

Abstract
One of the most frequent and serious complications to develop in septic patients is acute kidney injury (AKI), a disorder characterized by a rapid failure of the kidneys to adequately filter the blood, regulate ion and water balance, and generate urine. AKI greatly worsens the already poor prognosis of sepsis and increases cost of care. To date, therapies have been mostly supportive; consequently there has been little change in the mortality rates over the last decade. This is due, at least in part, to the delay in establishing clinical evidence of an infection and the associated presence of the systemic inflammatory response syndrome and thus, a delay in initiating therapy. A second reason is a lack of understanding regarding the mechanisms leading to renal injury, which has hindered the development of more targeted therapies. In this review, we summarize recent studies, which have examined the development of renal injury during sepsis and propose how changes in the peritubular capillary microenvironment lead to and then perpetuate microcirculatory failure and tubular epithelial cell injury. We also discuss a number of potential therapeutic targets in the renal peritubular microenvironment, which may prevent or lessen injury and/or promote recovery.
Keywords: sepsis, acute kidney injury, microcirculation, oxidative stress, peritubular capillary, tubular epithelium
1. Introduction
1.1 Sepsis and acute kidney injury
Sepsis is a condition characterized by a disseminated inflammatory response triggered by a bacterial, viral or fungal infection. The most recent statistics list sepsis as the 7th leading cause of all deaths in children 1–4 years of age and the 8th in adults 65–75 years of age1 but it is the major cause of death among critically ill patients. Each year approximately 750,000 patients in the United States (Hotchkiss and Karl, 2003) and 18 million people worldwide are affected (Marshall, et al., 2005). Mortality rates for sepsis range from 25% to 70% and are correlated with the presence of hypotension (shock) and the development of an associated single or multi-organ failure (Russell, 2006). To date, therapies have been mostly supportive; consequently there has been little change in the mortality rates over the last decade. This is due, at least in part, to the delay in establishing clinical evidence of an infection and the associated presence of the systemic inflammatory response syndrome (SIRS) and thus, a delay in initiating therapy (Remick, 2007; Stearns-Kurosawa, et al., 2011). A second reason is a lack of understanding regarding the mechanisms leading to the development of organ injury.
One of the most frequent and serious complications to develop in septic patients is acute kidney injury (AKI), a disorder characterized by a rapid failure of the kidneys to adequately filter the blood, regulate ion and water balance, and generate urine (Zarjou and Agarwal, 2011). AKI greatly worsens prognosis and increases cost of care. The incidence of AKI increases with the severity of sepsis (S. Heemskerk, et al., 2009) and estimates are that AKI develops within the first 24 hours in 64% of patients with severe sepsis and hypotension (Bagshaw, et al., 2009). Strikingly, the mortality rate for septic patients with AKI is approximately doubled compared with sepsis alone. Thus, protecting the kidney could significantly reduce morbidity and mortality in patients with severe sepsis. Unfortunately, treatment of sepsis-induced AKI has advanced little during the last several decades (Ricci, et al., 2011). This review will focus on recent studies, which suggest the therapeutic potential for targeting the renal microcirculatory microenvironment in treating or even preventing sepsis-induced AKI.
As mentioned earlier, effective therapy in the septic patient is hampered because therapy is usually begun only after the onset of symptoms (Russell, 2006). In fact, Kumar and co-investigators reviewed the medical records of 2,700 patients with septic shock between 1989 and 2004 and showed that only approximately 50% of the patients received adequate antibiotic treatment within the first six hours of hypotension and alarmingly, each hour of delay in initiating therapy decreased survival by 7.6% (Kumar, et al., 2006). Since the symptoms of SIRS are initiated by an infection but are driven by endogenous mediators such as cytokines (Lam and Ng, 2008; Mera, et al., 2011), treatments targeting cytokines have the potential for being effective but have not been successful clinically due, once again, to the delay in initiating therapy (Remick, 2007). Clearly, the time at which therapy is initiated has a profound impact on outcome and this is especially true with regard to the development of AKI (Dudley, 2004). Early goal-directed therapy (EGDT) (E. Rivers, et al., 2001), consisting of antibiotics, fluid resuscitation and hemodynamic support in an attempt to protect organ perfusion, is being evaluated as a systematic approach to supportive care and does improve survival compared to standard supportive therapy (E. P. Rivers, et al., 2008); however, mortality rates are still high even among adequately resuscitated patients (Lundy and Trzeciak, 2009; Otero, et al., 2006). Evidence-based guidelines for care developed through the Surviving Sepsis Campaign (Dellinger, et al., 2004) recommend approaching therapy in two phases: antibiotics and resuscitation within the first 6 hours and management within the first 24 hours (M. M. Levy, et al., 2010). Still, therapy is primarily supportive utilizing broad-spectrum antibiotics, fluid resuscitation, pressor agents, lung-protective ventilation, and if necessary, dialysis.
1.2 Renal microcirculatory failure
Animal and human studies along with clinical observations support the view that maintaining systemic pressure per se is not necessarily sufficient to maintain organ perfusion in the septic patient. Clinical findings indicate that the severity of microvascular dysfunction correlates with patient mortality (Sakr, et al., 2004; Vincent and De Backer, 2005) supporting the concept that maintaining the microcirculation is key to preserving organ function. In animal models the link between microcirculatory failure and organ injury is reasonably well established, at least for the renal microcirculation (Holthoff, et al., 2010; Z. Wang, et al., 2011; L. Wu and Mayeux, 2007). Nevertheless, because direct measurements of the microcirculation in humans is difficult and generally limited to the skin or sublingual microcirculation using sidestream dark-field imaging (Spanos, et al., 2010), while suggested, the value of preserving the microcirculation has not yet been directly proven in humans (Boerma and Ince, 2010). Studies using Doppler ultrasonography to monitor flow through the renal intralobular arteries in humans with sepsis did show that raising mean arterial pressure with norepinephrine to 75 mmHg (above the renal autoregulatory pressure) reduced the resistance index suggesting improved perfusion of the renal microcirculation. However, increasing pressure further to 85 mmHg did not result in additional improvement (Deruddre, et al., 2007). Hence, the goal of hemodynamic support need not be to completely restore mean arterial pressure but rather to elevate it enough to preserve the microcirculation (Boerma and Ince, 2010). One caveat with regard to the kidney is that autoregulatory systems controlling the microcirculation can limit overall perfusion even when systemic blood pressure is near normal, as described later. Suffice it to say, progress toward uncovering new specific therapeutic targets to treat or prevent sepsis-induced AKI requires a better understanding regarding the mechanistic relationships between the changes in the peritubular microcirculation and the development of renal tubular injury.
1.3. Animal models of sepsis
Significant advances have been made in understanding the development of renal injury during sepsis through the use of small and large animal models. Unfortunately, there are no current animal models, which fully replicate all of the complexities of human sepsis. One of the most frequently used models in rodents is the cecal ligation and puncture (CLP) model of polymicrobial peritonitis (Rittirsch, et al., 2009). Other models of sepsis such as administration of lipopolysaccharide (LPS) from the Gram negative bacterial cell wall and administration of live or killed bacteria have been used as well; however, the inflammatory response in LPS models is quite different from that initiated by live bacteria models and CLP in both the kinetics and magnitude of cytokine release (Miyaji, et al., 2003) as well as the role of the TLR4 receptor (Dear, et al., 2006; Kalakeche, et al., 2011). The severity of sepsis and, to some extent, the severity of AKI can be manipulated in each of these models by changing the dose of LPS or bacteria or by changing the size and/or number of cecal punctures. Sepsis models in larger animals such as sheep (Langenberg, et al., 2006; Ramchandra, et al., 2009) and pigs (Brandt, et al., 2009; Chvojka, et al., 2008) have been used and can exhibit hemodynamic changes that are more similar to human sepsis than most rodent models. Of course these models are largely impractical for mechanistic studies and are best used in pre-clinical evaluations of new therapies. The reader is directed to excellent reviews on animal models of sepsis, which discuss the advantages, disadvantages and limitations of each (Doi, et al., 2009; Dyson and Singer, 2009; Remick and Ward, 2005).
Whether or not changes in renal blood flow (RBF) in septic patients contribute to renal injury is still unclear. The primary reason for this is the scarcity of actual measurements of RBF in septic patients. Not surprisingly, measuring RBF in severely ill patients is rarely done. In the few patients where RBF has been measured, high variability in these measurements among patients hinder reliable conclusions regarding the state of RBF during the course of sepsis-induced AKI (Bradley, et al., 1976; Brenner, et al., 1990; Langenberg, et al., 2005). Consequently, the relationships between mean arterial pressure (MAP), RBF and the development of AKI in these critically ill patients are unknown. Unfortunately, animal studies have only added to the controversy regarding changes in RBF during sepsis. In hyperdynamic models of sepsis in larger animals where heart rate and cardiac output are increased, which more closely mimic what is observed in septic patients, RBF may increase, decrease or remain unchanged. For example, RBF increases over time in sheep following E. coli infusion (Langenberg, et al., 2006; Ramchandra, et al., 2009); however, in pigs subjected to autologous fecal peritonitis, a model more closely resembling polymicrobial peritonitis, RBF falls gradually coinciding with a decrease in MAP (Brandt, et al., 2009; Chvojka, et al., 2008).
In the most frequently studied rodent models of sepsis, systemic and renal hemodynamics, when reported, are variable and depend on the model used (Doi, et al., 2009; W Wang, et al., 2002), the severity of sepsis induced (Doi, et al., 2008; Dyson and Singer, 2009) and the age and species/strain of the animal (Doi, et al., 2008; Seely, et al., 2011; Yang, et al., 2002). In these murine and rat models of LPS and CLP where AKI has been documented, there is a decline in RBF, glomerular filtration rate (GFR) and peritubular capillary perfusion (Holthoff, et al., PMID: 215975863; Seely, et al., 2011; Tiwari, et al., 2005; W Wang, et al., 2003; L. Wu, Gokden, et al., 2007; Yasuda, et al., 2006). Most of the studies examining the renal microcirculation during sepsis have used aged mice where CLP does mimic some key features of severe septic shock in humans. These include release of cytokines with a similar magnitude and kinetic profile (Doi, et al., 2009; Miyaji, et al., 2003) and a progressive fall in MAP associated with increased systemic NO production (Villalpando, et al., 2006; Z. Wang, et al., PMID: 22119717), which is largely dependent on upregulation of iNOS (S. Heemskerk, et al., 2006; L. Wu, Tiwari, et al., 2007). The magnitude of renal injury and the time course of development following CLP is also similar to that observed in humans (Bagshaw, et al., 2009; S. Heemskerk, et al., 2006; Miyaji, et al., 2003; Murugan and Kellum, 2011; L. Wu, Gokden, et al., 2007). Still, a limitation of this model is that it does not always replicate the initial hyperdynamic circulation observed in patients with sepsis (Hollenberg, 2005; Wan, et al., 2008; Z. Wang, et al., PMID: 22119717).
We recently performed detailed time course studies on the changes in systemic hemodynamics, RBF, and cortical peritubular capillary perfusion in aged (40-week old) mice subjected to CLP and found that as early as 4 hours following CLP there was a dramatic fall in MAP, RBF and peritubular capillary perfusion (Z. Wang, et al., PMID: 22119717). At 6 hours following CLP there was a 50% drop in the percentage of continuously perfused cortical capillaries, a 65% drop in red blood cell velocity in those capillaries and a 65% drop in GFR (Holthoff, et al., PMID: 215975863);Wang, PMID: 22119717 #3736}. We also examined the effects of CLP in rat pups because, as mentioned earlier, sepsis-induced AKI is a major cause of death in children. RBF and peritubular capillary perfusion fall to a similar extent in rat pups (17 days old) (Seely, et al., 2011); however, unlike in aged mice, there is no fall in MAP, supporting the notion that sepsis-induced AKI is not simply due to a fall in systemic blood pressure.
The earliest functional change in the renal microcirculation thus far identified following induction of sepsis is an increase in microvascular permeability. At 2 hours post CLP leakage of Evans blue dye into the interstitium increases nearly 5-fold (Z. Wang, et al., PMID: 22119717). This increase is maintained through 6 hours (Z. Wang, et al., PMID: 22119717; Yasuda, et al., 2006) and improves only slightly through 24 hours (Yasuda, et al., 2006). Cytokines released during sepsis can activate and injure capillary endothelial cells (W. L. Lee and Slutsky, 2010) so the rapid development of renal microcirculatory leakage is likely due to the early release of cytokines (Miyaji, et al., 2003) and their actions on the peritubular capillaries (Cepinskas and Wilson, 2008). What has emerged from these studies is a series of events (damage to the capillary and decreased perfusion) resulting in a hypoxic pro-oxidant peritubular microenvironment favoring the generation of reactive oxygen species (ROS), reactive nitrogen species (RNS), and subsequent renal tubular epithelial injury (Kalakeche, et al., 2011; Z. Wang, et al., PMID: 22119717; L. Wu, Gokden, et al., 2007; L. Wu and Mayeux, 2007; L. Wu, Tiwari, et al., 2007; Yasuda, et al., 2006). In the following sections, we summarize recent evidence primarily from animal studies to suggest that therapy targeting the peritubular capillary microenvironment could reduce or even prevent sepsis-induced AKI in humans.
1.4. In vitro models of sepsis
In the past, most in vitro models of sepsis-induced renal injury use renal epithelial cell cultures treated with relatively high concentration of LPS and/or individual cytokines (Du, et al., 2006; Markewitz, et al., 1993; Tiwari, et al., 2006). These models can test the effects of specific proinflammatory agents or arbitrary combinations of LPS/cytokines, but the relevance to sepsis-induced renal injury is quite limited. We (Pathak, et al., 2012; Pathak and Mayeux, 2010) and others (Boulos, et al., 2003) have used cultured cells exposed to septic serum to more closely mimic the septic milieu endothelial and renal epithelial cells might be exposed to in vivo. Still, these models also fail to replicate the interactions between endothelial and epithelial cells (and other resident interstitial cells) in the renal peritubular microenvironment. A seemingly more relevant in vitro model for examining in the peritubular microenvironment during sepsis was devised using a coculture system of human microvascular endothelial and human proximal tubular epithelial cell lines. The transmigration of leukocytes across the endothelial-epithelial bilayer was enhanced by TNF-α and LPS (Bijuklic, et al., 2007). However, there appears to be differences in the mechanisms driving transmigration of inflammatory cells in this model system. The anti-inflammatory peptide, α-melanocyte-stimulating hormone, α-MSH, was more efficacious at inhibiting LPS-induced transmigration when compared to TNF-α. This same model system was used to examine the proximal tubular epithelial permeability barrier. Interestingly, the epithelial permeability barrier was enhanced when cocultured with endothelial cells, suggesting that the endothelial-derived extracellular matrix promotes the epithelial permeability barrier (Aydin, et al., 2008).
In vitro models using primary cultures or stable cell lines of either endothelial or tubular epithelial cells might seem at first glance to be irrelevant for the study of sepsis-induced AKI. However, these models do have a place in sepsis research because they can suggest therapeutic targets (Bijuklic, et al., 2007; Dauphinee and Karsan, 2006; El-Achkar, et al., 2006; Pathak, et al., 2012) and are more amenable to investigating cellular signaling pathways than animal models. In fact, results from in vitro models have suggested many of the therapeutic targets discussed in this review.
2. Anatomy of the renal microenvironment
2.1. Renal microcirculation
The kidney microcirculation is unique relative to other organs in that it contains two capillary beds, which have high flow and low resistance (Palmer, et al., 1992). Blood is delivered via the renal artery, which branches from the abdominal aorta and enters the kidney through two branches at the renal hilus. Within the kidney, interlobar arteries course between the renal pyramids and become arcuate arteries at the cortico-medullary junction. Interlobular arteries branch from the arcuate arteries and make their way through the cortex ending in pre-glomerular afferent arterioles feeding the glomeruli, the filtering unit of the nephron. The first capillary system lies within the glomerulus and is specialized to effectively filter the blood while maintaining a very low permeability for macromolecules such as plasma protein. The glomerular capillary bed is also unique in that it terminates into an arteriole rather than a venule. These arterioles exit the glomeruli as post-glomerular efferent arterioles and branch to become the second capillary system, the peritubular capillary network (Figure 1). Peritubular capillaries eventually course into the medulla. Vascular bundles in the outer medulla give rise to the descending vasa recta (Yuan and Pannabecker, 2010). These straight capillaries travel into the medulla then make hairpin turns paralleling the loops of Henle. The anatomical arrangements of these vessels establish the countercurrent exchange mechanism necessary for urinary concentration of salts. Venous drainage from the cortex and medulla converge at the cortico-medullary junction and drain through medullary rays toward the hilus. Generally, a single renal vein returns blood into the inferior vena cava.
Figure 1. Anatomy of the renal circulation.

The renal microcirculation is depicted illustrating the anatomical relationship between the renal tubules and the peritubular capillaries.
2.2. Renal epithelium
The renal tubules are established by an epithelial monolayer anchored to a basement membrane (Lemley and Kriz, 1991). The epithelial cells are specialized transporting cells with different functions and morphology depending on the type and function of the tubule. Apical junction complexes allow for both paracellular and transcellular transport between the tubular lumen containing the glomerular filtrate and the peritubular capillary. The primary driving force for active transcellular transport is the Na-K-ATPase located in the basolateral membrane. Mitochondria are concentrated near this membrane to provide the energy necessary to drive the transport pump.
Blood ultrafiltrate leaves the glomerulus through the proximal tubule (nearest to the glomerulus). The proximal tubules are located mostly in the cortex and can be divided ultrastructurally into S1, S2 and S3 segments with S1 located closest to the glomerulus and S3 descending into the outer medulla and transitioning into the thin descending limb of the loop of Henle. The thick ascending limbs of the loops of Henle transition into the distal convoluted tubules, which are located in close proximity to the proximal tubules. Collecting tubules arise from the distal convoluted tubules and descend into the medulla growing in size to become collecting ducts. These become the papillary ducts, leading to the minor then major calyx and eventually empting into the ureter at the renal pelvis.
The ability of the renal tubules to reabsorb and secrete properly requires a close association with the peritubular capillary network. The high metabolic rate of the renal tubular epithelium needed to maintain tubular transport of sodium requires an extremely high rate of oxygen usage and thus, oxygen delivery (Evans, et al., 2008). The unique anatomical arrangement of capillaries and tubules means that changes in the microcirculation will have a profound effect on renal function and underlie much of the susceptibility of the kidney to acute renal ischemic injury (reviewed in (Evans, et al., 2008)). Surprisingly however, the arterial-to-venous (AV) gradient across the kidney is lower than what would be expected given the high oxygen demand of the transporting epithelium. Despite the relatively small AV O2 gradient (overall O2 usage) there is a tremendous drop in PO2 in blood exiting the efferent arteriole and supplying the cortical tubules (Welch, et al., 2001). Consequently, the cortical tubules receive an adequate but minimal amount of O2 under normal physiological conditions. Recently, a mathematical model was derived using existing measurements of rat renal arterial and venous PO2 to predict AV oxygen shunting and the effects reduced RBF might have on oxygenation of the renal cortex and medulla (Gardiner, et al., 2011). The model suggests that AV oxygen shunting acts to stabilize cortical tissue PO2 during changes in arterial PO2. However, under conditions where RBF is severely reduced, AV shunting could contribute to the development of hypoxia in both cortical and medullary regions even if medullary perfusion is maintained.
