

Summary
While the microtubule-binding capacity of the protein tau has been known for many years, new functions of tau in signaling and cytoskeletal organization have recently emerged. In this review, we highlight these functions and the potential roles of tau in neurodegenerative disease. We also discuss the therapeutic potential of drugs targeting various aspects of tau biology.
A Microtubule-associated Protein Involved in Disease
The microtubule-associated protein tau was identified as a microtubule-assembly factor in the mid-1970s (Weingarten et al., 1975; Witman et al., 1976). Subsequently, hyperphosphorylated, insoluble, filamentous tau was shown to be the main component of neurofibrillary tangles (NFTs), a pathological hallmark of Alzheimer's disease (AD) (Grundke-Iqbal et al., 1986; Kondo et al., 1988; Lee et al., 1991; Nukina and Ihara, 1986; Wood et al., 1986). Neurodegenerative disorders with tau inclusions are referred to as tauopathies (Lee et al., 2001). These include AD; frontotemporal lobar degeneration with tau inclusions (FTLD-tau) such as Pick's disease, progressive supranuclear palsy and corticobasal degeneration; agyrophillic grain disease; some prion diseases; amyotrophic lateral sclerosis/parkinsonism–dementia complex, chronic traumatic encephalopathy; and some genetic forms of Parkinson's disease (Lee et al., 2001; Omalu et al.; Rajput et al., 2006; Santpere and Ferrer, 2009). Although associations per se cannot prove cause-effect relationships, tau inclusions are widely thought to contribute to the pathogenesis of these disorders because they occur in specific brain regions whose functions are altered by these conditions, and NFT formation correlates with the duration and progression of AD (Giannakopoulos et al., 2003; Ihara, 2001). Tau inclusions also appear to modulate the clinical features of other neurodegenerative diseases. In dementia with Lewy bodies, an α-synuclein disorder, accumulation of insoluble tau inclusions is associated with a more AD-like phenotype (Merdes et al., 2003).
Tau is expressed in the central and peripheral nervous system and, to a lesser extent, in kidney, lung and testis (Gu et al., 1996). It is most abundant in neuronal axons (Lee et al., 2001; Trojanowski et al., 1989), but can also be found in neuronal somatodendritic compartments (Tashiro et al., 1997) and in oligodendrocytes (Klein et al., 2002). Tau can be subdivided into four regions: an N-terminal projection region, a proline-rich domain, a microtubule-binding domain (MBD), and a C-terminal region (Mandelkow et al., 1996). Alternative splicing around the N-terminal region and MBD generates six main isoforms in adult human brain (Goedert et al., 1989). Tau isoforms are named by how many microtubule binding repeat sequences are expressed (termed R) and by which N-terminal exons are included (termed N) (Figure 1). For example, 3R tau has three microtubule binding repeat sequences, while 4R tau has four due to inclusion of exon 10. 0N tau includes no N-terminal exons, 1N tau exon 2, and 2N tau exons 2 and 3 (Lee et al., 2001). Tau mutations are numbered by their location in 4R2N human tau (Lee et al., 2001). Six additional isoforms are formed by alternative splicing around exon 6, resulting in a total of 12 tau isoforms expressed in brain (Wei and Andreadis, 1998), although these additional splice variants have not yet been widely studied.
Figure 1. Tau Structure and Function.

Tau is an intrinsically disordered protein that can be alternatively spliced at N terminal exons (N1, 2) and the microtubule repeat domains (R). The domains of tau bind many different types of molecules, suggesting a central role in signaling pathways and cytoskeletal organization. The diversity of tau binding partners is highlighted in Table 1. N-term, N-terminus; C-term, C-terminus; SH3, protein SH3 domain
Physiological tau has an intrinsically disordered structure and is subject to a complex array of post-translational modifications. Many serine and threonine residues on tau are phosphorylated by a variety of kinases in both physiological and pathological conditions (for a comprehensive table of tau phosphorylation and corresponding kinases, see http://cnr.iop.kcl.ac.uk/hangerlab/tautable). Tau is also post-translationally modified by tyrosine phosphorylation (Lee et al., 2004), acetylation (Cohen et al., 2011; Min et al., 2010), crosslinking by transglutaminase (Wilhelmus et al., 2009), glycation (Ledesma et al., 1994), isomerization (Miyasaka et al., 2005b), nitration (Reyes et al., 2008), sumoylation (Dorval and Fraser, 2006), O-GlcNAcylation (Arnold et al., 1996), and ubiquitination (Cripps et al., 2006). The diversity of these modifications suggests that tau is highly regulated.
Tau can bind to the outside and, possibly, also the inside of microtubules, with its N- and C-terminal regions projecting outward (Kar et al., 2003; Santarella et al., 2004). Its N-terminal region can associate with the cell membrane, likely as part of a membrane-associated complex (Figure 1), and regulate the spacing between microtubules (Al-Bassam J et al., 2002; Frappier et al., 1994; Maas et al., 2000). Its proline-rich domain includes many phosphorylation sites (Augustinack et al., 2002; Biernat et al., 1992) and can bind to SH3 domains of other proteins (Reynolds et al., 2008), including the tyrosine kinase Fyn (Lee et al., 1998). Tau's ability to bind microtubules depends on the MBD and on adjacent regions (Gustke et al., 1994). The tandem repeat sequences within the MBD are thought to directly bind microtubules through their positive net charge, which interacts with negatively charged residues in tubulin (Jho et al., 2010; Kar et al., 2003; Lee et al., 1988). Phosphorylation of tau regulates its binding to microtubules and is also associated with tau aggregation in disease. Phosphorylation of tau in and around the MBD may neutralize the positive charge (Jho et al., 2010) and alter the conformation of the MBD of tau (Fischer et al., 2009), detaching tau from microtubules. The detached tau accumulates in neuronal cell bodies and neurites, forming insoluble filaments and, ultimately, NFTs (Lee et al., 2001; von Bergen et al., 2005). The MBD also contains PHF6 (VQIVYK) and PHF6* (VQIINK), critical sequences that can assume the beta-sheet structures necessary for tau aggregation and formation of pathological inclusions (von Bergen et al., 2001, 2005)
Evidence for Multiple Functions of Tau
Although tau has been studied ever more intensely in recent years, its precise functions and roles have, if anything, become more mysterious. Some of its activities are known in great molecular detail, but were established in rather reductionist paradigms, and their in vivo significance remains uncertain. Other functions were revealed by analysis of tau knockout mice, but the precise mechanisms are poorly understood. A major advantage of tau knockout models is that they can reveal unique functions of tau that are not redundant with the functions of other proteins. For example, tau reduction prevents behavioral deficits in several models of AD (see below), suggesting that unique functions of tau are important in the pathogenesis of this condition. It remains controversial whether animal models with high levels of tau overexpression can provide relevant insights into human conditions in which such overexpression does not occur. However, the accumulation and abnormal distribution of hyperphosphorylated and aggregated tau in these models does simulate key aspects of human tauopathies. Concerns may also be raised about the relevance of studies investigating tau in nonneuronal cells. Although neurons are probably the most relevant cell type to study in relation to tauopathies, some tauopathies are associated with tau pathology in glial cells (Higuchi et al., 2005) and the proteins that interact with tau in different cell types likely overlap.
Tau has numerous binding partners (Table 1), including signaling molecules, cytoskeletal elements and lipids, suggesting that it is a multifunctional protein. Indeed, tau can bind to and affect cytoskeletal components and regulate signaling pathways by acting as a protein scaffold for signaling complexes; tau binding also activates or inhibits several enzymes.
Table 1. Partial List of Tau Binding Partners.
| Tau Binding Partners | Function of Binding Partner | References |
|---|---|---|
| ApoE3 | Lipid carrier | Fleming et al., 1996; Strittmatter et al., 1994 |
| Beta tubulin | Cytoskeleton | Kar et al., 2003 |
| cSrc | Src-family kinase | Lee et al., 1988; Reynolds et al., 2008 |
| F-actin | Cytoskeleton | Fulga et al., 2007 |
| Fgr | Src-family kinase | Reynolds et al., 2008 |
| Fyn | Src-family kinase | Lee et al., 1988; Reynolds et al., 2008 |
| Growth factor receptor-bound protein 2 (Grb2) | Adaptor protein for growth factor signaling | Reynolds et al., 2008 |
| Lck | Src-family kinase | Lee et al., 1988 |
| p85α | Regulatory subunit of PI3K | Reynolds et al., 2008 |
| Phosphatidyl inositol | Signaling lipid | Surridge and Burns, 1994 |
| Phosphatidylinositol bisphosphate | Signaling lipid | Flanagan et al., 1997 |
| PLCγ | Cleaves phospholipids into signaling molecules | Hwang et al., 1996; Jenkins and Johnson, 1998; Reynolds et al., 2008 |
Cytoskeletal Binding Functions
The most extensively described activity of tau—binding to microtubules—occurs in vitro and in vivo. In fact, the majority of tau in the cell is bound to microtubules. In cell-free conditions, this microtubule binding activity promotes microtubule assembly and stability (Weingarten et al., 1975). However, in cell culture, tau co-localizes with those microtubules that are most dynamic and most susceptible to drug-induced depolymerization (Kempf et al., 1996). Moreover, the population of tau-bound microtubules has the highest basal turnover rate of any microtubule population, both in rat primary neuronal culture and in mouse hippocampus in vivo (Fanara et al., 2010), raising doubts about the essential role of tau in microtubule stabilization postulated on the basis of in vitro findings. In addition, knockdown of tau by siRNA is not lethal to primary neurons in culture and does not decrease the number of microtubules or their polymerization state (King et al., 2006; Qiang et al., 2006). Thus, microtubule stabilization may not be a critical function of tau in vivo.
The in vivo functions of tau appear to overlap with those of MAP1B, another microtubule-associated protein found in axons. Complete ablation of tau by homologous recombination does not significantly impair longevity or most critical brain functions, but clearly worsens the MAP1B knockout phenotype of premature mortality and brain dysgenesis (Takei et al., 2000). Knockout of both tau and MAP1B results in severe brain dysgenesis and is lethal within the first month of life. Assuming that this phenotype relates to the microtubule-binding activities of tau and MAP1B, which is uncertain, it is reasonable to speculate that MAP1B is more important for microtubule stabilization than tau and that their overlapping functions are critical for postnatal brain maturation. However, because of the early lethality, it is impossible to draw firm conclusions from the double-knockout phenotype on the functions of tau and MAP1B in the adult or aging brain.
In principle, tau's binding to microtubules could regulate axonal transport. Tau can interfere with the binding of motor proteins to microtubules (Dixit et al., 2008; Ebneth et al., 1998), and there is a gradient of tau along the axon; the highest levels are closest to the synapse (Mandell and Banker, 1996). This distribution might facilitate the detachment of motor proteins from their cargo near the presynaptic terminal, increasing axonal transport efficiency (Dixit et al., 2008). However, ablation of tau does not alter axonal transport in primary neuronal culture (Vossel et al., 2010) or in vivo (Yuan et al., 2008), making an essential role of tau in this physiological function less likely.
Tau can also bind to and bundle actin filaments (Fulga et al., 2007; He et al., 2009; Kotani et al., 1985), activities mediated primarily by its MBD (Farias et al., 2002; Yu and Rasenick, 2006) and assisted by the adjacent proline-rich domain (He et al., 2009) (Figure 1). It is possible that tau connects microtubule and actin filament networks (Farias et al., 2002).
