB-468 cells on treatment with 5 µM of 34 (data not shown). Cyclin D1 was not inhibited in HCC-827 cells. However, survivin was reduced in the lung line (Figure 4E) and the breast line (data not shown).
Stat3 phosphorylation is inhibited in other tumor cell lines
A panel of cell lines was tested for the inhibition of Stat3 phosphorylation by 34 (Figure 5). Melanoma lines MeWo and A375 and the ovarian cancer line HEY have no or very little basal pStat3 levels. However, these cell lines are very responsive to IL-6, which induces high levels of Stat3 phosphorylation on Tyr705. After 1.5 hour exposure to prodrugs, cells were stimulated with IL-6. As shown in Figure 5, 34 inhibited pStat3 formation but slightly higher concentrations were required to completely abrogate phosphorylation. The constitutively activated Stat3 in SKOV3ip ovarian cancer cells was completely inhibited at 1 µM. At 5 µM, 34 inhibited pStat3 in 10 min in HCC-827 non-small-cell lung cancer cells (Figure 4D). The effect lasted for at least 4 h at at 24 h pStat3 had not returned to pretreatment levels (Figure 4E).
Figure 5.

Inhibition of Stat3 phosphorylation by prodrug 34 in ovarian cancer (SKOV3-ip, Hey) and melanoma (MeWo and A375) cell lines. For each pair of blots, the upper is pStat3 and the lower is Stat3.
Prodrugs are weakly cytotoxic to cultured cell lines
Compound 34 and the diastereomeric pairs 35, 36 and 37, 38 were assayed for cytotoxicity to MDA-MB-468 cells using the MTT assay at 72 h. As shown in Figure 6A, 34 and diastereoismeric pair 37 and 38 exhibited IC50 values of ca 30 µM. Prodrug 35 was more potent, with an IC50 value of 10–15 µM. Interestingly, 36, containing the opposite stereoisomer of mPro that does not inhibit pStat3 formation until 25 µM, also inhibited growth at 10–15 µM. Because both diastereoisomers inhibited growth at equal concentrations, and 34, 37, and 38 were not inhibitory until 30 µM, we can not conclude that the observed cytotoxicity of 35 was mediated through its effects on Stat3 inhibition.
Figure 6.

Effect of prodrugs on the survival of cultured cancer cell lines. A. MDA-MB-468 cells were treated with a single dose of the indicated compounds for 72 h. B. MDA-MB-468 cells were treated daily with 34 and 35 for three days. Cell viability in panels A and B was determined by MTT assays. C. Cancer cell lines were treated daily with 34 for three days. D. Cancer cell lines were treated daily with 40 for three days. Cell viability in panels C and D was determined by SRB assays.
Knowing that pStat3 levels recover after 8 h, the experiment was repeated with daily dosing of 34 and 35 (Figure 6B). There was little change in the survival curves. Similar studies were conducted with daily treatment of MCF-7 breast cancer cells, which do not harbor constitutively phosphorylated Stat3, and on SKOV3-ip ovarian cancer cells and HCC-827 lung cancer cells, both of which have constitutively phosphorylated Stat3 (Figure 6C). In all of these lines, 34 elicited very weak cytotoxicities, with IC50 values >30 µM. The most sensitive cell line was MDA-MB-468, and intermediate sensitivity was observed in HCC-827 cells. Both MCF7 and SKVO3-ip cells were equally insensitive. Thus no strong correlation between cytotoxicity and constitutive Stat3 phosphorylation was observed. Note that the concentrations of prodrugs in these experiments are much higher than those required to completely inhibit the phosphorylation of Tyr705 of Stat3. Additional cancer cell lines harboring constitutive Stat3 phosphorylation, melanoma cells MeWO and A375, and NSCLC cells H1299, H1819, H520 H528, and A549, all showed <20% inhibition at 5–10 µM of 34 and 35 (data not shown), concentrations that completely abrogate pStat3 levels.
To assess the effect of the phosphonate group on cytotoxicity, compound 40, which retained diethyl protection on the phosphonate oxygens, was examined (Figure 6D). Trialkylphosphates and dialkylphosphonates are known to be biologically stable51 and indeed at 25 µM, the highest concentration examined, this compound had no effect on Stat3 phosphorylation in MDA-MB-468 cells (data not shown). Growth inhibition was not apparent until well above 50 µM. These results suggest that the observed cytotoxicities of 34 were not due to the β-methylcinnamate, Haic, or 4-aminopentamide moieties, but rather to the phosphonate group.
Discussion
In this report we show that peptidomimetic phosphopeptide prodrugs targeting the SH2 domain of Stat3 can potently inhibit the phosphorylation of Stat3 in intact tumor cells. Compounds 34, 35, and 37 are some of the highest potency SH2 domain-targeted compounds reported to date, as regards to inhibiting their target. The β-methyl group on the cinnamide-based pTyr mimic resulted in 2–3 fold increases in affinity, and slight enhancement for inhibition of Stat3 phosphorylation in intact cells. Two central dipeptide scaffolds, Haic, and Nle-mPro, were evaluated and found to behave identically in potency for Stat3 inhibition in intact breast tumor cells. The C-terminus of the peptide was quite important. Even though the methyl group resulted in lower affinity than the benzylcarbamoyl group for the isolated protein (Table 1), the former resulted in much greater potency in intact cells. The C-terminal ethyl benzyl ether of 35 likely produces off-target cytotoxicity, since 36 exhibited the same degree of growth inhibition but it was 20–25 fold less potent at inhibiting Stat3 phosphorylation. In addition, in intact cells, incorporation of the glutamine mimic, 4-aminopentamide, into either of the Haic or Nle-mPro scaffolds, resulted in higher potency inhibition of Stat3 phosphorylation than 2-aminoethyl urea and 2-aminoethylcarbamate, two surrogates that increased affinity for Stat3 protein. Two POM esters are required for efficient inhibition of Stat3 phosphorylation. This is consistent with observations that negatively charged compounds are not cell permeable.
Selectivity of inhibitors for SH2 domains in intact cells has not received much attention presumably because there have not been many reported cell-permeable antagonists of these domains. Our prodrugs were selective for the SH2 domain of Stat3 in breast tumor cells at ten times the concentration that completely inhibited Stat3 phosphorylation. The fact that the prodrugs do not inhibit PI3K and Src function is not surprising, since the SH2 domains of these proteins accommodate the hydrophobic amino acids Met and Ile and their analogs at position pY+3, respectively.52, 53 At this position, our inhibitors have hydrophilic glutamine mimics which would not bind in the hydrophobic pockets of p85 and Src. The 3° structures of the SH2 domains of Stat333 and Stat554 are remarkably similar.34 However, their amino acid sequences are dissimilar in the peptide binding regions which would account for the difference in binding. It has been observed that the IL-6 response includes weak and transient activation of Stat1 (reviewed by Regis et al.64). Reciprocally, IFNγ promotes weak stimulation of Stat3. Indeed Gerhartz et al. showed that Stat1 could be recruited to pTyr-Xxx-Pro-Gln sequences on the IL-6 co-receptor, gp130, centered on Tyr905 and Tyr915.55 Our peptidomimetics are derived from the former binding site. The SH2 domains of Stat1 and Stat3 are highly similar both in sequence and in 3° structure.34 Therefore, cross-reactivity for these two proteins both by biological stimulation and by our peptidomimetics is not surprising. However, since these Stats are activated by different cytokines and growth factors, it remains to be seen if the reduced inhibition of Stat1 is significant. Although this is not an exhaustive survey of SH2 domains the results are very encouraging. Selectivity for Stat3 over Stat1 and Stat5 can not be achieved by inhibitors of the JAK kinases. Thus our compounds are the most selective inhibitors of Stat3 phosphorylation reported to date.
The lack of cytotoxicity of our prodrugs and as well as small molecule, ATP-competitive JAK2 inhibitors7, 8, at concentrations that completely inhibit Tyr705 phosphorylation, runs counter to the hypothesis that pStat3 is essential for tumor cell growth and that inhibition of Stat3 results in apoptosis in cultured cells.5, 56–58 It is possible that knockdown of total Stat3 with antisense or siRNA abrogates the recently reported mitochondrial function of this protein10, 11 and co-transcriptional activity of U-Stat3 which may contribute to the apoptosis reported by these treatments. Our results and those of Kreiss et al.7 and Hedvat et al.8 suggest that survival assays of cancer cell lines grown in two-dimensional cell cultures on plastic dishes are not effective models of the efficacy of Stat3 inhibitors.