3. Control of the Renal Microcirculation
3.1. Renal blood flow
The chief function of the kidney, filtration of plasma and formation of urine, dictates that RBF, and in particular blood flow in the renal cortex, is much greater than that which would be necessary to meet the metabolic requirements of the kidney. At only approximately 2% of body mass, the kidneys receive a disproportional amount of cardiac output (~25%) for their size to drive glomerular filtration, the first step in removal of metabolic waste products and xenobiotics as well as in the maintenance of fluid and electrolyte balance. Consequently, it is critically important that filtration be maintained at a steady and optimal rate despite transient fluctuations in the delivery of blood to the kidneys. To ensure adequate GFR, RBF is tightly regulated primarily through two mechanisms: the myogenic response and tubuloglomerular feedback (TGF) (Cupples and Braam, 2007; Iliescu, et al., 2008; Just, 2007).
Keeping in mind that RBF is the rate of kidney perfusion (e.g., ml/min) and not simply the rate of blood delivery, factors which limit (or promote) intrarenal perfusion also regulate RBF. The primary regulators of RBF are the tone of the preglomerular afferent arterioles and intralobular arteries. In turn, GFR is controlled through changes in glomerular hydrostatic pressure mediated primarily through changes in tone of the afferent and efferent arterioles, since they are arranged in series. However, tubular damage can reduce GFR by increasing the hydrostatic pressure opposing filtration. Changes in afferent and efferent arteriolar tone are also involved in the regulation of peritubular capillary perfusion and medullary blood flow (Pallone, et al., 1998).
3.2. Peritubular capillary network
Proper reabsorption of water and electrolytes is dependent on optimal blood flow through specific regions of the kidney. Consequently, blood flow in the cortex and medulla is regulated differently (Damkjaer, et al., 2010). Cortical peritubular capillaries are fed by efferent arterioles originating from cortical glomeruli while medullary peritubular capillaries are fed by efferent arterioles from cortico-medullary glomeruli. Tone in the afferent and efferent arterioles is regulated through complex interactions between vasodilators such as nitric oxide (NO) and prostaglandin E2 (PGE2) and vasoconstrictors such as endothelin, angiotensin II, and adenosine (Bauerle, et al., 2011; Guan, et al., 2007; Hansen and Schnermann, 2003; Pallone, et al., 2003). However, different receptor subtypes for some of these vasoactive autacoids can mediate vasoconstriction or vasodilation. For example, vasodilation mediated by adenosine A2 receptor activation present on both afferent and efferent arterioles moderates A1 receptor-mediated vasoconstriction and the TGF response (Al-Mashhadi, et al., 2009; Carlstrom, et al., 2010; Feng and Navar, 2010; Nishiyama, et al., 2001). Also, PGE2 causes vasodilation at lower, more physiological concentrations but vasoconstriction at higher concentrations in the afferent arteriole (Tang, et al., 2000). This biphasic response appears to be mediated by E-prostanoid (EP) receptors EP4 and EP3, respectively.
It is still unclear how or even if flow through afferent or efferent arterioles is regulated differently based on the anatomical location of the glomeruli. However, there is evidence to suggest there might be differences in receptor coupling and/or species differences at least with regard to the vasoactive peptide, endothelin (Fenhammar, et al., 2011; Inscho, et al., 2005; Nitescu, Grimberg, Ricksten, et al., 2008; Pollock, et al., 2005; Schildroth, et al., 2011) as well as region-specific interactions between vasodilators and vasoconstrictors. For example, PGE2 moderates the vasoconstrictor activity of endothelin in the outer medullary descending vasa recta suggesting an interactive feedback control of medullary perfusion (Silldorff, et al., 1995).
4. Therapeutic targets in the peritubular microenvironment
4.1. Vasoactive regulators of the renal microcirculation
The roles of vascular endothelin ETA and ETB receptors in control of RBF and the renal microcirculation are complex and likely dependent on the type of sepsis model being studied. Plasma endothelin-1 levels are elevated in animals treated with LPS (Fenhammar, et al., 2011; Nitescu, Grimberg, Ricksten, et al., 2008) and in patients with sepsis where the levels correlate with the severity of illness (Pittet, et al., 1991). These findings suggest that endothelin may be playing a detrimental role. However, activation of ETB by endogenous endothelin appears to help maintain RBF in normotensive LPS-induced sepsis in the rat (Nitescu, Grimberg, Ricksten, et al., 2008). This is presumably due, at least in part, to a vasodilatory effect of ETB activation at the level of the efferent arteriole and/or its role in regulating distribution of flow within the kidney (Inscho, et al., 2005). Still, while it is possible to unmask an apparent ETB-mediated decrease in renal vascular resistance, the physiological or pathophysiological relevance of this effect remains unclear (Goddard, et al., 2004; Just, et al., 2008; Nitescu, Grimberg, Ricksten, et al., 2008). ETA receptors also play a role in regulating distribution of flow within the kidney. For example, in a normotensive porcine LPS model, blockade of ETA receptors did not preserve RBF but did preserve medullary flow without affecting cortical flow (Fenhammar, et al., 2011). One caveat with selective ETA blockers is that they can cause a significant fall in MAP during sepsis (Fenhammar, et al., 2011; Nitescu, Grimberg, Ricksten, et al., 2008), which could limit their therapeutic usefulness. Interestingly, when both ETA and ETB receptors are blocked with the nonselective endothelin receptor antagonist, bosentan, survival is increased in the murine CLP model (Iskit, et al., 2004). Also, another nonselective antagonist, tezosentan, used in a hypotensive porcine LPS model, did not decrease MAP further yet decreased renal vascular resistance and increased renal cortical blood flow (Fenhammar, et al., 2008).
Understanding how activation of ETA and ETB receptors might regulate the renal microcirculation during sepsis is far from clear. Nevertheless, the ability of endothelin to regulate renal vascular resistance and redirect intrarenal blood flow (Fenhammar, et al., 2011; Millar and Thiemermann, 2002; Sullivan, et al., 2009) support the notion that endothelin receptors should be evaluated further as potential therapeutic targets to help preserve the renal microcirculation during sepsis.
Tone in the afferent and efferent arterioles are regulated differently through selective expression of receptors and their signaling pathways (Chilton, et al., 2008; Loutzenhiser and Loutzenhiser, 2000; Nagahama, et al., 2000; Pollock, et al., 2005; Poulsen, et al., 2011), setting up the possibility for selectively manipulating tone in either vessel. For example, angiotensin II constricts both the afferent and efferent arterioles through activation of angiotensin AT1 receptors (Harrison-Bernard, et al., 2006; Paxton, et al., 1993); however, the signaling pathways leading to constriction appear to be different in the two vessels (Nagahama, et al., 2000). In the afferent arteriole angiotensin II-induced constriction is primarily mediated by inositol triphosphate (IP3) and L-type voltage-dependent calcium channels whereas constriction in the efferent arteriole is primarily mediated by store-operated calcium influx and protein kinase C (PKC) (Loutzenhiser and Loutzenhiser, 2000; Nagahama, et al., 2000). Also, products of cyclooxygenase released in response to angiotensin II moderate the vasoconstriction in the afferent arteriole but not in the efferent arteriole (Tang, et al., 2000). As with targeting endothelin receptors, targeting the renin-angiotensin system or angiotensin receptors during sepsis could negatively impact the systemic circulation. Still, the therapeutic potential of this approach was demonstrated with the AT1 receptor antagonist candesartan, where a dose, which did not decrease MAP, improved RBF and intrarenal PO2 in a rat model of LPS-induced sepsis (Nitescu, Grimberg, and Guron, 2008).
The potential for targeting adenosine receptors in sepsis-induced AKI has not specifically been tested yet. Adenosine receptors A1, 2A,2B,3 are expressed in the kidney; however, their anatomical locations and whether expression might be altered during sepsis are not completely understood (Bauerle, et al., 2011). Circumstantial evidence for a role of adenosine in sepsis-induced AKI comes from the findings that adenosine levels, as measured by microdialysis, increase in the kidney during LPS infusion and that the A1 antagonist FK352 can ameliorate the decline in RBF (Nishiyama, et al., 1999). Adenosine levels can rise in the kidney due to hypoxia and cellular stress, both of which increase intracellular and extracellular adenosine formation and decrease adenosine degradation (Bauerle, et al., 2011; Ramakers, et al., 2011). Dissecting out the role of adenosine specifically on the renal microcirculation during sepsis has been difficult because of the complex interactions between adenosine receptors not just in the kidney but also as regulators of the immune response. What is clear is that the model of sepsis used to evaluate the role of adenosine is critically important because adenosine can modulate inflammation and the inflammatory responses are different in LPS and live bacterial models such as CLP (reviewed in (Ramakers, et al., 2011). Also, the use of global adenosine receptor knockout mice to evaluate the role of adenosine in the kidney or any organ is problematic, again because of the influence of adenosine on the immune system (Gallos, et al., 2005; H. T. Lee, et al., 2006; Nemeth, et al., 2006). Pharmacological studies with available selective adenosine receptor antagonist and agonists could be used to address these issues. Unfortunately, these agents have been administered, thus far, either at the time of induction of sepsis or within a few hours of induction (Inoue, et al., 2011; H. T. Lee, et al., 2006; Nemeth, et al., 2006), but still within the time frame of the initial inflammatory response. This complicates the interpretation of the findings with respect to selective microcirculatory effects.
There has been a great deal of interest recently regarding the potential benefits of the calcium sensitizer and ATP-sensitive K+ (KATP) channel agonist levosimendan on systemic hemodynamics and even on the microcirculation during severe sepsis. The therapeutic potential of levosimendan as a single therapy or in combination with other inotropes or pressor agents is still controversial because its effects appear to be related to the model used. In porcine LPS infusion models of endotoxemia, levosimendan showed little improvements (Chew, et al., 2011; Cunha-Goncalves, et al., 2009); however, levosimendan attenuated the increase in pulmonary resistance, improved portal blood flow, and improved urine output following infusion of live E. coli in a porcine model (Garcia-Septien, et al., 2010). In the rat subjected to single injection of LPS, levosimendan did reduce serum creatinine, a marker of renal injury (Zager, et al., 2006). On the other hand, in the peritonitis models of sepsis, levosimendan was shown to be quite protective in sheep (Rehberg, et al., 2010) and in the rat (Scheiermann, et al., 2009). In addition to providing a level of cardiovascular support, levosimendan appears capable of also improving sublingual microvascular flow in patients with severe sepsis (Morelli, et al., 2010). Additional studies are needed to specifically address whether the KATP agonist activity of levosimendan could decrease renal vascular resistance since afferent and efferent arterioles express KATP channels (Ikenaga, et al., 2000; Reslerova and Loutzenhiser, 1995) and other inward rectifier K+ channels (Chilton, et al., 2008), which when activated could improve peritubular capillary perfusion.
Under conditions of severe sepsis and septic shock, the normally efficient autoregulation of RBF and GFR can fail if systemic blood pressure falls below the renal autoregulatory pressure. Nevertheless, sepsis-induced AKI cannot be explained by hemodynamic failure alone (prerenal-AKI), which supports the emerging view that renal microcirculatory failure and direct tubular injury, even after correction of systemic hemodynamics, contribute significantly to sepsis-induced AKI in humans (Wan, et al., 2008). Endothelial injury and loss of vascular reactivity (Lundy and Trzeciak, 2009; Zanotti-Cavazzoni, et al., 2009) are features of sepsis that can result in localized areas of microcirculatory hypoperfusion and hypoxia (Lundy and Trzeciak, 2009), which can lead to tubular epithelial injury. Still, care must be taken when targeting the microvasculature with vasoactive agents because the tubular epithelium may also express receptors for some of these agents. For example, receptors for angiotensin (Paxton, et al., 1993), adenosine (Di Sole, 2008) and endothelin (Wendel, et al., 2006) are expressed by the tubular epithelium. Consequently, agonists/antagonists could unpredictably be detrimental to tubular function. Moreover, the effects of targeting the microcirculation with agents that could impact systemic circulation will be difficult to predict since cardiovascular autoregulation is altered in these very ill patients and the generation of other vasoactive agents such as NO can impact cardiovascular responses (Boerma and Ince, 2010). Nevertheless, the unique anatomy of the renal microcirculation suggests that targeting the microcirculation to preserve/restore perfusion would not only lessen injury but also promote organ recovery (Ince, 2005; Le Dorze, et al., 2009), as discussed later.
4.2. Sphingosine-1-phosphate
Increased microvascular permeability is a key feature of sepsis, which can have detrimental effects on the microcirculation. Damage to the endothelial barrier can serve as an initiator of the activation of platelets and leukocytes, which can further damage the capillary permeability barrier and impede flow. Increases in microvascular permeability can also allow substances in the blood normally excluded to have direct contact with parenchymal cells.
There is growing evidence to suggest that sphingosine-1-phosphate (S1P), a lipid mediator derived from ceramide and sphingosine, plays a role in regulation of microvascular permeability. Two kinases, sphingosine kinase-1 (SphK1) and SphK2 phosphorylate sphingosine and are essentially the rate limiting steps in the generation of S1P. SphK1 and SphK2 are regulated differently and may have different cellular locations, depending on the cell type (Wattenberg, 2010). SphK1 activity is upregulated by hypoxia (Ader, et al., 2009; Schwalm, et al., 2008), ischemia-reperfusion (IR) in the kidney (Jo, et al., 2009; Park, et al., 2011), and during sepsis in inflammatory cells and lung (Puneet, et al., 2010; Wadgaonkar, et al., 2009). The regulation of SphK2 is less understood. It does not appear to be activated by hypoxia, at least in endothelial cells in vitro (Schwalm, et al., 2008), but it is upregulated in the liver by ischemia (Shi, et al., 2012). SphK2 is also upregulated in the lung in vivo by LPS but to a lesser extent compared to SphK1 (Wadgaonkar, et al., 2009). Nevertheless, evidence is accumulating to suggest that SphK2, located on the inner mitochondrial membrane, controls mitochondrial S1P generation, which can act to stabilize mitochondrial respiration (Strub, et al., 2011) and perhaps protect against mitochondrial injury during hypoxia.
S1P acts on the G-protein coupled receptors S1PR1–5 expressed on numerous cell types, reviewed recently by Edmonds (Edmonds, et al., 2011). Vascular endothelial cells, vascular smooth muscle cells and cardiac myocytes express S1PR1, S1PR2 or S1PR3 in various combinations, while S1PR4 is expressed mainly in immune cells and S1PR5 is expressed in cells of the nervous system (Takuwa, et al., 2008). S1PR1 are coupled to Gi (Garcia, et al., 2001), while S1PR2 and S1PR3 are coupled to Gi, Gq, and G12/13 (Murakami, et al., 2010; Sanchez, et al., 2007). The list of pharmacological tools for studying the roles of SphKs and S1PRs is growing (French, et al., 2010; Murakami, et al., 2010) and was recently reviewed (Huwiler and Pfeilschifter, 2008; Marsolais and Rosen, 2009). It is now possible to use selective agonists and antagonists of S1PRs and inhibitors of SphKs to study their roles in physiology and pathophysiology. One caveat is that targeting SphKs can also alter the levels of ceramide and its downstream signaling (Maceyka, et al., 2005; Wattenberg, 2010). Consequently, the effects of SphK inhibition will likely be organ and cell type specific and difficult to predict.
In the vasculature, S1P is an important regulator of microvessel development (Allende, et al., 2003) and the endothelial permeability barrier (M. J. Lee, et al., 1999). Stimulation of endothelial S1PR1 enhances the permeability barrier through the activation of Rac GTPase (Garcia, et al., 2001) leading to assembly of adheren and tight junctions, cytoskeletal reorganization and focal adhesion formation (reviewed in (L. Wang and Dudek, 2009)). In contrast, stimulation of S1PR2 decreases activation of Rac and activates Rho GTPase, leading to ROCK- and PTEN-dependent disruption of adherens junctions and increased paracellular permeability (Sanchez, et al., 2007). Less is known regarding the role of S1PR3 in microvascular permeability; however, S1PR3 activation can cause vascular smooth muscle contraction (Murakami, et al., 2010).
In renal IR injury, the roles of SphK2 and SphK1 are far from resolved. The absence of functional global SphK2 was found to be associated with a worsening of renal IR injury in one study (Jo, et al., 2009) but associated with a slight improvement in another (Park, et al., 2011). These opposite findings may be explained by differences in the IR models used and/or differences in the strains of knockout mice tested. In an elegant set of experiments Park et al. (Park, et al., 2011) used global SphK1 knockout mice, lentiviral gene delivery of SphK1 to the kidney, and selective antagonism to examine the roles of SphK1 and S1PR1 in renal IR injury. They presented convincing evidence that activation of SphK1 is protective through generation of S1P and stimulation of S1PR1. To directly address the role of tubular epithelial S1PR1 in renal IR, Bajwa et al (Bajwa, et al., 2010) selectively depleted S1PR1 in the mouse proximal tubular epithelium. These conditional knockout mice were more sensitive to renal IR injury. Furthermore, treatment with the S1PR1 agonist SEW2871 protected these mice from renal IR injury, establishing a role for tubular epithelial S1PR1 independent of lymphocyte S1PR1. Interestingly, renal epithelial cells express S1PR1–3 and S1PR1 is the most sensitive to upregulation by LPS (Bajwa, et al., 2010). Moreover, SEW2871 blocks LPS-induced tubular epithelial cell death in vitro (Bajwa, et al., 2010).
In a compelling set of experiments, Wadgaonkar et al. (Wadgaonkar, et al., 2009) demonstrated that SphK1 and SphK2 have distinct roles in sepsis-induced lung injury. They showed that global SphK1−/− mice were more susceptible to LPS-induced lung microvascular injury than wild-type mice or SphK2−/− mice. Moreover, SphK1 delivered to the lungs by adenoviral vector attenuated LPS-induced lung injury while delivery of SphK2 not only failed to protect but actually increased lung microvascular injury.
Platelets are a major source of extracellular S1P. Platelet activation and disseminated coagulation are common during sepsis and likely contribute to reduced microvascular perfusion (Tyml, 2011), so it is reasonable to assume S1P could be released in the microcirculation during sepsis and interact with capillary endothelial S1PRs. In an elegant set of in vitro and in vivo studies, Lee et al. (J. F. Lee, et al., 2009) showed that cremaster muscle venules, and senescent (but not young) endothelial cells in cultures express both S1PR1 and S1PR2 and that it is the relative levels of S1PR1 and S1PR2 activation that regulate microvascular permeability. Perfusion with S1P did not alter histamine-induced increase in vascular permeability. However, when S1PR2 were blocked with JTE-013, S1P reduced permeability elicited by histamine and when S1PR1 (and S1PR3) were blocked with VPC 23019, S1P increased the permeability elicited by histamine. Importantly, activation of S1PR1 with SEW2871 inhibited histamine-induced permeability. Zhang and coworkers (G. Zhang, et al., 2010) reported a similar protective effect of S1PR1 activation against bradykinin and platelet activation factor-induced increases in hydraulic conductivity in perfused rat mesenteric venules; however, they found no protective effect of S1PR2 blockade, suggesting the regulation of vessel permeability in capillaries and venules may be different.