Modulation of Signaling Pathways through Scaffolding
Tau could also act as a protein scaffold, and regulation of its binding partners may alter signaling pathways. For example, tau modulates the activity of Src family kinases. In mouse brain tissues, tau co-immunoprecipitates with both the tyrosine kinase Fyn and the scaffolding protein PSD-95, and in the absence of tau, Fyn can no longer traffic into postsynaptic sites in dendrites (Figure 2) (Ittner et al., 2010). The authors speculated that tau normally tethers Fyn to PSD-95/NMDA receptor signaling complexes. Although very little tau is normally present in dendrites, it may be enough to ensure proper localization of postsynaptic components (Ittner et al., 2010). Similarly, tau acts as a protein scaffold in oligodendrocytes, connecting Fyn and microtubules to enable process extension (Klein et al., 2002). In cell culture, tau binds to and activates both cSrc and Fyn and facilitates cSrc-mediated actin rearrangements following platelet-derived growth factor stimulation (Sharma et al., 2007).
Figure 2. Endogenous Tau Exists in Dendrites.
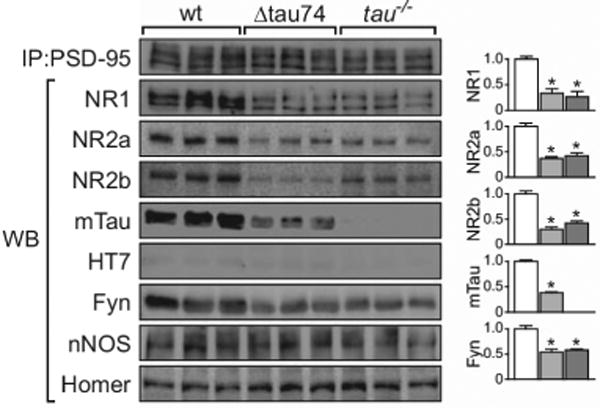
Tau immunoprecipitates with PSD95, a postsynaptic protein, and regulates the association of PSD-95 with NMDAR subunits and the tyrosine kinase Fyn. Overexpression of a human N-terminal truncation of tau (Δtau74, light grey) or knocking out tau (tau−/−, dark grey) reduced the amount of NR1, NR2a, NR2b, and Fyn within PSD95 complexes. From Ittner et al., 2010 with permission.
Tau may also regulate signaling cascades that control neurite extension, although this is a somewhat controversial area. Some investigators have reported a defect in neurite extension in tau knockout neurons in vitro (Dawson et al., 2001), whereas others found no such defect despite observing decreased numbers of microtubules (Harada et al., 1994). Knockdown of tau by siRNA decreased the length of axons (Qiang et al., 2006) but not the number of microtubules (King et al., 2006; Qiang et al., 2006), and overexpression of tau promoted neurite extension in cell culture (Brandt et al., 1995). These effects may relate to tau's ability to thwart microtubule-severing proteins (Qiang et al., 2006), but could also involve facilitation of nerve growth factor (NGF) signaling.
In PC12 cells, overexpression of full-length tau was associated with normal neurite extension and an increased number of neurites per cell, whereas overexpression of the N-terminus of tau suppressed NGF-induced neurite extension (Brandt et al., 1995). Thus, increased levels of tau may enhance NGF function, whereas the N-terminus of tau may impair NGF signaling, possibly by a dominant-negative mechanism. Enhancement of NGF signaling by tau may involve increased association of tau with actin filaments, which occurs after stimulation with NGF and is mainly mediated by the MBD (Yu and Rasenick, 2006) rather than the N-terminus. In PC12 cells, tau facilitates signaling through receptors for NGF and epidermal growth factor (EGF), thereby increasing activity in the mitogen-activated protein kinase (MAPK) pathway (Leugers and Lee, 2010). Stimulation of PC12 cells with NGF or EGF causes tau phosphorylation at T231, a modification necessary for the growth factor-induced activation of the Ras-MAPK pathway (Leugers and Lee, 2010), nicely illustrating the functional significance of a single tau phosphorylation site. As tau is not known to directly interact with growth factor receptors, it may facilitate signaling by binding to adapter proteins such as Grb2 (Reynolds et al., 2008). The enhancement of growth factor signaling by increased tau expression may explain why several forms of chemotherapy-naive cancer cells overexpress tau (Rouzier et al., 2005; Souter and Lee, 2009).
Other Roles of Tau in Signaling Pathways
Tau binds phospholipase C (PLC) γ in human neuroblastoma (SH-SY5Y) cells (Jenkins and Johnson, 1998). Under cell-free conditions and in the presence of unsaturated fatty acids, tau activates PLCγ independently of the tyrosine phosphorylation usually required to activate this enzyme (Hwang et al., 1996). At high tau concentrations, this activation does not require fatty acids (Hwang et al., 1996) and may involve binding of tau to both the enzyme and the substrates phosphatidylinositol (Surridge and Burns, 1994) or phosphatidylinositol 4,5-bisphosphate (Flanagan et al., 1997), which could facilitate the cleavage reaction. Activation of PLCγ by tau was particularly facilitated by arachidonic acid (Hwang et al., 1996). Arachidonic acid is released from phospholipids by cytosolic phospholipase A2, whose activity in the brain is increased in AD patients and related mouse models (Sanchez-Mejia et al., 2008). Thus, increased levels of tau and arachidonic acid may jointly increase signaling through the PLCγ pathway in AD.
Tau can also act as a direct enzyme inhibitor. For example, it can bind to and inhibit histone deacetylase-6 (Perez et al., 2009), which deacetylates tubulin and may regulate microtubule stability (Perez et al., 2009). Thus, tau may affect microtubule stability by a mechanism independent of tubulin binding, although reports regarding the levels of acetylated tubulin in tau knockout mice vary (Perez et al., 2009; Rapoport et al., 2002).
Tau also appears to participate in the cellular response to heat shock. During heat shock of neurons, tau bound DNA and facilitated DNA repair, and tau knockout neurons showed increased DNA damage (Sultan A et al., 2010). However, when cultured neurons were allowed to recover from heat shock, tau knockout actually protected against heat shock-induced cell damage, as determined by measurements of neurite length and lactate dehydrogenase release (Miao et al., 2010). Compared with wildtype neurons, tau knockout neurons showed a delayed and prolonged activation of Akt and less GSK3β activity during recovery from heat shock (Miao et al., 2010), suggesting that the protective effect of tau knockout may be upstream of Akt/GSK3β phosphorylation. In sensory neurons of C. elegans, overexpression of 4R0N tau decreased the response to touch, and this phenotype was exacerbated by heat shock when tested after a recovery period of 24 hours (Miyasaka et al., 2005a). These results suggest that tau has a role in the cellular response to heat shock, both during the insult and in the subsequent recovery phase.
Adult Neurogenesis
Tau affects adult neurogenesis. Three-repeat tau is expressed and highly phosphorylated in adult-born granule cells in the dentate gyrus (Bullmann et al., 2007; Hong et al., 2009). In one strain of tau knockout mice, adult neurogenesis was found to be severely reduced (Hong et al., 2009). However, tau does not appear to be needed for embryonic neurogenesis, as tau knockout mice have grossly normal brain anatomy. The functional significance of adult neurogenesis is a topic of intense study and debate (Zhao et al., 2008). Notably, adult tau knockout mice showed no deficits in a variety of learning and memory paradigms (Dawson et al., 2010; Ittner et al., 2010; Roberson et al., 2007, 2011).
Mechanisms by Which Tau Contributes to Disease
As mentioned above, tau probably fulfills multiple functions and may contribute to neuropathogenesis in multiple ways. In principle, this might include both gain- and loss-of-function effects, although the latter mechanism has recently been called into question by several lines of experimental evidence. Furthermore, tau does not act alone. For example, in AD it appears to enable the pathogenic effects of both Aβ and apolipoprotein E4 (apoE4) (Andrews-Zwilling et al., 2010; Ittner et al., 2010; Roberson et al., 2007, 2011).
Loss of Function
A prominent theory about tau pathology was that disease phenotypes were caused by loss of tau function due to hyperphosphorylation and sequestration of soluble tau (Zhang et al., 2005). In many tauopathies, tau is hyperphosphorylated, which releases tau from microtubules, apoE (Strittmatter et al., 1994), Src (Bhaskar et al., 2005), and possibly other binding partners. Although it is conceivable that this process results in loss of specific tau functions, increased phosphorylation of tau per se is probably not detrimental, as it occurs naturally during hibernation (Arendt et al., 2003) and fetal development (Yu et al., 2009). Although phosphorylated tau from AD brains may seed the aggregation of control human tau (Alonso et al., 1996), we are unaware of any evidence that tau aggregation actually lowers levels of soluble tau in vivo.
Recent experimental studies have shown more directly that loss of tau function is an unlikely cause of neurodegeneration and neuronal dysfunction. In our opinion, longevity and behavioral functions are among the most compelling outcome measures for the evaluation of biologically meaningful functions affecting the central nervous system. Complete ablation of tau in knockout mice does not cause premature mortality or major neurological deficits (Dawson et al., 2001; Harada et al., 1994; Ikegami et al., 2000; Roberson et al., 2007, 2011; Yuan et al., 2008). Four independent tau knockout lines have been established and most of them have normal behavior throughout most of their lives (Table 2). Only one of these lines was reported to have motor deficits, hyperactivity in the open field test, and learning impairments in contextual fear conditioning at 10–11 weeks of age (Ikegami et al., 2000). Using fear conditioning to assess learning and memory in hyperactive mice is problematic because the hyperactivity confounds the interpretation of diminished freezing (Rudy et al., 2004). Because tau ablation has so little impact on neural functions, two of the tau knockout lines were actually generated as indicator tools for neuron-specific expression of EGFP (Tucker et al., 2001) or Cre (Muramatsu et al., 2008). Although axonal abnormalities have been reported in the cingulate cortex and genu of the corpus callosum of 10- and 12-month-old tau knockout mice, these mice showed no behavioral deficits in the rotarod test, Morris water maze, or radial arm water maze (Dawson et al., 2010). To our knowledge, no deficits of any kind have been identified in hemizygous knockout mice, which have roughly half normal tau levels (Ikegami et al., 2000; Roberson et al., 2007).