At concentrations greater than 25 µM, ∼50-fold that which inhibited Stat3 phosphorylation, the prodrugs did indeed exhibit cytoxicity. The non-hydrolyzable bis ethyl ester indicated that growth inhibition was due to the phosphonate group. At 25 µM, two hour treatment with 34 inhibited phosphorylation of FAK, Akt, and Stat5 in MDA-MB-468 cells indicating off-target effects (Figure S2). It is unknown whether other pathways are perturbed by high concentrations of our phosphopeptide mimetics. It appears that at the high concentrations of the bis-POM prodrugs that result in cytotoxicity, the phosphonate group is reacting non-selectively with other SH2 domains and possibly phosphotyrosine binding domains or the active sites of protein tyrosine phosphatases which might impact survival, even of cells such as MCF-7 which do not harbor constitutively phosphorylated Stat3.
At 5 µM, ∼10-fold the concentration at which pStat3 is inhibited, the prodrugs reported here did not reduce the expression of the canonical downstream genes Bcl-xl in the MDA-MB-468 breast cancer line or cyclin D1 in this or the NSCLC line, HCC-827. Other transcription factors and pathways, for example NF-κB59 and PI3K/Akt,60, 61 are involved in the expression of these genes. We conclude that inhibition of Stat3 phosphorylation alone is not sufficient to inhibit downstream gene expression and that disruption of more than one transcripition factor may be required. At high concentration of prodrug, the observed off-target inhibition of other pathways would make attributing reduction in the expression of canonical downstream genes solely to inhibition of Stat3 Tyr705 phosphorylation tenuous.
In summary, the availability of these highly potent and selective inhibitors of Stat3 phosphorylation have allowed dissection of pathways downstream of this key effector molecule from off-target, cytotoxic responses. Evaluation and development of 34 and analogues as potential anti-tumor agents in tumor xenograft and tumor microenvironment models is in progress, and will be reported under separate cover.
Methods
Nα-protected amino acids were purchased from NovaBiochem, ChemImpex, or Anaspec. HOBt was from ChemImpex. Anhydrous DMF for amino acid solutions was from Aldrich. Other solvents were reagent grade and were used without further purification. NMR spectra were obtained on either a Bruker DPX 300 MHz spectrometer or a Bruker DRX 500 MHz spectrometer. Fmoc-Glu-NHBn was prepared as described by Coleman et al.27 4-(di-tert-butoxyphosphoryloxy)-cinnamic acid was synthesized as described in Mandal et al.29 (R)-4-(9-fluorenylmethoxycarbonlyamino)-pentanoate, 4-nitrophenyl 2-(9-fluorenylmethoxycarbonlyamino)ethyl carbamate, 4-nitrophenyl 2-(9-fluorenyloxycarbonlyamino)ethylcarbonate, and (4R,5S)-4-(9-fluorenyloxycarbonlyamino)-5-benzyloxyhexanoate were prepared as described by Mandal et al.30 Racemic Fmoc-cis-3,4-methanoproline was purchased from EMD Biosciences (Novabiochem). Haic was synthesized as described in Mandal et al.29 Peptides were assayed for affinity to Stat3 using fluorescence polarization as described by Coleman et al.27 Stat3 was expressed and purified as described.62 For the synthesis of phosphopeptides, Rink resin with a loading of 0.6 mmol/gm was employed. For the synthesis of prodrugs, Rink resin with a loading of 1.2 mmol/gm was used. Resins were obtains from Advanced Chemtech, Inc. Antibodies used in the western blots are described in a table in the supporting information.
General Procedure for the synthesis of phosphopeptides and peptidomimetics, 4–19
Solid phase syntheses were carried out manually using commercially available Rink resin. Resin, 0.2 gm, was placed in a manual reactor and swollen and washed with 5 × 10 mL of DMF/CH2Cl2. Fmoc groups were removed with 3 × 6 mL of 20% piperidine/DMF for 5 min each. For coupling, three-fold excesses of Fmoc-amino acids, DIC, and HOBt were used in 8–10 mL of DMF/CH2Cl2 and were allowed to proceed until resin samples tested negative with ninhydrin tests. 4-Nitrophenyl 2-(9-fluorenylmethoxycarbonlyamino)ethyl carbamate and 4-nitrophenyl 2-(9-fluorenyloxycarbonlyamino)ethylcarbonate were coupled to Rink resin by addition of 3 eq plus 3 eq of DIEA in 8–10 mL of DMF/CH2Cl2 until ninhydrin tests were negative.28 For Fmoc-Haic, Fmoc-cis-3,4-methanoproline, and phosphorylated cinnamic acid derivatives, couplings were performed with 1.5–2 equivalents each of acid, DIC and HOBt in DMF/CH2Cl2 overnight or until ninhydrin tests were negative. After coupling and deprotection steps, resins were washed with 5 × 10 mL of DMF/CH2Cl2. On completion of the peptide chain, resins were washed with CH2Cl2 (3 × 10 mL) and were treated with TFA:TIS:H2O (95:2.5:2.5).63 (3 × 5 mL) for 15 min each. The combined filtrates sat at rt for 1–2 h and the volumes were reduced in vacuo. Peptides were precipitated in ice cold Et2O, collected by centrifugation, and washed 2 × more with the same solvent and centifiged. After drying, peptides were purified by reverse phase HPLC on a Rainin Rabbit HPLC or a Varian Dynamax HPLC using a Phenomenex Luna C18(2) 10 µM 2.1 × 25 cm column. Gradients of MeCN in H2O or MeCN in 0.01 M NH4OAc (pH 6.5) at 10–20 mL/min were employed. For phosphopeptides, solvents contained 0.1% TFA. For prodrugs, no TFA was used in the mobile phase. Peptides were tested for purity by reverse phase HPLC on a Hewlett Packard 1090 HPLC or an Agilent 1100 HPLC using a Phenomenex Luna C18(2) 5µM 4.6 × 250 mm column. A gradient of 0–40% MeCN/30 min was used for posphopeptides and peptide intermediates. For prodrugs the gradient was 10–80% MeCN/30 min. Phosphopeptides and prodrug intermediates were dried in vacuo over P2O5 at 37° for 24 h prior to use.27 All compounds were >95% pure (HPLC) before evaluation. Purities, yields, and mass spectral characteristics of phosphopeptides and prodrugs are provided in the supporting information.
Synthesis of 4-diethylphosphoryloxy acetophenone, 21
To an ice cold stirred solution of 4-hydroxyacetophenone (2.0 g, 14.7 mmol) and TEA (4.1 mL, 29.4 mmol) in 30 mL of CH2Cl2 under argon, diethylchlorophosphate (2.5 mL, 17.6 mmol) was added dropwise. The mixture was stirred overnight and was quenched by the addition of 30 mL of 5% aqueous HCl. The layers were separated and the aqueous phase was extracted with CH2Cl2 (2 × 30 mL). The combined organic layers were washed with brine and dried (MgSO4). The solvent was removed under vacuum and the crude product was purified by silica gel column chromatography eluting with EtOAc-hexanes. Yield 3.6 gm (90%). 1H NMR (CDCl3, 300 MHz) δ1.31–1.38 (m, 6H), 2.58 (s, 3H), 4.17–4.28 (m, 4H), 7.31 (d, J = 8.7 Hz, 2H), 7.97 (d, J = 8.7 Hz, 2H). 13C NMR (CDCl3, 75.0 MHz) δ 16.0, 26.5, 64.8, 82.5, 119.9, 130.3, 133.9, 154.4, 196.6. HRMS (M+H) calc, 273.0892; found, 273.0965.
