

Abstract
Inhibitors of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel are predicted to slow cyst enlargement in polycystic kidney disease and reduce intestinal fluid loss in secretory diarrheas. Screening of ~110000 small synthetic and natural compounds for inhibition of halide influx in CFTR-expressing epithelial cells yielded a new class of pyrimido-pyrrolo-quinoxalinedione (PPQ) CFTR inhibitors. Testing of 347 analogues established structure–activity relationships. The most potent compound, 7,9-dimethyl-11-phenyl-6-(5-methylfuran-2-yl)-5,6-dihydro-pyrimido-[4′,5′-3,4]pyrrolo[1,2-a]quinoxaline-8,10-(7H,9H)-dione, PPQ-102, completely inhibited CFTR chloride current with IC50 ~90 nM. The PPQs, unlike prior CFTR inhibitors, are uncharged at physiological pH, and therefore not subject to membrane potential-dependent cellular partitioning or block efficiency. Patch-clamp analysis confirmed voltage-independent CFTR inhibition by PPQ-102 and showed stabilization of the channel closed state. PPQ-102 prevented cyst expansion and reduced the size of preformed cysts in a neonatal kidney organ culture model of polycystic kidney disease. PPQ-102 is the most potent CFTR inhibitor identified to date.
Introduction
The cystic fibrosis transmembrane conductance regulator (CFTRa) gene encodes a cAMP-regulated chloride channel expressed in epithelial cells in the airways, intestine, testis, and other tissues.1,2 Mutations in CFTR cause the hereditary disease cystic fibrosis. CFTR is excessively active in enterotoxin-mediated secretory diarrheas such as cholera and traveler’s diarrhea. CFTR inhibitors are predicted to reduce intestinal fluid loss in secretory diarrheas,3–5 as well as slow renal cyst expansion in polycystic kidney disease (PKD), where fluid accumulation in renal cysts is CFTR-dependent.6–13 There is interest in the identification of CFTR inhibitors for these clinical indications and as research tools to study disease pathogenesis in cystic fibrosis by blocking CFTR function in ex vivo human tissues and animal models.
Older CFTR inhibitors, including glibenclamide, diphenylamine-2-carboxylate (DPC), and 5-nitro-2-(3-phenylpropylamino)benzoate (NPPB) (Figure 1A) are nonselective in their action and have low potency. One study reported strong CFTR inhibition by α-aminoazaheterocyclic-methylglyoxal adducts,14 although CFTR inhibition was not subsequently confirmed.15 We identified two classes of improved CFTR inhibitors by high-throughput screening. The thiazolidinone CFTRinh-172 (Figure 1A) acts from the cytoplasmic side of the plasma membrane to block CFTR chloride conductance with IC50 ~ 0.3–3 µM depending on cell type and membrane potential.16 Patch-clamp analysis indicated a voltage-independent channel block mechanism in which CFTRinh-172 stabilizes the channel closed state;17 CFTR mutagenesis suggested CFTRinh-172 interaction at arginine-347 located near the cytoplasmic entrance of the CFTR pore.18 CFTRinh-172 has low toxicity, undergoes renal excretion with minimal metabolism, and accumulates in the intestine by enterohepatic recirculation.19 A second compound class, the glycine hydrazides (GlyH-101, Figure 1A), inhibit CFTR with IC50 ~ 5 µM.20 Patch-clamp analysis showed inward rectifying chloride current following GlyH-101 application with rapid channel flicker, indicating an external pore occlusion mechanism. Nonabsorbable conjugates of glycine hydrazides with polyethylene glycols21,22 and lectins4 inhibited CFTR when added at the mucosal surface and had improved potency compared to GlyH-101. Cell culture and animal models provided proof-of-concept for the potential utility of thiazolidinones and glycine hydrazides in secretory diarrheas3,16,19–22 and PKD.23
Figure 1.

Discovery of pyrimido-pyrrolo-quinoxalinedione (PPQ) CFTR inhibitors. (A) Chemical structures of CFTR inhibitors. Shown are the original CFTR inhibitors DPC, NPPB, and glibenclamide, the thiazolidinone CFTRinh-172, the glycine hydrazide GlyH-101, and new compound PPQ-102. (B) Representative screening data. Cell-based screening done in 96-well plates containing FRT cells expressing human CFTR and the YFP halide sensor YFP-H148Q/I152L. CFTR was maximally stimulated by an agonist mixture and CFTR-mediated iodide influx was measured as YFP fluorescence quenching. Fluorescence data from individual wells shown in the absence of agonists and in the presence of agonists for the negative control (DMSO vehicle alone), positive control (10 µM CFTRinh-172), and test compounds (at 25 µM).
Although CFTRinh-172 has been used extensively to block CFTR chloride conductance in cells and tissues, its low aqueous solubility is a potential concern, as is its membrane-potential dependent partitioning across cell membranes, which reduces its potency in cells because of their interior negative membrane potential. Thiazolidinone analogues with improved water solubility were synthesized,24 although they had reduced CFTR inhibition potency compared to CFTRinh-172 and retained the negative charge that reduces their accumulation in cytoplasm. The glycine hydrazides, including their macromolecular conjugates, also suffer from reduced CFTR inhibition potency at physiological interior-negative membrane potentials, but for a different reason. Glycine hydrazides produce strongly inwardly rectifying CFTR currents, with reduced inhibition potency at interior-negative membrane potential as a consequence of their electrostatic expulsion from the CFTR pore.20,22 Here, we screened synthetic and natural small molecules to identify new classes of CFTR inhibitors. We report here the discovery, structure–activity analysis, and characterization of pyrimido-pyrrolo-quinoxalinediones (PPQs), which are the first uncharged, and thus membrane-potential insensitive, CFTR inhibitors, and are the most potent small-molecule CFTR inhibitors identified to date.