The physiological roles of renal epithelial S1PR1–3 are still unclear; however, the S1PR1 antagonist W146 blocked the increases in sodium excretion and medullary blood flow produced by an infusion of the nonselective S1P agonist FTY720 into the renal medulla (Zhu, et al., 2011). Whether these effects are mediated exclusively by epithelial S1PRs or a combination of microvascular and epithelial S1PRs remains to be determined. As discussed earlier, renal microvascular leakage begins within hours following CLP in rat pups (Seely, et al., 2011) and in aged mice (Z. Wang, et al., PMID: 22119717). We have found that the S1PR1 agonist SEW2871 can significantly reduce microvascular leakage in the kidney in aged mice subjected to CLP (unpublished observations). Consequently, selective agonists or antagonists could be effective pharmacological tools to help maintain the renal microvascular permeability barrier during sepsis.
4.3. Oxidant generation
It has been recognized for some time that sepsis is associated with oxidative stress (Macdonald, et al., 2003) as evidenced, in part, by increased oxidative stress markers in severely ill septic patients (Goode, et al., 1995). Hence, oxidative stress is an additional hallmark of sepsis suggested to link microvascular failure and parenchymal cell injury (Boueiz and Hassoun, 2009; Galley, 2010; Spanos, et al., 2010; Tyml, 2011). While not yet demonstrated in septic patients, animal studies indicate that changes in overall flow or even just redistribution of flow through the renal microcirculation during sepsis result in areas of localized hypoxia (Z. Wang, et al., PMID: 22119717; Yasuda, et al., 2006). Moreover, localized areas with decreased perfusion are correlated spatially with tubular epithelium under redox stress (Z. Wang, et al., PMID: 22119717; L. Wu and Mayeux, 2007).
Importantly, reduced peritubular capillary perfusion is not a requirement for sepsis-induced renal epithelial oxidative stress. In a remarkable set of experiments, Kalakeche and co-workers (Kalakeche, et al., 2011) utilized 2-photon microscopy of the kidney in live mice to reveal that the S1 segment of the proximal tubule internalizes LPS through a TLR4-dependent process, which then causes oxidant production in neighboring S2 segments. The authors propose the cross-talk between the S1 and S2 segments may be due to cytokines released from the S1 segment, which in turn causes oxidant generation in the downstream S2 segment but this hypothesis needs to be validated. Our laboratory has utilized in vitro models of cultured mouse renal epithelial cells exposed to serum from septic (CLP-treated) mice and found that CLP serum caused increased oxidant production and mitochondrial injury compared to cells treated with serum collected from non-septic (sham surgery) mice (Pathak, et al., 2012; Pathak and Mayeux, 2010). Collectively, these findings show that the renal epithelium can be directly injured by blood-born factors released during sepsis, which reach the tubular epithelium from the glomerular filtrate or from peritubular capillary leakage into the interstitial space. Importantly, human septic serum was shown to inhibit cultured endothelial cell mitochondrial respiration (Boulos, et al., 2003). So while reduced microvascular perfusion is not required for oxidant production, at least in the kidney, studies are needed to specifically address whether hypoxia might augment mitochondrial damage and oxidant production leading to more renal epithelial and endothelial cell injury.
An increase in nitric oxide (NO) synthesis through activation of constitutive nitric oxide synthases (NOS) and induction of inducible NOS (iNOS) is another key feature of sepsis, which contributes to hemodynamic failure as well as NO-derived RNS. NO synthesis via iNOS is increased in the kidney of septic patients and is proposed to contribute to the development of AKI (S. Heemskerk, et al., 2009; S. Heemskerk, et al., 2006; S Heemskerk, et al., 2004). Induction of iNOS in the kidney also occurs in rodents during sepsis (Seely, et al., 2011; L. Wu, Gokden, et al., 2007; L. Wu, Tiwari, et al., 2007) and in the tubular epithelium in in vitro models of sepsis (Pathak, et al., 2012; Pathak and Mayeux, 2010), where iNOS-derived NO contributes to the generation of RNS by the renal tubules.
As discussed earlier, oxidative stress has been suggested to contribute to organ injury during sepsis. Key oxidants thought to be important are the O2-derived ROS superoxide radical [O2.−], hydroxyl radical [.OH] and hydrogen peroxide [H2O2], and the NO-derived RNS peroxynitrite [ONOO−], the product of the reaction between superoxide and NO. It is important to note that superoxide is the proximal oxidant species, and is involved in the formation of other oxidants (Figure 2). Multiple, diverse cellular systems produce superoxide, but levels generally are kept in the nanomolar range by superoxide dismutases (SOD). One major source of superoxide production is within the mitochondria via the respiratory chain (Crouser, 2004; Galley, 2010). Other intracellular sources of superoxide include its production via NADPH oxidase (Griendling, et al., 1994; W. Wang, et al., 1994; Zulueta, et al., 1995) and even NOS expressed by the renal tubular epithelium (Traylor and Mayeux, 1997).
Figure 2.

Schematic showing the synthesis of oxidant derived from NO and superoxide.
MnSOD (sometimes referred to as SOD2) is a major antioxidant in the mitochondria. Its main function is to catalyze the dismutation of superoxide, which is continually generated (~1–2% of total oxygen consumption) via uncoupling in the electron transport chain and from other mitochondrial sources (Poyton, et al., 2009). MnSOD is a nuclear-encoded protein that is transported into the mitochondria via an amino-terminal targeting sequence, and is subsequently cleaved to form its native homotetrameric structure of 96 kDa. Studies with MnSOD knockout mice provide unequivocal evidence that MnSOD is essential for life, a finding that presumably extends to humans (Lebovitz, et al., 1996; Y. Li, et al., 1995). Our laboratory demonstrated a 50% reduction in MnSOD activity in kidneys exposed to warm or cold IR (Cruthirds, et al., 2003; Saba, et al., 2008). Moreover, cold ischemia can disrupt mitochondrial complex I and II activities and increase mitochondrial superoxide production (Mitchell, et al., 2011). Whether or not SOD activities are altered in the kidney during sepsis remains unclear. Protein levels of Cu-Zn SOD (SOD1) (Leach, et al., 1998) and extracellular SOD (SOD3) (W Wang, et al., 2003) and the activity of total SOD (Mitra, et al., 2007) are decreased in the kidney following LPS-induced sepsis and we have found that the activity of MnSOD in the kidney is decreased within a few hours following CLP (unpublished data), which could be a result of peroxynitrite inactivation (MacMillan-Crow, et al., 1996; MacMillan-Crow and Thompson, 1999).
It is also important to recognize that superoxide is charged and hence its diffusion is limited, whereas NO is uncharged and readily diffuses through membranes. Consequently, the diffusion of NO into a microenvironment where superoxide is produced would result in peroxynitrite formation (Beckman and Crow, 1993; Huie and Padmaja, 1993). In this regard, peroxynitrite formation could be considered to be more a function of local superoxide production than of NO concentration per se. Unlike NO and superoxide, peroxynitrite reactivity leaves a “footprint,” nitrated proteins, which can be detected immunologically. Our laboratories have previously implicated peroxynitrite in sepsis (L. Wu, Gokden, et al., 2007; L. Wu and Mayeux, 2007; Zhang, Walker, Hinson, et al., 2000; Zhang, Walker, and Mayeux, 2000) and IR renal injury (Cruthirds, et al., 2003; L. M. Walker, et al., 2000; L. M. Walker, et al., 2001).
With all of the apparent detrimental effects NO could exert during sepsis, targeting NO would seem to be a logical therapeutic approach. However, non-selective inhibition of constitutive NOS and iNOS worsened some hemodynamic parameters and actually increased mortality in septic patients (Lopez, et al., 2004). Preclinical studies targeting iNOS-derived NO with iNOS selective inhibitors suggest that this approach can improve renal function and increase survival by preserving the beneficial effects of constitutively generated NO (reviewed in (S. Heemskerk, et al., 2009); however, not all iNOS inhibitors exhibit the same predicted beneficial effects (Cuzzocrea, et al., 2006; W Wang, et al., 2003). The iNOS inhibitor, L-N6 -(1-iminoethyl)-lysine (L-NIL), has been frequently tested in both LPS and CLP model of sepsis-induced AKI and is generally effective at improving serum markers of AKI in rodent models of LPS- and CLP-induced AKI (Tiwari, et al., 2005; L. Wu, Gokden, et al., 2007; Zhang, Walker, and Mayeux, 2000) and improving peritubular capillary perfusion (Tiwari, et al., 2005; L. Wu, Gokden, et al., 2007). L-NIL also reduces RNS generation by the renal tubules during sepsis (L. Wu, Gokden, et al., 2007; L. Wu and Mayeux, 2007; Zhang, Walker, and Mayeux, 2000), suggesting that iNOS-derived NO contributes significantly to the development of oxidative stress in the kidney.
Superoxide is another logical target and there are effective scavengers, which have been tested in animal models. These agents may have added benefits because they are not completely selective for superoxide and can scavenge other ROS and RNS. One such compound is tempol, a nitroxide scavenger recently reviewed thoroughly by Wilcox (Wilcox, 2010). Tempol has been shown to improve renal injury in both the LPS (Leach, et al., 1998; W Wang, et al., 2003) and CLP (Liaw, et al., 2005) models of sepsis. Ethyl pyruvate is another more generalized ROS scavenger with greater selectivity for hydrogen peroxide and hydroxyl radical than superoxide (Kao and Fink, 2010). Both ethyl pyruvate (Miyaji, et al., 2003) and a more stable analogue, methyl-2-acetamidoacrylate, (Leelahavanichkul, et al., 2008) protected the kidney from CLP-induced AKI. Importantly, those researchers use a clinically relevant dosing paradigm – therapy begun only after the onset of symptoms – by starting treatment with these agents 6 or 12 hours post CLP. Two other more generalized anti-inflammatory agents, AP214, an α-melanocyte-stimulating hormone analogue (Doi, et al., 2008), and chloroquine (Yasuda, et al., 2008) also protected against CLP-induced renal injury even with delayed treatment and also improved survival.
Until recently, evidence of superoxide generation in the peritubular microenvironment during sepsis was only inferred from the protective effects of scavengers and the presence of nitrated proteins, indicating peroxynitrite formation. Kalakeche et al. (Kalakeche, et al., 2011) used dihydroethidium (DHE), a putative selective probe for superoxide to demonstrate superoxide generation in renal tubules within hours following LPS administration using intravital microscopy. We used intravital microscopy and the mitochondria-targeted superoxide selective probe MitoSox Red (a derivative of DHE) to look for superoxide generation in the renal tubules following CLP (Z. Wang, et al., PMID: 22119717) and found superoxide generation occurred as early as 4 hours post CLP surgery, which correlated temporally with the decline in peritubular capillary perfusion but was delayed relative to the development of capillary leakage.
We also used the RNS-selective probe dihydrorhodamine 123, which is oxidized to rhodamine preferentially by peroxynitrite and possibly by other RNS and ROS but not by superoxide (Crow, 2000; Gomes, et al., 2006) to detect RNS generation using intravital microscopy (Figure 3). CLP also resulted in a time-dependent increase in rhodamine fluorescence in renal tubules that, like superoxide generation, was spatially correlated to tubules bordered by capillaries with reduced perfusion and confirmed by immunolocalization of nitrotyrosine-protein adducts in the tubular epithelium (Z. Wang, et al., 2011; Z. Wang, et al., PMID: 22119717; L. Wu, Gokden, et al., 2007; L. Wu and Mayeux, 2007). Since superoxide and peroxynitrite are generated by the renal tubules during sepsis, it is reasonable to assume that there may be therapeutic potential in targeting these oxidants. Metalloporphyrins, which are superoxide dismutase (SOD) mimetics and peroxynitrite scavengers have been tested in sepsis models and have been shown to reduce vascular dysfunction (Cuzzocrea, et al., 2006; Nin, et al., 2011; Zingarelli, et al., 1997), preserve mitochondrial energetics in the heart and diaphragm (Nin, et al., 2004), and preserve GFR and RBF (W Wang, et al., 2003) when animals were pretreated with these agents. We treated mice with the SOD mimetic and peroxynitrite scavenger, Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin, tetratosylate, hydroxide (MnTMPyP) 6 hours after CLP, a time when RBF, GFR, and capillary perfusion are already greatly reduced and a time when superoxide and RNS generation are already greatly elevated (Z. Wang, et al., PMID: 22119717). Even with this delayed treatment protocol, MnTMPyP blocked subsequent oxidant generation and actually reversed the decline in RBF, GFR, capillary perfusion, and preserved tubular morphology at 18 hours, all without increasing MAP. Importantly, delayed treatment with MnTMPyP also prolonged survival. To more directly address the role of peroxynitrite, we tested the peroxynitrite scavenger resveratrol (Holthoff, et al., 2010) and found that it also reversed the decline RBF, GFR, capillary perfusion, and preserved tubular morphology even when using a delayed, clinically relevant dosing protocol. Moreover, resveratrol was also capable of acutely (within 30 minutes) improving RBF and GRF (Holthoff, et al., PMID: 215975863), while MnTMPyP could not (Z. Wang, et al., PMID: 22119717). This finding suggests resveratrol may have reduced renal vascular resistance since resveratrol is known to activate potassium channels (Gojkovic-Bukarica, et al., 2008; Novakovic, et al., 2006), which are present in both afferent and efferent arterioles (Chilton, et al., 2008) as discussed earlier. As with MnTMPyP, delayed therapy with resveratrol also prolonged survival in mice subjected to CLP (Holthoff, et al., PMID: 215975863). However, the dual action of resveratrol, reducing renal vascular resistance and peroxynitrite scavenging activity, could offer added clinical benefits.
Figure 3. Imaging of RNS generation in renal tubules with intravital microscopy.

Shown are representative images of perfusion (A) and rhodamine fluorescence (B) captured from videos of the same field of view from the kidney of a live 40-week-old mouse 16 hours after CLP. Arrows indicate capillaries with no perfusion. A pseudocolored image of B is shown in C to highlight changes in pixel intensity. Intense regions of rhodamine fluorescence are localized to discrete regions of the tubular epithelium bordered by capillaries with reduced perfusion (from (L. Wu, Gokden, et al., 2007) used with permission).
MitoQuinone or MitoQ comprises the ubiquinone antioxidant moiety of coenzyme Q10 covalently attached to a lipophilic triphenylphosphonium cation, which directs the compound to the mitochondria (Murphy and Smith, 2007). Because of the large mitochondrial membrane potential, MitoQ can accumulate within mitochondria up to 1,000 fold, compared to non-targeted antioxidants. MitoQ was shown to reduce renal and liver injury in a rodent model of bacteria sepsis (Lowes, et al., 2008) and protect against damage observed following rodent cardiac IR (Adlam, et al., 2005). The selective uptake of MitoQ into mitochondria should greatly enhance its efficacy and specificity while decreasing potential side effects. For more information on publications that have tested experimental treatments for mitochondrial damage during sepsis please refer the review by Dare et al. (Dare, et al., 2009).
In addition to directly injuring the tubular epithelium, oxidants can negatively impact the peritubular microenvironment in a number of other ways. For example, ROS and RNS can activate latent matrix metalloproteinases MMPs (Gu, et al., 2002; Okamoto, et al., 2001) causing damage to tubular and capillary basement membranes, as discussed later. Peroxynitrite generated in the microvascular microenvironment can also directly damage the endothelial permeability barrier (Knepler, et al., 2001; F. Wu and Wilson, 2009) promoting leukocyte infiltration and allowing plasma constituents to enter the interstitial space and contact the tubular epithelium (Boueiz and Hassoun, 2009; Sutton, 2009). Superoxide and perhaps peroxynitrite are reported to mediate, at least in part, the renal vasoconstrictor responses to endothelin and angiotensin II as well as basal tone (Just, et al., 2008; Madrid, et al., 1997; Matavelli, et al., 2009).
4.4. Mitochondrial dysfunction
As discussed earlier, it has been suggested that oxidative stress might play a causal role in mitochondrial dysfunction in sepsis (Dare, et al., 2009; R. J. Levy, 2007). Pro-inflammatory mediators as well as oxidative stress impair the function of mitochondrial respiratory complexes and also cause structural damage to mitochondrial lipids, proteins and DNA, thereby impairing cellular ATP production (Galley, 2011). Human skeletal muscle biopsies show that ATP and mitochondrial complex I activity is lowered in patients with severe sepsis (Brealey, et al., 2002; Svistunenko, et al., 2006). The majority of these publications have shown sepsis induced cardiac and hepatic mitochondrial dysfunction leading to organ injury, but the role of mitochondrial dysfunction in sepsis induced AKI is not as well understood. A very recent paper demonstrated that renal mitochondrial electron transport complexes I and IV were decreased 18 hours after CLP (Tran, et al., 2011).
Mitochondrial electron complex inactivation is known to increase oxidant production; however, the molecular events, which induce mitochondrial complex damage are not well understood. The reader is directed to Figure 4 for a schematic showing the complexes involved with generation of the proton gradient (H+ within the intermembrane space) and mitochondrial electron transport. It is well established that superoxide is generated at multiple complexes including I and III (reviewed in (Turrens, 2003)). Thus, it is clear that mitochondrial damage can lead to increased mitochondrial superoxide production and under conditions where MnSOD is inactive superoxide accumulates, leading to more ROS and RNS formation resulting in further injury. What is unclear is the cause of the initial mitochondrial injury and whether is occurs late enough in the development of sepsis-induced AKI to be a clinically relevant target.
Figure 4.
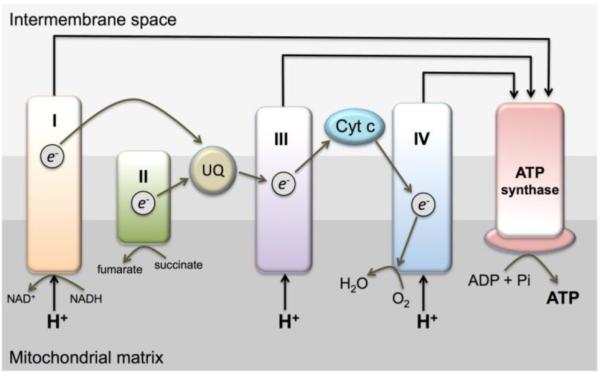
Depiction of the mitochondrial electron transport chain.
One hypothesis currently being tested is that complex IV (cytochrome oxidase) may serve as a mitochondrial sensor for the extent of damage taking place during sepsis (Chvojka, et al., 2010; R. J. Levy, et al., 2004). The idea is that when complex IV is inhibited, ATP production is reduced, which may put mitochondria in a `hibernation' mode that reduces the energy demand of the particular organ under stress. Furthermore, this period may allow for adaptive mechanisms to allow recovery or repair of mitochondria, which could in turn lead to improved organ function (Chvojka, et al., 2010; Ruggieri, et al., 2010). This theory could even explain why in many cases overt histological damage is not observed during sepsis, especially with regard to the kidney (Hotchkiss, et al., 1999).