Synthesis of tert-butyl 3-(4-diethylphosphoryloxyphenyl)butenoate, 22
nBuLi in hexane (4.0 mL of 2.5 M, 9.5 mmol) was added carefully to dry tBuOH (10 mL) via a syringe under argon atmosphere. After 30 min, a solution of tert-butyl diethylphosphonoacetate (2.00 g, 8 mmol) in 10 mL of dry tBuOH was added at room temperature and the solution was stirred for 1.0 h. A solution of 21 (2 g, 7.4 mmol) in 5 mL of tBuOH was added and the mixture stirred overnight at room temperature. The reaction was quenched with 30 mL of saturated NH4Cl (aq) and extracted with ether (4 × 40 mL). The combined organic extracts were washed with water (2 × 20 mL), brine (1 × 30 mL) and dried (MgSO4). The solvent was removed and the crude product was purified by silica gel column chromatography, eluting with 20% EtOAc-hexane (v/v), yielding 2.0 g (74%) of 22. 1H NMR (CDCl3, 300 MHz) δ1.32–1.37 (m, 6H), 1.51 (s, 9H), 2.50 (s, 3H), 4.16–4.27 (m, 4H), 6.0 (s, 1H), 7.2 (d, J = 8.7Hz, 2H), 7.43( d, J = 8.7Hz, 2H). 13C NMR (CDCl3, 75.0 MHz) δ 16.0,16.1, 17.6, 26.2, 28.3, 64.7, 64.83, 80.1, 115.3, 119.1, 119.8, 127.7, 130.7, 139.3, 151.0, 152.7, 161.8, 166.3, 196.9. Anal. Calc’d for C18H27O6P: C, 58.37; H, 7.35; O, 25.92; P, 8.36: Found C, 58.62; H, 7.33. HRMS (M+H) calc, 371.1624; found, 371.1558.
Synthesis of [2E] 3-(4-diethylphosphoryloxyphenyl) butenoic acid, 23
Compound 22 (1.0 g) was treated with 10 mL of TFA: CH2Cl2 (95:5) for 1.0 h. The solvents were removed in vacuo, and residual TFA was removed by the addition and evaporation of toluene (3 × 5 mL). Compound 23 was used without further purification. 1H NMR (CDCl3, 300 MHz) δ 1.34–1.39 (m, 6H), 2.51 (s, 3H), 6.13 (s, 1H), 7.22 (d, J = 8.7 Hz, 2H), 7.47 (d, J = 8.7Hz, 2H).
Synthesis of pentachlorophenyl (E) 3-(4-diethoxyphosphorylphenyl) but-2-enoate, 24
A solution of 23 (2.00g, 6.4mmol), pentachlorophenol (1.9g, 7.0mmol), DCC (1.6g, 7.7 mmol) and DMAP (0.08g, 0.64mmol) in 100 mL of ethyl acetate was stirred at room temperature for 24h. The mixture was filtered through celite and the solvent removed in vacuuo. The crude product was purified by silica gel chromatography eluting with 25% ethyl acetate-hexanes to give 2.6g (72%) of 24 as white solid. 1H NMR (CDCl3, 300 MHz) δ 1.26–1.32 (m, 6H), 2.54 (d, J = 1.2Hz, 3H), 4.12–4.21 (m, 4H), 6.34 (d, J = 1.2 Hz, 1H), 7.2 (d, J = 8.7 Hz, 2H), 7.49 (d, J = 8.7 Hz, 2H). 13C NMR (CDCl3, 75.0 MHz) δ 16.0, 16.1, 18.4, 64.8, 64.8, 68.9, 113.4, 120.2, 120.3, 128.0, 128.1, 131.2, 131.9, 137.8, 141.9, 144.3, 152.1, 160.6, 161.9. HRMS (M+H) calc, 560.9362; found, 560.7184.
Synthesis of pentachlorophenyl (E)-3-(4-phosphorylphenyl) but-2-enoate, 25
Iodotrimethylsilane (2.0 mL, 14.2 mmol) in 5 mL of dry CH2Cl2 was added dropwise to a solution of 24 (2.0 g, 3.55 mmol) and bis(trimethylsilyl)trifluoroacetamide (1.8 mL, 7.1 mmol) in 20 mL of dry CH2Cl2 at 0°C under argon. Stirring was continued for 1 h at 0°C and 1 h at room temperature. The solution was concentrated in vacuo. The residue was treated with 20 mL MeCN/H2O (9:1) and 5 drops of conc. HCl for 30 min and the solvents were removed in vacuo. Toluene (5 mL) was added and evaporated twice. On addition of Et2O solids separated, which were collected by filtration and washed with the same solvent to give 1.6 g of 25 as a white powder (88%) which was used without further purification. 1H NMR (DMSO-d6, 300 MHz) δ 2.6 (s, 3H), 6.6 (s, 1H), 7.24 (d, J = 8.7 Hz, 2H), 7.75 (d, J = 8.7 Hz, 2H). HRMS (M+H) calc, 503.8658; found, 503.5797.
Synthesis of βMpCinn-Leu-Pro-Apa, 8
Rink resin (0.2 g, 0.15 mmol) was swollen in DMF/CH2Cl2 (1:1) and was washed with 2 × 10 mL of the same solvent. The Fmoc group was removed by treatment with 20% piperidine in DMF (3 × 5 min). Coupling of 4-(9-fluorenylmethyloxycarbonylamino) pentanoic acid30 was accomplished with 3-fold excesses of amino acid, PyBop, HOBt and DIPEA in 10 mL of DMF/CH2Cl2 (1:1). For proline and leucine, three-fold excesses of Fmoc-amino acids, DIC, and HOBt were used. The final coupling was carried out with a two-fold excess of pentachlorophenyl-3-methyl-4-phosphoryloxycinnamate (25), triethylamine and HOBt in 10 mL of DMF/CH2Cl2. After completion of the synthesis, the resin was washed with 3 × 10 mL of DMF/CH2Cl2 followed by CH2Cl2 (3 × 10 mL). The resins was cleaved with three treatments of 10 mL of TFA:TIS:H2O (95:2.5:2.5) for 10 min each and the combined TFA solutions were removed in vacuum, stripped remainder TFA off by addition of toluene (2 × 5mL) and addition of ether resulted off-white precipitate. The solid was collected by centrifugation, dried, and purified by reverse phase HPLC to get 34 mg of 8. ES-MS (M+H) calc, 567.26; found, 567.25.
Synthesis of βMpCinn-Leu-Pro-AEC, 9
Rink resin (0.2 g, 0.15 mmol) was swollen in DMF/CH2Cl2 (1:1) and was washed with 2 × 10 mL of the same solvent. The Fmoc group was removed by treatment with 20% piperidine in DMF (3 × 5 min). Coupling of p-nitrophenyl 2-(9-fluorenylmethoxycarbonylamino)ethyl carbonate30 was accomplished with 3-fold excesses of carbonate, HOBt and DIPEA in 10 mL of DMF/CH2Cl2 (1:1). For coupling of Fmoc-proline and Fmoc-leucine, three-fold excesses of Fmoc-amino acids, DIC, and HOBt were used. The final coupling was carried out with two-fold excess of pentachlorophenyl-3-methyl-4-phosphoryloxycinnamate (25), triethylamine and HOBt in 10 mL of DMF/CH2Cl2. After all coupling, resins were washed with 3 × 10 mL of DMF/CH2Cl2 followed by CH2Cl2 (3 × 10 mL). Resins were cleaved with three treatments of 10 mL of TFA:TIS:H2O (95:2.5:2.5) for 10 min each and the combined filtrates were removed in vacuo, stripped of residual TFA by addition and evaporation of toluene (2 × 5mL) and addition of ether resulted off-white precipitate. The solid was collected by centrifugation, dried, and purified by reverse phase HPLC to get 22 mg of 9. HRMS (M+H) calc, 555.2220; found, 555.2186.
Synthesis of βMpCinn-Leu-Pro-AEU, 10
Rink resin (0.2 g, 0.15 mmol) was swollen in DMF/CH2Cl2 (1:1) and was washed with 2 × 10 ml of the same solvent. The Fmoc group was removed by treatment with 20% piperidine in DMF for 5 min (repeated 3 times). Coupling of p-nitrophenyl 2-(9-fluorenylmethoxycarbonyl) aminoethyl urea30 was accomplished with 3-fold excesses of urea, HOBt and DIPEA in 10 mL of DMF/CH2Cl2 (1:1). For coupling of Fmoc-proline and Fmoc-leucine, three-fold excesses of Fmoc-amino acids, DIC, and HOBt were used in 10 mL of DMF/CH2Cl2 (1:1). The final coupling was carried out with two fold excess of pentachlorophenyl-3-methyl-4’-phosphoryloxy cinnamate (23), triethylamine and HOBt in 10 mL of DMF/CH2Cl2. After all couplings resins were washed with 3 × 10 mL of DMF/CH2Cl2 followed by only CH2Cl2 (3 × 10 mL). Resins were cleaved with three treatments of 10 mL of TFA:TIS:H2O (95:2.5:2.5) for 10 min each and combined TFA solutions were removed in vacuum, stripped remainder TFA off by addition of toluene (2 × 5 mL) and addition of ether resulted off-white precipitate. The solid was collected by centrifugation, dried, and purified by reverse phase HPLC to get 26 mg of 10. HRMS (M+H) calc, 555.2186; found, 554.2402.