Results
Screening of collections of synthetic and natural compounds was carried out to identify new classes of CFTR inhibitors with improved properties over known inhibitors. A cell-based fluorescence assay was used in which CFTR inhibitors were identified by reduced iodide influx in FRT cells coexpressing human CFTR and a YFP halide sensor, in which CFTR was maximally activated by a mixture of agonists having different activating mechanisms. Inhibition of iodide influx was seen as reduced YFP fluorescence quenching in response to rapid iodide addition to each well of 96-well plates. On the basis of prior knowledge of the small percentage of active CFTR inhibitors identified from screening of random compounds, primary screening was done at 25 µM concentration with test compounds preincubated for 15 min prior to assay.
Figure 1B shows examples of YFP fluorescence data in negative control (vehicle-only) and positive control (10 µM CFTRinh-172) wells, and wells containing test compounds, showing data for two active compounds. Of 54 compounds giving > 50% CFTR inhibition at 25 µM, rescreening and electrophysiological measurements indicated three compounds with > 50% CFTR inhibition at 5 µM. Several active compounds were related to previously identified CFTR inhibitors, and a new class of pyrimido-pyrrolo-quinoxalinedione (PPQ) compounds was identified. The structure of the PPQ analogue with greatest CFTR inhibition potency, PPQ-102, is shown in Figure 1A. The PPQs are not related structurally to known CFTR or ion channel inhibitors, and unlike prior CFTR inhibitors they are uncharged at physiological pH.
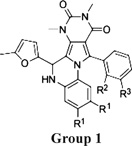
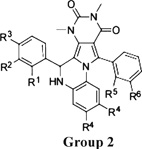
Structure–activity analysis was done to identify the most potent PPQ-class CFTR inhibitors for further characterization and biological testing. Of 347 commercially available PPQ analogues screened using the fluorescence platereader assay, 54 compounds inhibited CFTR-mediated iodide influx by > 50%at 25 µM (structures and activity data for analogues provided in Supporting Information). Table 1 summarizes CFTR inhibition data for some compounds. Apparent IC50 values obtained using the fluorescence platereader assay are semiquantitative because of the dilution procedure used but useful for comparative purposes. Figure 2A summarizes the structural determinants for CFTR inhibition deduced from the functional data. Analogues containing a 5-methyl furanyl moiety (PPQ-101 to PPQ-105) showed greater inhibition potencies than unsubstituted furanyl and thiophene analogues (group 1, Table 1). Compounds containing other heterocycles such as 1H-benzimidazole-2-yl and chromenone in place of furan were inactive. Analogues containing phenyls, PPQ-201 to PPQ-203, PPQ-209, and PPQ-210 (group 2, Table 1) were mildly less potent than the furan analogues PPQ-101 and PPQ-102. All active analogues contained a phenyl ring (substituted or unsubstituted) at the 2-position of pyrrole. Methyl substituents on this phenyl ring increased CFTR inhibition potency, as seen in PPQ-101, 102, 103, and PPQ-215, 202, 213. Aromatization of PPQ-102 to PPQ-102b (Figure 2B), which removes the stereocenter resulting in a planar structure, abolished CFTR inhibition activity.
Table 1.
CFTR Inhibition by PPQ Analoguesa
 |
 |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Group 1 | R1 | R2 | R3 | % inhibition at 25/5 µM |
IC50app (µM) |
||||
| PPQ-101 | H | H | CH3 | 98/82 | 0.7 | ||||
| PPQ-102 | H | H | H | 97/87 | 0.8 | ||||
| PPQ-103 | H | CH3 | H | 97/86 | 0.8 | ||||
| PPQ-104 | CH3 | H | H | 90/84 | 0.8 | ||||
| PPQ-105 | CH3 | F | H | 87/80 | 1.5 | ||||
| Group 2 | R1 | R2 | R3 | R4 | R5 | R6 | |||
| PPQ-201 | F | F | H | H | H | H | 93/72 | 1.2 | |
| PPQ-202 | H | NO2 | H | H | H | H | 90/60 | 1.2 | |
| PPQ-203 | H | H | OH | CH3 | H | H | 99/80 | 1.5 | |
| PPQ-204 | H | OCH3 | H | H | H | H | 86/63 | 1.7 | |
| PPQ-205 | F | H | H | H | H | H | 81/61 | 2 | |
| PPQ-206 | H | H | H | H | H | H | 92/61 | 2.5 | |
| PPQ-207 | H | H | OH | H | H | H | 86/64 | 4.5 | |
| PPQ-208 | OCH3 | H | OH | H | H | H | 56/5 | 100 | |
| PPQ-209 | H | H | OH | H | H | CH3 | 94/71 | 1.7 | |
| PPQ-210 | H | OCH3 | H | H | H | CH3 | 98/73 | 1.7 | |
| PPQ-211 | H | OCH3 | H | H | F | H | 97/62 | 2 | |
| PPQ-212 | F | NO2 | H | H | H | CH3 | 94/61 | 2 | |
| PPQ-213 | H | NO2 | H | H | CH3 | H | 96/64 | 2 | |
| PPQ-214 | H | Cl | H | H | F | H | 98/70 | 2.5 | |
| PPQ-215 | F | H | H | H | H | CH3 | 80/67 | 2.5 | |
| PPQ-216 | H | OCH3 | H | H | CH3 | H | 94/62 | 3 | |
| PPQ-217 | H | CH3 | H | H | CH3 | H | 100/46 | 3 | |
| PPQ-218 | H | H | H | H | CH3 | H | 97/51 | 5 | |
| PPQ-219 | H | F | H | H | H | CH3 | 93/53 | 8 | |
IC50app is an apparent IC50 determined from concentration–inhibition data from the fluorescence platereader assay.
Figure 2.