Multiple copies of mitochondrial DNA (mtDNA) are present in each mitochondrion and remain in close proximity to the electron transport chain, where ROS are generated. Consequently, they are key targets of ROS-mediated damage within mitochondria. The mtDNA encode for two ribosomal RNAs, 22 transfer RNAs, and 13 electron transport chain proteins. In addition, mtDNA have limited repair ability and replication of cells with damaged mtDNA is not inhibited by cell-cycle checkpoint control mechanisms. Thus, damage to mtDNA can result in accumulation of mutations and the production of dysfunctional electron transport proteins, which in turn may produce more ROS and initiate a damaging cycle. It has been demonstrated that mtDNA copy number is reduced in the liver and heart following LPS administration, and that this is accompanied by increased oxidative stress (Suliman, Carraway, and Piantadosi, 2003; Suliman, et al., 2004). The extent of mtDNA damage in CLP-mediated sepsis remains unknown.
4.5. Metalloproteinases
Basement membranes in the kidney are not simply anchors for the basal plasma membranes of parenchymal cells. They also serve a number of critical functions including helping to establish polarity of the tubular epithelium, which are attached to the tubular basement membrane (TBM) and functioning as part of the permeability-selective barrier of the glomerulus through attachments of the endothelial cells and podocytes to the glomerular basement membrane (GBM). The TBM is a specialized form of the extracellular matrix composed primarily of type IV collagen, laminin, proteoglycans, nidogen, and fibronectin (Abrahamson and Leardkamolkarn, 1991). It is the relative activities of the matrix-digesting MMPs and their endogenous inhibitors, the tissue inhibitors of metalloproteinases (TIMPs) that regulate destruction and subsequent remodeling of the extracellular matrix (Catania, et al., 2007). Consequently, deregulation of the equilibrium between synthesis of the extracellular matrix and its degradation can lead to tubulointerstitial fibrosis and promote the progression of chronic kidney disease (Ronco and Chatziantoniou, 2008; Zheng, et al., 2009). Extracellular matrix (ECM) proteins are not the only substrates for the MMPs. Peptide growth factors, cytokines and chemokines, and cell-adhesion molecules are additional potential substrates. Hence, MMPs can directly and indirectly influence a variety of pathological processes (reviewed in (Page-McCaw, et al., 2007)).
The 24 mammalian MMPs are members of a zinc-containing subfamily (metzincin) of metalloproteinases that can degrade structural components of the extracellular matrix. Members of the metzincin family include the serralysins, astacins, ADAMs (A Disintegrin And Metalloproteinase), and matrixins (the MMPs). MMPs can be classified based on their structural and substrate specificity into five groups: collagenases, gelatinases, stromelysins, minimal or matrilysins, and membrane types. Latent or proforms of MMPs become activated through proteolytic cleavage of the prodomain or non-proteolytic activation through disruption of the zinc-thiol interaction between the pro- and active domains followed by autolysis to the active form (Ra and Parks, 2007). This latter mechanism can occur through allosteric interactions or through modification of the critical thiol by oxidants such as peroxynitrite and via S-nitrosylation as demonstrated for MMP-1, -8 and -9 (Gu, et al., 2002; Okamoto, et al., 2001). This method of activation may be especially relevant in sepsis-induced AKI because oxidants are generated in the peritubular microenvironment during sepsis, as discussed later.
Much of what is known regarding the consequences of damage to the ECM and TBM comes from studies of renal IR injury (Catania, et al., 2007; Molitoris and Marrs, 1999). The second-generation tetracycline antibiotic, minocycline, is an inhibitor of MMPs among its other activities (Dejonckheere, et al., 2011) and not only protects the renal tubular epithelium from IR injury in rats and mice (Kelly, et al., 2004; Kunugi, et al., 2011) but also reduces microvascular leakage (Sutton, et al., 2005). To directly address the roles of MMP-2 and MMP-9, MMP-2−/− and MMP-9−/− mice have been studied following renal IR. Interestingly, only the MMP-2−/− mice showed reduced functional and morphological renal injury at 24 hours post IR (Kunugi, et al., 2011; S. Y. Lee, et al., 2011). Renal IR injury in MMP-9−/− mice was similar to that in wild-type controls in the acute phase of injury but at four weeks after IR injury there appeared to be a preservation or regeneration of the peritubular microvascular density suggesting that MMP-9 activation may have a negative impact on peritubular capillary maintenance (S. Y. Lee, et al., 2011).
The susceptibility of MMP knockout mice to sepsis-induced organ injury has not been tested so the importance of specific MMPs in the development of renal microvascular failure and AKI during sepsis remains unanswered. Nevertheless, there is circumstantial evidence to suggest that MMPs and other metalloproteinases should be evaluated as possible therapeutic targets. The MMP-9 activity is increased in the plasma of LPS-treated rats and the MMP inhibitor doxycycline reduces the aortic hyporeactivity associated with endotoxemia (Cena, et al., 2010). Certain MMPs also appear in the plasma of septic patients. Notably, protein levels for MMP-3, -7, -8 and -9 but not MMP-2 (Lauhio, et al., 2011; Muhl, et al., 2011; Yazdan-Ashoori, et al., 2011) have been reported to be elevated in patients with severe sepsis upon admission to the ICU. Moreover, plasma levels of MMP-8 were significantly higher in non-surviving patients than in survivors (Yazdan-Ashoori, et al., 2011). Still, cytokines and the associated inflammatory response can induce MMP synthesis and release. Since MMPs can be pro- or anti-inflammatory (Page-McCaw, et al., 2007), it is difficult to interpret of the consequences of changes in plasma levels of MMPs alone (Yassen, et al., 2001).
Additional findings, which suggest activation of metalloproteinases may be playing a role in sepsis-induced AKI come from evaluation of tubular morphology and the appearance in the urine of the tubular injury biomarker, kidney injury molecule-1 (KIM-1). Sepsis-induced tubular injury is characterized by loss of brush-border membrane, tubular swelling and epithelial cell detachment with measurable apoptosis but generally no or very mild necrosis (Guo, et al., 2004; Miyaji, et al., 2003; Tiwari, et al., 2005; L. Wu, Gokden, et al., 2007). KIM-1 is a membrane glycoprotein released from the tubular epithelium by metalloproteinases (Bailly, et al., 2002; Han, et al., 2002) and appears in the urine of patients with ischemic, nephrotoxic and sepsis-induced AKI (Liangos, et al., 2007), suggesting the activation of renal metalloproteinases in these forms of AKI.
The perivascular matrix is composed primarily of collagen IV, a substrate of MMP-2 and MMP-9 as well as other metalloproteinases such as the meprins (Kaushal, et al., 1994; Sterchi, et al., 2008). What is significant with regard to the kidney is that meprin A, a hetero-oligomeric protein composed of both α- and β-subunits, is estimated to comprise as much as 5% of the proximal tubule brush-border membrane protein (Craig, et al., 1987; Kaushal, et al., 1994). In addition to acting on collagen IV, laminin, fibronectin, and nitogen (Kaushal, et al., 1994; P. D. Walker, et al., 1998) meprin can also degrade the cytoskeletal proteins, villin and actin, present in the brush-border membrane (Ongeri, et al., 2011). Meprins are the only astacins metalloproteinases that can function attached to the membrane as well as extracellularly (Sterchi, et al., 2008). Consequently, they can exert local and perhaps more remote effects. This property is especially relevant with regard to sepsis because meprin A acts on pro-IL-1β to generate active IL-1β, a key pro-inflammatory cytokine released early during sepsis (Ebong, et al., 1999; P. Li, et al., 1995). Actinonin, a naturally occurring hydroxamate found in actinomycetes and a potent inhibitor of meprin A (Kruse, et al., 2004), protects against renal IR in rats (Carmago, et al., 2002) and cisplatin-induced nephrotoxicity in mice (Herzog, et al., 2007). Actinonin administered at the time of CLP is also protective against sepsis-induced AKI (Holly, et al., 2006). Moreover, meprin-α knockout mice are protected against LPS-induced AKI (Yura, et al., 2009). While these reports suggested a role for meprin in sepsis-induced tubular injury they did not address the mechanism of protection. As discussed earlier, a delayed treatment paradigm is the mostly clinically relevant approach to establishing therapeutic potential of a particular agent. This is especially relevant for the evaluation of actinonin since when administered at the time of the induction of sepsis it inhibits the generation of active IL-1 β and thereby reduces the inflammatory response (Z. Wang, et al., 2011; Yura, et al., 2009). To address more directly whether actinonin could protect the kidney during sepsis, Wang et al (Z. Wang, et al., 2011) administered actinonin at 7 hours after CLP in mice. At this time TNF-α and IL-1β were already elevated (Miyaji, et al., 2003; Pathak and Mayeux, 2010) and peritubular capillary perfusion was already compromised (Z. Wang, et al., 2011). Actinonin not only preserved tubular morphology and renal function but also reversed the decline in peritubular capillary perfusion. Agents targeting meprin A could offer real advantages in treating sepsis because they would target both the early IL-1β generation and the later tubular injury. Nevertheless, these findings only support the notion that activation of meprin and perhaps other metalloproteinases participate in the development of tubular injury during sepsis. Further studies are needed using other inhibitors to better evaluate the metalloproteinases as therapeutic targets.
Since peritubular capillaries and tubular epithelium are held together in close contact by the ECM it could be expected that damage to either or both could lead to cross-talk with interstitial cells such as fibroblasts and dendritic cells (Kaissling and Le Hir, 2008; Lemley and Kriz, 1991) through the generation of signaling molecules derived from the ECM (Basile, et al., 2004; Pitera, et al., 2004). Thus, the challenge becomes identifying which signaling pathways augment injury and which might facilitate recovery.
5. Therapies targeting renal recovery
Agents that have been shown to be effective when administered after the onset of renal injury suggest that targeting recovery may also have an important place in therapy. Data from the CLP model suggest that preventing the progression of microcirculatory failure (Doi, et al., 2008) or tubular injury (Holthoff, et al., PMID: 215975863; Miyaji, et al., 2003; Z. Wang, et al., 2011; Z. Wang, et al., PMID: 22119717) may actually allow the kidney to recover. The mechanism of recovery of the microcirculation is unclear but may be related to the protection against oxidant-induced tubular epithelial damage. As the epithelial cells become injured, release mediators, and the tubules swell, peritubular capillary perfusion will likely be impeded further. This could augment local hypoxia and oxidant generation to exacerbate tubular injury and perpetuate capillary dysfunction (Holthoff, et al., PMID: 215975863; Z. Wang, et al., PMID: 22119717; Yasuda, et al., 2006). The mechanism of recovery of the tubular epithelium is also unclear but likely related to reduced oxidative stress, improved mitochondrial function, and preservation of the extracellular matrix (Holthoff, et al., PMID: 215975863; Miyaji, et al., 2003; Z. Wang, et al., 2011; Z. Wang, et al., PMID: 22119717).
Autophagy is a process by which a cell degrades and recycles its own nonessential components. This process can be triggered by variety of stressors including those produced by oxidants or limited nutrients. Consequently, autophagy is generally considered an adaptive response, which could help preserve organ function acutely and allow for recovery later; however, overstimulation of autophagy can lead to cell death (Y. C. Hsieh, et al., 2009), which could delay recovery. Watanabe and coworkers (Watanabe, et al., 2009) recently presented convincing morphological evidence of autophagy in liver biopsies from septic patients. It is unknown if autophagy occurs in the kidneys of septic patients or septic animals. Jiang et al. (Jiang, et al., 2010) used pharmacological inhibitors of autophagy and showed that renal IR injury was significantly worsened when autophagy was blocked. Kimura et al. (Kimura, et al., 2011) confirmed the protective role of autophagy in renal IR injury using conditional Atg5 gene deletion to block autophagy in the renal proximal tubule. They found that these mice developed a more severe renal injury. Together these studies suggest that renal epithelial autophagy may be an adaptive response during periods of hypoxia and/or oxidative stress; however, the mechanism of how autophagy might preserve renal function or promote recovery is unknown.
There is increasing interest in the notion that mitochondrial repair and biogenesis may play important roles in the recovery of AKI (Funk, et al., 2010; Rasbach and Schnellmann, 2007; Weinberg, 2011) since protecting the mitochondria could promote tubular regeneration as suggested in the recovery of renal IR injury (Szeto, et al., 2011). Proliferation of rat proximal tubule epithelium (S3 segments) can occur relatively rapidly after injury due to a large fraction (~40%) of epithelial cells in the G1 phase (Vogetseder, et al., 2008). In an elegant set of experiments Humphreys et al. (Humphreys, et al., 2011) showed that injured and dedifferentiated proximal tubule epithelial cells, rather than uninjured bystanders or progenitor cells, proliferate to repopulate the tubular epithelium following renal IR injury.
The safe removal of damaged mitochondrial (mitophagy) as well as synthesis of new mitochondria (biogenesis) are important events that can lead to recovery from septic organ damage. This has been shown to occur in the liver (Bartz, et al., 2011; Suliman, Carraway, Welty-Wolf, et al., 2003; Sweeney, et al., 2010) and heart (Lancel, et al., 2009); however, the role of mitophagy/biogenesis has not been well studied in sepsis-induced AKI. A recent paper by Tran et al. (Tran, et al., 2011) showed that mitochondrial biogenesis was important for potential recovery of renal function following LPS-induced sepsis. Specifically, this group determined that PPARγ coactivator 1α (PGC-1α), a critical regulator of mitochondrial biogenesis, needs to be induced in order to achieve recovery. Others have shown that increased mitochondrial ROS production leads to mitochondrial damage but also appears to induce mitochondrial biogenesis or repair (R. J. Levy and Deutschman, 2007; Reynolds, et al., 2009; Suliman, Carraway, Welty-Wolf, et al., 2003). Interestingly, iNOS appears to be necessary for efficient hepatic mitochondrial biogenesis to take place following LPS mediated sepsis (Reynolds, et al., 2009).
It would be logical to attempt to induce both mitophagy and biogenesis following sepsis as a possible therapeutic strategy. A recent paper using a CLP model showed that rapamycin (the classic mTOR inhibitor and autophagy inducer) provided cardioprotection (C. H. Hsieh, et al., 2011). However, rapamycin was given only 1 hour following CLP, a more delayed treatment would enhance the therapeutic relevance of these findings. In addition, resveratrol and carbon monoxide releasing compounds have been shown to induce mitochondrial biogenesis (Csiszar, et al., 2009; Lagouge, et al., 2006; Lancel, et al., 2009). As mentioned earlier, our laboratory recently demonstrated that resveratrol provided significant protection in a murine CLP model of severe sepsis (Holthoff, et al., PMID: 215975863), studies are underway to determine the possible role of mitochondrial biogenesis had on the protection observed. Other studies have documented protection against organ mediated septic damage with resveratrol but have not evaluated the role of mitochondrial biogenesis as a possible mechanism of action (Kolgazi, et al., 2006; Sebai, et al., 2008; Sebai, et al., 2011).
In a relatively mild sepsis model of peritonitis induced by a S. aureus-impregnated fibrin clot, mitochondrial biogenesis in the liver was shown to help restore oxidative metabolism and suggested to be a prosurvival mechanism to recover organ function (Haden, et al., 2007). This study raises the possibility that mitochondrial biogenesis or at least preservation of mitochondrial function could be an important therapeutic approach in the treatment of sepsis. Along similar lines, there is also growing interest in the role of sirtuin-1 (SIRT1), an NAD+-dependent histone deacetylase and its possible role in inflammatory conditions such as sepsis (Hu, et al., 2011; Z. Zhang, et al., 2010). SIRT1 levels decline rapidly in the liver following LPS administration in mice and this is accompanied by an increase in the autophagy indicator LC3-II (Z. Zhang, et al., 2010). Interestingly, SIRT1 levels increase and LC3-II levels decrease in the recovery phase, which is associated with a restoration of ATP levels in the liver. Chronic treatment with resveratrol was able to block the early changes in both SIRT1 and LC3-II and preserve liver ATP levels, suggesting a link between the protective effects of resveratrol and modulation of SIRT1 and autophagy, as detailed in a recent review (Chung, et al., 2010). Unfortunately, the acute effects of resveratrol were not tested in this sepsis model and the kidney was not examined for evidence of autophagy.
The ability of resveratrol to reverse peritubular capillary dysfunction and preserve renal function as discussed earlier (Holthoff, et al., PMID: 215975863) could be a result of resveratrol's ability to acutely scavenge RNS and then facilitate peritubular capillary endothelial cell and epithelial cell recovery. This could occur through interactions with SIRT1 leading to upregulation of MnSOD and catalase (Chung, et al., 2010). This would reduce mitochondrial oxidant generation and possibly induce mitochondria biogenesis (Beeson, et al., 2010; Csiszar, et al., 2009; Funk, et al., 2010; Ungvari, et al., 2009). Resveratrol has been considered a direct activator of SIRT1 but recent evidence disputes this (Pacholec, et al., 2010). Nevertheless, much of the renal protective activities attributed to chronic resveratrol dosing appear to be mediated, at least in part, by SIRT1 (He, et al., 2010; Kim, et al., 2011). Studies are needed to specifically examine the acute effects of resveratrol and agents like it on autophagy and mitochondrial biogenesis in the recovery phase of sepsis-induced AKI.
6. Conclusions
It is clear from both clinical observations and animal studies that sepsis-induced AKI is not due to hemodynamic failure alone (prerenal-AKI). It is now generally accepted that renal microcirculatory failure and direct tubular injury contribute significantly to sepsis-induced AKI in humans (Wan, et al., 2008) and that targeting the early hemodynamic failure with the goal to maintain organ perfusion (M. M. Levy, et al., 2010) is not always effective in preserving organ function because the effects of the initial hemodynamic insult may be difficult to prevent or reverse (Stearns-Kurosawa, et al., 2011). Impaired microcirculation (depicted as reduced flow and increased permeability in Figure 5) is a very early event in sepsis-induced AKI leading to subsequent mitochondrial damage and a pro-oxidant peritubular microenvironment, which promotes tubular epithelial injury. Targeting these later events is key because therapies targeting the initial capillary injury alone may have limited therapeutic value. As discussed in this review there have been studies that have specifically addressed the therapeutic potential of delayed treatments designed to break the cycle of injury (Figure 6) by targeting oxidants and metalloproteinases to allow the renal microcirculation and tubular epithelium to recover and restore renal function. Another very intense area of research is to identify agents that can promote repair through induction of autophagy/mitophagy as well as mitochondrial biogenesis, which could serve to limit sepsis-mediated renal injury.
Figure 5. Changes in the peritubular capillary microenvironment during sepsis.

Depicted in A is the normal peritubular capillary microenvironment. The early impaired microcirculation with increased capillary permeability and decrease capillary perfusion are depicted in B. Subsequent tubular epithelial damage is depicted in C.
Figure 6. Sepsis-induced cycle of injury in the peritubular microenvironment.

Green arrows indicate events, which may occur too rapidly to be clinically relevant therapeutic targets. Red arrows indicate possible targets in the peritubular microenvironment, which could break the cycle of injury and allow recovery of the renal microcirculation and tubular function.