Synthesis of tert-butyl (E) 3-(4-iodophenyl)but-2-enoate, 27
A solution of tert-butyl diethylphosphonoacetate (10.0 g, 39.6 mmol) in 30 mL of dry THF was added slowly to a freshly prepared solution of 19 mL of 2.5 M (hexanes) lithium tert-butoxide and tBuOH (30 mL) and stirred for 1h. A solution of 4-iodoacetophenone (26) (9.0 g, 36.6 mmol) in 20 mL of dry THF was added to the reaction mixture and stirring was continued overnight. The solvents were removed under vacuum. The residue was dissolved in 400 mL of ether and was washed with water (2 × 30 mL) followed by brine (30 mL) and dried over MgSO4. After filtration and evaporation of the solvent, the crude product was then purified by silica gel column chromatography, eluting with 1% EtOAc in hexane. The product 27 was obtained as an oil (10.0 g, 79% yield). 1H NMR (CDCl3, 300 MHz) δ 1.515 (s, 9H), 2.48 (s, 3H), 6.03 (s, 1H), 7.18 (d, 2H, J =8.4 Hz), 7.68 (d, 2H, J = 8.4 Hz). 13C NMR (CDCl3, 75.0 MHz) δ 17.5, 28.3, 80.2, 94.6, 119.5, 128.0, 137.6, 141.9, 152.6, 166.1. Anal. Calcd. for C14H17IO2: C, 48.85; H, 4.98; I, 36.87; O, 9.3 : Found C, 49.40; H, 5.00, I, 35.97. HRMS (M+H) calc, 345.0351; found, 345.0319.
Synthesis of tert-butyl (2E) 3-[4-[(diethoxyphosphinyl)difluoromethyl]phenyl]-but-2-enoate, 28
To a solution of diethyl bromodifluoromethylphosphonate (6.45 g, 24.1 mmol) in dry DMF (100 mL), cadmium powder (5.41 g, 48.2 mmol) was added. The suspension was stirred for 8 h under argon. Unreacted cadmium was removed by filtration under argon and the filtrate was treated with CuCl (2.86 g, 28.9 mmol) and 27 (5.00 g, 15.1 mmol) at room temperature for 24h. Et2O, 400 mL, was added and the mixture was stirred for 5 min and filtered. The organic layer was washed with saturated NH4Cl (2 × 40 mL) and water (4 × 40 mL), dried (MgSO4) and evaporated to give an oily residue. The crude product was purified by silica gel column chromatography with 40% EtOAc-hexane (v/v) as the eluent to give 4.0 g (68%) of 28 as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ1.3–1.35 (m, 6H), 1.52 (s, 9H), 2.54 (s, 3H), 4.13–4.28 (m, 4H), 6.07 (s, 1H) 7.53 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H). 13C NMR (CDCl3, 75.0 MHz) δ 16.3, 16.4, 17.7, 28.3, 64.8, 64.9, 80.3, 120.4, 126.4, 145.0, 152.7, 166.0. 19F NMR (CDCl3, 282.0 MHz) δ −108.57 (d, J = 115.6 Hz, 2F). 31P NMR (CDCl3, 202.0 MHz) δ 6.15 (t, J = 115.1 Hz, 1P). Anal.Calcd for C19H27F2O5P: C, 56.43; H, 6.73; F, 9.4; O, 19.78; P, 7.66: Found C, 56.13; H, 6.73; F, 9.15. HRMS (M+H) calc, 405.1642; found, 405.1698.
Synthesis of pentachlorophenyl (2E)-3-[4-[(diethoxyphosphinyl)difluoromethyl]phenyl]-but-2-enoate, 29a
A solution of 28 (4.00 g, 9.9 mmol) in 5 mL dry CH2Cl2 was treated with 20 mL of TFA for 1 h at room temperature. The TFA was removed in vacuo and residual acid was removed by addition and evaporation of toluene (2 × 10 mL). The crude cinnamic acid derivative (3.5 g, 9.8 mmol), pentachlorophenol (2.8 g, 10.7 mmol), DCC (3.0 g, 14.7 mmol) and DMAP (0.120 g, 0.98 mmol) in 100 mL of EtOAc was stirred at room temperature for 24 h. The mixture was filtered through celite and the solvent removed in vacuuo. The crude product was purified by silica gel chromatography eluting with 25% EtOAc-hexanes to give 5.1 g (84%) of 29a as white solid. 1H NMR (CDCl3, 300 MHz) δ 1.34–1.4 (m, 6H), 2.67 (s, 3H), 4.18–4.32 (m, 4H), 6.5 (s, 1H), 7.65–7.72 (m, 4H). 13C NMR (CDCl3, 75.0 MHz) δ16.3, 16.4, 18.5, 64.8, 64.9, 114.9, 126.56, 126.58, 126.65, 126.71, 126.74, 126.8, 127.93, 131.4, 132.0, 143.63, 144.24, 160.5, 161.73. 19F NMR (CDCl3, 282.0 MHz) δ −108.8 (d, J = 112.8 Hz, 2F). Anal. Calcd for C21H18Cl5F2O5P: C, 42.28; H, 3.04; Cl, 29.71; F, 6.37: Found C, 42.48; H, 3.15; Cl, 29.45; F, 6.25. HRMS (M+H) calc, 594.9381; found, 594.9357.
Synthesis of pentachlorophenyl (2E)-3-[4-(phosphoryldifluoromethyl)phenyl]-but-2-enoate, 30a
Iodotrimethylsilane (2.0 mL, 13.4 mmol) in 5 mL of dry CH2Cl2 was added dropwise to a solution of 29a (2.0 g, 3.35 mmol) and bis(trimethylsilyl)trifluoroacetamide (1.8 mL, 6.8 mmol ) in 20 mL of dry CH2Cl2 at 0°C under argon. Stirring was continued for 1h at 0°C and 1h at room temperature. The solution was concentrated in vacuo. The residue was taken up in 20 mL MeCN/H2O/AcOH (8:1:1), stirred for 45min and concentrated in vacuo. Toluene (5 mL) was added and evaporated twice. On addition of ether solids separated, which were collected by filtration and washed with the same solvent to give 1.6 g of 30a as a white powder (89%). It was used directly in the next step with no purification. HRMS (M+H) calc, 538.8755; found, 538.8773.
Synthesis of pentachlorophenyl (2E)-3-[4-[[bis[(2,2-dimethyl-1-oxopropoxy)methoxy]phosphinyl]difluoromethyl]phenyl]-but-2-enoate, 31a
NaOH (144 mg, 3.6 mmol) in 2 mL of H2O was added dropwise to a stirred suspension of 30a (1 g, 1.9 mmol) in 5 mL of H2O. When the mixture became clear (pH∼9), AgNO3 (807 mg, 4.75 mmol) was added. After 2 h at 4°C the gray precipitate was collected by filtration, dried, and pulverized in a mortar and pestle. The powder was suspended in dry toluene (10 mL) and pivyloxymethyl iodide (1.4 g, 5.7 mmol) was added and stirred for 48 h at room temperature. After filtration the solvent was removed in vacuo and the crude product was purified by silica gel column chromatography eluting with 30% EtOAc-hexanes to give colorless sticky liquid of 31a (0.9 g, 64%). 1H NMR (CDCl3, 500 MHz) δ 1.23 (s, 18H), 2.65 (s, 3H), 5.66–5.76 (m, 4H), 6.47 (s, IH), 7.66 (m, 4H). 13C NMR (CDCl3, 125 MHz) δ18.5, 26.8, 38.8, 82.4, 82.5, 115.1, 126.6, 126.7, 126.8, 127.9, 131.4, 132.0, 144.0, 144.2, 160.3, 161.7, 176.5. 19F NMR (CDCl3, 282.0 MHz) δ −109.22 (d, J = 124.0 Hz, 2F). 31P NMR (CDCl3, 202.0 MHz) δ 4.81 (t, J = 123.2 Hz, 1P). Anal.Calcd for C29H30Cl5F2O9P: C, 45.31; H, 3.93; Cl, 23.06; F, 4.94: Found C, 45.12; H, 3.93; Cl, 22.90; F, 5.08. HRMS (M+H) calc, 767.0116; found, 767.0124.