Table 2. Behavioral Tests in Tau Knockout Mice.
| Mouse Line | Age | Behavioral Test | Result |
|---|---|---|---|
| Tau Knockout Line 1 (Harada et al., 1994) | 10–11 weeks | Balance Beam | Impaired motor function (Ikegami et al., 2000) |
| 10–11 weeks | Contextual Fear Conditioning | Impaired learning/memory (Ikegami et al., 2000) | |
| 10–11 weeks | Open Field | Hyperactive (Ikegami et al., 2000) | |
| 10–11 weeks | Wire Hang | Impaired motor function (Ikegami et al., 2000) | |
| Tau Knockout Line 2 (Dawson et al., 2001) | 4–7 months, 12–16 months | Elevated Plus Maze | Normal anxiety and exploration (Roberson et al., 2007) |
| 4-7 months | Morris Water Maze | Normal learning/memory (Roberson et al., 2007) | |
| 10–12 months | Normal learning/memory (Dawson et al., 2010) | ||
| 10–12 months | Rotor Rod | Normal motor function (Dawson et al., 2010) | |
| 10–12 months | Radial Arm Water Maze | Normal learning/memory (Dawson et al., 2010) | |
| 4–7 months | Y-maze | Normal activity (Roberson et al., 2007) | |
| Tau Knockout/EGFP Knockin Line (Tucker et al., 2001) | 4.5–7.5 months | Elevated Plus Maze | Normal anxiety and exploration (Roberson et al., 2011) |
| 4.5–7.5 months | Novel Object Recognition | Normal learning/memory (Roberson et al., 2011) | |
| 8 months | T Maze | Normal learning/memory (Ittner et al., 2010) | |
| Tau Knockout/Cre Knockin Line (Muramatsu et al., 2008) | – | None | – |
Based on electrophysiological recordings in acute hippocampal slices, tau knockout mice and wildtype controls have similar NMDA/AMPA receptor currents, synaptic transmission strength, and short-term as well as long-term synaptic plasticity (Roberson et al., 2011; Shipton et al., 2011). Surprisingly, tau knockout mice are more resistant to seizures elicited by disinhibition, excitotoxins, or amyloid-β (Aβ) peptides than wildtype mice (Figure 3A) (Ittner et al., 2010; Roberson et al., 2007, 2011). Compared with wildtype controls, neurons in hippocampal slices from tau knockout mice are more resistant to disinhibition-induced bursting activity (Figure 3B), which may be due, at least in part, to an increased frequency of spontaneous inhibitory postsynaptic currents in tau knockout mice (Roberson et al., 2011). These findings suggest that tau has a complex role in regulating neural network activity and that tau reduction could prevent aberrant neuronal excitability, synchrony, or both.
Figure 3. Tau Reduction Suppresses Drug-induced Seizures in Mice and Neuronal Bursting Activity in Acute Hippocampal Slices.
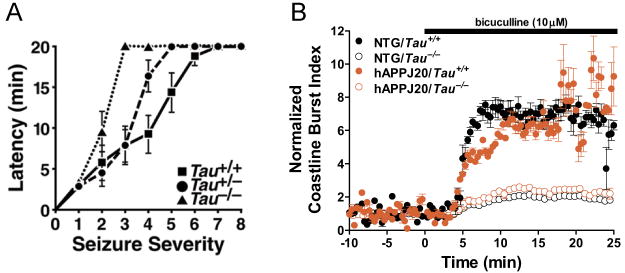
A) Partial or complete reduction of tau in mice without hAPP expression delayed the onset and reduced the severity of seizures induced by pentylenetetrazol, a GABAA receptor antagonist (Roberson et al., 2007). B) Tau knockout reduced aberrant neuronal discharges in acute hippocampal slices after disinhibition with the GABAA receptor antagonist bicuculline, as illustrated by measurements of the coastline burst index (Roberson et al., 2011). NTG, no hAPP expression; hAPPJ20, hAPP mice from line J20
The resistance of tau knockout mice to seizures may also relate to alterations in brain oscillatory patterns. Tau knockout mice have decreased peak frequency of theta waves in the hippocampus and decreased coherence of gamma waves in the frontal cortex (Cantero et al.). The potential effects of these alterations on Aβ-induced dysrythmias and cognitive abnormalities remain to be determined.
In conventional tau knockout mice, other MAPs might compensate for tau loss, particularly MAP1A and MAP1B. However, no changes in MAP1A, MAP1B or MAP2 protein levels were detected in 12-month-old adult tau knockout mice (Dawson et al., 2001). To evaluate the safety of tau reduction strategies for therapeutic purposes more conclusively, tau needs to be reduced in adult mice after brain development and maturation are complete, and such experiments are in progress. In cultured cells, acute knockdown of tau did not affect the stability or polymerization state of microtubules (King et al., 2006; Qiang et al., 2006), and reducing tau levels in brains of 3-month-old wildtype mice for 12 weeks by methylene blue administration caused no behavioral deficits in the rotarod test or Morris water maze (O'Leary et al., 2010). Thus, it is unlikely that loss of tau function is an important cause of neuronal dysfunction or degeneration in AD and related conditions. In addition, the findings summarized above suggest that partial reduction of tau may be well tolerated and could effectively protect the brain against Aβ, epileptogenesis and excitotoxicity.
Tau Functions Enabling Pathogenesis
In transgenic mice, wildtype levels of tau are required for Aβ and apoE4 to cause neuronal, synaptic and behavioral deficits (Andrews-Zwilling et al., 2010; Ittner et al., 2010; King et al., 2006; Rapoport et al., 2002; Roberson et al., 2007, 2011; Vossel et al., 2010). However, whether Aβ and apoE4 contribute to AD-related cognitive decline through the same or distinct tau-dependent mechanism(s) remains to be determined. Acute exposure of neuronal cultures to Aβ led to hyperphosphorylation (De Felice et al., 2008) and mislocalization of tau into dendritic spines (Zempel et al., 2010), which, at least in some dendrites, was associated with spine collapse and dendritic degeneration. As tau phosphorylation releases tau from many of its binding partners, it is tempting to speculate that tau is initially hyperphosphorylated in AD to reduce its function, in an effort to counteract Aβ-induced neuronal dysfunction. With time, though, this compensatory mechanism fails because hyperphosphorylated tau becomes detrimental at concentrations sufficient to form toxic tau aggregates.
While tau is abnormally phosphorylated in apoE4 transgenic mice (Brecht et al., 2004), we have so far found no evidence of abnormal phosphorylation or aggregation of tau in hAPP-J20 mice, whose robust Aβ-dependent neuronal and behavioral deficits were prevented by reduction of wildtype murine tau (Table 3 and Figure 4) (Roberson et al., 2007, 2011). While we continue to search for a direct pathogenic tau mediator and a pathogenic mislocalization of tau in hAPP-J20 mice, the above findings raise the possibility that physiological functions of tau, rather than an abnormal tau gain of function, permit Aβ and other AD-related factors to elicit aberrant neuronal excitation (Ittner et al., 2010; Roberson et al., 2007, 2011), abnormalities in axonal transport (Vossel et al., 2010), and impairment of inhibitory interneurons (Andrews-Zwilling et al., 2010) (Figure 5). Notably, even partial tau reduction improved longevity and cognitive functions in hAPP-J20 mice (Figure 4) (Roberson et al., 2007). Tau knockout also improved longevity and cognitive functions in APP23 mice and in both lines markedly increased resistance to seizures in mice with or without hAPP (Ittner et al., 2010; Roberson et al., 2007, 2011). For unclear reasons, tau reduction was not beneficial in the Tg2576 hAPP mouse model (Dawson et al., 2010).
Table 3. Electrophysiological Abnormalities of hAPP-J20 Mice Prevented by Tau Knockout.
(Roberson et al., 2011).Similar results were observed after exposure of hippocampal slices to recombinant Aβ aggregates (Shipton et al., 2011).
| Electrophysiological Measure* | Tau−/− Mice** | hAPP/Tau+/+ Mice** | hAPP/Tau−/− Mice*** |
|---|---|---|---|
| Action Potential-driven | Normal | ↓ | Normal |
| IPSC Frequency | |||
| Coastline Burst Index | ↓ | ↑ | ↓ |
| EEG epileptiform activity (cortex) | Normal | ↑ | Normal |
| eEPSC Amplitude | Normal | ↑ | Normal |
| eIPSC Amplitude | Normal | ↓ | Normal |
| Long-term Potentiation | Normal | ↓ | Normal |
| mEPSC Frequency | Normal | ↓ | Normal |
| mIPSC Frequency | Normal | ↑ | Normal |
| NMDAR/AMPAR Ratio | Normal | ↓ | Normal |
| Paired Pulse Ratio | Normal | ↓ | Normal |
| sIPSC Frequency | ↑ | ↓ | ↑ |
| Synaptic Strength (CA1) | Normal | Normal |
EPSC, excitatory post-synaptic current; IPSC, inhibitory post-synaptic current; e, evoked; m, miniature; s, spontaneous
Recorded at perforant path togranule cell synapsein dentate gyrusunless indicated otherwise. All recordings were obtained in acute hippocampal slices.
Relative to Tau+/+ mice
Relative to hAPP/Tau+/+ mice
Figure 4. Tau Reduction Does Not Change Amyloid Pathology But Prevents Aβ-dependent Cognitive and Functional Neuronal Deficits.
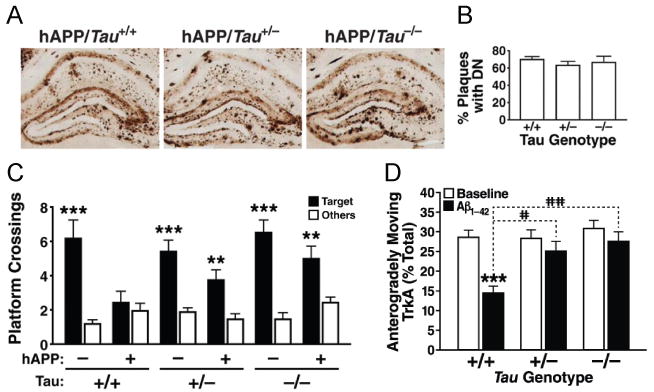
Tau reduction did not affect (A) Aβ deposition or (B) the number of plaques with dystrophic neurites (DN) in hAPP-J20 mice. However, even partial tau reduction prevented (C) memory deficits in the Morris water maze (72 hour probe trial) in hAPP-J20 mice (Roberson et al., 2007) and (D) Aβ oligomer-induced axonal transport deficits in primary hippocampal neurons, although it had no effect on axonal transport at baseline (Vossel et al., 2010).
Figure 5. Potential Mechanisms of Tau-dependent Aβ Toxicity.

Experiments in which neurons were acutely exposed to Aβ suggest that Aβ can trigger tau-mediated neurotoxicity by enhancing tau phosphorylation, which in turn directs pathogenic tau species into dendritic spines where they exert adverse effects (left). In hAPP mice, though, in which neurons are chronically exposed to elevated Aβ levels, it has so far been impossible to find clear evidence for a similar process. Nonetheless, Aβ-induced neuronal dysfunction in these models strictly depends on the presence of tau, raising the possibility that physiological functions of tau permit Aβ to cause neuronal dysfunction (right). Such functions may involve the intraneuronal trafficking of factors that regulate synaptic activity at the pre- or postsynaptic level.
The tyrosine kinase Fyn appears to be important in the development of Aβ- and tau-dependent neuronal deficits. Neuronal overexpression of Fyn sensitizes hAPP mice to Aβ-induced neuronal, synaptic, and cognitive deficits (Chin et al., 2004, 2005) that are prevented by knocking out tau in hAPP-J9/FYN doubly transgenic lines (Roberson et al., 2011). Tau knockout prevented behavioral deficits in the Morris water maze and elevated plus maze of hAPP/FYN mice and premature mortality in two separate lines of hAPP/FYN mice (Roberson et al., 2011). In addition, tau knockout prevented spontaneous epileptic activity in hAPP/FYN mice and hAPP-J20 mice (Table 3) (Roberson et al., 2011). This striking antiepileptic effect could result from the reduction of tau in axons, dendrites, or both. Although tau knockout did not affect axonal transport at baseline (Vossel et al., 2010; Yuan et al., 2008), it precluded Aβ-induced deficits in the axonal transport of cargoes that could affect neuronal excitability (Figure 5D) (Vossel et al., 2010). Tau is also required for Fyn to gain access to and phosphorylate the NR2B subunit of dendritic NMDA receptors (Ittner et al., 2010). Consistent with our hypothesis that tau reduction protects against Aβ by preventing neuronal overexcitation (Roberson et al., 2007), targeted perturbation of the NR/PSD-95 interaction, which prevents excitotoxicity, also prevented premature mortality and memory deficits in APP23 mice (Ittner et al., 2010). These findings suggest that modulating tau, its interaction with Fyn, or key proteins involved in or affected by this interaction may be of therapeutic benefit in AD.