Finally, it is likely that agents found to protect the kidney during sepsis would also have beneficial effects in other organs as well because oxidative stress and microvascular dysfunction are not unique to the kidney (Bateman, et al., 2003; Vincent and De Backer, 2005) and kidney injury can have a profound impact on other organs (Okusa, 2010).
Acknowledgments
Sources of Funding Work in the authors' laboratories is supported by grants from the NIH/NIDDK (DK0759911 and DK078936). The preparation of this manuscript was supported by the UAMS Translational Research Institute and the NIH/NCRR grant UL1 RR029884.
Abbreviations
- AKI
acute kidney injury
- RBF
renal blood flow
- MAP
mean arterial pressure
- GFR
glomerular filtration rate
- LPS
lipopolysaccharide
- CLP
cecal ligation and puncture
- MMP
matrix metalloproteinase
- S1P
sphingosine-1-phosphate
- S1PR
sphingosine-1-phosphate receptor
- SphK
sphingosine kinase
- SOD
superoxide dismutase
- iNOS
inducible nitric oxide synthase
- NO
nitric oxide
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- IR
ischemia-reperfusion
- SIRT1
sirtuin-1
- MnTMPyP
Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin, tetratosylate, hydroxide (MnTMPyP)
- TNF- α
tumor necrosis factor alpha
- α-MSH
alpha-melanocyte stimulating hormone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement The authors declare that there are no conflicts of interest.
U.S. Census Bureau, Statistical Abstract of the United States: 2011.
References
- Abrahamson DR, Leardkamolkarn V. Development of kidney tubular basement membranes. Kidney International. 1991;39:382–393. doi: 10.1038/ki.1991.50. [DOI] [PubMed] [Google Scholar]
- Ader I, Malavaud B, Cuvillier O. When the sphingosine kinase 1/sphingosine 1-phosphate pathway meets hypoxia signaling: new targets for cancer therapy. Cancer Research. 2009;69:3723–3726. doi: 10.1158/0008-5472.CAN-09-0389. [DOI] [PubMed] [Google Scholar]
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB Journal. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- Al-Mashhadi RH, Skott O, Vanhoutte PM, Hansen PB. Activation of A(2) adenosine receptors dilates cortical efferent arterioles in mouse. Kidney International. 2009;75:793–799. doi: 10.1038/ki.2008.684. [DOI] [PubMed] [Google Scholar]
- Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102:3665–3667. doi: 10.1182/blood-2003-02-0460. [DOI] [PubMed] [Google Scholar]
- Aydin S, Signorelli S, Lechleitner T, Joannidis M, Pleban C, Perco P, Pfaller W, Jennings P. Influence of microvascular endothelial cells on transcriptional regulation of proximal tubular epithelial cells. American Journal of Physiology Cell Physiology. 2008;294:C543–C554. doi: 10.1152/ajpcell.00307.2007. [DOI] [PubMed] [Google Scholar]
- Bagshaw SM, Lapinsky S, Dial S, Arabi Y, Dodek P, Wood G, Ellis P, Guzman J, Marshall J, Parrillo JE, Skrobik Y, Kumar A. Acute kidney injury in septic shock: clinical outcomes and impact of duration of hypotension prior to initiation of antimicrobial therapy. Intensive Care Medicine. 2009;35:871–881. doi: 10.1007/s00134-008-1367-2. [DOI] [PubMed] [Google Scholar]
- Bailly V, Zhang Z, Meier W, Cate R, Sanicola M, Bonventre JV. Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. Journal of Biological Chemistry. 2002;277:39739–39748. doi: 10.1074/jbc.M200562200. [DOI] [PubMed] [Google Scholar]
- Bajwa A, Jo SK, Ye H, Huang L, Dondeti KR, Rosin DL, Haase VH, Macdonald TL, Lynch KR, Okusa MD. Activation of sphingosine-1-phosphate 1 receptor in the proximal tubule protects against ischemia reperfusion injury. Journal of the American Society of Nephrology. 2010;21:955–965. doi: 10.1681/ASN.2009060662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz RR, Suliman HB, Fu P, Welty-Wolf K, Carraway MS, MacGarvey NC, Withers CM, Sweeney TE, Piantadosi CA. Staphylococcus aureus sepsis and mitochondrial accrual of the 8-oxoguanine DNA glycosylase DNA repair enzyme in mice. American Journal of Respiratory and Critical Care Medicine. 2011;183:226–233. doi: 10.1164/rccm.200911-1709OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile DP, Fredrich K, Weihrauch D, Hattan N, Chilian WM. Angiostatin and matrix metalloprotease expression following ischemic acute renal failure. American Journal of Physiology Renal Physiology. 2004;286:F893–F902. doi: 10.1152/ajprenal.00328.2003. [DOI] [PubMed] [Google Scholar]
- Bateman RM, Sharpe MD, Ellis CG. Bench-to-bedside review: microvascular dysfunction in sepsis--hemodynamics, oxygen transport, and nitric oxide. Critical Care. 2003;7:359–373. doi: 10.1186/cc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerle JD, Grenz A, Kim JH, Lee HT, Eltzschig HK. Adenosine generation and signaling during acute kidney injury. Journal of the American Society of Nephrology. 2011;22:14–20. doi: 10.1681/ASN.2009121217. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Crow JP. Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochemical Society Transactions. 1993;21:330–334. doi: 10.1042/bst0210330. [DOI] [PubMed] [Google Scholar]
- Beeson CC, Beeson GC, Schnellmann RG. A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Analytical Biochemistry. 2010;404:75–81. doi: 10.1016/j.ab.2010.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijuklic K, Jennings P, Kountchev J, Hasslacher J, Aydin S, Sturn D, Pfaller W, Patsch JR, Joannidis M. Migration of leukocytes across an endothelium-epithelium bilayer as a model of renal interstitial inflammation. American Journal of Physiology Cell Physiology. 2007;293:C486–C492. doi: 10.1152/ajpcell.00419.2006. [DOI] [PubMed] [Google Scholar]
- Boerma EC, Ince C. The role of vasoactive agents in the resuscitation of microvascular perfusion and tissue oxygenation in critically ill patients. Intensive Care Medicine. 2010 doi: 10.1007/s00134-010-1970-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boueiz A, Hassoun PM. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvascular Research. 2009;77:26–34. doi: 10.1016/j.mvr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Boulos M, Astiz ME, Barua RS, Osman M. Impaired mitochondrial function induced by serum from septic shock patients is attenuated by inhibition of nitric oxide synthase and poly(ADP-ribose) synthase. Critical Care Medicine. 2003;31:353–358. doi: 10.1097/01.CCM.0000050074.82486.B2. [DOI] [PubMed] [Google Scholar]
- Bradley VE, Shier MR, Lucas CE, Rosenberg IK. Renal hemodynamic response to furosemide in septic and injured patients. Surgery. 1976;79:549–554. [PubMed] [Google Scholar]
- Brandt S, Regueira T, Bracht H, Porta F, Djafarzadeh S, Takala J, Gorrasi J, Borotto E, Krejci V, Hiltebrand LB, Bruegger LE, Beldi G, Wilkens L, Lepper PM, Kessler U, Jakob SM. Effect of fluid resuscitation on mortality and organ function in experimental sepsis models. Critical Care. 2009;13:R186. doi: 10.1186/cc8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- Brenner M, Schaer GL, Mallory DL, Suffredini AF, Parrillo JE. Detection of renal blood flow abnormalities in septic and critically ill patients using a newly designed indwelling thermodilution renal vein catheter. Chest. 1990;98:170–179. doi: 10.1378/chest.98.1.170. [DOI] [PubMed] [Google Scholar]
- Carlstrom M, Wilcox CS, Welch WJ. Adenosine A(2) receptors modulate tubuloglomerular feedback. American Journal of Physiology Renal Physiology. 2010;299:F412–F417. doi: 10.1152/ajprenal.00211.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmago S, Shah SV, Walker PD. Meprin, a brush-border enzyme, plays an important role in hypoxic/ischemic acute renal tubular injury in rats. Kidney International. 2002;61:959–966. doi: 10.1046/j.1523-1755.2002.00209.x. [DOI] [PubMed] [Google Scholar]
- Catania JM, Chen G, Parrish AR. Role of matrix metalloproteinases in renal pathophysiologies. American Journal of Physiology Renal Physiology. 2007;292:F905–F911. doi: 10.1152/ajprenal.00421.2006. [DOI] [PubMed] [Google Scholar]
- Cena JJ, Lalu MM, Cho WJ, Chow AK, Bagdan ML, Daniel EE, Castro MM, Schulz R. Inhibition of matrix metalloproteinase activity in vivo protects against vascular hyporeactivity in endotoxemia. American Journal of Physiology Heart Circulation Physiology. 2010;298:H45–H51. doi: 10.1152/ajpheart.00273.2009. [DOI] [PubMed] [Google Scholar]
- Cepinskas G, Wilson JX. Inflammatory response in microvascular endothelium in sepsis: role of oxidants. Journal of Clinical Biochemistry and Nutrition. 2008;42:175–184. doi: 10.3164/jcbn.2008026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew MS, Hawthorne WJ, Bendall J, Whereat S, Huang S, Ting I, Simond D, McLean A. No beneficial effects of levosimendan in acute porcine endotoxaemia. Acta Anaesthesiology Scandinavia. 2011;55:851–861. doi: 10.1111/j.1399-6576.2011.02436.x. [DOI] [PubMed] [Google Scholar]
- Chilton L, Loutzenhiser K, Morales E, Breaks J, Kargacin GJ, Loutzenhiser R. Inward rectifier K(+) currents and Kir2.1 expression in renal afferent and efferent arterioles. Journal of the American Society of Nephrology. 2008;19:69–76. doi: 10.1681/ASN.2007010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Yao H, Caito S, Hwang JW, Arunachalam G, Rahman I. Regulation of SIRT1 in cellular functions: role of polyphenols. Archives of Biochemistry and Biophysics. 2010;501:79–90. doi: 10.1016/j.abb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvojka J, Sykora R, Karvunidis T, Radej J, Krouzecky A, Novak I, Matejovic M. New developments in septic acute kidney injury. Physiological Research. 2010;59:859–869. doi: 10.33549/physiolres.931936. [DOI] [PubMed] [Google Scholar]
- Chvojka J, Sykora R, Krouzecky A, Radej J, Varnerova V, Karvunidis T, Hes O, Novak I, Radermacher P, Matejovic M. Renal haemodynamic, microcirculatory, metabolic and histopathological responses to peritonitis-induced septic shock in pigs. Critical Care. 2008;12:R164. doi: 10.1186/cc7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig SS, Reckelhoff JF, Bond JS. Distribution of meprin in kidneys from mice with high- and low-meprin activity. American Journal of Physiology. 1987;253:C535–C540. doi: 10.1152/ajpcell.1987.253.4.C535. [DOI] [PubMed] [Google Scholar]
- Crouser ED. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion. 2004;4:729–741. doi: 10.1016/j.mito.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Crow JP. Peroxynitrite scavenging by metalloporphyrins and thiolates. Free Radical Biology and Medicine. 2000;28:1487–1494. doi: 10.1016/s0891-5849(00)00249-5. [DOI] [PubMed] [Google Scholar]
- Cruthirds DL, Novak L, Akhi KM, Sanders PW, Thompson JA, MacMillan-Crow LA. Mitochondrial targets of oxidative stress during renal ischemia/reperfusion. Archives of Biochemistry and Biophysics. 2003;412:27–33. doi: 10.1016/s0003-9861(03)00039-0. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Pinto JT, Ballabh P, Zhang H, Losonczy G, Pearson K, de Cabo R, Pacher P, Zhang C, Ungvari Z. Resveratrol induces mitochondrial biogenesis in endothelial cells. American Journal of Physiology Heart Circulation Physiology. 2009;297:H13–H20. doi: 10.1152/ajpheart.00368.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha-Goncalves D, Perez-de-Sa V, Grins E, Dahm PL, Thorne J, Blomquist S. Inotropic support during experimental endotoxemic shock: part I. The effects of levosimendan on splanchnic perfusion. Anesthesia and Analgesia. 2009;109:1568–1575. doi: 10.1213/ane.0b013e3181af3fe3. [DOI] [PubMed] [Google Scholar]
- Cupples WA, Braam B. Assessment of renal autoregulation. American Journal of Physiology Renal Physiology. 2007;292:F1105–F1123. doi: 10.1152/ajprenal.00194.2006. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Di Paola R, Esposito E, Macarthur H, Matuschak GM, Salvemini D. A role for nitric oxide-mediated peroxynitrite formation in a model of endotoxin-induced shock. Journal of Pharmacology and Experimental Therapeutics. 2006;319:73–81. doi: 10.1124/jpet.106.108100. [DOI] [PubMed] [Google Scholar]
- Damkjaer M, Vafaee M, Moller ML, Braad PE, Petersen H, Hoilund-Carlsen PF, Bie P. Renal cortical and medullary blood flow responses to altered NO availability in humans. American Journal of Physiology Regulatory Integrative and Comparative Physiol. 2010;299:R1449–R1455. doi: 10.1152/ajpregu.00440.2010. [DOI] [PubMed] [Google Scholar]
- Dare AJ, Phillips AR, Hickey AJ, Mittal A, Loveday B, Thompson N, Windsor JA. A systematic review of experimental treatments for mitochondrial dysfunction in sepsis and multiple organ dysfunction syndrome. Free Radical Biology and Medicine. 2009;47:1517–1525. doi: 10.1016/j.freeradbiomed.2009.08.019. [DOI] [PubMed] [Google Scholar]
- Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Laboratory Investigation. 2006;86:9–22. doi: 10.1038/labinvest.3700366. [DOI] [PubMed] [Google Scholar]
- Dear JW, Yasuda H, Hu X, Hieny S, Yuen PS, Hewitt SM, Sher A, Star RA. Sepsis-induced organ failure is mediated by different pathways in the kidney and liver: Acute renal failure is dependent on MyD88 but not renal cell apoptosis. Kidney International. 2006;69:832–836. doi: 10.1038/sj.ki.5000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejonckheere E, Vandenbroucke RE, Libert C. Matrix metalloproteinases as drug targets in ischemia/reperfusion injury. Drug Discovery Today. 2011;16:762–778. doi: 10.1016/j.drudis.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, Cohen J, Gea-Banacloche J, Keh D, Marshall JC, Parker MM, Ramsay G, Zimmerman JL, Vincent JL, Levy MM. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Critical Care Medicine. 2004;32:858–873. doi: 10.1097/01.ccm.0000117317.18092.e4. [DOI] [PubMed] [Google Scholar]
- Deruddre S, Cheisson G, Mazoit JX, Vicaut E, Benhamou D, Duranteau J. Renal arterial resistance in septic shock: effects of increasing mean arterial pressure with norepinephrine on the renal resistive index assessed with Doppler ultrasonography. Intensive Care Medicine. 2007;33:1557–1562. doi: 10.1007/s00134-007-0665-4. [DOI] [PubMed] [Google Scholar]
- Di Sole F. Adenosine and renal tubular function. Current Opinion in Nephrology and Hypertension. 2008;17:399–407. doi: 10.1097/MNH.0b013e32830321e1. [DOI] [PubMed] [Google Scholar]
- Doi K, Hu X, Yuen PS, Leelahavanichkul A, Yasuda H, Kim SM, Schnermann J, Jonassen TE, Frokiaer J, Nielsen S, Star RA. AP214, an analogue of alpha-melanocyte-stimulating hormone, ameliorates sepsis-induced acute kidney injury and mortality. Kidney International. 2008;73:1266–1274. doi: 10.1038/ki.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. Journal of Clinical Investigation. 2009;119:2868–2878. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Guan Q, Diao H, Yin Z, Jevnikar AM. Nitric oxide induces apoptosis in renal tubular epithelial cells through activation of caspase-8. American Journal of Physiology Renal Physiology. 2006;290:F1044–F1054. doi: 10.1152/ajprenal.00341.2005. [DOI] [PubMed] [Google Scholar]
- Dudley C. Maximizing renal preservation in acute renal failure. BJUInternational. 2004;94:1202–1206. doi: 10.1046/j.1464-410x.2004.05129.x. [DOI] [PubMed] [Google Scholar]
- Dyson A, Singer M. Animal models of sepsis: why does preclinical efficacy fail to translate to the clinical setting? Critical Care Medicine. 2009;37:S30–S37. doi: 10.1097/CCM.0b013e3181922bd3. [DOI] [PubMed] [Google Scholar]
- Ebong S, Call D, Nemzek J, Bolgos G, Newcomb D, Remick D. Immunopathologic alterations in murine models of sepsis of increasing severity. Infection and Immunity. 1999;67:6603–6610. doi: 10.1128/iai.67.12.6603-6610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds Y, Milstien S, Spiegel S. Development of small-molecule inhibitors of sphingosine-1-phosphate signaling. Pharmacology and Therapeutics. 2011;132:352–360. doi: 10.1016/j.pharmthera.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Achkar TM, Huang X, Plotkin Z, Sandoval RM, Rhodes GJ, Dagher PC. Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. American Journal of Physiology Renal Physiology. 2006;290:F1034–F1043. doi: 10.1152/ajprenal.00414.2005. [DOI] [PubMed] [Google Scholar]
- Evans RG, Gardiner BS, Smith DW, O'Connor PM. Intrarenal oxygenation: unique challenges and the biophysical basis of homeostasis. American Journal of Physiology Renal Physiology. 2008;295:F1259–F1270. doi: 10.1152/ajprenal.90230.2008. [DOI] [PubMed] [Google Scholar]
- Feng MG, Navar LG. Afferent arteriolar vasodilator effect of adenosine predominantly involves adenosine A2B receptor activation. American Journal of Physiology Renal Physiology. 2010;299:F310–F315. doi: 10.1152/ajprenal.00149.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenhammar J, Andersson A, Forestier J, Weitzberg E, Sollevi A, Hjelmqvist H, Frithiof R. Endothelin receptor a antagonism attenuates renal medullary blood flow impairment in endotoxemic pigs. PLoS One. 2011;6:e21534. doi: 10.1371/journal.pone.0021534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenhammar J, Andersson A, Frithiof R, Forestier J, Weitzberg E, Sollevi A, Hjelmqvist H. The endothelin receptor antagonist tezosentan improves renal microcirculation in a porcine model of endotoxemic shock. Acta Anaesthesiology Scandinavia. 2008;52:1385–1393. doi: 10.1111/j.1399-6576.2008.01768.x. [DOI] [PubMed] [Google Scholar]
- French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. Journal of Pharmacology and Experimental Therapeutics. 2010;333:129–139. doi: 10.1124/jpet.109.163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk JA, Odejinmi S, Schnellmann RG. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. Journal of Pharmacology and Experimental Therapeutics. 2010;333:593–601. doi: 10.1124/jpet.109.161992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galley HF. Bench-to-bedside review: Targeting antioxidants to mitochondria in sepsis. Crit Care. 2010;14:230. doi: 10.1186/cc9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. British Journal of Anaesthesia. 2011;107:57–64. doi: 10.1093/bja/aer093. [DOI] [PubMed] [Google Scholar]
- Gallos G, Ruyle TD, Emala CW, Lee HT. A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. American Journal of Physiology Renal Physiology. 2005;289:F369–F376. doi: 10.1152/ajprenal.00470.2004. [DOI] [PubMed] [Google Scholar]
- Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. Journal of Clinical Investigation. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Septien J, Lorente JA, Delgado MA, de Paula M, Nin N, Moscoso A, Sanchez-Ferrer A, Perez-Vizcaino F, Esteban A. Levosimendan increases portal blood flow and attenuates intestinal intramucosal acidosis in experimental septic shock. Shock. 2010;34:275–280. doi: 10.1097/SHK.0b013e3181cd8c5b. [DOI] [PubMed] [Google Scholar]
- Gardiner BS, Smith DW, O'Connor PM, Evans RG. A mathematical model of diffusional shunting of oxygen from arteries to veins in the kidney. American Journal of Physiology Renal Physiology. 2011;300:F1339–F1352. doi: 10.1152/ajprenal.00544.2010. [DOI] [PubMed] [Google Scholar]
- Goddard J, Johnston NR, Hand MF, Cumming AD, Rabelink TJ, Rankin AJ, Webb DJ. Endothelin-A receptor antagonism reduces blood pressure and increases renal blood flow in hypertensive patients with chronic renal failure: a comparison of selective and combined endothelin receptor blockade. Circulation. 2004;109:1186–1193. doi: 10.1161/01.CIR.0000118499.69469.51. [DOI] [PubMed] [Google Scholar]
- Gojkovic-Bukarica L, Novakovic A, Kanjuh V, Bumbasirevic M, Lesic A, Heinle H. A role of ion channels in the endothelium-independent relaxation of rat mesenteric artery induced by resveratrol. Journal of Pharmacological Sciences. 2008;108:124–130. doi: 10.1254/jphs.08128fp. [DOI] [PubMed] [Google Scholar]
- Gomes A, Fernandes E, Lima JL. Use of fluorescence probes for detection of reactive nitrogen species: a review. Journal of Fluorescence. 2006;16:119–139. doi: 10.1007/s10895-005-0030-3. [DOI] [PubMed] [Google Scholar]
- Goode HF, Cowley HC, Walker BE, Howdle PD, Webster NR. Decreased antioxidant status and increased lipid peroxidation in patients with septic shock and secondary organ dysfunction. Critical Care Medicine. 1995;23:646–651. doi: 10.1097/00003246-199504000-00011. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circulation Research. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Guan Z, Osmond DA, Inscho EW. P2X receptors as regulators of the renal microvasculature. Trends in Pharmacological Science. 2007;28:646–652. doi: 10.1016/j.tips.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Guo R, Wang Y, Minto AW, Quigg RJ, Cunningham PN. Acute renal failure in endotoxemia is dependent on caspase activation. Journal of the American Society of Nephrology. 2004;15:3093–3102. doi: 10.1097/01.ASN.0000145530.73247.F5. [DOI] [PubMed] [Google Scholar]
- Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, Piantadosi CA. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. American Journal of Respiratory and Critical Care Medicine. 2007;176:768–777. doi: 10.1164/rccm.200701-161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney International. 2002;62:237–244. doi: 10.1046/j.1523-1755.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- Hansen PB, Schnermann J. Vasoconstrictor and vasodilator effects of adenosine in the kidney. American Journal of Physiology Renal Physiology. 2003;285:F590–F599. doi: 10.1152/ajprenal.00051.2003. [DOI] [PubMed] [Google Scholar]
- Harrison-Bernard LM, Monjure CJ, Bivona BJ. Efferent arterioles exclusively express the subtype 1A angiotensin receptor: functional insights from genetic mouse models. American Journal of Physiology Renal Physiology. 2006;290:F1177–F1186. doi: 10.1152/ajprenal.00265.2005. [DOI] [PubMed] [Google Scholar]
- He W, Wang Y, Zhang MZ, You L, Davis LS, Fan H, Yang HC, Fogo AB, Zent R, Harris RC, Breyer MD, Hao CM. Sirt1 activation protects the mouse renal medulla from oxidative injury. Journal of Clinical Investigation. 2010;120:1056–1068. doi: 10.1172/JCI41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk S, Masereeuw R, Russel FG, Pickkers P. Selective iNOS inhibition for the treatment of sepsis-induced acute kidney injury. Nature Reviews Nephrology. 2009;5:629–640. doi: 10.1038/nrneph.2009.155. [DOI] [PubMed] [Google Scholar]
- Heemskerk S, Pickkers P, Bouw MP, Draisma A, van der Hoeven JG, Peters WH, Smits P, Russel FG, Masereeuw R. Upregulation of renal inducible nitric oxide synthase during human endotoxemia and sepsis is associated with proximal tubule injury. Clinical Journal of the American Society of Nephrology. 2006;1:853–862. doi: 10.2215/CJN.00490206. [DOI] [PubMed] [Google Scholar]
- Heemskerk S, Pickkers P, van Eijk L, van der Hoeven H, Peters W, Russel F, Masereeuw R. Upregulation of renal inducible NO synthase and proximal tubule damage during human endotoxemia. Critical Care. 2004;8(suppl 1):P197. doi: 10.2215/CJN.00490206. [DOI] [PubMed] [Google Scholar]
- Herzog C, Seth R, Shah SV, Kaushal GP. Role of meprin A in renal tubular epithelial cell injury. Kidney International. 2007;71:1009–1018. doi: 10.1038/sj.ki.5002189. [DOI] [PubMed] [Google Scholar]
- Hollenberg SM. Mouse models of resuscitated shock. Shock. 2005;24(Suppl 1):58–63. doi: 10.1097/01.shk.0000191415.02085.48. [DOI] [PubMed] [Google Scholar]
- Holly MK, Dear JW, Hu X, Schechter AN, Gladwin MT, Hewitt SM, Yuen PS, Star RA. Biomarker and drug-target discovery using proteomics in a new rat model of sepsis-induced acute renal failure. Kidney International. 2006;70:496–506. doi: 10.1038/sj.ki.5001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthoff JH, Wang Z, Seely KA, Gokden N, Mayeux PR. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney International. doi: 10.1038/ki.2011.347. PMID: 215975863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthoff JH, Woodling KA, Doerge DR, Burns ST, Hinson JA, Mayeux PR. Resveratrol, a dietary polyphenolic phytoalexin, is a functional scavenger of peroxynitrite. Biochemical Pharmacology. 2010;80:1260–1265. doi: 10.1016/j.bcp.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. New England Journal of Medicine. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Critical Care Medicine. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- Hsieh CH, Pai PY, Hsueh HW, Yuan SS, Hsieh YC. Complete induction of autophagy is essential for cardioprotection in sepsis. Annals of Surgery. 2011;253:1190–1200. doi: 10.1097/SLA.0b013e318214b67e. [DOI] [PubMed] [Google Scholar]
- Hsieh YC, Athar M, Chaudry IH. When apoptosis meets autophagy: deciding cell fate after trauma and sepsis. Trends in Molecular Medicine. 2009;15:129–138. doi: 10.1016/j.molmed.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Liu J, Wang J, Liu Q. The controversial links among calorie restriction, SIRT1, and resveratrol. Free Radical Biology and Medicine. 2011;51:250–256. doi: 10.1016/j.freeradbiomed.2011.04.034. [DOI] [PubMed] [Google Scholar]
- Huie RE, Padmaja S. The reaction of NO with superoxide. Free Radical Research Communications. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- Humphreys BD, Czerniak S, Dirocco DP, Hasnain W, Cheema R, Bonventre JV. Repair of injured proximal tubule does not involve specialized progenitors. Proceedings of the National Academy of Science USA. 2011;108:9226–9231. doi: 10.1073/pnas.1100629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huwiler A, Pfeilschifter J. New players on the center stage: sphingosine 1-phosphate and its receptors as drug targets. Biochemical Pharmacology. 2008;75:1893–1900. doi: 10.1016/j.bcp.2007.12.018. [DOI] [PubMed] [Google Scholar]
- Ikenaga H, Bast JP, Fallet RW, Carmines PK. Exaggerated impact of ATP-sensitive K(+) channels on afferent arteriolar diameter in diabetes mellitus. Journal of the American Society of Nephrology. 2000;11:1199–1207. doi: 10.1681/asn.v1171199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliescu R, Cazan R, McLemore GR, Jr., Venegas-Pont M, Ryan MJ. Renal blood flow and dynamic autoregulation in conscious mice. American Journal of Physiology Renal Physiology. 2008;295:F734–F740. doi: 10.1152/ajprenal.00115.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince C. The microcirculation is the motor of sepsis. Critical Care. 2005;9(Suppl 4):S13–S19. doi: 10.1186/cc3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Tanaka H, Sumi Y, Woehrle T, Chen Y, Hirsh MI, Junger WG. A3 adenosine receptor inhibition improves the efficacy of hypertonic saline resuscitation. Shock. 2011;35:178–183. doi: 10.1097/SHK.0b013e3181f221fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inscho EW, Imig JD, Cook AK, Pollock DM. ETA and ETB receptors differentially modulate afferent and efferent arteriolar responses to endothelin. British Journal of Pharmacology. 2005;146:1019–1026. doi: 10.1038/sj.bjp.0706412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskit AB, Senel I, Sokmensuer C, Guc MO. Endothelin receptor antagonist bosentan improves survival in a murine caecal ligation and puncture model of septic shock. European Journal of Pharmacology. 2004;506:83–88. doi: 10.1016/j.ejphar.2004.10.038. [DOI] [PubMed] [Google Scholar]
- Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. American Journal of Pathology. 2010;176:1181–1192. doi: 10.2353/ajpath.2010.090594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo SK, Bajwa A, Ye H, Vergis AL, Awad AS, Kharel Y, Lynch KR, Okusa MD. Divergent roles of sphingosine kinases in kidney ischemia-reperfusion injury. Kidney International. 2009;75:167–175. doi: 10.1038/ki.2008.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just A. Mechanisms of renal blood flow autoregulation: dynamics and contributions. American Journal of Physiology Regulatory Integrative and Comparative Physiol. 2007;292:R1–R17. doi: 10.1152/ajpregu.00332.2006. [DOI] [PubMed] [Google Scholar]
- Just A, Whitten CL, Arendshorst WJ. Reactive oxygen species participate in acute renal vasoconstrictor responses induced by ETA and ETB receptors. American Journal of Physiology Renal Physiology. 2008;294:F719–F728. doi: 10.1152/ajprenal.00506.2007. [DOI] [PubMed] [Google Scholar]
- Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochemistry and Cell Biology. 2008;130:247–262. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, Sandoval RM, Dagher PC. Endotoxin Uptake by S1 Proximal Tubular Segment Causes Oxidative Stress in the Downstream S2 Segment. Journal of the American Society of Nephrology. 2011;22:1505–1516. doi: 10.1681/ASN.2011020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao KK, Fink MP. The biochemical basis for the anti-inflammatory and cytoprotective actions of ethyl pyruvate and related compounds. Biochemical Pharmacology. 2010;80:151–159. doi: 10.1016/j.bcp.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Kaushal GP, Walker PD, Shah SV. An old enzyme with a new function: purification and characterization of a distinct matrix-degrading metalloproteinase in rat kidney cortex and its identification as meprin. Journal of Cell Biology. 1994;126:1319–1327. doi: 10.1083/jcb.126.5.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly KJ, Sutton TA, Weathered N, Ray N, Caldwell EJ, Plotkin Z, Dagher PC. Minocycline inhibits apoptosis and inflammation in a rat model of ischemic renal injury. American Journal of Physiology Renal Physiology. 2004;287:F760–F766. doi: 10.1152/ajprenal.00050.2004. [DOI] [PubMed] [Google Scholar]
- Kim DH, Jung YJ, Lee JE, Lee AS, Kang KP, Lee S, Park SK, Han MK, Lee SY, Ramkumar KM, Sung MJ, Kim W. SIRT1 activation by resveratrol ameliorates cisplatin-induced renal injury through deacetylation of p53. American Journal of Physiology Renal Physiology. 2011;301:F427–F435. doi: 10.1152/ajprenal.00258.2010. [DOI] [PubMed] [Google Scholar]
- Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y. Autophagy Protects the Proximal Tubule from Degeneration and Acute Ischemic Injury. Journal of the American Society of Nephrology. 2011;22:902–913. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepler JL, Jr., Taher LN, Gupta MP, Patterson C, Pavalko F, Ober MD, Hart CM. Peroxynitrite causes endothelial cell monolayer barrier dysfunction. American journal of physiology. Cell physiology. 2001;281:C1064–1075. doi: 10.1152/ajpcell.2001.281.3.C1064. [DOI] [PubMed] [Google Scholar]
- Kolgazi M, Sener G, Cetinel S, Gedik N, Alican I. Resveratrol reduces renal and lung injury caused by sepsis in rats. Journal of Surgical Research. 2006;134:315–321. doi: 10.1016/j.jss.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Kruse MN, Becker C, Lottaz D, Kohler D, Yiallouros I, Krell HW, Sterchi EE, Stocker W. Human meprin alpha and beta homo-oligomers: cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochemical Journal. 2004;378:383–389. doi: 10.1042/BJ20031163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, Suppes R, Feinstein D, Zanotti S, Taiberg L, Gurka D, Cheang M. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Critical Care Medicine. 2006;34:1589–1596. doi: 10.1097/01.CCM.0000217961.75225.E9. [DOI] [PubMed] [Google Scholar]
- Kunugi S, Shimizu A, Kuwahara N, Du X, Takahashi M, Terasaki Y, Fujita E, Mii A, Nagasaka S, Akimoto T, Masuda Y, Fukuda Y. Inhibition of matrix metalloproteinases reduces ischemia-reperfusion acute kidney injury. Laboratory Investigation. 2011;91:170–180. doi: 10.1038/labinvest.2010.174. [DOI] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Lam HS, Ng PC. Biochemical markers of neonatal sepsis. Pathology. 2008;40:141–148. doi: 10.1080/00313020701813735. [DOI] [PubMed] [Google Scholar]
- Lancel S, Hassoun SM, Favory R, Decoster B, Motterlini R, Neviere R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. Journal of Pharmacology and Experimental Therapeutics. 2009;329:641–648. doi: 10.1124/jpet.108.148049. [DOI] [PubMed] [Google Scholar]
- Langenberg C, Bellomo R, May C, Wan L, Egi M, Morgera S. Renal blood flow in sepsis. Critical Care. 2005;9:R363–R374. doi: 10.1186/cc3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenberg C, Wan L, Egi M, May CN, Bellomo R. Renal blood flow in experimental septic acute renal failure. Kidney International. 2006;69:1996–2002. doi: 10.1038/sj.ki.5000440. [DOI] [PubMed] [Google Scholar]
- Lauhio A, Hastbacka J, Pettila V, Tervahartiala T, Karlsson S, Varpula T, Varpula M, Ruokonen E, Sorsa T, Kolho E. Serum MMP-8, -9 and TIMP-1 in sepsis: High serum levels of MMP-8 and TIMP-1 are associated with fatal outcome in a multicentre, prospective cohort study. Hypothetical impact of tetracyclines. Pharmacological Research. 2011;64:590–594. doi: 10.1016/j.phrs.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Le Dorze M, Legrand M, Payen D, Ince C. The role of the microcirculation in acute kidney injury. Current Opinion in Critical Care. 2009;15:503–508. doi: 10.1097/MCC.0b013e328332f6cf. [DOI] [PubMed] [Google Scholar]
- Leach M, Frank S, Olbrich A, Pfeilschifter J, Thiemermann C. Decline in the expression of copper/zinc superoxide dismutase in the kidney of rats with endotoxic shock: effects of the superoxide anion radical scavenger, tempol, on organ injury. British Journal of Pharmacology. 1998;125:817–825. doi: 10.1038/sj.bjp.0702123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr., Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proceedings of the National Academy of Science USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. American Journal of Physiology Regulatory Integrative and Comparative Physiol. 2006;291:R959–R969. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- Lee JF, Gordon S, Estrada R, Wang L, Siow DL, Wattenberg BW, Lominadze D, Lee MJ. Balance of S1P1 and S1P2 signaling regulates peripheral microvascular permeability in rat cremaster muscle vasculature. American Journal of Physiology Heart Circulation Physiology. 2009;296:H33–H42. doi: 10.1152/ajpheart.00097.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha'afi RI, Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–312. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- Lee SY, Horbelt M, Mang HE, Knipe NL, Bacallao RL, Sado Y, Sutton TA. MMP-9 gene deletion mitigates microvascular loss in a model of ischemic acute kidney injury. American Journal of Physiology Renal Physiology. 2011;301:F101–F109. doi: 10.1152/ajprenal.00445.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WL, Slutsky AS. Sepsis and endothelial permeability. New England Journal of Medicine. 2010;363:689–691. doi: 10.1056/NEJMcibr1007320. [DOI] [PubMed] [Google Scholar]
- Leelahavanichkul A, Yasuda H, Doi K, Hu X, Zhou H, Yuen PS, Star RA. Methyl-2-acetamidoacrylate, an ethyl pyruvate analog, decreases sepsis-induced acute kidney injury in mice. American Journal of Physiology Renal Physiology. 2008;295:F1825–F1835. doi: 10.1152/ajprenal.90442.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemley KV, Kriz W. Anatomy of the renal interstitium. Kidney International. 1991;39:370–381. doi: 10.1038/ki.1991.49. [DOI] [PubMed] [Google Scholar]
- Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC, Bion J, Schorr C, Artigas A, Ramsay G, Beale R, Parker MM, Gerlach H, Reinhart K, Silva E, Harvey M, Regan S, Angus DC. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Critical Care Medicine. 2010;38:367–374. doi: 10.1097/CCM.0b013e3181cb0cdc. [DOI] [PubMed] [Google Scholar]
- Levy RJ. Mitochondrial dysfunction, bioenergetic impairment, and metabolic down-regulation in sepsis. Shock. 2007;28:24–28. doi: 10.1097/01.shk.0000235089.30550.2d. [DOI] [PubMed] [Google Scholar]
- Levy RJ, Deutschman CS. Cytochrome c oxidase dysfunction in sepsis. Critical Care Medicine. 2007;35:S468–S475. doi: 10.1097/01.CCM.0000278604.93569.27. [DOI] [PubMed] [Google Scholar]
- Levy RJ, Vijayasarathy C, Raj NR, Avadhani NG, Deutschman CS. Competitive and noncompetitive inhibition of myocardial cytochrome C oxidase in sepsis. Shock. 2004;21:110–114. doi: 10.1097/01.shk.0000108400.56565.ab. [DOI] [PubMed] [Google Scholar]
- Li P, Allen H, Banerfii S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J. Mice deficient in IL-b-converting enzyme are defective in production of mature IL-1b and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Liangos O, Perianayagam MC, Vaidya VS, Han WK, Wald R, Tighiouart H, MacKinnon RW, Li L, Balakrishnan VS, Pereira BJ, Bonventre JV, Jaber BL. Urinary N-acetyl-beta-(D)-glucosaminidase activity and kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. Journal of the American Society of Nephrology. 2007;18:904–912. doi: 10.1681/ASN.2006030221. [DOI] [PubMed] [Google Scholar]