Synthesis of 4-nitrophenyl (2E)-3-[4-[(diethoxyphosphinyl)difluoromethyl]phenyl]-but-2-enoate, 29b
A solution of 28 (4.00 g, 9.9 mmol) in 5mL dry CH2Cl2 was treated with 20 mL of trifluoroacetic acid for 1 h at room temperature. The TFA was removed in vacuo and residual acid was removed by addition and evaporation of toluene (2 × 10 mL). The crude cinnamic acid derivative (3.5 g, 10.0 mmol), p-nitrophenol (1.7g, 12.0 mmol) and DCC (3.0 g, 14.7 mmol) in 100 mL of EtOAc were stirred at room temperature for 24h. The mixture was filtered through celite and the solvent removed in vacuuo. The crude product was purified by silica gel chromatography eluting with 25% EtOAc in hexanes to give 3.8 g (80%) of 29b as white solid. 1H NMR (CDCl3, 300 MHz) δ 1.32–1.4 (m, 6H), 2.66 (s, 3H), 4.16–4.3 (m, 4H), 6.37 (s, 1H), 7.35 (d, J= 9.0 Hz, 4H), 7.6–7.7 (m, 4H), 8.3 (d, J = 9.0Hz, 2H). 13C NMR (CDCl3, 75.0 MHz) δ16.3, 16.4, 18.4, 64.8, 64.9, 116.3, 122.5, 125.2, 126.5, 126.6, 126.7, 126.8, 143.9, 145.2, 155.5, 159.2, 163.5. 19F NMR (CDCl3, 282.0 MHz) δ −108.8 (d, J = 112.8 Hz, 2F). 31P NMR (CDCl3, 202.0 MHz) δ 5.94 (t, J = 113.1 Hz, 1P). Anal.Calcd for C21H22F2NO7P: C, 53.74; H, 4.72; F, 8.1; N, 2.98; O, 23.86; P, 6.6: Found C, 53.84; H, 4.72; F, 8.12; N, 3.12.
Synthesis of 4-nitrophenyl (2E)-3-[4-(phosphoryldifluoromethyl)phenyl]-but-2-enoate, 30b
Iodotrimethylsilane (2.5 mL, 17.0 mmol) in 10mL of dry CH2Cl2 was added dropwise to a solution of 27b (2.0 g, 4.26 mmol) in 20 mL of dry CH2Cl2 at 0°C under argon. Stirring was continued for 1h at 0°C and 1h at room temperature. The solution was concentrated in vacuo. The residue was taken up in 20 mL MeCN/H2O/AcOH (8:1:1), stirred for 45min and concentrated in vacuo. Toluene (5 mL) was added and evaporated twice. On addition of Et2O solids separated, which were collected by filtration and washed with the same solvent to give 1.5 g of 28b as a white powder (85%), which was used without purification. 1H NMR (DMSO-d6, 300 MHz) δ 2.6 (s, 3H), 6.53 (s, 3H), 7.53 (d, J = 10.2Hz, 2H), 7.61 (d, J= 8.1Hz, 2H), 7.80 (d, J= 8.1Hz, 2H), 8.32 (d, J = 10.2 Hz, 2H). 19F NMR (DMSO-d6, 282.0 MHz) δ −1108.4 (d, J = 104.3 Hz, 2F).
Synthesis of p-Nitrophenyl (2E)-3-[4-[[bis[(2,2-dimethyl-1-oxopropoxy)methoxy]phosphinyl]difluoromethyl]phenyl]-but-2-enoate (31b)
NaOH (174 mg, 4.3 mmol) in 2 mL of water was added dropwise to a stirred suspension of 30b (1.0 g, 2.4 mmol) in 5 mL of water. When the mixture became clear (pH∼9), AgNO3 (910 mg, 5.32 mmol) was added. After 2h at 4°C the gray precipitate was collected by filtration, dried, and pulverized in a mortar and pestle. The powder was suspended in dry toluene (10 mL) and pivaloyloxymethyl iodide (1.8 g, 7.2 mmol) was added and stirred for 48 h at room temperature. After filtration the solvent was removed in vacuo and the crude product was purified by silica gel column chromatography eluting with 30% EtOAc-hexanes to give 31b (0.9 g, 58%) as an oil. 1H NMR (CDCl3, 300 MHz) δ 1.24 (s, 18H), 2.66 (s, 3H), 5.66–5.8 (m, 4H), 6.38 (s, 1H), 7.36 (d, J = −7.72 (m, 4H). 19F NMR (CDCl3, 282.0 MHz) δ −108.8 (d, J = 112.8 Hz, 2F). Anal.Calcd for C29H34F2NO11P: C, 54.29; H, 5.34; F, 5.92; N, 2.18; O, 27.43; P, 4.83: Found C, 54.00; H, 5.47; F, 6.10; N, 2.20.
Synthesis of βMF2Pm(POM2)Cinn-Haic-Apa, 34
Method A
Rink resin (0.3 g, 0.225 mmol) was swollen in DMF/CH2Cl2 (1:1) and was washed with 2 × 5 mL of the same solvent. The Fmoc group was removed by treatment with 20% piperidine in DMF for 3 min (repeated 3 times). For coupling of the next two amino acids, Fmoc-(R)-4-aminopentanoic acid and Fmoc-Haic-OH, three-fold excesses of the Fmoc-amino acids, PyBOP, and HOBt were used along with six-fold excesses of DIPEA in 4 mL of DMF/CH2Cl2. After assembly of the amino acid chain, the Fmoc group was removed by treatment with 20% piperidine in DMF and the resins were washed with 3 × 10 mL of DMF/CH2Cl2 (1:1). Cleavage was accomplished with three treatments of the resins with 5 mL of TFA:TIS:H2O (95:2.5:2.5) for 10 min each. The solvents were removed in vacuo and residual acid was removed by addition and evaporation of toluene (3 × 5 mL). Et2O was added and the precipitate was collected by centrifugation. The crude product was purified by reverse phase HPLC using a gradient of MeCN in H2O. HRMS (M + H) calc, 345.1927; found, 345.1101. Pure H-Haic-NHCH(CH3)CH2CH2CONH2 (100 mg, 0.29 mmol), 31b (0.223 g, 0.29 mmol), dry and distilled DIPEA (0.1 mL, 0.58 mmol) and HOBt (0.045 g, 0.29 mmol) in 4 mL of dry N-methylpyrrolidone and CH2Cl2 (1:1) were mixed together and stirred for two h. The reaction was monitored by HPLC. After completion, the solvent was removed and the crude product was purified by reverse phase HPLC using a gradient of MeCN in H2O to yield 27 mg of 34. 1H NMR (Acetonitrile-d3 500 MHz) δ 1.1 (d, J = 6.5 Hz, 3H), 1.2 (s, 18H), 1.6–1.73 (m, 2H), 2.03–2.26 (m, 8H), 2.52 (s, 3H), 3.0–3.17 (m, 2H), 3.2–3.26 (m, 1H), 3.38 (m, 1H), 3.8 (m, 1H), 4.5 (m, 1H), 5.03 (m, 1H), 5.46 (s, 1H), 5.62–5.7 (m, 4H), 6.2 (s, 1H), 6.36 (s, 1H), 6.74 (d, J = 8.0 Hz, 1H), 7.00 (m, 1H), 7.09–7.11 (m, 2H), 7.32 (d, J = 6.5 Hz, 1H), 7.58 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H). 13C NMR (Acetonitrile-d3 125 MHz) δ 0.2, 0.3, 0.5, 0.6, 0.8, 16.4, 20.1, 26.0, 29.6, 31.1, 31.7, 31.9, 38.4, 44.9, 53.3, 61.45, 82.8, 121.5, 123.0, 124.2, 126.4, 126.6, 129.5, 133.2, 138.7, 145.6, 148.7, 165.5, 170.3, 170.4, 174.8, 176.4. HRMS (M+H) calc 847.3495 found 847.3489.