The interaction between Fyn and tau may also contribute to FTLD. Several forms of FTLD-mutant tau and pseudohyperphosphorylated tau bind Fyn more tightly than wildtype tau (Bhaskar et al., 2005), which may increase neuronal Fyn activity. Furthermore, Fyn binds more tightly to 3R0N tau than 4R0N tau (Bhaskar et al., 2005), implying that FTLD mutations that alter tau splicing could also alter the activity or localization of Fyn. In mice overexpressing P301L 4R0N tau under the mouse prion promoter (JNPL3 model), phosphorylation of tau at Y18 by Fyn increases simultaneously with tau hyperphosphorylation on serine/theronine sites before the onset of behavioral deficits (Bhaskar et al., 2010).
The physiological actin-bundling function of tau may also contribute to pathology. Filamentous actin inclusions, closely resembling Hirano bodies in AD, were found in Drosophila models overexpressing wildtype or R406W 4R0N tau and in mice overexpressing P301L 4R0N tau under the TRE promoter with the tet-off element under the CaMKII promoter (rTg4510 model) (Fulga et al., 2007). Knocking out or destabilizing actin filaments in the Drosophila models prevented tau-induced degeneration (Fulga et al., 2007), implicating alterations in actin dynamics as a mediator of tau toxicity.
Abnormal Gain of Function
The largest amount of work in this field has focused on tau phosphorylation and aggregation. Tau is highly phosphorylated in fetal brain without eliciting toxicity and is also phosphorylated on many sites in adult brain, albeit with lower frequency (Matsuo et al., 1994; Yu et al., 2009). Tau is transiently hyperphosphorylated during hibernation without long-term harm to neural networks (Arendt et al., 2003). Tau phosphorylation is also markedly increased in response to various stressors. In humans, tau becomes hyperphosphorylated and aggregated after head trauma, following earlier increases in APP expression, axonal swelling and microtubule disruption (Gentleman et al., 1993; Omalu et al.). Tau also becomes hyperphosphorylated in mouse brain in response to hypothermia and experimental insulin-dependent diabetes (Planel et al., 2007). In cell culture and brain slice models of neuronal injury, tau is hyperphosphorylated during recovery from heat shock (Miao et al., 2010), in response to Aβ oligomer treatment (De Felice et al., 2008; Zempel et al., 2010), hypoxia, and glucose deprivation (Burkhart et al., 1998). In cell culture, ATP, glutamate, hydrogen peroxide, serum deprivation and Aβ oligomer treatment all cause tau mislocalization into dendrites (Zempel et al., 2010), a process that is likely triggered by hyperphosphorylation-induced dissociation of tau from microtubules and cell membranes. Thus, increased phosphorylation and redistribution of tau may be common responses to neuronal stress.
Findings in Drosophila models suggest that tau phosphorylation may cause neurotoxicity in a combinatorial fashion rather than through the modification of individual phosphorylation sites and involves the folding of tau into an abnormal conformation resembling tau conformations found in AD (Steinhilb et al., 2007). Hyperphosphorylated tau has a tighter, more folded conformation and an increased propensity to aggregate (Jeganathan et al., 2008), as does tau with mutations found in FTLD (Lee et al., 2001). In C. elegans, overexpression of wildtype or mutant 4R1N tau causes axonal degeneration and an uncoordinated phenotype indicative of neuronal dysfunction (Kraemer et al., 2003). The extent of phosphorylation was similar across mutant and wildtype tau lines, but more insoluble tau was found in the former (Kraemer et al., 2003). Worms overexpressing mutant tau that formed aggregates had a more severe phenotype (Kraemer et al., 2003).
Although filamentous tau inclusions are a pathologic hallmark of tauopathies, experimental evidence suggests that filamentous tau may not be responsible for neuronal dysfunction. In a regulatable P301L 4R0N tau transgenic mouse (rTg4510 model), inhibiting tau production after filamentous tau inclusions formed reversed behavioral deficits in the Morris water maze, even though inclusion formation progressed (SantaCruz et al., 2005). Acute tau reduction by methylene blue treatment in this model improved memory scores in correlation with the reduction of soluble tau in the brain but did not alter the number or length of tau fibrils or the amount of Sarkosyl-insoluble tau compared to untreated transgenic mice (O'Leary et al., 2010). In other mouse lines, tet-off transgenes were regulated by the CaMKII promoter to express either the 4R microtubule repeat domain of human tau with a deletion of lysine 280 (termed TauRD), which is highly prone to aggregation, or the TauRD construct with an additional two mutations (I277P/I308P) that prevent TauRD aggregation (Mocanu et al., 2008). The proaggregation transgenic mouse, which formed hyperphosphorylated tau inclusions containing TauRD and endogenous mouse tau, developed synaptic loss (Mocanu et al., 2008), memory deficits and electrophysiological deficits (Sydow et al., 2011). In contrast, the anti-aggregation transgenic mouse showed none of these abnormalities. Turning off the transgene in the proaggregation mouse reversed behavioral and electrophysiological deficits without eliminating insoluble tau aggregates, which were composed entirely of endogenous mouse tau after the transgene had been turned off for 4 months (Sydow et al., 2011). These data highlight that tau aggregation causes toxicity, possibly through the formation of tau oligomers.
This conclusion is also supported by studies using in vivo two-photon microscopy demonstrating that very few neurons containing tau inclusions in regulatable rTg4510 transgenic mice have caspase activation or membrane disruption (de Calignon et al., 2009, 2010). Decreasing the levels of soluble tau reduced caspase activation in inclusion-positive neurons without affecting the number or size of tau inclusions (de Calignon et al., 2010), implicating soluble tau, not tau inclusions, in the activation of proapoptotic pathways. Neurons with or without tau inclusions in this regulatable P301L tau model showed similar electrophysiological deficits, relative to wildtype neurons (Rocher et al., 2010). Studies in young transgenic flies overexpressing wildtype or mutant 4R0N tau constructs also indicated that toxicity was conferred by soluble tau species, possibly dimers (Feuillette et al., 2010).
Collectively, these studies suggest that tau inclusions are not very toxic and that neuronal toxicity is caused by a smaller, soluble aggregate of a specific conformation of tau. Tau oligomers have been identified in in vitro and in vivo models as well as in AD brains (Berger et al., 2007; Maeda et al., 2007; Sahara et al., 2008). In regulatable P301L 4R0N transgenic mice (rTg4510 model), the extent of memory deficits correlated with the level of putative tau oligomers (Berger et al., 2007).
Tau can also be cleaved in various places by caspase-3, calpain, and cathepsin L, and several of the resulting fragments are thought to increase tau aggregation. In primary neurons exposed to Aβ, calpain generates a 17-kDa tau fragment. However, the toxicity and in vivo relevance of this fragment are debated; its presence is variable in both control and AD brains (Garg et al., 2011; Park and Ferreira, 2005) and it appears to be absent from brains of hAPP-J20 mice (Roberson et al., 2007). Aβ treatment of cortical neurons causes caspase cleavage of tau at Asp421, and cleavage at this site facilitates the formation of tau aggregates in cell-free conditions (Gamblin et al., 2003). Caspase activation precedes formation of filamentous tau inclusions in P301L 4R0N tau transgenic mice (rTg4510 model), raising the possibility that caspase cleavage is important for aggregation of FTLD-mutant tau in vivo (de Calignon et al., 2010). In an inducible cell culture model overexpressing the microtubule repeat domain of tau missing K280, cytosolic cleavage by unknown proteases generated putative tau oligomers associated with lysosomal membranes and inhibited chaperone-mediated autophagy; smaller fragments produced by cathepsin L seeded tau aggregation (Wang et al., 2009).
Tau may also exert toxic effects from the extracellular milieu (Gomez-Ramos et al., 2006). The death of degenerating neurons or extrusion of tau from living cells containing tau aggregates (Frost et al., 2009) may result in the release of pathogenic tau species into the extracellular space, where they may adversely affect neighboring cells. For example, a peptide in the C-terminus of tau (amino acids 391–407) increased intracellular calcium concentrations by activating the muscarinic receptors M1 and M3 (Gomez-Ramos et al., 2008). Tau aggregates released from cells were taken up by and triggered tau aggregation within co-cultured cells that had no pre-existing tau aggregates (Frost et al., 2009).
Injection of insoluble P301S human 4R0N tau from transgenic mouse brainstem extracts into the hippocampus of transgenic mice expressing wildtype 4R2N human tau under the mouse Thy1.2 promoter caused intraneuronal formation of wildtype 4R2N human tau inclusions in the hippocampus that spread along synaptic connections to distant brain regions (Clavaguera et al., 2009). However, we are unaware of any evidence that this transfer of tau aggregation causes neuronal dysfunction or neurodegeneration. Injecting soluble P301S tau into the transgenic mice or insoluble P301S tau into nontransgenic mice failed to cause extensive pathology.
Thus, artificial introduction of insoluble tau into the brain parenchyma triggers propagation of tau pathology along neuronal pathways, but only in the presence of the correct tau template. It is unknown whether the potential progression of AD from one brain region to another (Braak and Braak, 1997) depends on similar processes, the presynaptic release of Aβ (Harris et al., 2010), or other mechanisms.
Do FTLD Mutations Simulate Tau Pathology in AD?
Interestingly, mutations in the tau gene cause FTLD disorders such as progressive supranuclear palsy, corticobasal degeneration, and frontotemporal dementia, but never AD. While the clinical spectrum associated with the many rare tau mutations varies, most FTLD disorders differ from AD both in the types of tau inclusions and in the brain regions affected (Mann et al., 2001), indicating a possible divergence in the roles of tau in these conditions. The trigger for increased phosphorylation and aggregation of tau is also likely different in AD and FTLD.
Two recent studies set out to compare the consequences of overexpressing wildtype human 4R2N tau versus P301L-mutant human 4R2N tau in transgenic mice. Each group generated two mouse lines with approximately matched tau expression levels and patterns directed by the Thy1 promoter (Terwel et al., 2005) or the CaMKII promoter (Kimura et al., 2010). In both studies, P301L tau mice differed from wildtype tau mice in tau phosphorylation patterns and in that P301L tau aggregated more readily than wildtype tau, consistent with previous findings (von Bergen et al., 2001). Remarkably, in both studies, behavior was impaired earlier in wildtype tau transgenic mice than in P301L tau transgenic mice, even though tau was similarly expressed under the same promoter and only P301L mutant tau transgenic mice had tau aggregates. Thy1-wildtype-tau mice had early motor impairments and axonopathy, whereas Thy1-P301L-tau mice had late motor impairments, insoluble tau inclusions and no axonopathy (Terwel et al., 2005). CaMKII-wildtype-tau mice had earlier memory deficits in the Morris water maze and synaptic loss than CaMKII-P301L tau mice, but tau inclusions and neuronal loss were observed only in CaMKII-P301L tau mice (Kimura et al., 2010). These findings suggest that P301L tau causes neuronal loss through its propensity to aggregate and, possibly, to form toxic oligomeric tau species, whereas wildtype human tau may cause early neuronal and cognitive dysfunction without neuronal cell death, possibly by enhancing normal functions of tau or altering the regulation of endogenous tau.