Structure–activity analysis and synthesis of PPQ-102. (A) Summary of structure–activity analysis, showing the structural determinants for PPQ-102 inhibition of CFTR chloride conductance. (B) Synthesis of PPQ-102 and PPQ-102b. Reagents and conditions: (a) Me2SO4, NaOH, 40 °C, 4 h, 43%; (b) PhCOCl, ZnCl2, toluene, reflux, 6 h, 28%; (c) Br2, CHCl3, rt, 2 h, 57%; (d) N-(2-aminophenyl)acetamide, microwave, 170 °C, 1 h, 51%; (e) HCl, reflux, 6 h, 67%; (f) 5-Me-furan-2-carbaldehyde, 170 °C, 10 min, 43%; (g) KMnO4, Me2CO, 1 h, 40%.
The most potent CFTR inhibitor confirmed by electrophysiological testing, PPQ-102, was resynthesized, confirmed, and further characterized. Synthesis of PPQ-102 was achieved in six steps (Figure 2B). Commercially available 6-methyluracil 1 was methylated using dimethylsulfate to produce 1,3,6-trimethyluracil 2, which upon Friedel–Crafts acylation using zinc chloride as a catalyst yielded 5-benzoyl-1,3,6-trimethylpyrimidine-2,4(1H,3H)-dione 3 as a white powder. Bromination of 3 followed by reaction with N-(2-aminophenyl)-acetamide generated amino-protected intermediate 5. The acetamido function of 5 was hydrolyzed, and the resultant intermediate 6 was cyclocondensed with 5-methylfurfural to yield yellowish–white racemic PPQ-102. Aromatic compound PPQ-102b, which lacks a stereocenter, was prepared from PPQ-102 by oxidation with potassium permanganate.
Short-circuit current analysis of CFTR chloride conductance was done to study compound potency, reversibility, and specificity. Figure 3A (left) shows PPQ-102 inhibition of chloride current in CFTR-expressing FRT cells following CFTR stimulation by the cAMP agonist CPT-cAMP. Measurements were done in the presence of a transepithelial chloride gradient and following basolateral membrane permeabilization with amphotericin B, so that measured current is a direct, quantitative measure of CFTR chloride conductance. PPQ-102 inhibition of CFTR was approximately 100% at higher concentrations, with IC50 ~90 nM (Figure 3A, right). Inhibition occurred over several minutes at low PPQ-102 concentrations, suggesting an intracellular site of action. Inhibition was reversible, as seen by complete restoration of CFTR chloride current after 30 min incubation with 2 µM PPQ-102 followed by 10 min washout (data not shown).
Figure 3.

CFTR inhibition by PPQ-102. (A) Apical membrane current measured in CFTR-expressing FRT cells in the presence of a transepithelial chloride gradient and with amphotericin B permeabilization of the basolateral membrane. CFTR was activated by CPT-cAMP (100 µM), with increasing concentrations of PPQ-102 added as shown. (left) Original recording; (right) dose–response (SE n = 4). (B) Measurements as in (A), but with apigenin (100 µM) or IBMX (100 µM) as agonists. Representative of three sets of experiments. (C) Short-circuit current measured in T84 (left) and human bronchial airway epithelial cells (right). CFTR was maximally activated by 10 µM forskolin and 100 µM IBMX (“forsk”). Current in the absence of inhibitor indicated as “control”. (D) Calcium-activated chloride channels were activated by UTP (100 µM) in cystic fibrosis (CFTR-deficient) human bronchial epithelial cells, with PPQ-102 added as indicated. ENaC was inhibited by amiloride (10 µM). (E) Cellular cAMP assayed in CHO-K1 cells under basal conditions and after 20 µM forskolin (SE n = 4, differences with PPQ-102 not significant).
Figure 3B shows PPQ-102 inhibition of CFTR chloride current following CFTR activation by apigenin, a flavone-type CFTR agonist that acts by direct CFTR binding, and IBMX, a phosphodiesterase inhibitor that also binds directly to CFTR. The mildly reduced PPQ-102 potency in response to these agonists, compared to a pure cAMP agonist (CPT-cAMP) that activates CFTR by a physiological phosphorylation mechanism, is consistent with PPQ-102 action at nucleotide binding domain(s) on the intracellular CFTR surface. Figure 3C shows PPQ-102 inhibition of short-circuit current in (nonpermeabilized) human intestinal (T84) and bronchial cells following maximal CFTR activation by forskolin and IBMX. CFTR inhibition was near 100% at 10 µM PPQ-102 with IC50 well below 1 µM.
PPQ-102 did not inhibit calcium-activated chloride channels or cellular cAMP production. Figure 3D shows little inhibition of UTP-induced chloride currents in cystic fibrosis human bronchial cells by 10 or 20 µM PPQ-102. Figure 3E shows no significant effect of 10 µM PPQ-102 on basal or forskolin-stimulated cAMP production.
Whole-cell membrane current was measured by patch-clamp in CFTR-expressing FRT cells (Figure 4A, left). Stimulation by 10 µM forskolin produced a membrane current of 172 ± 39 pA/pF (n = 4) at + 100 mV (total membrane capacitance 13 ± 1 pF). PPQ-102 at 0.5 µM gave ~65% inhibition of CFTR chloride current. Figure 4A (right) shows an approximately linear current–voltage relationship for CFTR, as found previously.1,2 The CFTR current–voltage relationship remained linear after PPQ-102 addition, indicating a voltage-independent block mechanism, as expected for an uncharged inhibitor. Cell-attached patch recordings were done to examine single-channel CFTR function (Figure 4B). Addition of 10 µM forskolin and 100 µM IBMX to the bath resulted in CFTR channel opening. CFTR unitary conductance was 7 pS at +80mV. Application of 1 µM PPQ-102 did not change unitary conductance but reduced channel activity markedly, as seen by the less frequent channel openings (Figure 4B, left). Channel open probability (Po) was reduced from 0.50 ± 0.04 to 0.14 ± 0.03. Mean channel open time did not significantly change, but mean channel closed time was greatly increased (Figure 4B, right). These results suggest that PPQ-102 inhibits CFTR by an altered channel gating mechanism, with stabilization of the channel closed state.