Method B
To a stirred solution of TFA.H-Haic-NHCH(CH3)CH2CH2CONH2 (0.050 g, 0.11 mmol), N-methylmorpholine (0.036 mL, 0.33 mmol) and DMAP (0.005g, 0.033 mmol) in 3 mL of dry NMP, was added a solution of 31a (0.085 g, 0.11 mmol) in 2 mL of dry MeCN under inert atmosphere. The reaction was monitored by HPLC. After completion, about 1 h, the reaction mixture was concentrated under vacuum then purified by reverse phase HPLC using MeCN-water system. Yield: 0.070g (76%) of 34. HRMS (M + H) calcd 847.3495, found 847.3489.
Synthesis of Synthesis of βMF2PmCinn-Haic-Apa, 34-NP
Rink resin (0.15 g, 0.18 mmol) was swollen in DMF/CH2Cl2 (1:1) and was washed with 2 × 10 mL of the same solvent. The Fmoc group was removed by treatment with 20% piperidine in DMF (3 × 5 min). For coupling of 4-(9-fluorenylmethyloxycarbonylamino) pentanoic acid,30 3-fold excesses of amino acid, PyBop, HOBt and DIPEA in 10 mL of DMF/CH2Cl2 (1:1) were used. Coupling of Fmoc-Haic-OH was done with two-fold excesses of Fmoc-amino acid, DIC, and HOBt. The final coupling was carried out with two-fold excess of 30a, Et3N and HOBt in 10 mL of DMF/CH2Cl2. After all coupling resins were washed with 3 × 10 mL of DMF/CH2Cl2 followed by only CH2Cl2 (3 × 10 mL). Resins were cleaved with TFA:TIS:H2O (95:2.5:2.5) (3 × 10 mL) for 10 min each and combined TFA solutions were removed in vacuum, stripped remainder TFA off by addition of toluene (2 × 5 mL) and addition of ether resulted off-white precipitate. The solid was collected by centrifugation, dried, and purified by reverse phase HPLC to get 34 mg of desired material. HRMS (M+H) calc, 619.2133; found, 619.2139.
Synthesis of βMF2Pm(POM)Cinn-Haic-Apa, 34-MP
To a stirred solution of TFA·H-Haic-NHCH(CH3)CH2CH2CONH2 (0.050 g, 0.11 mmol), HOBt·H2O (0.019 g, 0.12 mmol) and DIPEA (0.04 mL, 0.22 mmol) in 2 mL of DMF, was added a solution of 31a (0.085 g, 0.11 mmol) in 2 mL of CH2Cl2 under inert atmosphere. The reaction was monitored by HPLC. After completion, about 1 h, the reaction mixture was concentrated under vacuum and triturated with hexane-ether. The solid residue (0.089 g) was purified by reverse phase HPLC using MeCN-water system. Yield: 0.027 g (33%) 34-MP. HRMS (M + H) calcd 733.2814, found 733.2814 and 0.016g (22%) 34. HRMS (M + H) calcd 847.3495, found 847.3489.
Inhibition of Stat3 tyrosine 705 phosphorylation in tumor cells
MDA-MB-468 breast tumor cells (4 × 105) were plated in 6-well culture dishes in DMEM media containing 10% FCS and were allowed to grow overnight. Prodrugs were prepared as 10 mM stock solutions in DMSO immediately before use and aliquots were added to the culture media to give the correct final concentrations. After 2 h the cells were washed with ice cold phosphate buffered saline. Washed cells were treated with lysis buffer (50 mM Hepes, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM sodium pyrophosphate, 10% glycerol, 1% Triton X-100, 1 mM PMSF, 1 mM Na3VO4, 10 µg/mL leupeptin and 10 µg/mL aprotinin). Cell-free detergent extracts were centrifuged at 15,000 rpm in a microcentriguge for 30 min at 4°C and the protein concentrations of the supernatants determined. Aliquots containing 12 µg of protein were separated on 8% SDS-PAGE and were transferred to PVDF filters. The filters were blocked with 5% bovine serum albumin and were probed with pStat3Y705 antibody followed by secondary antibody, whose signal was detected with an enhanced chemiluminescence kit (ECL, Amersham, Chicago, IL). Filters were stripped with stripping buffer (62.5 mM Tris, pH 6.8, 2% SDS, and 0.1 M 2-mercaptoethanol) at 50°C for 30 min. Filters were then probed with total Stat3 antibody and visualized with chemiluminescence as above. SKOV3-ip cells were cultured in McCoy's 5A medium at 3 × 105 cells/well. Inhibition of pStat3 was assayed identically as in the case of MDA-MB-468 cells. Hey ovarian tumor cells and MeWo and A375 melanoma cells were cultured at a density of 3 × 105 cells/well in RPMI1640 media. After overnight culture and media change, cells were treated with increasing concentrations of 32 for 2 h. IL-6 (10 ng/mL) was added at the last 30 min of incubations. Cells were lysed and pStat3 and total Stat3 were determined as above.
Effect of prodrug on the phosphorylation of Stat3, Stat5 and Akt in response to Epidermal Growth Factor stimulation
MDA-MB-468 cells were prepared and prodrugs were added to the culture media to give the correct final concentrations as above. After 1.5 h epidermal growth factor (EGF) was added at 100 ng/mL. After 30 minutes cells were collected and lysed and proteins were separated by PAGE and transferred to three PVDF filters as above. The first filter was blocked with 5% bovine serum albumin and probed for total and phosphoStat3 as above. The second filter was probed for phosphoSer473Akt and total Akt using appropriate antibodies and similar detection procedures to those used for Stat3. The third filter was probed for phosphoTyr699 Stat5 and total Stat5 using appropriate antibodies and similar detection procedures to those used for Stat3.
Effect of Prodrugs on the phosphorylation of Stat1
MDA-MB-468 cells were prepared and prodrugs were added to the culture media to give the correct final concentrations as above. After 1.5 h interferon γ was added at 25 ng/mL. After 30 minutes cells were collected and lysed and proteins were separated by PAGE and transferred to PVDF filters as above. Filters were probed for phosphoTyr701 Stat1 and total Stat1 using appropriate antibodies and similar detection procedures to those used for Stat3.
Effect of Prodrugs on the phosphorylation of focal adhesion kinase
MDA-MB-468 cells were prepared and prodrugs (10 mM/DMSO) were added to the culture media to give the correct final concentrations as above. After 2 h cells were lysed and total FAK and pTyr861FAK was assayed by western blots as described above.
Inhibition of growth of MDA-MB-468 breast cancer cells
MDA-MB-468 cells were cultured in DMEM with 10% FBS. Cells were plated into 96 well plates (1500 cells per well) in triplicate. The next day the media was changed. Immediately before use, prodrugs were dissolved in ethanol to a concentration of 10 mM. This stock solution was then diluted to appropriate concentrations for addition to the wells containing the MDA-MB-468 cells. MTT assays were performed at 72 h. These assays were run three times.
For daily treatment, cells were plated and treated with prodrugs as above. At 24 and 48 h, prodrug was added to the same media. Total EtOH concentration was less than 1% in all wells. Cell viability was determined with the MTT assay.
Immunofluorescence microcroscopy
MDA-MB-468 cells were plated onto slides and treated with DMSO or 34 (5 µM). After 2h, cells were fixed in 4% paraformaldehyde, and permeabilized using 0.5% Triton X-100. Washed cells were blocked with 3% BSA and incubated with antibodies against pTyr705Stat3. After washing, cells were incubated with secondary antibodies conjugated with Alexa Fluor 594 (Molecular Probe, Invitrogen). Finally, slides were mounted and examined using confocal microscopy (Olympus FluoView 500 or 1000, Olympus, Inc., Melville, NY). All images were obtained with the same microscope settings.