Another recent study compared transgenic mice expressing P301L human 4R0N tau (rTg4510 model) or wildtype human 4R0N tau directed by the TRE promoter and tTA (tet-off) directed by the CaMKII promoter. Both lines showed deficits in the Morris water maze; however, the deficits worsened with aging in the P301L tau line but not in the wildtype tau line (Hoover et al., 2010). As in the lines described above, neurodegeneration was identified only in the P301L tau line but not in the wildtype tau line. In primary cultures, neurons expressing P301L tau showed tau in dendritic spines more frequently and had greater reductions of miniature excitatory postsynaptic potentials (mEPSCs) and dendritic GluR1, GluR2/3 and NR1 levels than neurons expressing wildtype tau (Hoover et al., 2010). Tau phosphorylation was required for tau to enter into dendritic spines and to impair mEPSCs in transfected primary rat neurons (Hoover et al., 2010).
In slice cultures, wildtype human 3R0N tau and R406W human 4R2N tau each enhanced Aβ-induced neuronal cell death in the hippocampal CA3 region, whereas P301L human 4R2N tau did not (Tackenberg and Brandt, 2009). A 3R0N tau mutant that prevents phosphorylation and a 3R0N tau mutant that mimics hyperphosphorylation showed no synergistic neurotoxic effect with Aβ, implying that dynamic tau phosphorylation may be required for the enhancement of Aβ toxicity by tau (Tackenberg and Brandt, 2009). These results are consistent with findings indicating that tau requires phosphorylation to enter the dendritic spine in order to affect synaptic function (Hoover et al., 2010) and that Aβ oligomers acutely increase phosphorylation and mislocalization of wildtype tau into dendritic spines (Zempel et al., 2010).
It may be that P301L tau is already phosphorylated and present in dendritic spines, and therefore no further toxicity is seen in the presence of Aβ oligomers. Interestingly, the adverse effects of wildtype tau were dependent on the activity of both NMDAR and GSK3β, whereas the effects of R406W tau were dependent only on NMDAR activity, suggesting partly distinct mechanisms of toxicity (Tackenberg and Brandt, 2009). These results imply that AD-relevant pathogenic mechanisms and therapeutic interventions might be missed in FTLD-mutant transgenic mice due to the overriding effect of the FTLD mutations.
Treatments Targeting Tau
Treatments targeting various aspects of tau biology are under intense investigation. Inhibitors of tau phosphorylation and aggregation and microtubule stabilizers are already in clinical trials for people with MCI, AD, or FTLD (Table 4), while tau reduction strategies are still in preclinical stages of development. As we learn more about how tau expression is regulated and about tau's involvement in cell signaling and cytoskeletal organization, additional approaches are likely to emerge.
Table 4. Tau-targeted Therapies in Clinical Trials*.
| Target | Drug | Diagnosis | Goal Sample Size | Treatment Duration | Stage | Status/Outcome |
|---|---|---|---|---|---|---|
| Tau phosphorylation by GSK3β | Lithium ± Divalproex | AD | 35 | 6 weeks | Phase II | Completed, not yet published |
| Lithium | MCI | 80 | 2 years | Phase II | Ongoing | |
| Lithium | PSP, CBD | 17 | 28 weeks | Phase I/II | Completed, not yet published | |
| Lithium | Mild AD | 71 | 10 weeks | RCT | Completed. No effect on lymphocyte GSK3 activity or CSF p-tau (Hampel et al., 2009) | |
| Microtubule stabilization/tau phosphorylation | NAP, AL-108 | MCI | 120 | 12 weeks | Phase II | Completed, not yet published |
| PSP | 300 | 52 weeks | Phase II/III | Recruiting | ||
| FTLD-tau, PSP, CBD | 12 | 12 weeks (pilot) | Phase II | Ongoing | ||
| Tau aggregation | Methylene blue** | AD | 321 | 50 weeks | Phase II | Completed, not yet published |
PSP, progressive supranuclear palsy; CBD, corticobasal degeneration; RCT, randomized placebo-controlled trial; CSF, cerebrospinal fluid; p-tau, phosphorylated tau
Based on www.clinicaltrials.gov and PubMED search
May also reduce soluble tau levels.
Tau phosphorylation inhibitors
The function and aggregation of tau appear to be regulated by phosphorylation, as reviewed above. Of the numerous tau kinases implicated in AD pathogenesis, the most widely studied are GSK-3β, CDK5, MARK, and MAPK (Augustinack et al., 2002; Mi and Johnson, 2006). Lithium, which inhibits GSK-3β and is used to treat bipolar disorder, improved behavior and reduced tau pathology in transgenic mice overexpressing P301L human 4R0N tau (JNPL3 model) (Noble et al., 2005). However, because lithium has multiple targets, the rescue observed may not have been solely due to a reduction in GSK3β activity. Lithium also has a narrow safety margin (Grandjean and Aubry, 2009). In addition, reduction of GSK-3β impairs NMDAR-mediated long-term depression (Peineau et al., 2007) and memory consolidation (Kimura et al., 2008), raising concerns about potential side effects of GSK-3β inhibitors. In a similar vein, CDK5 inhibitors prevent Aβ-induced hyperphosphorylation of tau and cell death in culture (Alvarez et al., 1999; Zheng Y L et al., 2005), but CDK5 is essential for multiple cell signaling pathways and adult neurogenesis, limiting its appeal as a tau-targeting therapy for AD. However, CDK5 and p25, a truncated form of the CDK5 subunit p35, also promote neurodegeneration through mechanisms that are independent of tau phosphorylation, involving inhibition of histone deacetylase 1 (HDAC1) and aberrant expression cell cycle genes (Kim et al., 2008), raising possibilities for additional therapeutic intervention.
Tau aggregation inhibitors
In vitro, tau aggregation is induced by polyanionic compounds such as RNA (Kampers et al., 1996), heparin (Crowe et al., 2007; Goedert et al., 1996; Perez et al., 1996) and lipid micelles (Chirita et al., 2003). Many of the drugs that block the aggregation of tau also block the pathological aggregation of other proteins under cell-free conditions, including Aβ and α-synuclein (Masuda et al., 2006), suggesting that they might be of benefit in diverse proteinopathies. Some tau aggregation inhibitors are effective in Neuro2A cell lines overexpressing a 4R tau microtubule repeat domain fragment with a K280 deletion, which promotes its aggregation (Pickhardt et al., 2005). In human AD patients, the phenothiazine methylene blue showed some promise for slowing disease progression in a phase II clinical trial conducted for 1 year (Gura, 2008). Methylene blue was originally thought to inhibit tau-tau interactions (Wischik et al., 1996), but it may also reduce soluble tau through other mechanisms (O'Leary et al., 2010) as it is known to have many targets (Schirmer et al.). Phase III trials with a newer formulation of methylene blue (LMTX) are planned (Wischik).
The immunosuppressant FK506 reduces microgliosis and tau aggregation in transgenic mice overexpressing P301S human 4R1N tau under the mouse prion promoter (PS19 model) (Yoshiyama et al., 2007). Since FK506 affects diverse signaling pathways in many cell types, it may act directly on neurons or influence the neuronal environment by modulating microglial activation. Inhibition of tau aggregation may also be mediated by direct binding of tau to the FK506 binding protein 52 (Chambraud et al., 2010).
As discussed above, it is far from certain that filamentous tau is actually toxic. Indeed, it is not known which tau assembly or conformation is responsible for tau-dependent neuronal dysfunction and degeneration. Not surprisingly, it is equally uncertain whether the abundance of this entity is lowered by any of the available tau aggregation blockers. In fact, some tau aggregation inhibitors enhance the formation of potentially toxic tau oligomers (Taniguchi et al., 2005). This scenario is reminiscent of the current state of anti-Aβ treatment, where it is also unclear whether any of the anti-Aβ strategies that have undergone or are currently in clinical trials significantly reduce the abundance of Aβ oligomers in human brain tissues, which are suspected to be the main mediators of Aβ-induced neuronal dysfunction (Ashe and Zahs, 2010; Cheng et al., 2007; Sakono and Zako, 2010; Shankar et al., 2008).
Reduction of overall tau levels
In mice, partial reduction of tau during early development is well tolerated, increases resistance to chemically induced seizures, and markedly diminishes Aβ- and ApoE-induced neuronal and cognitive impairments in vivo (Andrews-Zwilling et al., 2010; Ittner et al., 2010; Roberson et al., 2007, 2011). Assuming ongoing experiments confirm that reduction of overall tau levels is efficacious and safe also when initiated in adult and old animals with AD-related pathologies, tau could be targeted directly with RNAi approaches in patients with AD. Alternatively, tau levels could be reduced indirectly by targeting molecules that regulate the expression or clearance of tau.
Tau is thought to be degraded via the ubiquitin-proteasome and lysosomal pathways. The ubiquitin ligase for tau was identified as the C-terminus of HSP70-interacting protein (CHIP) (Hatakeyama et al., 2004; Petrucelli et al., 2004; Shimura et al., 2004). Reduction of CHIP levels increased the accumulation of tau aggregates in P301L human 4R0N tau mice (JNPL3 model), and CHIP levels are reduced in AD brains (Sahara et al., 2005). Furthermore, as its name suggests, CHIP works in combination with heat shock proteins to regulate tau degradation (Dickey et al.,2001); levels of heat shock protein 90(Hsp90) correlate inversely with the levels of soluble tau and tau oligomers (Sahara et al., 2007b).
In AD brains, tau is hyperacetylated, which should increase its half-life (Min et al., 2010), alter its microtubule binding and enhance aggregation (Cohen et al., 2011). Because both acetylation and ubiquitination target lysine residues, acetylation of tau by the acetyl transferase p300 inhibits ubiquitination and stabilizes tau (Min et al., 2010). In addition, acetylation of the 6 amino acid motif VQIINK (PHF6*) inhibits the binding of tau to microtubules and enhances tau aggregation (Cohen et al., 2011). This motif is critical for the formation of tau oligomers and filaments (Sahara et al., 2007a; von Bergen et al., 2001). Thus, the combination of a tau acetylation inhibitor and a ubiquitination-proteasome enhancer might synergize to lower the level of pathogenic tau species.
Larger aggregates of tau are not likely to be accessible to the proteasome but can be degraded by the lysosomal pathway, in which autophagosomes engulf the aggregates and fuse with lysosomes. In cells overexpressing the microtubule repeat domain of tau with a deletion of K280, aggregated tau is removed by the lysosomal pathway (Wang et al., 2009). In slice culture, inhibition of the lysosomal pathway produces NFT-like tau deposition (Bi et al., 1999). The lysosomal pathway of tau degradation is also involved in Niemann-Pick type C (NPC) disease, an autosomal recessive disorder associated with neurological symptoms and NFT formation in the brain (Auer et al., 1995). NPC disease is caused by a loss of function of NPC1, a lysosomal trafficking protein (Pacheco and Lieberman, 2008), suggesting that tau is degraded in lysosomes and that lysosomal dysfunction leads to tau accumulation. Consistent with this notion, phosphorylated tau is increased in the brains of NPC1-deficient mice and of NPC patients (Bu et al., 2002). However, crossbreeding of NPC1-deficient mice with tau knockout mice worsened the phenotype (Pacheco et al., 2009), suggesting that the role of tau in this disease is complex. The autophagic-lysosomal pathway has also been interrogated in a mouse model of tauopathy with parkinsonism overexpressing human 4R2N tau under the mouse Thy1 promoter with deletion of parkin (PK−/−/TauVLW) (Rodriguez-Navarro et al., 2010). Treatment of 3-month-old PK−/−/TauVLW mice with trehalose, an mTOR-independent autophagy activator, for 2.5 months prevented dopaminergic neuron loss in the ventral midbrain, reduced phosphorylated tau and total tau in the striatum and limbic system, prevented brain astrogliosis and improved motor and cognitive behavior. Biochemical and electron microscopy data suggested that the protective effects of trehalose were mediated, at least in part, by autophagy activation (Rodriguez-Navarro et al., 2010).