- Liaw WJ, Chen TH, Lai ZZ, Chen SJ, Chen A, Tzao C, Wu JY, Wu CC. Effects of a membrane-permeable radical scavenger, Tempol, on intraperitoneal sepsis-induced organ injury in rats. Shock. 2005;23:88–96. doi: 10.1097/01.shk.0000145937.70085.89. [DOI] [PubMed] [Google Scholar]
- Lopez A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, Brockway M, Anzueto A, Holzapfel L, Breen D, Silverman MS, Takala J, Donaldson J, Arneson C, Grove G, Grossman S, Grover R. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Critical Care Medicine. 2004;32:21–30. doi: 10.1097/01.CCM.0000105581.01815.C6. [DOI] [PubMed] [Google Scholar]
- Loutzenhiser K, Loutzenhiser R. Angiotensin II-induced Ca(2+) influx in renal afferent and efferent arterioles: differing roles of voltage-gated and store-operated Ca(2+) entry. Circulation Research. 2000;87:551–557. doi: 10.1161/01.res.87.7.551. [DOI] [PubMed] [Google Scholar]
- Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radical Biology and Medicine. 2008;45:1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Lundy DJ, Trzeciak S. Microcirculatory dysfunction in sepsis. Critical Care Clinics. 2009;25:721–731. doi: 10.1016/j.ccc.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Macdonald J, Galley HF, Webster NR. Oxidative stress and gene expression in sepsis. British Journal of Anaesthesia. 2003;90:221–232. doi: 10.1093/bja/aeg034. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, Jr., Milstien S, Spiegel S. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. Journal of Biological Chemistry. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- MacMillan-Crow LA, Crow JP, Kerby JK, Beckman JS. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proceedings of the National Academy of Science USA. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan-Crow LA, Thompson JA. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Archives of Biochemistry and Biophysics. 1999;366:82–88. doi: 10.1006/abbi.1999.1202. [DOI] [PubMed] [Google Scholar]
- Madrid MI, Garcia-Salom M, Tornel J, de Gasparo M, Fenoy FJ. Interactions between nitric oxide and angtiotensin II on renal cortical and papillary blood flow. Hypertension. 1997;30:1175–1182. doi: 10.1161/01.hyp.30.5.1175. [DOI] [PubMed] [Google Scholar]
- Markewitz BA, Michael JR, Kohan DE. Cytokine-induced expression of a nitric oxide synthase in rat renal tubule cells. Journal of Clinical Investigation. 1993;91:2138–2143. doi: 10.1172/JCI116439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC, Vincent JL, Guyatt G, Angus DC, Abraham E, Bernard G, Bombardier C, Calandra T, Jorgensen HS, Sylvester R, Boers M. Outcome measures for clinical research in sepsis: a report of the 2nd Cambridge Colloquium of the International Sepsis Forum. Critical Care Medicine. 2005;33:1708–1716. doi: 10.1097/01.ccm.0000174478.70338.03. [DOI] [PubMed] [Google Scholar]
- Marsolais D, Rosen H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nature Reviews Drug Discovery. 2009;8:297–307. doi: 10.1038/nrd2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matavelli LC, Kadowitz PJ, Navar LG, Majid DS. Renal hemodynamic and excretory responses to intra-arterial infusion of peroxynitrite in anesthetized rats. American Journal of Physiology Renal Physiology. 2009;296:F170–F176. doi: 10.1152/ajprenal.90487.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mera S, Tatulescu D, Cismaru C, Bondor C, Slavcovici A, Zanc V, Carstina D, Oltean M. Multiplex cytokine profiling in patients with sepsis. APMIS. 2011;119:155–163. doi: 10.1111/j.1600-0463.2010.02705.x. [DOI] [PubMed] [Google Scholar]
- Millar CG, Thiemermann C. Carboxy-PTIO, a scavenger of nitric oxide, selectively inhibits the increase in medullary perfusion and improves renal function in endotoxemia. Shock. 2002;18:64–68. doi: 10.1097/00024382-200207000-00012. [DOI] [PubMed] [Google Scholar]
- Mitchell T, Rotaru D, Saba H, Smith RA, Murphy MP, MacMillan-Crow LA. The mitochondria-targeted antioxidant mitoquinone protects against cold storage injury of renal tubular cells and rat kidneys. Journal of Pharmacology and Experimental Therapeutics. 2011;336:682–692. doi: 10.1124/jpet.110.176743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Bansal S, Wang W, Falk S, Zolty E, Schrier RW. Erythropoietin ameliorates renal dysfunction during endotoxaemia. Nephrology Dialysis Transplantation. 2007;22:2349–2353. doi: 10.1093/ndt/gfm216. [DOI] [PubMed] [Google Scholar]
- Miyaji T, Hu X, Yuen PS, Muramatsu Y, Iyer S, Hewitt SM, Star RA. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney International. 2003;64:1620–1631. doi: 10.1046/j.1523-1755.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- Molitoris BA, Marrs J. The role of cell adhesion molecules in ischemic acute renal failure. American Journal of Medicine. 1999;106:583–592. doi: 10.1016/s0002-9343(99)00061-3. [DOI] [PubMed] [Google Scholar]
- Morelli A, Donati A, Ertmer C, Rehberg S, Lange M, Orecchioni A, Cecchini V, Landoni G, Pelaia P, Pietropaoli P, Van Aken H, Teboul JL, Ince C, Westphal M. Levosimendan for resuscitating the microcirculation in patients with septic shock: a randomized controlled study. Critical Care. 2010;14:R232. doi: 10.1186/cc9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhl D, Nagy B, Woth G, Falusi B, Bogar L, Weber G, Lantos J. Dynamic changes of matrix metalloproteinases and their tissue inhibitors in severe sepsis. Journal of Critical Care. 2011;26:550–555. doi: 10.1016/j.jcrc.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Murakami A, Takasugi H, Ohnuma S, Koide Y, Sakurai A, Takeda S, Hasegawa T, Sasamori J, Konno T, Hayashi K, Watanabe Y, Mori K, Sato Y, Takahashi A, Mochizuki N, Takakura N. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: investigation based on a new S1P3 receptor antagonist. Molecular Pharmacology. 2010;77:704–713. doi: 10.1124/mol.109.061481. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annual Review of Pharmacology and Toxicology. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- Murugan R, Kellum JA. Acute kidney injury: what's the prognosis? Nature Reviews Nephrology. 2011;7:209–217. doi: 10.1038/nrneph.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahama T, Hayashi K, Ozawa Y, Takenaka T, Saruta T. Role of protein kinase C in angiotensin II-induced constriction of renal microvessels. Kidney International. 2000;57:215–223. doi: 10.1046/j.1523-1755.2000.00822.x. [DOI] [PubMed] [Google Scholar]
- Nemeth ZH, Csoka B, Wilmanski J, Xu D, Lu Q, Ledent C, Deitch EA, Pacher P, Spolarics Z, Hasko G. Adenosine A2A receptor inactivation increases survival in polymicrobial sepsis. Journal of Immunology. 2006;176:5616–5626. doi: 10.4049/jimmunol.176.9.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nin N, Cassina A, Boggia J, Alfonso E, Botti H, Peluffo G, Trostchansky A, Batthyany C, Radi R, Rubbo H, Hurtado FJ. Septic diaphragmatic dysfunction is prevented by Mn(III)porphyrin therapy and inducible nitric oxide synthase inhibition. Intensive Care Medicine. 2004;30:2271–2278. doi: 10.1007/s00134-004-2427-x. [DOI] [PubMed] [Google Scholar]
- Nin N, El-Assar M, Sanchez C, Ferruelo A, Sanchez-Ferrer A, Martinez-Caro L, Rojas Y, de Paula M, Hurtado J, Esteban A, Lorente JA. Vascular Dysfunction in Sepsis: Effects of the Peroxynitrite Decomposition Catalyst MnTMPyP. Shock. 2011;36:165–161. doi: 10.1097/SHK.0b013e31821e50de. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Inscho EW, Navar LG. Interactions of adenosine A1 and A2a receptors on renal microvascular reactivity. American Journal of Physiology Renal Physiology. 2001;280:F406–F414. doi: 10.1152/ajprenal.2001.280.3.F406. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Miura K, Miyatake A, Fujisawa Y, Yue W, Fukui T, Kimura S, Abe Y. Renal interstitial concentration of adenosine during endotoxin shock. European Journal of Pharmacology. 1999;385:209–216. doi: 10.1016/s0014-2999(99)00716-5. [DOI] [PubMed] [Google Scholar]
- Nitescu N, Grimberg E, Guron G. Low-dose candesartan improves renal blood flow and kidney oxygen tension in rats with endotoxin-induced acute kidney dysfunction. Shock. 2008;30:166–172. doi: 10.1097/shk.0b013e31815dd780. [DOI] [PubMed] [Google Scholar]
- Nitescu N, Grimberg E, Ricksten SE, Herlitz H, Guron G. Endothelin B receptors preserve renal blood flow in a normotensive model of endotoxin-induced acute kidney dysfunction. Shock. 2008;29:402–409. doi: 10.1097/shk.0b013e3181454118. [DOI] [PubMed] [Google Scholar]
- Novakovic A, Bukarica LG, Kanjuh V, Heinle H. Potassium channels-mediated vasorelaxation of rat aorta induced by resveratrol. Basic Clinical Pharmacology and Toxicology. 2006;99:360–364. doi: 10.1111/j.1742-7843.2006.pto_531.x. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Akaike T, Sawa T, Miyamoto Y, van der Vliet A, Maeda H. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. Journal of Biological Chemistry. 2001;276:29596–29602. doi: 10.1074/jbc.M102417200. [DOI] [PubMed] [Google Scholar]
- Okusa MD. The changing pattern of acute kidney injury: from one to multiple organ failure. Contributions to Nephrology. 2010;165:153–158. doi: 10.1159/000313754. [DOI] [PubMed] [Google Scholar]
- Ongeri EM, Anyanwu O, Reeves WB, Bond JS. Villin and actin in the mouse kidney brush border membrane bind to and are degraded by meprins: This interaction contributes to injury in ischemia reperfusion. American Journal of Physiology Renal Physiology. 2011;301:F871–F882. doi: 10.1152/ajprenal.00703.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero RM, Nguyen HB, Huang DT, Gaieski DF, Goyal M, Gunnerson KJ, Trzeciak S, Sherwin R, Holthaus CV, Osborn T, Rivers EP. Early goal-directed therapy in severe sepsis and septic shock revisited: concepts, controversies, and contemporary findings. Chest. 2006;130:1579–1595. doi: 10.1378/chest.130.5.1579. [DOI] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, Qiu X, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. Journal of Biological Chemistry. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nature reviews. Molecular Cell Biology. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallone TL, Silldorff EP, Turner MR. Intrarenal blood flow: microvascular anatomy and the regulation of medullary perfusion. Clinical and Experimental Pharmacololgy and Physiology. 1998;25:383–392. doi: 10.1111/j.1440-1681.1998.tb02220.x. [DOI] [PubMed] [Google Scholar]
- Pallone TL, Zhang Z, Rhinehart K. Physiology of the renal medullary microcirculation. American Journal of Physiology Renal Physiology. 2003;284:F253–F266. doi: 10.1152/ajprenal.00304.2002. [DOI] [PubMed] [Google Scholar]
- Palmer RMJ, Andrews T, Foxwell NA, Moncada S. Glucocorticoids do not affect the induction of a novel calcium-dependent nitric oxide synthase in rabbit chondrocytes. Biochemical and Biophysical Research Communication. 1992;188:209–215. doi: 10.1016/0006-291x(92)92371-4. [DOI] [PubMed] [Google Scholar]
- Park SW, Kim M, D'Agati VD, Lee HT. Sphingosine kinase 1 protects against renal ischemia-reperfusion injury in mice by sphingosine-1-phosphate(1) receptor activation. Kidney International. 2011;80:1315–1327. doi: 10.1038/ki.2011.281. [DOI] [PubMed] [Google Scholar]
- Pathak E, MacMillan-Crow LA, Mayeux PR. Role of mitochondrial oxidants in an in vitro model of sepsis-induced renal injury. Journal of Pharmacology and Experimental Therapeutics. 2012;340:192–201. doi: 10.1124/jpet.111.183756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak E, Mayeux PR. In vitro model of sepsis-induced renal epithelial reactive nitrogen species generation. Toxicological Sciences. 2010;115:475–481. doi: 10.1093/toxsci/kfq058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxton WG, Runge M, Horaist C, Cohen C, Alexander RW, Bernstein KE. Immunohistochemical localization of rat angiotensin II AT1 receptor. American Journal of Physiology Renal Physiology. 1993;264:F989–F995. doi: 10.1152/ajprenal.1993.264.6.F989. [DOI] [PubMed] [Google Scholar]
- Pitera JE, Woolf AS, Gale NW, Yancopoulos GD, Yuan HT. Dysmorphogenesis of kidney cortical peritubular capillaries in angiopoietin-2-deficient mice. American Journal of Pathology. 2004;165:1895–1906. doi: 10.1016/S0002-9440(10)63242-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittet JF, Morel DR, Hemsen A, Gunning K, Lacroix JS, Suter PM, Lundberg JM. Elevated plasma endothelin-1 concentrations are associated with the severity of illness in patients with sepsis. Annals of Surgery. 1991;213:261–264. doi: 10.1097/00000658-199103000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock DM, Jenkins JM, Cook AK, Imig JD, Inscho EW. L-type calcium channels in the renal microcirculatory response to endothelin. American Journal of Physiology Renal Physiology. 2005;288:F771–F777. doi: 10.1152/ajprenal.00315.2004. [DOI] [PubMed] [Google Scholar]
- Poulsen CB, Al-Mashhadi RH, Cribbs LL, Skott O, Hansen PB. T-type voltage-gated calcium channels regulate the tone of mouse efferent arterioles. Kidney International. 2011;79:443–451. doi: 10.1038/ki.2010.429. [DOI] [PubMed] [Google Scholar]
- Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends in Endocrinology and Metabolism. 2009;20:332–340. doi: 10.1016/j.tem.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Puneet P, Yap CT, Wong L, Lam Y, Koh DR, Moochhala S, Pfeilschifter J, Huwiler A, Melendez AJ. SphK1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science. 2010;328:1290–1294. doi: 10.1126/science.1188635. [DOI] [PubMed] [Google Scholar]
- Ra HJ, Parks WC. Control of matrix metalloproteinase catalytic activity. Matrix Biology. 2007;26:587–596. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakers BP, Riksen NP, van der Hoeven JG, Smits P, Pickkers P. Modulation of innate immunity by adenosine receptor stimulation. Shock. 2011;36:208–215. doi: 10.1097/SHK.0b013e318225aee4. [DOI] [PubMed] [Google Scholar]
- Ramchandra R, Wan L, Hood SG, Frithiof R, Bellomo R, May CN. Septic shock induces distinct changes in sympathetic nerve activity to the heart and kidney in conscious sheep. American Journal of Physiology Regulatory Integrative and Comparative Physiol. 2009;297:R1247–R1253. doi: 10.1152/ajpregu.00437.2009. [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant injury. Journal of Biological Chemistry. 2007;282:2355–2362. doi: 10.1074/jbc.M608009200. [DOI] [PubMed] [Google Scholar]
- Rehberg S, Ertmer C, Vincent JL, Spiegel HU, Kohler G, Erren M, Lange M, Morelli A, Seisel J, Su F, Van Aken H, Traber DL, Westphal M. Effects of combined arginine vasopressin and levosimendan on organ function in ovine septic shock. Critical Care Medicine. 2010;38:2016–2023. doi: 10.1097/CCM.0b013e3181ef4694. [DOI] [PubMed] [Google Scholar]
- Remick DG. Pathophysiology of sepsis. American Journal of Pathology. 2007;170:1435–1444. doi: 10.2353/ajpath.2007.060872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remick DG, Ward PA. Evaluation of endotoxin models for the study of sepsis. Shock. 2005;24(Suppl 1):7–11. doi: 10.1097/01.shk.0000191384.34066.85. [DOI] [PubMed] [Google Scholar]
- Reslerova M, Loutzenhiser R. Divergent mechanisms of ATP-sensitive K+ channel-induced vasodilation in renal afferent and efferent arterioles. Evidence of L-type Ca2+ channel-dependent and -independent actions of pinacidil. Circulation Research. 1995;77:1114–1120. doi: 10.1161/01.res.77.6.1114. [DOI] [PubMed] [Google Scholar]
- Reynolds CM, Suliman HB, Hollingsworth JW, Welty-Wolf KE, Carraway MS, Piantadosi CA. Nitric oxide synthase-2 induction optimizes cardiac mitochondrial biogenesis after endotoxemia. Free Radical Biology and Medicine. 2009;46:564–572. doi: 10.1016/j.freeradbiomed.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci Z, Polito A, Ronco C. The implications and management of septic acute kidney injury. Nature Reviews Nephrology. 2011;7:218–225. doi: 10.1038/nrneph.2011.15. [DOI] [PubMed] [Google Scholar]
- Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nature Protocols. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, Peterson E, Tomlanovich M. Early goal-directed therapy in the treatment of severe sepsis and septic shock. New England Journal of Medicine. 2001;345:1368–1377. doi: 10.1056/NEJMoa010307. [DOI] [PubMed] [Google Scholar]
- Rivers EP, Coba V, Whitmill M. Early goal-directed therapy in severe sepsis and septic shock: a contemporary review of the literature. Current Opinion in Anesthesiology. 2008;21:128–140. doi: 10.1097/ACO.0b013e3282f4db7a. [DOI] [PubMed] [Google Scholar]
- Ronco P, Chatziantoniou C. Matrix metalloproteinases and matrix receptors in progression and reversal of kidney disease: therapeutic perspectives. Kidney International. 2008;74:873–878. doi: 10.1038/ki.2008.349. [DOI] [PubMed] [Google Scholar]
- Ruggieri AJ, Levy RJ, Deutschman CS. Mitochondrial dysfunction and esuscitation in sepsis. Critical Care Clinics. 2010;26:567–575. doi: 10.1016/j.ccc.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA. Management of sepsis. New England Journal of Medicine. 2006;355:1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- Saba H, Munusamy S, Macmillan-Crow LA. Cold preservation mediated renal injury: involvement of mitochondrial oxidative stress. Renal Failure. 2008;30:125–133. doi: 10.1080/08860220701813327. [DOI] [PubMed] [Google Scholar]
- Sakr Y, Dubois MJ, De Backer D, Creteur J, Vincent JL. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Critical Care Medicine. 2004;32:1825–1831. doi: 10.1097/01.ccm.0000138558.16257.3f. [DOI] [PubMed] [Google Scholar]
- Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arteriosclerosis Thrombosis Vascular Biology. 2007;27:1312–1318. doi: 10.1161/ATVBAHA.107.143735. [DOI] [PubMed] [Google Scholar]
- Scheiermann P, Ahluwalia D, Hoegl S, Dolfen A, Revermann M, Zwissler B, Muhl H, Boost KA, Hofstetter C. Effects of intravenous and inhaled levosimendan in severe rodent sepsis. Intensive Care Medicine. 2009;35:1412–1419. doi: 10.1007/s00134-009-1481-9. [DOI] [PubMed] [Google Scholar]
- Schildroth J, Rettig-Zimmermann J, Kalk P, Steege A, Fahling M, Sendeski M, Paliege A, Lai EY, Bachmann S, Persson PB, Hocher B, Patzak A. Endothelin type A and B receptors in the control of afferent and efferent arterioles in mice. Nephrology Dialysis Transplantation. 2011;26:779–789. doi: 10.1093/ndt/gfq534. [DOI] [PubMed] [Google Scholar]