Supplementary Material
Acknowledgements
Our gratitude to Manish Shanker and Ailing W. Scott for technical assistance. We acknowledge the National Cancer Institute (CA096652), the MDACC SPORE in ovarian Cancer P50 CA083639, and the CTT/TI-3D Chemistry & Molecularly-Targeted Therapeutic Development Grant Program for support of this work. We also acknowledge the NCI Cancer Center Support Grant CA016672 for the support of our NMR facility and the Translational Chemistry Core Facility for mass spectrometry and X-ray crystallography. Lung tumor and melanoma analyses were supported by the UT SPORE in lung cancer P50 CA070907 and the MDACC SPORE in Melanoma, P50 CA093459.
Abbreviations
- Aec
2-aminoethylcarbamate
- Aeu
2-aminoethylurea
- Apa
(R)-4-aminopentamide
- DIEA
diisopropylethylamine
- DIPCDI
diisopropylcarbodiimide
- EGF
epidermal growth factor
- Fmoc
9-fluorenylmethoxycarbonyl
- Haic
5-(amino)-1,2,4,5,6,7-hexahydro-4-oxo-(2S,5S)-azepino[3,2,1-hi]indole-2-carboxylic acid
- HOBt
1-hydroxybenzotriazole
- IL-6
interleukin 6
- JAK
Janus kinase
- mPro
cis-3,4-methanoproline
- MMP-2
matrix metalloproteinase-2
- pCinn
4-phosphoryloxy cinnamide
- βMpCinn
β-methyl pCinn or [2E] 3-(4-phosphoryloxyphenyl)-2-butenamide
- βMF2PmCinn
[2E] 3-[4-[(phosphinyl)difluoromethyl]phenyl]-2-butenamide
- PDGF
platelet derived growth factor
- POM
pivaloyloxymethyl
- PyBOP
1H-Benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate
- SAR
structure activity relationship
- SH2 domain
Src homology 2 domain
- Stat3
signal transducer and activator of transcription 3
- TES
triethylsilane
- TIS
triisopropylsilane
- U-Stat3
unphosphorylated Stat3
- VEGF
vascular endothelial growth factor
Footnotes
Supporting Information Available. Table of peptide characterization. Table of prodrug characterization. Table of antibodies used in Western Blots. Figure of the crystal structure of iodophenylbut-2-enoyl leucine tert-butyl ester and NOE pattern of peptidomimetics 3-NP and 34-NP. NME spectra of 3-NP and 34-NP. Figure of inhibition of FAK, Akt, Stat3 and Stat5 phosphorylation by 34 at high concentration.
References
- 1.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell. Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 2.Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R, Huang S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23:3550–3560. doi: 10.1038/sj.onc.1207383. [DOI] [PubMed] [Google Scholar]
- 3.Wei D, Le X, Zheng L, Wang L, Frey JA, Gao AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL, Xie K. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene. 2003;22:319–329. doi: 10.1038/sj.onc.1206122. [DOI] [PubMed] [Google Scholar]
- 4.Chen Z, Han ZC. STAT3: a critical transcription activator in angiogenesis. Med. Res. Rev. 2008;28:185–200. doi: 10.1002/med.20101. [DOI] [PubMed] [Google Scholar]
- 5.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat. Rev. Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 6.Thompson JE, Cubbon RM, Cummings RT, Wicker LS, Frankshun R, Cunningham BR, Cameron PM, Meinke PT, Liverton N, Weng Y, DeMartino JA. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg. Med. Chem. Lett. 2002;12:1219–1223. doi: 10.1016/s0960-894x(02)00106-3. [DOI] [PubMed] [Google Scholar]
- 7.Kreis S, Munz GA, Haan S, Heinrich PC, Behrmann I. Cell density dependent increase of constitutive signal transducers and activators of transcription 3 activity in melanoma cells is mediated by Janus kinases. Mol. Cancer Res. 2007;5:1331–1341. doi: 10.1158/1541-7786.MCR-07-0317. [DOI] [PubMed] [Google Scholar]
- 8.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, Wang L, Ye M, McEachern K, Chen H, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18:443–451. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- 10.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Nikolovska-Coleska Z, Yang CY, Gomez C, Gao W, Krajewski K, Jiang S, Roller P, Wang S. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via 'click chemistry'. Bioorg. Med. Chem. Lett. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]
- 13.Dourlat J, Valentin B, Liu WQ, Garbay C. New syntheses of tetrazolylmethylphenylalanine and O-malonyltyrosine as pTyr mimetics for the design of STAT3 dimerization inhibitors. Bioorg. Med. Chem. Lett. 2007;17:3943–3946. doi: 10.1016/j.bmcl.2007.04.107. [DOI] [PubMed] [Google Scholar]
- 14.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003;63:3923–3930. [PubMed] [Google Scholar]
- 15.Shao H, Xu X, Mastrangelo MA, Jing N, Cook RG, Legge GB, Tweardy DJ. Structural requirements for signal transducer and activator of transcription 3 binding to phosphotyrosine ligands containing the YXXQ motif. J. Biol. Chem. 2004;279:18967–18973. doi: 10.1074/jbc.M314037200. [DOI] [PubMed] [Google Scholar]
- 16.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, Yi H, Wang S. Structure-Based Design of Conformationally Constrained, Cell-Permeable STAT3 Inhibitors. ACS Med. Chem. Lett. 2010;1:85–89. doi: 10.1021/ml100010j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fletcher S, Singh J, Zhang X, Yue P, Page BD, Sharmeen S, Shahani VM, Zhao W, Schimmer AD, Turkson J, Gunning PT. Disruption of transcriptionally active Stat3 dimers with non-phosphorylated, salicylic acid-based small molecules: potent in vitro and tumor cell activities. Chembiochem. 2009;10:1959–1964. doi: 10.1002/cbic.200900172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez C, Bai L, Zhang J, Nikolovska-Coleska Z, Chen J, Yi H, Wang S. Design, synthesis, and evaluation of peptidomimetics containing Freidinger lactams as STAT3 inhibitors. Bioorg. Med. Chem. Lett. 2009;19:1733–1736. doi: 10.1016/j.bmcl.2009.01.091. [DOI] [PubMed] [Google Scholar]
- 20.Gunning PT, Katt WP, Glenn M, Siddiquee K, Kim JS, Jove R, Sebti SM, Turkson J, Hamilton AD. Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: structural recognition of STAT SH2 domains. Bioorg. Med. Chem. Lett. 2007;17:1875–1878. doi: 10.1016/j.bmcl.2007.01.077. [DOI] [PubMed] [Google Scholar]
- 21.Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schrock C, Sebti SM, Jove R, Hamilton AD, Turkson J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem. Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 22.Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, Haura E, Sebti S, Hamilton AD, Jove R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer. Ther. 2004;3:261–269. [PubMed] [Google Scholar]
- 23.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 24.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu X, Kasembeli MM, Jiang X, Tweardy BJ, Tweardy DJ. Chemical probes that competitively and selectively inhibit Stat3 activation. PLoS One. 2009;4:e4783. doi: 10.1371/journal.pone.0004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coleman DRIV, Kaluarachchi K, Ren Z, Chen X, McMurray JS. Solid phase synthesis of phosphopeptides incorporating 2,2-dimethyloxazolidine pseudoproline analogs: evidence for trans Leu-Pro peptide bonds in Stat3 inhibitors. Int. J. Pept. Res. Ther. 2008;14:1–9. [Google Scholar]
- 27.Coleman DRt, Ren Z, Mandal PK, Cameron AG, Dyer GA, Muranjan S, Campbell M, Chen X, McMurray JS. Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J. Med. Chem. 2005;48:6661–6670. doi: 10.1021/jm050513m. [DOI] [PubMed] [Google Scholar]