Tau degradation can also be enhanced by specific activation of the immune system. Active immunization targeting phosphorylated tau reduced filamentous tau inclusions and neuronal dysfunction in transgenic mice overexpressing K257T/P301S human 4R0N tau under the rat tau promoter or P301L human 4R0N tau (JNPL3 model) (Asuni et al., 2007; Boimel et al., 2010). The mechanism by which intracellular proteins, including tau and α-synuclein, are cleared by immunization is not known but may involve lysosomal degradation (Masliah et al., 2005; Sigurdsson, 2008, 2009). Once antibodies enter into brain, they could be taken up by receptor-mediated endocytosis and activate autophagy (Sigurdsson, 2009) or interact with tau in the extracellular matrix. Extracellular tau in cerebrospinal fluid (CSF) is used in combination with other biomarkers to diagnose AD (Trojanowski et al., 2010); phosphorylated tau and total tau ratios in the CSF can also predict disease severity (Wallin et al., 2010). Extracellular tau could come from the death of neurons or be released from live cells (Kim et al., 2010). If there is an equilibrium between intracellular and extracellular tau, clearance of tau/antibody complexes from the extracellular space may ultimately lower intracellular tau levels (Brody and Holtzman, 2008; Sigurdsson, 2009).
Microtubule stabilizers
Microtubule disruption has been observed in several models of AD and FTLD, including transgenic mice overexpressing wildtype human 0N3R tau under the mouse prion promoter (T44 model) (Zhang et al., 2005) or P301S human 4R1N tau (PS19 model) (Yoshiyama et al., 2007), and wildtype neuronal cultures exposed to Aβ oligomers (King et al., 2006; Zempel et al., 2010). Some FTDP-17 tau mutations (Hong et al., 1998) and tau hyperphosphorylation (Alonso et al., 1994; Merrick et al., 1997) reduce the binding of tau to microtubules. Although tau overexpression seems to be associated with destabilization of microtubules, it is unclear whether this phenomenon is always pathogenic and whether it results from a loss- or gain-of-function of tau. Indeed, tau is necessary for Aβ-induced microtubule disassembly in vitro (King et al., 2006), suggesting that tau is actually required for microtubule destabilization. A loss-of-function mechanism seems also unlikely because tau knockout mice have a rather benign phenotype, and tau reduction protects neurons from Aβ-induced impairments ex vivo (King et al., 2006; Rapoport et al., 2002; Shipton et al., 2011; Vossel et al., 2010) and in vivo (Ittner et al., 2010; Roberson et al., 2007, 2011).
Despite these caveats regarding underlying mechanism, microtubule stabilizers have shown promise in preclinical and clinical trials for AD. For example, paclitaxel prevented Aβ-induced toxicity in vitro (Zempel et al., 2010) as well as axonal transport deficits and motor impairments in transgenic mice overexpressing wildtype human 0N3R tau (T44 model) (Zhang et al., 2005). Epothilone D, which has better blood-brain barrier permeability, improved microtubule density and cognition in P301S human 4R1N tau mice (PS19 model) (Brunden et al., 2010). The peptide NAP stabilizes microtubules (Divinski et al., 2006) and reduces tau hyperphosphorylation (Vulih-Shultzman et al., 2007), suggesting that microtubule-stabilizing compounds can have more than one mechanism of action. NAP can be administered intranasally and showed some promise in a phase II clinical trial (Gozes et al., 2009).
Conclusions
While tau has long been implicated in neurodegenerative conditions, its functions in the adult brain and the precise mechanisms by which it contributes to neuronal dysfunction and degeneration in these disorders remain to be elucidated. A flurry of recent publications has challenged major dogmas in this field, including the notion that filamentous tau aggregates are the most pernicious forms of tau, that loss of tau function plays a major role in the pathogenesis of tauopathies, that tau enters dendritic spines only under pathological circumstances and that the adverse activities of tau aggregates are restricted to intracellular compartments. Provocative discoveries suggest that tau regulates neuronal excitability and that it is required for Aβ and other excitotoxins to cause neuronal deficits, aberrant network activity and cognitive decline. Indeed, tau has ‘graduated’ from a putative microtubule stabilizer to a multifunctional protein with many interacting signaling networks and to a master regulator of the intracellular trafficking of organelles and molecules involved in synaptic functions at the pre- and postsynaptic level. The hunt has also been intensified for the most pathogenic forms of tau, some of which have been traced into dendritic spines, and more has been learned about the complex posttranslational modification of tau, particularly acetylation, which appears to regulate the ubiquitination, turnover and aggregation of tau. These and other findings are providing critical guidance in the development of better treatments for tauopathies aimed at tau itself, tau regulators or factors mediating its putative functions. Identifying the functions and precise roles of tau in neurodegenerative disorders will likely require the analysis of conditional knockout models and the clinical evaluation of pertinent drugs with well defined modes of action.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Bassam J, Ozer RS, Safer D, Halpain S, Milligan RA. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol. 2002;157:1187–1196. doi: 10.1083/jcb.200201048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- Alvarez A, Toro R, Cáceres A, Maccioni RB. Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett. 1999;459:421–426. doi: 10.1016/s0014-5793(99)01279-x. [DOI] [PubMed] [Google Scholar]
- Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L, Huang Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30:13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, Harkany T, Holzer M, Hartig W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23:6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem. 1996;271:28741–28744. doi: 10.1074/jbc.271.46.28741. [DOI] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer IA, Schmidt ML, Lee VM, Curry B, Suzuki K, Shin RW, Pentchev PG, Carstea ED, Trojanowski JQ. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta Neuropathol. 1995;90:547–551. doi: 10.1007/BF00318566. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol (Berl) 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K, Hobbs GA, Yen SH, Lee G. Tyrosine phosphorylation of tau accompanies disease progression in transgenic mouse models of tauopathy. Neuropathol Appl Neurobiol. 2010;36:462–477. doi: 10.1111/j.1365-2990.2010.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K, Yen SH, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005;280:35119–351125. doi: 10.1074/jbc.M505895200. [DOI] [PubMed] [Google Scholar]
- Bi X, Zhou J, Lynch G. Lysosomal protease inhibitors induce meganeurites and tangle-like structures in entorhinohippocampal regions vulnerable to Alzheimer's disease. Exp Neurol. 1999;158:312–327. doi: 10.1006/exnr.1999.7087. [DOI] [PubMed] [Google Scholar]
- Biernat J, Mandelkow EM, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010;224:472–485. doi: 10.1016/j.expneurol.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Brandt R, Leger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol. 1995;131:1327–1340. doi: 10.1083/jcb.131.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, Wyss-Coray T, Buttini M, Mucke L, Mahley RW, Huang Y. Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24:2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008 doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, Iba M, James MJ, Xie SX, Ballatore C, et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010;30:13861–13866. doi: 10.1523/JNEUROSCI.3059-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu B, Klunemann H, Suzuki K, Li J, Bird T, Jin LW, Vincent I. Niemann-Pick disease type C yields possible clue for why cerebellar neurons do not form neurofibrillary tangles. Neurobiol Dis. 2002;11:285–297. doi: 10.1006/nbdi.2002.0551. [DOI] [PubMed] [Google Scholar]
- Bullmann T, de Silva R, Holzer M, Mori H, Arendt T. Expression of embryonic tau protein isoforms persist during adult neurogenesis in the hippocampus. Hippocampus. 2007;17:98–102. doi: 10.1002/hipo.20255. [DOI] [PubMed] [Google Scholar]
- Burkhart KK, Beard DC, Lehman RA, Billingsley ML. Alterations in tau phosphorylation in rat and human neocortical brain slices following hypoxia and glucose deprivation. Exp Neurol. 1998;154:464–472. doi: 10.1006/exnr.1998.6899. [DOI] [PubMed] [Google Scholar]
- Cantero JL, Moreno-Lopez B, Portillo F, Rubio A, Hita-Yanez E, Avila J. Role of tau protein on neocortical and hippocampal oscillatory patterns. Hippocampus. doi: 10.1002/hipo.20798. In press. [DOI] [PubMed] [Google Scholar]
- Chambraud B, Sardin E, Giustiniani J, Dounane O, Schumacher M, Goedert M, Baulieu EE. A role for FKBP52 in Tau protein function. Proc Natl Acad Sci USA. 2010;107:2658–2663. doi: 10.1073/pnas.0914957107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng I, Scearce-Levie K, Legleiter J, Palop J, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe K, Muchowski P, Mucke L. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24:4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirita CN, Necula M, Kuret J. Anionic micelles and vesicles induce tau fibrillization in vitro. J Biol Chem. 2003;278:25644–25650. doi: 10.1074/jbc.M301663200. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. 2006;281:10825–10838. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]
- Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VM. High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochem Biophys Res Commun. 2007;358:1–6. doi: 10.1016/j.bbrc.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Cantillana V, Jansen M, Wang H, Vitek MP, Wilcock DM, Lynch JR, Laskowitz DT. Loss of tau elicits axonal degeneration in a mouse model of Alzheimer's disease. Neuroscience. 2010;169:516–531. doi: 10.1016/j.neuroscience.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Spires-Jones TL, Pitstick R, Carlson GA, Hyman BT. Tangle-bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009;68:757–761. doi: 10.1097/NEN.0b013e3181a9fc66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, et al. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divinski I, Holtser-Cochav M, Vulih-Schultzman I, Steingart RA, Gozes I. Peptide neuroprotection through specific interaction with brain tubulin. J Neurochem. 2006;98:973–984. doi: 10.1111/j.1471-4159.2006.03936.x. [DOI] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorval V, Fraser PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 2006;281:9919–9924. doi: 10.1074/jbc.M510127200. [DOI] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow EM, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer's disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanara P, Husted KH, Selle K, Wong PY, Banerjee J, Brandt R, Hellerstein MK. Changes in microtubule turnover accompany synaptic plasticity and memory formation in response to contextual fear conditioning in mice. Neuroscience. 2010;168:167–178. doi: 10.1016/j.neuroscience.2010.03.031. [DOI] [PubMed] [Google Scholar]
- Farias GA, Munoz JP, Garrido J, Maccioni RB. Tubulin, actin, and tau protein interactions and the study of their macromolecular assemblies. J Cell Biochem. 2002;85:315–324. doi: 10.1002/jcb.10133. [DOI] [PubMed] [Google Scholar]
- Feuillette S, Miguel L, Frebourg T, Campion D, Lecourtois M. Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J Neurochem. 2010;113:895–903. doi: 10.1111/j.1471-4159.2010.06663.x. [DOI] [PubMed] [Google Scholar]
- Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, Mandelkow E, Zweckstetter M. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48:10047–10055. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- Flanagan LA, Cunningham CC, Chen J, Prestwich GD, Kosik KS, Janmey PA. The structure of divalent cation-induced aggregates of PIP2 and their alteration by gelsolin and tau. Biophys J. 1997;73:1440–1447. doi: 10.1016/S0006-3495(97)78176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming LM, Weisgraber KH, Strittmatter WJ, Troncoso JC, Johnson GV. Differential binding of apolipoprotein E isoforms to tau and other cytoskeletal proteins. Exp Neurol. 1996;138:252–260. doi: 10.1006/exnr.1996.0064. [DOI] [PubMed] [Google Scholar]
- Frappier TF, Georgieff IS, Brown K, Shelanski ML. tau Regulation of microtubule-microtubule spacing and bundling. J Neurochem. 1994;63:2288–2294. doi: 10.1046/j.1471-4159.1994.63062288.x. [DOI] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y. Cleavage of Tau by calpain in Alzheimer's disease: The quest for the toxic 17 kD fragment. Aging Neurobiol. 2011;32:1–14. doi: 10.1016/j.neurobiolaging.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. b-Amyloid precursor protein (bAPP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Gomez-Ramos A, Diaz-Hernandez M, Cuadros R, Hernandez F, Avila J. Extracellular tau is toxic to neuronal cells. FEBS Lett. 2006;580:4842–4850. doi: 10.1016/j.febslet.2006.07.078. [DOI] [PubMed] [Google Scholar]
- Gomez-Ramos A, Diaz-Hernandez M, Rubio A, Miras-Portugal MT, Avila J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci. 2008;37:673–681. doi: 10.1016/j.mcn.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Gozes I, Stewart A, Morimoto B, Fox A, Sutherland K, Schmeche D. Addressing Alzheimer's disease tangles: From NAP to AL-108. Curr Alzheimer Res. 2009;6:455–460. doi: 10.2174/156720509789207895. [DOI] [PubMed] [Google Scholar]
- Grandjean EM, Aubry JM. Lithium: Updated human knowledge using an evidence-based approach: Part III: Clinical safety. CNS Drugs. 2009;23:397–418. doi: 10.2165/00023210-200923050-00004. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- Gu Y, Oyama F, Ihara Y. Tau is widely expressed in rat tissues. J Neurochem. 1996;67:1235–1244. doi: 10.1046/j.1471-4159.1996.67031235.x. [DOI] [PubMed] [Google Scholar]
- Gura T. Hope in Alzheimer's fight emerges from unexpected places. Nat Med. 2008;14:894. doi: 10.1038/nm0908-894. [DOI] [PubMed] [Google Scholar]
- Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- Hampel H, Ewers M, Burger K, Annas P, Mortberg A, Bogstedt A, Frolich L, Schroder J, Schonknecht P, Riepe MW, et al. Lithium trial in Alzheimer's disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70:922–931. [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Harris JA, Devidze N, Verret L, Ho K, Hamto T, Lo I, Yu GQ, Palop JJ, Masliah E, Mucke L. Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2010;68:428–441. doi: 10.1016/j.neuron.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama S, Matsumoto M, Kamura T, Murayama M, Chui DH, Planel E, Takahashi R, Nakayama KI, Takashima A. U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat Tau and is involved in neurodegeneration of tauopathy. J Neurochem. 2004;91:299–307. doi: 10.1111/j.1471-4159.2004.02713.x. [DOI] [PubMed] [Google Scholar]
- He HJ, Wang XS, Pan R, Wang DL, Liu MN, He RQ. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. 2009;10:81. doi: 10.1186/1471-2121-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Zhang B, Forman MS, Yoshiyama Y, Trojanowski JQ, Lee VM. Axonal degeneration induced by targeted expression of mutant human tau in oligodendrocytes of transgenic mice that model glial tauopathies. J Neurosci. 2005;25:9434–9443. doi: 10.1523/JNEUROSCI.2691-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- Hong XP, Peng CX, Wei W, Tian Q, Liu YH, Yao XQ, Zhang Y, Cao FY, Wang Q, Wang JZ. Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus. 2009 doi: 10.1002/hipo.20712. [DOI] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SC, Jhon DY, Bae YS, Kim JH, Rhee SG. Activation of phospholipase C-gamma by the concerted action of tau proteins and arachidonic acid. J Biol Chem. 1996;271:18342–18349. doi: 10.1074/jbc.271.31.18342. [DOI] [PubMed] [Google Scholar]
- Ihara Y. PHF and PHF-like fibrils--cause or consequence? Neurobiol Aging. 2001;22:123–126. doi: 10.1016/s0197-4580(00)00200-1. [DOI] [PubMed] [Google Scholar]
- Ikegami S, Harada A, Hirokawa N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci Lett. 2000;279:129–132. doi: 10.1016/s0304-3940(99)00964-7. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Johnson GV. Tau complexes with phospholipase C-gamma in situ. Neuroreport. 1998;9:67–71. doi: 10.1097/00001756-199801050-00014. [DOI] [PubMed] [Google Scholar]
- Jho YS, Zhulina EB, Kim MW, Pincus PA. Monte carlo simulations of tau proteins: Effect of phosphorylation. Biophys J. 2010;99:2387–2397. doi: 10.1016/j.bpj.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–349. doi: 10.1016/s0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- Kar S, Fan J, Smith MJ, Goedert M, Amos LA. Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J. 2003;22:70–77. doi: 10.1093/emboj/cdg001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- Kempf M, Clement A, Faissner A, Lee G, Brandt R. Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J Neurosci. 1996;16:5583–5592. doi: 10.1523/JNEUROSCI.16-18-05583.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–817. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Lee S, Hall GF. Secretion of human tau fragments resembling CSF-tau in Alzheimer's disease is modulated by the presence of the exon 2 insert. FEBS Lett. 2010;584:3085–3088. doi: 10.1016/j.febslet.2010.05.042. [DOI] [PubMed] [Google Scholar]
- Kimura T, Fukuda T, Sahara N, Yamashita S, Murayama M, Mizoroki T, Yoshiike Y, Lee B, Sotiropoulos I, Maeda S, Takashima A. Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synaptic loss. J Biol Chem. 2010;285:38692–38699. doi: 10.1074/jbc.M110.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Yamashita S, Nakao S, Park JM, Murayama M, Mizoroki T, Yoshiike Y, Sahara N, Takashima A. GSK-3beta is required for memory reconsolidation in adult brain. PLoS One. 2008;3:e3540. doi: 10.1371/journal.pone.0003540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar β-amyloid. J Cell Biol. 2006;175:541–546. doi: 10.1083/jcb.200605187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci. 2002;2:698–707. doi: 10.1523/JNEUROSCI.22-03-00698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo J, Honda T, Mori H, Hamada Y, Miura R, Ogawara M, Ihara Y. The carboxyl third of tau is tightly bound to paired helical filaments. Neuron. 1988;1:827–834. doi: 10.1016/0896-6273(88)90130-4. [DOI] [PubMed] [Google Scholar]
- Kotani S, Nishida E, Kumagai H, Sakai H. Calmodulin inhibits interaction of actin with MAP2 and Tau, two major microtubule-associated proteins. J Biol Chem. 1985;260:10779–10783. [PubMed] [Google Scholar]
- Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci USA. 2003;100:9980–9985. doi: 10.1073/pnas.1533448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma MD, Bonay P, Colaco C, Avila J. Analysis of microtubule-associated protein tau glycation in paired helical filaments. J Biol Chem. 1994;69:21614–21619. [PubMed] [Google Scholar]
- Lee G, Cowan N, Kirschner M. The primary structure and heterogeneity of tau protein from mouse brain. Science. 1988;239:285–288. doi: 10.1126/science.3122323. [DOI] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111(Pt 21):3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci. 2004;24:2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Leugers CJ, Lee G. Tau potentiates nerve growth factor-induced mitogen-activated protein kinase signaling and neurite initiation without a requirement for microtubule binding. J Biol Chem. 2010;285:19125–19134. doi: 10.1074/jbc.M110.105387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas T, Eidenmuller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem. 2000;275:15733–15740. doi: 10.1074/jbc.M000389200. [DOI] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, Miyasaka T, Murayama S, Ikai A, Takashima A. Granular tau oligomers as intermediates of tau filaments. Biochemistry. 2007;46:3856–3861. doi: 10.1021/bi061359o. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Schweers O, Drewes G, Biernat J, Gustke N, Trinczek B, Mandelkow E. Structure, microtubule interactions, and phosphorylation of tau protein. Ann NY Acad Sci. 1996;777:96–106. doi: 10.1111/j.1749-6632.1996.tb34407.x. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Banker GA. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. 1996;16:5727–5740. doi: 10.1523/JNEUROSCI.16-18-05727.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM, McDonagh AM, Pickering-Brown SM, Kowa H, Iwatsubo T. Amyloid beta protein deposition in patients with frontotemporal lobar degeneration: Relationship to age and apolipoprotein E genotype. Neurosci Lett. 2001;304:161–164. doi: 10.1016/s0304-3940(01)01785-2. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, et al. Effects of α-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S, Goedert M, Hasegawa M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry. 2006;45:6085–6094. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O'Connor M, Trojanowski JQ, Lee VM. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- Merdes AR, Hansen LA, Jeste DV, Galasko D, Hofstetter CR, Ho GJ, Thal LJ, Corey-Bloom J. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology. 2003;60:1586–1590. doi: 10.1212/01.wnl.0000065889.42856.f2. [DOI] [PubMed] [Google Scholar]
- Merrick SE, Trojanowski JQ, Lee VM. Selective destruction of stable microtubules and axons by inhibitors of protein serine/threonine phosphatases in cultured human neurons. J Neurosci. 1997;17:5726–5737. doi: 10.1523/JNEUROSCI.17-15-05726.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi K, Johnson GV. The role of tau phosphorylation in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2006;3:449–463. doi: 10.2174/156720506779025279. [DOI] [PubMed] [Google Scholar]
- Miao Y, Chen J, Zhang Q, Sun A. Deletion of tau attenuates heat shock-induced injury in cultured cortical neurons. J Neurosci Res. 2010;88:102–110. doi: 10.1002/jnr.22188. [DOI] [PubMed] [Google Scholar]
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka T, Ding Z, Gengyo-Ando K, Oue M, Yamaguchi H, Mitani S, Ihara Y. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol Dis. 2005a;20:372–383. doi: 10.1016/j.nbd.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Miyasaka T, Watanabe A, Saito Y, Murayama S, Mann DM, Yamazaki M, Ravid R, Morishima-Kawashima M, Nagashima K, Ihara Y. Visualization of newly deposited tau in neurofibrillary tangles and neuropil threads. J Neuropathol Exp Neurol. 2005b;64:665–674. doi: 10.1097/01.jnen.0000173890.79058.1d. [DOI] [PubMed] [Google Scholar]
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schonig K, Bujard H, Mandelkow E, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–1160. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- Muramatsu K, Hashimoto Y, Uemura T, Kunii M, Harada R, Sato T, Morikawa A, Harada A. Neuron-specific recombination by Cre recombinase inserted into the murine tau locus. Biochem Biophys Res Commun. 2008;370:419–423. doi: 10.1016/j.bbrc.2008.03.103. [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nukina N, Ihara Y. One of the antigenic determinants of paired helical filaments is related to tau protein. J Biochem. 1986;99:1541–1544. doi: 10.1093/oxfordjournals.jbchem.a135625. [DOI] [PubMed] [Google Scholar]
- O'Leary JC, 3rd, Li Q, Marinec P, Blair LJ, Congdon EE, Johnson AG, Jinwal UK, Koren J, 3rd, Jones JR, Kraft C, et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, Fitzsimmons R. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy [CTE] in American athletes. Neurosurgery. doi: 10.1227/NEU.0b013e318212bc7b. [DOI] [PubMed] [Google Scholar]