Figure 4.

Patch-clamp analysis of PPQ-102 inhibition of CFTR. (A) (left) Whole-cell currents measured in CFTR-expressing FRT cells recorded at a holding potential at 0 mV, and pulsing to voltages between ± 100 mV in steps of 20 mV in the absence and presence of 500 nM PPQ-102. CFTR was stimulated by 10 µM forskolin. (right) Current/voltage (I/V) plot of mean currents. Representative of four sets of measurements. (B) (left) Single channel recordings in the cell-attached configuration. CFTR was activated by 10 µM forskolin and 100 µM IBMX. Pipette potential was +80 mV. (right) Effect of 1 µM PPQ-102 on CFTR channel open probability (Po), mean channel open time, and mean channel closed time (SE, n = 3–4, * P < 0.01). O, open; C, closed.
PPQ-102 was tested in an embryonic kidney culture model of polycystic kidney disease. Kidneys were removed from day 13.5 embryonic mice and maintained in organ culture where they continue to grow. Whereas kidneys do not form cysts under control conditions as seen by transmission light microscopy, multiple cysts form and progressively enlarge when the culture medium was supplemented with the CFTR agonist 8-Br-cAMP (Figure 5A, left). Inclusion of PPQ-102 in the culture medium did not affect kidney growth but remarkably reduced the number and size of renal cysts formed in the 8-BrcAMP-containing medium. Figure 5A (right) summarizes the percentage area occupied by cysts from studies done on many kidneys, showing ~60% inhibition of cyst formation by 0.5 µM PPQ-102 and near complete absence of cysts at 2.5 and 5 µM PPQ-102. In control studies in which 2.5 µM PPQ-102 was removed after 3 days in organ culture, cysts rapidly enlarged in the continued presence of 8-Br-cAMP (data not shown), indicating that the inhibition effect of PPQ-102 is reversible. Figure 5B shows representative hematoxylin and eosin-stained paraffin sections of control and 8-Br-cAMP-treated kidneys cultured for 4 days in the presence of indicated concentrations of PPQ-102. In agreement with the transmission light micrographs of intact kidneys, PPQ-102 remarkably reduced cyst size.
Figure 5.

PPQ-102 prevents and reverses renal cyst expansion in an embryonic kidney organ culture model of PKD. E13.5 embryonic kidneys were maintained in organ culture in defined medium. (A) Inhibition of cyst formation. (left) Transmission light micrographs of kidneys in culture. As indicated, the culture medium contained 0 or 100 µM 8-Br-cAMP and/or 0, 0.5, or 5 µM PPQ-102. (right) Summary of cyst volumes after 4 days in culture shown as the fractional kidney area occupied by cysts (SE, 6–8 kidneys, * P < 0.001 compared to +8-Br-cAMP, 0 µM PPQ-102). (B) Hematoxylin and eosin-staining of kidney paraffin sections after 4 days in culture in the presence of 0 or 100 µM 8-Br-cAMP and indicated concentrations of PPQ-102. Representative of studies on three kidneys for each condition. (C) Reversal of preformed renal cysts. (left) Transmission light micrographs of kidneys cultured in 8-Br-cAMP for 3 days, with 5 µM PPQ-102 added at day 3 (two kidneys shown per condition). Micrographs at the right show kidneys at day 5 that were not exposed to PPQ-102. (right) Summary of cyst volumes at day 5 (SE, six kidneys, * P < 0.001).
The ability of PPQ-102 to reduce fluid accumulation in preformed cysts was tested by adding PPQ-102 to the 8-Br-cAMP-containing medium after kidneys were cultured for 3 days in the presence of 8-Br-cAMP. Figure 5C shows remarkable reduction in cyst size over 1 and 2 days after inclusion of PPQ-102 in the culture medium.
Discussion and Conclusions
This paper reports the discovery and characterization of PPQ CFTR inhibitors, which are the most potent CFTR inhibitors identified to date and the first uncharged CFTR inhibitors. Prior CFTR inhibitors, and most chloride channel inhibitors in general, are negatively charged, which may be required for their competition with chloride for binding to key positively charged amino acids in the channel pores.25 As predicted for an uncharged inhibitor and confirmed by patch-clamp analysis, CFTR inhibition by PPQ-102 is voltage-independent, which, as explained in the Introduction, is advantageous to maintain CFTR inhibition potency in interior-negative cells. Single-channel analysis indicated that PPQ-102 stabilizes the CFTR channel closed state, which together with its neutral charge and relatively slow time course of inhibition suggests action at a site on the cytoplasmic-facing surface of CFTR distinct from its pore.
The most potent PPQ CFTR inhibitor identified by SAR analysis, PPQ-102, inhibited CFTR chloride conductance with IC50 ~ 90 nM. Screening of PPQ analogues revealed many active compounds having a wide range of potencies. In nonpermeabilized T84 and bronchial epithelial cells, which have a strong interior-negative membrane potential, the IC50 for CFTR inhibition by PPQ-102 of ≪1 µM was substantially better than that of 3–5 µM measured previously for CFTRinh-172 and GlyH-101.16,20 The SAR analysis, as reported in Table 1 and summarized in Figure 2A, showed that greatest CFTR inhibition activity required a 5-methyl furan ring, 3-methylphenyl moiety, and the pyrimido[4′,5′-3,4]pyrrolo[1,2-a]quinoxaline template. The furan moiety in many of the active compounds could be replaced by the relatively stable phenyl moiety with minimal loss of activity. Abolishing the chiral center in PPQ-102 by oxidation resulted in loss of inhibition activity. SAR analysis also indicated the need for a phenyl ring at the 2-position of pyrrole.