- Schwalm S, Doll F, Romer I, Bubnova S, Pfeilschifter J, Huwiler A. Sphingosine kinase-1 is a hypoxia-regulated gene that stimulates migration of human endothelial cells. Biochemical and Biophysical Research Communication. 2008;368:1020–1025. doi: 10.1016/j.bbrc.2008.01.132. [DOI] [PubMed] [Google Scholar]
- Sebai H, Ben-Attia M, Sani M, Aouani E, Ghanem-Boughanmi N. Protective effect of resveratrol on acute endotoxemia-induced nephrotoxicity in rat through nitric oxide independent mechanism. Free Radical Research. 2008;42:913–920. doi: 10.1080/10715760802555577. [DOI] [PubMed] [Google Scholar]
- Sebai H, Sani M, Aouani E, Ghanem-Boughanmi N. Cardioprotective effect of resveratrol on lipopolysaccharide-induced oxidative stress in rat. Drug Chemical Toxicology. 2011;34:146–150. doi: 10.3109/01480545.2010.494666. [DOI] [PubMed] [Google Scholar]
- Seely KA, Holthoff JH, Burns ST, Wang Z, Thakali KM, Gokden N, Rhee SW, Mayeux PR. Hemodynamic changes in the kidney in a pediatric rat model of sepsis-induced acute kidney injury. American Journal of Physiology Renal Physiology. 2011;301:F209–F217. doi: 10.1152/ajprenal.00687.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Rehman H, Ramshesh VK, Schuartz J, Liu Q, Krishnasamy Y, Zhang X, Lemasters JJ, Smith CD, Zhong Z. Sphingosine kinase-2 inhibition improves mitochondrial function and survival after hepatic ischemia-reperfusion. Journal of Hepatology. 2012;56:137–145. doi: 10.1016/j.jhep.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silldorff EP, Yang S, Pallone TL. Prostaglandin E2 abrogates endothelin-induced vasoconstriction in renal outer medullary descending vasa recta of the rat. Journal of Clinical Investigation. 1995;95:2734–2740. doi: 10.1172/JCI117976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanos A, Jhanji S, Vivian-Smith A, Harris T, Pearse RM. Early microvascular changes in sepsis and severe sepsis. Shock. 2010;33:387–391. doi: 10.1097/SHK.0b013e3181c6be04. [DOI] [PubMed] [Google Scholar]
- Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The Pathogenesis of Sepsis. Annual Review of Pathology. 2011;6:19–48. doi: 10.1146/annurev-pathol-011110-130327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterchi EE, Stocker W, Bond JS. Meprins, membrane-bound and secreted astacin metalloproteinases. Molecular Aspects of Medicine. 2008;29:309–328. doi: 10.1016/j.mam.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC, Maceyka M, Price MM, Chen Q, Simpson DC, Kordula T, Milstien S, Lesnefsky EJ, Spiegel S. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB Journal. 2011;25:600–612. doi: 10.1096/fj.10-167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Piantadosi CA. Postlipopolysaccharide oxidative damage of mitochondrial DNA. American Journal of Respiratory and Critical Care Medicine. 2003;167:570–579. doi: 10.1164/rccm.200206-518OC. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Welty-Wolf KE, Whorton AR, Piantadosi CA. Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. Journal of Biological Chemistry. 2003;278:41510–41518. doi: 10.1074/jbc.M304719200. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Welty-Wolf KE, Carraway M, Tatro L, Piantadosi CA. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovascular Research. 2004;64:279–288. doi: 10.1016/j.cardiores.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Sullivan JC, Wang B, Boesen EI, D'Angelo G, Pollock JS, Pollock DM. Novel use of ultrasound to examine regional blood flow in the mouse kidney. American Journal of Physiology Renal Physiology. 2009;297:F228–F235. doi: 10.1152/ajprenal.00016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton TA. Alteration of microvascular permeability in acute kidney injury. Microvascular Research. 2009;77:4–7. doi: 10.1016/j.mvr.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton TA, Kelly KJ, Mang HE, Plotkin Z, Sandoval RM, Dagher PC. Minocycline reduces renal microvascular leakage in a rat model of ischemic renal injury. American Journal of Physiology. 2005;288:F91–F97. doi: 10.1152/ajprenal.00051.2004. [DOI] [PubMed] [Google Scholar]
- Svistunenko DA, Davies N, Brealey D, Singer M, Cooper CE. Mitochondrial dysfunction in patients with severe sepsis: an EPR interrogation of individual respiratory chain components. Biochimica et Biophysica Acta. 2006;1757:262–272. doi: 10.1016/j.bbabio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Sweeney TE, Suliman HB, Hollingsworth JW, Piantadosi CA. Differential regulation of the PGC family of genes in a mouse model of Staphylococcus aureus sepsis. PLoS One. 2010;5:e11606. doi: 10.1371/journal.pone.0011606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY, Seshan SV. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. Journal of the American Society of Nephrology. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuwa Y, Okamoto Y, Yoshioka K, Takuwa N. Sphingosine-1-phosphate signaling and biological activities in the cardiovascular system. Biochimica et Biophysica Acta. 2008;1781:483–488. doi: 10.1016/j.bbalip.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Tang L, Loutzenhiser K, Loutzenhiser R. Biphasic actions of prostaglandin E(2) on the renal afferent arteriole : role of EP(3) and EP(4) receptors. Circulation Research. 2000;86:663–670. doi: 10.1161/01.res.86.6.663. [DOI] [PubMed] [Google Scholar]
- Tiwari MM, Brock RW, Kaushal GP, Mayeux PR. Disruption of renal peritubular blood flow in lipopolysaccharide-induced renal failure: role of nitric oxide and caspases. American Journal of Physiology Renal Physiology. 2005;289:F1324–F1332. doi: 10.1152/ajprenal.00124.2005. [DOI] [PubMed] [Google Scholar]
- Tiwari MM, Messer KJ, Mayeux PR. Inducible nitric oxide synthase and apoptosis in murine proximal tubule epithelial cells. Toxicological Sciences. 2006;91:493–500. doi: 10.1093/toxsci/kfj168. [DOI] [PubMed] [Google Scholar]
- Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, David S, Burstein D, Karumanchi SA, Stillman IE, Arany Z, Parikh SM. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. Journal of Clinical Investigation. 2011;121:4003–4014. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traylor LA, Mayeux PR. Nitric oxide generation mediates lipid A-induced oxidant injury in renal proximal tubules. Archives of Biochemistry and Biophysics. 1997;338:129–135. doi: 10.1006/abbi.1996.9840. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. Journal of Physiology. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyml K. Critical role for oxidative stress, platelets, and coagulation in capillary blood flow impairment in sepsis. Microcirculation. 2011;18:152–162. doi: 10.1111/j.1549-8719.2010.00080.x. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Labinskyy N, Mukhopadhyay P, Pinto JT, Bagi Z, Ballabh P, Zhang C, Pacher P, Csiszar A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. American Journal of Physiology Heart Circulation Physiology. 2009;297:H1876–H1881. doi: 10.1152/ajpheart.00375.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalpando S, Gopal J, Balasubramanyam A, Bandi VP, Guntupalli K, Jahoor F. In vivo arginine production and intravascular nitric oxide synthesis in hypotensive sepsis. American Journal of Clinical Nutrition. 2006;84:197–203. doi: 10.1093/ajcn/84.1.197. [DOI] [PubMed] [Google Scholar]
- Vincent JL, De Backer D. Microvascular dysfunction as a cause of organ dysfunction in severe sepsis. Critical Care. 2005;9(Suppl 4):S9–S12. doi: 10.1186/cc3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogetseder A, Picard N, Gaspert A, Walch M, Kaissling B, Le Hir M. Proliferation capacity of the renal proximal tubule involves the bulk of differentiated epithelial cells. American Journal of Physiology Cell Physiology. 2008;294:C22–C28. doi: 10.1152/ajpcell.00227.2007. [DOI] [PubMed] [Google Scholar]
- Wadgaonkar R, Patel V, Grinkina N, Romano C, Liu J, Zhao Y, Sammani S, Garcia JG, Natarajan V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. American Journal of Physiology Lung Cellular and Molecular Physiology. 2009;296:L603–L613. doi: 10.1152/ajplung.90357.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Walker PD, Imam SZ, Ali SF, Mayeux PR. Evidence for peroxynitrite formation in renal ischemia-reperfusion injury: studies with the inducible nitric oxide synthase inhibitor L-N6-(1-iminoethyl)-lysine. Journal of Pharmacology and Experimental Therapeutics. 2000;295:417–422. [PubMed] [Google Scholar]
- Walker LM, York JL, Imam SZ, Ali SF, Muldrew KL, Mayeux PR. Oxidative stress and reactive nitrogen species generation during renal ischemia. Toxicological Sciences. 2001;63:143–148. doi: 10.1093/toxsci/63.1.143. [DOI] [PubMed] [Google Scholar]
- Walker PD, Kaushal GP, Shah SV. Meprin A, the major matrix-degrading enzyme in the kidney produces a novel nitrogen fragment in vitro and in vivo. Kidney International. 1998;53:1673–1680. doi: 10.1046/j.1523-1755.1998.00949.x. [DOI] [PubMed] [Google Scholar]
- Wan L, Bagshaw SM, Langenberg C, Saotome T, May C, Bellomo R. Pathophysiology of septic acute kidney injury: what do we really know? Critical Care Medicine. 2008;36:S198–S203. doi: 10.1097/CCM.0b013e318168ccd5. [DOI] [PubMed] [Google Scholar]
- Wang L, Dudek SM. Regulation of vascular permeability by sphingosine 1-phosphate. Microvascular Research. 2009;77:39–45. doi: 10.1016/j.mvr.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Falk SA, Jittikanont S, Gengaro PE, Edelstein CL, Schrier RW. Protective effect of renal denervation on normotensive endotoxemia-induced acute renal failure in mice. American Journal of Physiology. 2002;283:F583–F587. doi: 10.1152/ajprenal.00270.2001. [DOI] [PubMed] [Google Scholar]
- Wang W, Jittikanont S, Falk SA, Li P, Feng L, Gengaro PE, Poole BD, Bowler RP, Day BJ, Crapo JD, Schrier RW. Interaction among nitric oxide, reactive oxygen species, and antioxidants during endotoxemia-related acute renal failure. American Journal of Physiology. 2003;284:F532–F537. doi: 10.1152/ajprenal.00323.2002. [DOI] [PubMed] [Google Scholar]
- Wang W, Suzuki Y, Tanigaki T, Rank DR, Raffin TA. Effect of the NADPH oxidase inhibitor apocynin on septic lung injury in guinea pigs. American Journal of Respiratory and Critical Care Medicine. 1994;150:1449–1452. doi: 10.1164/ajrccm.150.5.7952574. [DOI] [PubMed] [Google Scholar]
- Wang Z, Herzog C, Kaushal GP, Gokden N, Mayeux PR. Actinonin, a meprin A inhibitor, protects the renal microcirculation during sepsis. Shock. 2011;35:141–147. doi: 10.1097/SHK.0b013e3181ec39cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Holthoff JH, Seely KA, Pathak E, Spencer HJ, Gokden N, Mayeux PR. Development of oxidative stress in the peritubular capillary microenvironment mediates sepsis-induced renal microcirculatory failure and acute kidney injury. American Journal of Pathology. doi: 10.1016/j.ajpath.2011.10.011. PMID: 22119717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe E, Muenzer JT, Hawkins WG, Davis CG, Dixon DJ, McDunn JE, Brackett DJ, Lerner MR, Swanson PE, Hotchkiss RS. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Laboratory Investigation. 2009;89:549–561. doi: 10.1038/labinvest.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattenberg BW. Role of sphingosine kinase localization in sphingolipid signaling. World Journal of Biological Chemistry. 2010;1:362–368. doi: 10.4331/wjbc.v1.i12.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JM. Mitochondrial biogenesis in kidney disease. Journal of the American Society of Nephrology. 2011;22:431–436. doi: 10.1681/ASN.2010060643. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Baumgartl H, Lubbers D, Wilcox CS. Nephron pO2 and renal oxygen usage in the hypertensive rat kidney. Kidney International. 2001;59:230–237. doi: 10.1046/j.1523-1755.2001.00483.x. [DOI] [PubMed] [Google Scholar]
- Wendel M, Knels L, Kummer W, Koch T. Distribution of endothelin receptor subtypes ETA and ETB in the rat kidney. Journal of Histochemistry & Cytochemistry. 2006;54:1193–1203. doi: 10.1369/jhc.5A6888.2006. [DOI] [PubMed] [Google Scholar]
- Wilcox CS. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacology and Therapeutics. 2010;126:119–145. doi: 10.1016/j.pharmthera.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Wilson JX. Peroxynitrite-dependent activation of protein phosphatase type 2A mediates microvascular endothelial barrier dysfunction. Cardiovascular Research. 2009;81:38–45. doi: 10.1093/cvr/cvn246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Gokden N, Mayeux PR. Evidence for the role of reactive nitrogen species in polymicrobial sepsis-induced renal peritubular capillary dysfunction and tubular injury. Journal of the American Society of Nephrology. 2007;18:1807–1815. doi: 10.1681/ASN.2006121402. [DOI] [PubMed] [Google Scholar]
- Wu L, Mayeux PR. Effects of the inducible nitric oxide synthase inhibitor L-N6-(1-iminoethyl)-lysine on microcirculation and reactive nitrogen species generation in the kidney following lipopolysaccharide administration in mice. Journal of Pharmacology and Experimental Therapeutics. 2007;320:1061–1067. doi: 10.1124/jpet.106.117184. [DOI] [PubMed] [Google Scholar]
- Wu L, Tiwari MM, Messer KJ, Holthoff JH, Gokden N, Brock RW, Mayeux PR. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipopolysaccharide administration in mice. American Journal of Physiology Renal Physiology. 2007;292:F261–F268. doi: 10.1152/ajprenal.00263.2006. [DOI] [PubMed] [Google Scholar]
- Yang S, Chung CS, Ayala A, Chaudry IH, Wang P. Differential alterations in cardiovascular responses during the progression of polymicrobial sepsis in the mouse. Shock. 2002;17:55–60. doi: 10.1097/00024382-200201000-00010. [DOI] [PubMed] [Google Scholar]
- Yassen KA, Galley HF, Webster NR. Matrix metalloproteinase-9 concentrations in critically ill patients. Anaesthesia. 2001;56:729–732. doi: 10.1046/j.1365-2044.2001.02083.x. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Leelahavanichkul A, Tsunoda S, Dear JW, Takahashi Y, Ito S, Hu X, Zhou H, Doi K, Childs R, Klinman DM, Yuen PS, Star RA. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. American Journal of Physiology Renal Physiology. 2008;294:F1050–F1058. doi: 10.1152/ajprenal.00461.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Yuen PS, Hu X, Zhou H, Star RA. Simvastatin improves sepsis-induced mortality and acute kidney injury via renal vascular effects. Kidney International. 2006;69:1535–1542. doi: 10.1038/sj.ki.5000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdan-Ashoori P, Liaw P, Toltl L, Webb B, Kilmer G, Carter DE, Fraser DD. Elevated plasma matrix metalloproteinases and their tissue inhibitors in patients with severe sepsis. Journal of Critical Care. 2011;26:556–565. doi: 10.1016/j.jcrc.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Yuan J, Pannabecker TL. Architecture of inner medullary descending and ascending vasa recta: pathways for countercurrent exchange. American Journal of Physiology Renal Physiology. 2010;299:F265–F272. doi: 10.1152/ajprenal.00071.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yura RE, Bradley SG, Ramesh G, Reeves WB, Bond JS. Meprin A metalloproteases enhance renal damage and bladder inflammation after LPS challenge. American Journal of Physiology Renal Physiology. 2009;296:F135–F144. doi: 10.1152/ajprenal.90524.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zager RA, Johnson AC, Lund S, Hanson SY, Abrass CK. Levosimendan protects against experimental endotoxemic acute renal failure. American Journal of Physiology Renal Physiology. 2006;290:F1453–F1462. doi: 10.1152/ajprenal.00485.2005. [DOI] [PubMed] [Google Scholar]
- Zanotti-Cavazzoni SL, Guglielmi M, Parrillo JE, Walker T, Dellinger RP, Hollenberg SM. Fluid resuscitation influences cardiovascular performance and mortality in a murine model of sepsis. Intensive Care Medicine. 2009;35:748–754. doi: 10.1007/s00134-008-1360-9. [DOI] [PubMed] [Google Scholar]
- Zarjou A, Agarwal A. Sepsis and Acute Kidney Injury. Journal of the American Society of Nephrology. 2011;22:999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- Zhang C, Walker LM, Hinson JA, Mayeux PR. Oxidant stress in rat liver after lipopolysaccharide administration: effect of inducible nitric oxide synthase inhibition. Journal of Pharmacology and Experimental Therapeutics. 2000;293:968–972. [PubMed] [Google Scholar]
- Zhang C, Walker LM, Mayeux PR. Role of nitric oxide in lipopolysaccharide-induced oxidant stress in the rat kidney. Biochemical Pharmacology. 2000;59:203–209. doi: 10.1016/s0006-2952(99)00324-x. [DOI] [PubMed] [Google Scholar]
- Zhang G, Xu S, Qian Y, He P. Sphingosine-1-phosphate prevents permeability increases via activation of endothelial sphingosine-1-phosphate receptor 1 in rat venules. American Journal of Physiology Heart Circulation Physiology. 2010;299:H1494–H1504. doi: 10.1152/ajpheart.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lowry SF, Guarente L, Haimovich B. Roles of SIRT1 in the acute and restorative phases following induction of inflammation. Journal of Biological Chemistry. 2010;285:41391–41401. doi: 10.1074/jbc.M110.174482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Lyons JG, Tan TK, Wang Y, Hsu TT, Min D, Succar L, Rangan GK, Hu M, Henderson BR, Alexander SI, Harris DC. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. American Journal of Pathology. 2009;175:580–591. doi: 10.2353/ajpath.2009.080983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Xia M, Wang Z, Li PL, Li N. A novel lipid natriuretic factor in the renal medulla: sphingosine-1-phosphate. American Journal of Physiology Renal Physiology. 2011;301:F35–F41. doi: 10.1152/ajprenal.00014.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingarelli B, Day BJ, Crapo JD, Salzman AL, Szabo C. The potential role of peroxynitrite in the vascular contractile and cellular energetic failure in endotoxic shock. British Journal of Pharmacology. 1997;120:259–267. doi: 10.1038/sj.bjp.0700872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulueta JJ, Yu FS, Hertig IA, Thannickal VJ, Hassoun PM. Release of hydrogen peroxide in response to hypoxia-reoxygenation: role of an NAD(P)H oxidase-like enzyme in endothelial cell plasma membrane. American Journal of Respiratory Cell and Molecular Biology. 1995;12:41–49. doi: 10.1165/ajrcmb.12.1.7529030. [DOI] [PubMed] [Google Scholar]