- Pacheco CD, Elrick MJ, Lieberman AP. Tau deletion exacerbates the phenotype of Niemann-Pick type C mice and implicates autophagy in pathogenesis. Hum Mol Genet. 2009;18:956–965. doi: 10.1093/hmg/ddn423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco CD, Lieberman AP. The pathogenesis of Niemann-Pick type C disease: A role for autophagy? Expert Rev Mol Med. 2008;10:e26. doi: 10.1017/S146239940800080X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: An alternative mechanism by which tau mediates β-amyloid-induced neurodegeneration. J Neurosci. 2005;25:5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Perez M, Santa-Maria I, Gomez de Barreda E, Zhu X, Cuadros R, Cabrero JR, Sanchez-Madrid F, Dawson HN, Vitek MP, Perry G, et al. Tau--an inhibitor of deacetylase HDAC6 function. J Neurochem. 2009;109:1756–1766. doi: 10.1111/j.1471-4159.2009.06102.x. [DOI] [PubMed] [Google Scholar]
- Perez M, Valpuesta JM, Medina M, Montejo de Garcini E, Avila J. Polymerization of tau into filaments in the presence of heparin: The minimal sequence required for tau-tau interaction. J Neurochem. 1996;67:1183–1190. doi: 10.1046/j.1471-4159.1996.67031183.x. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Pickhardt M, Gazova Z, von Bergen M, Khlistunova I, Wang Y, Hascher A, Mandelkow EM, Biernat J, Mandelkow E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer's paired helical filaments in vitro and in cells. J Biol Chem. 2005;280:3628–3635. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, Yu WH, Luchsinger JA, Wadzinski B, Duff KE, Takashima A. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci. 2007;27:13635–13648. doi: 10.1523/JNEUROSCI.3949-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L, Yu W, Andreadis A, Luo M, Baas PW. Tau protects microtubules in the axon from severing by katanin. J Neurosci. 2006;26:3120–3129. doi: 10.1523/JNEUROSCI.5392-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. Proc Natl Acad Sci USA. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes JF, Reynolds MR, Horowitz PM, Fu Y, Guillozet-Bongaarts AL, Berry R, Binder LI. A possible link between astrocyte activation and tau nitration in Alzheimer's disease. Neurobiol Dis. 2008;31:198–208. doi: 10.1016/j.nbd.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds CH, Garwood CJ, Wray S, Price C, Kellie S, Perera T, Zvelebil M, Yang A, Sheppard PW, Varndell IM, et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J Biol Chem. 2008;283:18177–18186. doi: 10.1074/jbc.M709715200. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on Tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, Luebke JI. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol. 2010;223:385–393. doi: 10.1016/j.expneurol.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ, Solano RM, Gomez A, Perucho J, Cuervo AM, Garciade Yebenes J, Mena MA. Trehaloseameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39:423–438. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Rouzier R, Rajan R, Wagner P, Hess KR, Gold DL, Stec J, Ayers M, Ross JS, Zhang P, Buchholz TA, et al. Microtubule-associated protein tau: A marker of paclitaxel sensitivity in breast cancer. Proc Natl Acad Sci USA. 2005;102:8315–8320. doi: 10.1073/pnas.0408974102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy JW, Huff NC, Matus-Amat P. Understanding contextual fear conditioning: Insights from a two-process model. Neurosci Biobehav Rev. 2004;28:675–685. doi: 10.1016/j.neubiorev.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci. 2007a;25:3020–3029. doi: 10.1111/j.1460-9568.2007.05555.x. [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Takashima A. Tau oligomerization: a role for tau aggregation intermediates linked to neurodegeneration. Curr Alzheimer Res. 2008;5:591–598. doi: 10.2174/156720508786898442. [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Yoshiike Y, Mizoroki T, Yamashita S, Murayama M, Park JM, Saito Y, Murayama S, Takashima A. Molecular chaperone-mediated tau protein metabolism counteracts the formation of granular tau oligomers in human brain. J Neurosci Res. 2007b;85:3098–3108. doi: 10.1002/jnr.21417. [DOI] [PubMed] [Google Scholar]
- Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R, Murata S, Tanaka K, Takashima A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J Neurochem. 2005;94:1254–1263. doi: 10.1111/j.1471-4159.2005.03272.x. [DOI] [PubMed] [Google Scholar]
- Sakono M, Zako T. Amyloid oligomers: Formation and toxicity of Abeta oligomers. FEBS J. 2010;277:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mejia RO, Newman JW, Toh S, Y GQ, Zhou Y, Halabisky B, Cisse M, Scearce-Levie K, Cheng IH, Gan L, et al. Phospholipase A2 reduction ameliorates cognitve deficits in mouse model of Alzheimer's disease. Nat Neurosci. 2008;11:1311–1318. doi: 10.1038/nn.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarella RA, Skiniotis G, Goldie KN, Tittmann P, Gross H, Mandelkow EM, Mandelkow E, Hoenger A. Surface-decoration of microtubules by human tau. J Mol Biol. 2004;339:539–553. doi: 10.1016/j.jmb.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Santpere G, Ferrer I. LRRK2 and neurodegeneration. Acta Neuropathol. 2009;117:227–246. doi: 10.1007/s00401-008-0478-8. [DOI] [PubMed] [Google Scholar]
- Schirmer RH, Adler H, Pickhardt M, Mandelkow E. Lest we forget you - methylene blue …. Neurobiol Aging. doi: 10.1016/j.neurobiolaging.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VM, Litersky JM, Bhaskar K, Lee G. Tau impacts on growth-factor-stimulated actin remodeling. J Cell Sci. 2007;120:748–757. doi: 10.1242/jcs.03378. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. 2004;279:4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- Shipton OA, Leitz JR, Dworzak J, Acton CEJ, Tunbridge EM, Denk F, Dawson HN, Vitek MP, Wade-Martins R, Paulsen O, Vargas-Caballero M. Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer's disease and related tauopathies. J Alzheimers Dis. 2008;15:157–168. doi: 10.3233/jad-2008-15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM. Tau-focused immunotherapy for Alzheimer's disease and related tauopathies. Curr Alzheimer Res. 2009;6:446–450. doi: 10.2174/156720509789207930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souter S, Lee G. Microtubule-associated protein tau in human prostate cancer cells: Isoforms, phosphorylation, and interactions. J Cell Biochem. 2009;108:555–564. doi: 10.1002/jcb.22287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Fulga TA, Felch DL, Feany MB. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol Biol Cell. 2007;18:5060–5068. doi: 10.1091/mbc.E07-04-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R, Huang DY, Pericak-Vance M, Schmechel D, Roses AD. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:11183–11186. doi: 10.1073/pnas.91.23.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan A, Nesslany F, Violet M, Begard S, Loyens A, Talahari S, Mansuroglu Z, Marzin D, Sergeant N, Humez S, et al. Nuclear tau: A key player in neuronal DNA protection. J Biol Chem. 2010;286:4566–4575. doi: 10.1074/jbc.M110.199976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surridge CD, Burns RG. The difference in the binding of phosphatidylinositol distinguishes MAP2 from MAP2C and Tau. Biochemistry. 1994;33:8051–8057. doi: 10.1021/bi00192a009. [DOI] [PubMed] [Google Scholar]
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011;31:2511–2525. doi: 10.1523/JNEUROSCI.5245-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackenberg C, Brandt R. Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci. 2009;29:14439–14450. doi: 10.1523/JNEUROSCI.3590-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Cell Biol. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- Tashiro K, Hasegawa M, Ihara Y, Iwatsubo T. Somatodendritic localization of phosphorylated tau in neonatal and adult rat cerebral cortex. Neuroreport. 1997;8:2797–2801. doi: 10.1097/00001756-199708180-00029. [DOI] [PubMed] [Google Scholar]
- Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem. 2005;280:3963–3973. doi: 10.1074/jbc.M409876200. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Schuck T, Schmidt ML, Lee VM. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem. 1989;37:209–215. doi: 10.1177/37.2.2492045. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Vandeerstichele H, Korecka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, et al. Update on the biomarker core of the Alzheimer's Disease Neuroimaging Initiative subjects. Alzheimers Dement. 2010;6:230–238. doi: 10.1016/j.jalz.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- Vanmechelen E, Vanderstichele H, Davidsson P, Van Kerschaver E, Van Der Perre B, Sjogren M, Andreasen N, Blennow K. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: A sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett. 2000;285:49–52. doi: 10.1016/s0304-3940(00)01036-3. [DOI] [PubMed] [Google Scholar]
- von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta. 2005;1739:158–166. doi: 10.1016/j.bbadis.2004.09.010. [DOI] [PubMed] [Google Scholar]
- von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, Mandelkow E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J Biol Chem. 2001;276:48165–48174. doi: 10.1074/jbc.M105196200. [DOI] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Aβ-induced impairments in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulih-Shultzman I, Pinhasov A, Mandel S, Grigoriadis N, Touloumi O, Pittel Z, Gozes I. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther. 2007;323:438–449. doi: 10.1124/jpet.107.129551. [DOI] [PubMed] [Google Scholar]
- Wallin AK, Blennow K, Zetterberg H, Londos E, Minthon L, Hansson O. CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology. 2010;74:1531–1537. doi: 10.1212/WNL.0b013e3181dd4dd8. [DOI] [PubMed] [Google Scholar]
- Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei ML, Andreadis A. Splicing of a regulated exon reveals additional complexity in the axonal microtubule-associated protein tau. J Neurochem. 1998;70:1346–1356. doi: 10.1046/j.1471-4159.1998.70041346.x. [DOI] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad SciUSA. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmus MM, Grunberg SC, Bol JG, van Dam AM, Hoozemans JJ, Rozemuller AJ, Drukarch B. Transglutaminases and transglutaminase-catalyzed cross-links colocalize with the pathological lesions in Alzheimer's disease brain. Brain Pathol. 2009;19:612–622. doi: 10.1111/j.1750-3639.2008.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik C. TauRX Group Fact Sheet. Singapore: [Google Scholar]
- Wischik CM, Edwards PC, Lai RYK, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witman GB, Cleveland DW, Weingarten MD, Kirschner MW. Tubulin requires tau for growth onto microtubule initiating sites. Proc Natl Acad Sci USA. 1976;73:4070–4074. doi: 10.1073/pnas.73.11.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (t) Proc Natl Acad Sci USA. 1986;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Yu JZ, Rasenick MM. Tau associates with actin in differentiating PC12 cells. FASEB J. 2006;20:1452–1461. doi: 10.1096/fj.05-5206com. [DOI] [PubMed] [Google Scholar]
- Yu Y, Run X, Liang Z, Li Y, Liu F, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem. 2009;108:1480–1494. doi: 10.1111/j.1471-4159.2009.05882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J Neurosci. 2008;28:1682–1687. doi: 10.1523/JNEUROSCI.5242-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. A{beta} Oligomers Cause Localized Ca2+ Elevation, Missorting of Endogenous Tau into Dendrites, Tau Phosphorylation, and Destruction of Microtubules and Spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci USA. 2005;102:227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC. A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J. 2005;24:209–220. doi: 10.1038/sj.emboj.7600441. [DOI] [PMC free article] [PubMed] [Google Scholar]