Multiple lines of evidence support the application of CFTR inhibitors to slow cyst growth in autosomal dominant PKD (ADPKD): (a) CFTR is expressed strongly in epithelial cells lining cysts in ADPKD, (b) cystic fibrosis (CFTR-deficient) mice are resistant to cyst formation, (c) CFTR inhibitors block cyst formation in cell/organ culture and in vivo models, and (d) in some families affected with ADPKD and cystic fibrosis, individuals with both ADPKD and CF have less severe renal disease than those with ADPKD only.9,11 We used here an established neonatal kidney organ culture model to test the efficacy of PPQ-102 to reduce cyst volume. Intact kidney models to study cystogenesis, although technically more difficult than cell culture models such as MDCK cells grown in collagen gels, are superior in recapitulating native kidney anatomy and cellular phenotype.26 We found > 60% reduction in the size of developing cysts in kidneys cultured in medium containing 0.5 µM PPQ-102, and near complete absence of cysts at 2.5 µM PPQ-102, demonstrating substantially greater potency of PPQ-102 compared to the best thiazolidinone and glycine hydrazide CFTR inhibitors tested previously.23 The shrinking of preformed cysts by PPQ-102 supports the notion that renal cystogenesis involves a balance between active fluid secretion into and absorption from the cyst lumen. The efficacy of PPQ inhibitors to prevent and reverse cyst formation in vivo will require testing in PKD animal models and, ultimately, in clinical trials.
In conclusion, screening of >100000 chemically diverse small molecules yielded a novel class of potent, uncharged CFTR chloride channel inhibitors that were effective in reducing cyst size in an organ culture model of PKD. The PPQ inhibitors are likely to be effective as well in reducing intestinal fluid accumulation in enterotoxin-mediated secretory diarrheas, although we did not study their antidiarrheal properties because existing malonic acid hydrazide-type CFTR inhibitors have the advantage over PPQs of being nonabsorbed and active extracellularly. The high potency of PPQs should also make them useful, as CFTRinh-172 has been, in characterizing CFTR function in cellular and in vivo models.
Experimental Section
Cell Lines and Compounds
Fischer rat thyroid (FRT) cells coexpressing human wildtype CFTR and the halide indicator YFP-H148Q were generated as described.16 Cells were plated in 96-well black-walled microplates (Corning Costar) at a density of 20000 cells per well in Coon’s modified F12 medium supplemented with 5% fetal calf serum, 2 mM l-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin. Assays were done at 48 h after plating when cells were just confluent. Some measurements were made on T84 human intestinal epithelial cells and on primary cultures of human bronchial epithelial cells, obtained and grown as described.3,27 The compound collections used for screening included: ~105000 synthetic small molecules from ChemDiv (San Diego, CA) and Asinex (San Diego, CA), and ~7500 purified natural compounds from Analyticon (Potsdam, Germany), Timtek (Newark, NJ), and Biomol (Plymouth Meeting, PA). Compounds were maintained as DMSO stock solutions. Structure–activity analysis was done on analogues purchased from ChemDiv and Asinex.
Screening Procedures
Assays were done using an automated screening platform (Beckman) equipped with FluoStar fluorescence plate readers (BMG Lab Technologies) as described.16 Each well of a 96-well plate was washed three times in PBS (300 µL/wash), leaving 50 µL of PBS. Ten µL of a CFTR-activating cocktail (5 µM forskolin, 100 µM IBMX, 25 µM apigenin in PBS) was added, and after 5 min test compounds (0.5 µL of 1 mM DMSO solution) were added to each well at 25 µM final concentration. After 15 min, 96-well plates were transferred to a plate reader for fluorescence assay. Each well was assayed individually for CFTR-mediated iodide influx by recording fluorescence continuously (200 ms per point) for 2 s (baseline) and then for 10 s after rapid addition of 160 µL of isosmolar PBS in which 137 mM chloride was replaced by iodide. The initial rate of iodide influx was computed from fluorescence data by nonlinear regression. Each plate contained negative (DMSO vehicle) and positive (10 µM CFTRinh-172) controls.
Short-Circuit Current
Snapwell inserts containing CFTR-expressing FRT cells, T84 cells, or human bronchial epithelial cells were mounted in Ussing chambers. For FRT cells, the hemichambers were filled with 5 mL of 75 mM NaCl and 75 mM Na gluconate (apical) and 150 mM NaCl (basolateral) (pH 7.3), and the basolateral membrane was permeabilized with 250 µg/mL amphotericin B. For bronchial epithelial cells and T84 cells, both hemichambers contained a Krebs-bicarbonate solution. Hemichambers were continuously bubbled with air (FRT cells) or 5% CO2 in air (bronchial and T84 cells) and maintained at 37 °C. Short-circuit current was recorded continuously using a DVC-1000 voltage clamp (World Precision Instruments) using Ag/AgCl electrodes and 3 M KCl agar bridges.
Patch-Clamp Analysis
Whole cell recordings were done on FRT cells stably expressing human wild-type CFTR. The pipet solution contained 140 mM N-methyl d-glucamine chloride (NMDG-Cl), 5 mM EGTA, 1 mM MgCl2, 1 mM Tris-ATP, and 10 mM HEPES (pH 7.2), and the bath solution contained 140 mM N-methyl d-glucamine chloride, 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4). Experiments were done at room temperature (22–25 °C). Pipettes were pulled from borosilicate glass and had resistances of 3–5 MΩ after fire polishing. Seal resistances were typically between 3 and 10 GΩ. After establishing the whole-cell configuration, CFTR was activated by adding forskolin and 3-isobutyl-1-methylxanthine (IBMX). Whole cell currents were elicited by applying hyperpolarizing and depolarizing voltage pulses from a holding potential of 0 mV to potentials between −100 and +100 mV in steps of 20 mV. Current was filtered at 5 kHz and digitized and analyzed using an AxoScope 10.0 system and a Digidata 1440A AC/DC converter. The single channel characteristics of CFTR were analyzed in the cell-attached configuration using fire-polished pipettes with a resistance of 10–15 MΩ. The pipet solution contained (in mM): 140 NMDG-Cl, 1 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES (pH 7.4), and the bath solution contained 140 KCl, 1 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES (pH 7.4). Recordings were performed at room temperature using an Axopatch-200B (Axon Instruments). The voltage and current data were low-pass filtered at 1 kHz and stored for later analysis. Single channel data were digitally filtered at 25 Hz and analyzed using Clampfit 10.0 software (Axon Instruments).