- 28.Mandal PK, Heard PA, Ren Z, Chen X, McMurray JS. Solid-phase synthesis of Stat3 inhibitors incorporating O-carbamoylserine and O-carbamoylthreonine as glutamine mimics. Bioorg. Med. Chem. Lett. 2007;17:654–656. doi: 10.1016/j.bmcl.2006.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandal PK, Limbrick D, Coleman DR, Dyer GA, Ren Z, Birtwistle JS, Xiong C, Chen X, Briggs JM, McMurray JS. Conformationally constrained peptidomimetic inhibitors of signal transducer and activator of transcription 3: evaluation and molecular modeling. J. Med. Chem. 2009;52:2429–2442. doi: 10.1021/jm801491w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mandal PK, Ren Z, Chen X, Xiong C, McMurray JS. Structure-affinity relationships of glutamine mimics incorporated into phosphopeptides targeted to the SH2 domain of signal transducer and activator of transcription 3. J. Med. Chem. 2009;52:6126–6141. doi: 10.1021/jm901105k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg. Med. Chem. Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- 32.Mandal PK, Liao WS, McMurray JS. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org. Lett. 2009;11:3394–3397. doi: 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 34.McMurray JS. Structural basis for the binding of high affinity phosphopeptides to Stat3. Biopolymers. 2008;90:69–79. doi: 10.1002/bip.20901. [DOI] [PubMed] [Google Scholar]
- 35.Burke TR, Jr, Smyth MS, Otaka A, Nomizu M, Roller PP, Wolf G, Case R, Shoelson SE. Nonhydrolyzable phosphotyrosyl mimetics for the preparation of phosphatase-resistant SH2 domain inhibitors. Biochemistry. 1994;33:6490–6494. doi: 10.1021/bi00187a015. [DOI] [PubMed] [Google Scholar]
- 36.Farquhar D, Khan S, Srivastva DN, Saunders PP. Synthesis and antitumor evaluation of bis[(pivaloyloxy)methyl] 2'-deoxy-5-fluorouridine 5'-monophosphate (FdUMP): a strategy to introduce nucleotides into cells. J. Med. Chem. 1994;37:3902–3909. doi: 10.1021/jm00049a009. [DOI] [PubMed] [Google Scholar]
- 37.Qabar MN, Urban J, Kahn M. A facile solution and solid phase synthesis of phosphotyrosine mimetic L-4-[diethylphosphono(difluoromethyl)]phenylalanine (F2Pmp(EtO)2) derivatives. Tetrahedron. 1997;53:11171–11178. [Google Scholar]
- 38.Clegg W, Elsegood MRJ, Jackson RFW, Fraser JL, Emsden LJ. N-(2,3,5,6-Tetrachloropyrid-4-yl)cinnamide. Acta Cryst. 1997;C53:797–799. [Google Scholar]
- 39.Iwamoto T, Kashino S, Haisa M. Structure of Cinnamide. Acta Cryst. 1989;C45:1110–1112. [Google Scholar]
- 40.Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267:1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- 41.Wiederkehr-Adam M, Ernst P, Muller K, Bieck E, Gombert FO, Ottl J, Graff P, Grossmuller F, Heim MH. Characterization of phosphopeptide motifs specific for the Src homology 2 domains of signal transducer and activator of transcription 1 (STAT1) and STAT3. J. Biol. Chem. 2003;278:16117–16128. doi: 10.1074/jbc.M300261200. [DOI] [PubMed] [Google Scholar]
- 42.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–1279. [PubMed] [Google Scholar]
- 43.Ladbury JE. Protein-protein recognition in phosphotyrosine-mediated intracellular signaling. Protein Reviews. 2005;3:165–184. (Proteomics and Protein-Protein Interactions) [Google Scholar]
- 44.Ladbury JE, Arold S. Searching for specificity in SH domains. Chem. Biol. 2000;7:R3–R8. doi: 10.1016/s1074-5521(00)00067-3. [DOI] [PubMed] [Google Scholar]
- 45.Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, Zinner R, Hung MC, Steck P, Siminovitch K, Mills GB. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034–7045. doi: 10.1038/sj.onc.1203183. [DOI] [PubMed] [Google Scholar]
- 46.Nicholson KM, Streuli CH, Anderson NG. Autocrine signalling through erbB receptors promotes constitutive activation of protein kinase B/Akt in breast cancer cell lines. Breast Cancer Res. Treat. 2003;81:117–128. doi: 10.1023/A:1025765215765. [DOI] [PubMed] [Google Scholar]
- 47.Calalb MB, Zhang X, Polte TR, Hanks SK. Focal adhesion kinase tyrosine-861 is a major site of phosphorylation by Src. Biochem. Biophys. Res. Commun. 1996;228:662–668. doi: 10.1006/bbrc.1996.1714. [DOI] [PubMed] [Google Scholar]
- 48.Laird AD, Li G, Moss KG, Blake RA, Broome MA, Cherrington JM, Mendel DB. Src family kinase activity is required for signal tranducer and activator of transcription 3 and focal adhesion kinase phosphorylation and vascular endothelial growth factor signaling in vivo and for anchorage-dependent and -independent growth of human tumor cells. Mol. Cancer Ther. 2003;2:461–469. [PubMed] [Google Scholar]
- 49.Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin. Cancer Res. 2005;11:6924–6932. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
- 50.Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, Gelovani JG, Kim SJ, Wang Z, Gallick GE. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68:3323–3333. doi: 10.1158/0008-5472.CAN-07-2997. [DOI] [PubMed] [Google Scholar]
- 51.Schultz C. Prodrugs of biologically active phosphate esters. Bioorg. Med. Chem. 2003;11:885–898. doi: 10.1016/s0968-0896(02)00552-7. [DOI] [PubMed] [Google Scholar]
- 52.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Neel BG, B BR, Fajardof JE, Chouf MM, Hanafusa H, Schaffhausen B, Cantley LC. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 53.Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T, Ren R, Baltimore D, Ratnofsky S, Feldman RA, Cantley LC. Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol. Cell. Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neculai D, Neculai AM, Verrier S, Straub K, Klumpp K, Pfitzner E, Becker S. Structure of the unphosphorylated STAT5a dimer. J. Biol. Chem. 2005;280:40782–40787. doi: 10.1074/jbc.M507682200. [DOI] [PubMed] [Google Scholar]
- 55.Gerhartz C, Heesel B, Sasse J, Hemmann U, Landgraf C, Schneider-Mergener J, Horn F, Heinrich PC, Graeve L. Differential activation of acute phase response factor/STAT3 and STAT1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. I. Definition of a novel phosphotyrosine motif mediating STAT1 activation. J. Biol. Chem. 1996;271:12991–12998. doi: 10.1074/jbc.271.22.12991. [DOI] [PubMed] [Google Scholar]
- 56.Costantino L, Barlocco D. STAT 3 as a Target for Cancer Drug Discovery. Curr. Med. Chem. 2008;15:834–843. doi: 10.2174/092986708783955464. [DOI] [PubMed] [Google Scholar]
- 57.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 58.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 59.Dong QG, Sclabas GM, Fujioka S, Schmidt C, Peng B, Wu T, Tsao MS, Evans DB, Abbruzzese JL, McDonnell TJ, Chiao PJ. The function of multiple IkappaB : NF-kappaB complexes in the resistance of cancer cells to Taxol-induced apoptosis. Oncogene. 2002;21:6510–6519. doi: 10.1038/sj.onc.1205848. [DOI] [PubMed] [Google Scholar]
- 60.Harnois C, Demers MJ, Bouchard V, Vallee K, Gagne D, Fujita N, Tsuruo T, Vezina A, Beaulieu JF, Cote A, Vachon PH. Human intestinal epithelial crypt cell survival and death: Complex modulations of Bcl-2 homologs by Fak, PI3-K/Akt-1, MEK/Erk, and p38 signaling pathways. J. Cell. Physiol. 2004;198:209–222. doi: 10.1002/jcp.10399. [DOI] [PubMed] [Google Scholar]
- 61.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 62.Ren Z, Mao X, Mertens C, Krishnaraj R, Qin J, Mandal PK, Romanowski MJ, McMurray JS, Chen X. Crystal structure of unphosphorylated STAT3 core fragment. Biochem. Biophys. Res. Commun. 2008;374:1–5. doi: 10.1016/j.bbrc.2008.04.049. [DOI] [PubMed] [Google Scholar]
- 63.Pearson DA, Blanchette M, Baker ML, Guindon CA. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Letters. 1989;30:2739–2742. [Google Scholar]
- 64.Regis G, Pensa S, Boselli D, Novelli F, Poli V. Ups and downs: the STAT1:STAT3 seesaw of Interferon and gp130 receptor signalling. Semin. Cell. Dev. Biol. 2008;19:351–359. doi: 10.1016/j.semcdb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 65.DeLano WL. The PyMOL Molecular Graphics System; version 1.1. DeLano Scientific LLC; 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.