Embryonic Organ Culture Model of PKD
Mouse embryos were obtained at embryonic day 13.5 (E13.5). Metanephroi were dissected and placed on transparent Falcon 0.4 mm diameter porous cell culture inserts as described.23 To the culture inserts was added DMEM/Ham’s F-12 nutrient medium supplemented with 2 mM l-glutamine, 10 mM HEPES, 5 µg/mL insulin, 5 µg/mL transferrin, 2.8 nM selenium, 25 ng/mL prostaglandin E, 32 pg/mL T3, 250 U/mL penicillin, and 250 µg/mL streptomycin. Kidneys were maintained in a 37 °C humidified CO2 incubator for up to 8 days. Culture medium containing 100 µM 8-Br-cAMP, with or without CFTR inhibitor, was replaced (in the lower chamber) every 12 h. In some studies, CFTR inhibitor was added at 3 days after 8-Br-cAMP to test its efficacy in reversing preformed cysts. Kidneys were photographed using a Nikon inverted microscope (Nikon TE 2000-S) equipped with 2× objective lens, 520 nm bandpass filter, and high-resolution CCD camera. Percentage cyst area was calculated as total cyst area divided by total kidney area.
Synthesis Procedures
1H and 13C NMR spectra were obtained in deuterated dimethyl sulfoxide (DMSO-d6) using a 400-MHz Varian spectrometer referenced to DMSO. Mass spectrometry was done using a Waters LC/MS system (Alliance HT 2790+ZQ, HPLC: Waters model 2690, Milford, MA). Flash chromatography was done using EM silica gel (230–400 mesh), and thin-layer chromatography was done on Merk silica gel 60 F254 plates (Darmstadt, Germany). Microwave reactions were performed in a Biotage Initiator (0.5–2 mL vials) with target temperature reached within 30 s at ~55 W. Melting points are uncorrected. Purity to > 98% was confirmed by LCMS.
1,3,6-Trimethyl-1H,3H-pyrimidine-2,4-dione (2).27
Dimethylsulfate (106 g, 80 mL, 844 mmol) was added dropwise to a solution of 2,4-dihydroxy-6-methylpyrimidine (30 g, 238 mmol) in 280 mL of 4 N NaOH at 40 °C. After stirring for 4 h at 40 °C, the reaction mixture was neutralized with careful addition of acetic acid and extracted three times with 100 mL of ethyl acetate. Combined organics were dried over MgSO4 and concentrated in vacuo to yield a white solid. Recrystallization from ethanol yielded 2 (15.8 g, 43%); mp 113–114 °C. 1H NMR (DMSO-d6): δ 5.58 (s, 1H), 3.26 (s, 3H), 3.09 (s, 3H,), 2.19 (s, 3H,). MS (ES+) (m/z): [M + 1]+ calculated for C7H11N2O2, 155.17, found 155.93. This compound matched the analytical data as reported.27
5-Benzoyl-1,3,6-trimethylpyrimidine-2,4(1H,3H)-dione (3)
A mixture of 1,3,6-trimethyl-compound 2 (12.3 g, 80 mmol), benzoyl chloride (11.5 g, 9.5 mL, 82 mmol), and anhydrous zinc chloride (10.8 g, 79 mmol) in toluene (100 mL) was heated to reflux for 6 h. The mixture was poured over ice (200 g), and the separated toluene layer was concentrated in vacuo. The crude residue was purified by flash chromatography to yield 3 (5.8 g, 28%); mp 132–134 °C. MS (ES+) (m/z): [M+1]+ calculated for C14H15N2O3, 259.28, found 259.09. This compound matched analytical data as reported.28
5-Benzoyl-6-(bromomethyl)-1,3-dimethylpyrimidine-2,4(1H,-3H)-dione (4).28,29
To a solution of 3 (2.61 g, 10.1 mmol) in chloroform (13 mL) was added bromine (1.62 g, 0.52 mL, 20.3 mmol in 3 mL chloroform) dropwise over 30 min at room temperature. The reaction mixture was further stirred for 1 min at room temperature before concentrated in vacuo. The crude reaction mixture was purified by flash chromatography to yield 4 (1.96 g, 57%); mp 164–167 °C. MS (ES+) (m/z): [M + 1]+ calculated for C14H14BrN2O3, 338.18, found 337.15, 338.93. This compound matched analytical data as reported.28,29
N-(2-(1,3-dimethyl-2,4-dioxo-5-phenyl-3,4-dihydro-1H-pyrrolo[3,4-d]pyrimidin-6(2H)-yl)phenyl)acetamide (5)
A mixture of N-(2-aminophenyl)acetamide (315 mg, 2.1 mmol), bromo-compound 4 (680 mg, 2 mmol), triethylamine (200 mg, 280 µL, 2 mmol), and ethanol (2 mL) was microwave-heated at 170 °C for 1 h (2–5 mL vial, pressure 13 bar). The shiny white crystalline mass was filtered, washed, and recrystallized from ethanol to give 5 (392 mg, 51% yield); mp > 250 °C. 1H NMR (DMSO-d6): δ 9.17 (s, 1H), 7.69 (d, 1H, pyrrole CH, J = 8.06 Hz), 7.33–7.14 (m, 6H), 7.10–6.94 (m, 3H), 3.31 (s, 3H), 3.17 (s, 3H), 1.87 (s, 3H). MS (ES+) (m/z): [M + 1]+ calculated for C22H21N4O3, 389.43, found 389.19.
6-(2-Aminophenyl)-1,3-dimethyl-5-phenyl-1H-pyrrolo[3,4-d]-pyrimidine-2,4(3H,6H)-dione (6)
Acetamide compound 5 (200 mg, 0.5 mmol) was refluxed in hydrochloric acid (4 N, 10 mL) for 6 h. The reaction mixture was evaporated under vacuum, and the residue was dissolved in water and neutralized to give 6 (120 mg, 67%) as a white precipitate; mp > 300 °C. 1H NMR (DMSO-d6): δ 7.32–7.26 (m, 2H), 7.23–7.15 (m, 3H), 7.00 (t, 1H, J = 7.32 Hz), 6.87 (s, 1H), 6.80 (d, 1H, J = 7.69), 6.70 (d, 1H, J = 8.06 Hz), 6.41 (t, 1H, J = 7.32 Hz), 5.00 (s, 2H), 3.29 (s, 3H), 3.16 (s, 3H). MS (ES+) (m/z): [M + 1]+ calculated for C20H19N4O2, 347.39, found 347.10.
7,9-Dimethyl-11-phenyl-6-(5-methylfuran-2-yl)-5,6-dihydropyrimido[4′,5′-3,4]pyrrolo[1,2-a]quinoxaline-8,10-(7H,9H)-dione (PPQ-102)
A mixture of 5-methylfuran-2-carbaldehyde (32 mg, 29 µL, 0.29 mmol), compound 6 (101 mg, 0.29 mmol), and ethanol (1 mL) were heated in a microwave reactor at 170 °C for 10 min. A white product was isolated, washed, and recrystallized from ethanol to afford 51 mg of PPQ-102 (42% yield); mp > 300 °C. 1H NMR (DMSO-d6): δ 7.41 (broad m, 4H), 6.95 (d, 2H, J = 8.42 Hz), 6.90–6.83 (m, 2H), 6.29 (d, 2 H, J = 2.93 Hz), 6.08 (d, 1H, J = 2.19 Hz), 5.80 (d, 1H, J = 2.93 Hz), 5.69 (d,1H, J = 2.93 Hz), 3.50 (s, 3H), 3.12 (s, 3H), 2.11 (s, 3H). 13C NMR (DMSO): 159.2, 153.1, 151.9, 151.9, 139.2, 131.5, 129.6, 129.4, 128.8, 126.9, 124.3, 122.8, 120.6, 118.2, 117.6, 111.9, 108.9, 107.1, 105.2, 47.8, 32.3, 28.2, 13.9. HRMS (ES+) (m/z): [M + 1]+ calculated for C26H23N4O3, 439.1765, found, 439.1771.
N-(2-(1,3-Dimethyl-2,4-dioxo-5-phenyl-3,4-dihydro-1H-pyrrolo[3,4-d]pyrimidin-6(2H)-yl)phenyl)-5-methylfuran-2-carboxamide (PPQ-102b)
To a solution of PPQ-102 (12 mg, 27 µmol in 2 mL acetone) was added dropwise a saturated solution of potassium permanganate (13 mg, 80 µmol, 200 µL). The reaction mixture was stirred for 1 h at room temperature and filtered. The residue was processed by standard methods,30 and the acetone solution was evaporated to yield PPQ-102b (5 mg, 40%). MS (ES+) (m/z): [M + 1]+ calculated for C26H21N4O3, 437.47, found, 437.12.
Supplementary Material
Acknowledgment
Supported by NIH grants DK86125, HL73856, DK72517, EB00415, HL59198, DK35124 and EY13574, and Research Development Program and Drug Discovery grants from the Cystic Fibrosis Foundation.
Footnotes
Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; CFTR, cystic fibrosis transmembrane conductance regulator; CPT-cAMP, chlorophenylthio-cAMP; IBMX, isobutylmethylxanthine, PKD, polycystic kidney disease; PPQ, pyrimido-pyrrolo-quinoxalinedione; YFP, yellow fluorescent protein.
Supporting Information Available: Analytical data for synthesized compounds and list of PPQ analogues tested and their CFTR inhibition activities. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol. Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 2.Gadsby DC, Vergani P, Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiagarajah J, Broadbent T, Hsieh E, Verkman AS. Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR inhibitor. Gastroenterology. 2004;126:511–519. doi: 10.1053/j.gastro.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Sonawane N, Zhao D, Zegarra-Moran O, Galietta LJV, Verkman AS. Lectin conjugates as potent, nonabsorbable CFTR inhibitors for reducing intestinal fluid secretion in cholera. Gastroenterology. 2007;132:1234–1244. doi: 10.1053/j.gastro.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 5.Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol. Rev. 2002;82:245–289. doi: 10.1152/physrev.00026.2001. [DOI] [PubMed] [Google Scholar]
- 6.Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J. Am. Soc. Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 7.Brill SR, Ross KE, Davidow CJ, Ye M, Grantham JJ, Caplan MJ. Immunolocalization of ion transport proteins in human autosomal dominant polycystic kidney epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10206–10211. doi: 10.1073/pnas.93.19.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat. Clin. Pract. Nephrol. 2006;2:40–55. doi: 10.1038/ncpneph0070. [DOI] [PubMed] [Google Scholar]
- 9.O’Sullivan DA, Torres VE, Gabow PA, Thibodeau SN, King BF, Bergstralh EJ. Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 1998;32:976–983. doi: 10.1016/s0272-6386(98)70072-1. [DOI] [PubMed] [Google Scholar]
- 10.Xu N, Glockner JF, Rossetti S, Babovich-Vuksanovic D, Harris PC, Torres VE. Autosomal dominant polycystic kidney disease coexisting with cystic fibrosis. J. Nephrol. 2006;19:529–534. [PubMed] [Google Scholar]
- 11.Cotton CU, Avner ED. PKD and CF: an interesting family provides insight into the molecular pathophysiology of polycystic kidney disease. Am. J. Kidney Dis. 1998;32:1081–1083. doi: 10.1016/s0272-6386(98)70088-5. [DOI] [PubMed] [Google Scholar]
- 12.Davidow CJ, Maser RL, Rome LA, Calvet JP, Grantham JJ. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. 1996;50:208–218. doi: 10.1038/ki.1996.304. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Findlay IA, Sheppard DN. The relationship between cell proliferation, Cl− secretion, and renal cyst growth: a study using CFTR inhibitors. Kidney Int. 2004;66:1926–1938. doi: 10.1111/j.1523-1755.2004.00967.x. [DOI] [PubMed] [Google Scholar]
- 14.Routaboul C, Norez C, Melin P, Molina MC, Boucherle B, Bossard F, Noel S, Robert R, Gauthier C, Becq F, Decout JL. Discovery of α-aminoazaheterocycle-methylglyoxal adducts as a new class of high-affinity inhibitors of cystic fibrosis transmembrane conductance regulator chloride channels. J. Pharmacol. Exp. Ther. 2007;322:1023–1035. doi: 10.1124/jpet.107.123307. [DOI] [PubMed] [Google Scholar]
- 15.Sonawane ND, Zegarra-Moran O, Namkung W, Galietta LJV, Verkman AS. α-Aminoazaheterocyclic-methylglyoxyl adducts do not inhibit cystic fibrosis transmembrane conductance chloride channel activity. J. Pharm. Exp. Ther. 2008;325:529–535. doi: 10.1124/jpet.107.132357. [DOI] [PubMed] [Google Scholar]
- 16.Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera-toxin induced intestinal fluid secretion. J. Clin. Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taddei A, Folli C, Zegarra-Moran O, Fanen P, Verkman AS, Galietta LJ. Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett. 2004;558:52–56. doi: 10.1016/S0014-5793(04)00011-0. [DOI] [PubMed] [Google Scholar]
- 18.Caci E, Caputo A, Hinzpeter A, Arous N, Fanen P, Sonawane ND, Verkman AS, Ravazzolo R, Zegarra-Moran O, Galietta LJV. Evidence for direct CFTR inhibition by CFTRinh-172 based on arginine 347 mutagenesis. Biochem. J. 2008;413:135–142. doi: 10.1042/BJ20080029. [DOI] [PubMed] [Google Scholar]
- 19.Sonawane ND, Muanprasat C, Nagatani R, Song Y, Verkman AS. In vivo pharmacology and antidiarrheal efficacy of a thiazolidinone CFTR inhibitor in rodents. J. Pharm. Sci. 2005;94:134–143. doi: 10.1002/jps.20228. [DOI] [PubMed] [Google Scholar]
- 20.Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJV, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure–activity analysis and in vivo efficacy. J. Gen. Physiol. 2004;124:125–137. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sonawane ND, Hu J, Muanprasat C, Verkman AS. Luminally-active, nonabsorbable CFTR inhibitors as potential therapy to reduce intestinal fluid losses in cholera. FASEB J. 2006;20:130–132. doi: 10.1096/fj.05-4818fje. [DOI] [PubMed] [Google Scholar]
- 22.Sonawane N, Zhao D, Zegarra-Mora O, Galietta LJ, Verkman AS. Nanomolar CFTR inhibition by pore-occluding divalent polyethylene glycol-malonic acid hydrazides. Chem. Biol. 2008;15:718–728. doi: 10.1016/j.chembiol.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2008;19:1300–1310. doi: 10.1681/ASN.2007070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonawane N, Verkman AS. Thiazolidinone CFTR inhibitors with improved water solubility identified by structure–activity analysis. Bioorg. Med. Chem. 2008;16:8187–8195. doi: 10.1016/j.bmc.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat. Rev. Drug Discovery. 2009;8:153–171. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magenheimer BS, St. John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, Maser RL, Grantham JJ, Calvet JP. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na+, K+, 2Cl− co-transporter-dependent cystic dilation. J. Am. Soc. Nephrol. 2006;17:3424–3437. doi: 10.1681/ASN.2006030295. [DOI] [PubMed] [Google Scholar]
- 27.Azas N, Rathelot P, Djekou S, Delmas F, Gellis A, Di Giorgio C, Vanelle P, Timon-David P. Antiparasitic activity of highly conjugated pyrimidine-2,4-dione derivatives. Farmaco. 2003;58:1263–1270. doi: 10.1016/j.farmac.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 28.Tsupak EB, Shevchenko MA, Pozharskii AF, Tkachenko YN. Pyrollopyrimidines. 5. Interaction of 6-amino-1,3-dimethylpyrrolo[3,4-d]-pyrimidin-2,4(1H,3H)-diones with 1,3-diketones. Khim. Geterotsikl. Soedin. 2003;7:1096–1102. [Google Scholar]
- 29.Tsupak EB, Shevchenko MA. [3,4]-annulated pyrroles 1. Polynuclear heterocyclic systems based on pyrrolo[3,4-d]pyrimidine-2,4-dione. Russ. Chem. Bull. 2006;55:2265–2270. [Google Scholar]
- 30.Oels R, Storer R, Young DW. Reinvestigation of synthesis of 3-dimethylallyl-4-hydroxy-2-quinolones—novel route to tetracyclic heteroaromatic-compounds. J. Chem. Soc. Perkin Trans. 1977;23:2546–2551. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.