

1. Introduction
The use of proteins for therapeutic applications has increased dramatically in the last several decades. There has been a tremendous increase in the number of approved drugs derived from recombinant proteins and monoclonal antibodies since the approval of recombinant insulin in 1982 (Figure 1).1 In addition, the number of approved indications is almost four times the number of approved drugs in 2005. Sales of protein drugs in 2008 are estimated to be around 71 billion USD.2 Thus, many proteins are currently being investigated for use as therapeutic agents for different diseases (i.e., cancer, autoimmune diseases). Their development as potential drugs, however, has been hampered by difficulties in formulating them into therapeutic agents with long shelf lives. Most proteins are physically and chemically unstable. This instability is a major barrier for researchers in developing stable therapeutic products. Factors that influence the stability of a protein can be divided into two categories: (1) intrinsic factors derived from the inherent physicochemical properties of the protein (e.g., primary, secondary, tertiary, and quaternary structures) and (2) the extrinsic factors derived from the environment of the protein such as pH, temperature, buffer, ionic strength, and excipients. Because physical degradation (e.g., conformational changes, aggregation and precipitation, etc) is one of the major problems in formulating protein therapeutics, many studies have been done to elucidate factors that influence physical stability of proteins. Recently, Middaugh et al. developed an empirical phase diagram based approach for the rapid evaluation of protein physical stability.3–5 The chemical degradation of proteins seen in some amino acids (i.e., Asn, Asp, and Met) has been extensively studied and reviewed.6–15 On the other hand, the effect of other amino acids such as Cysteine and Cystine on the physical and chemical stability of proteins has not been extensively reviewed. Thus, this review focuses on the role of Cys residue in the structure and stability of proteins.
Figure 1.
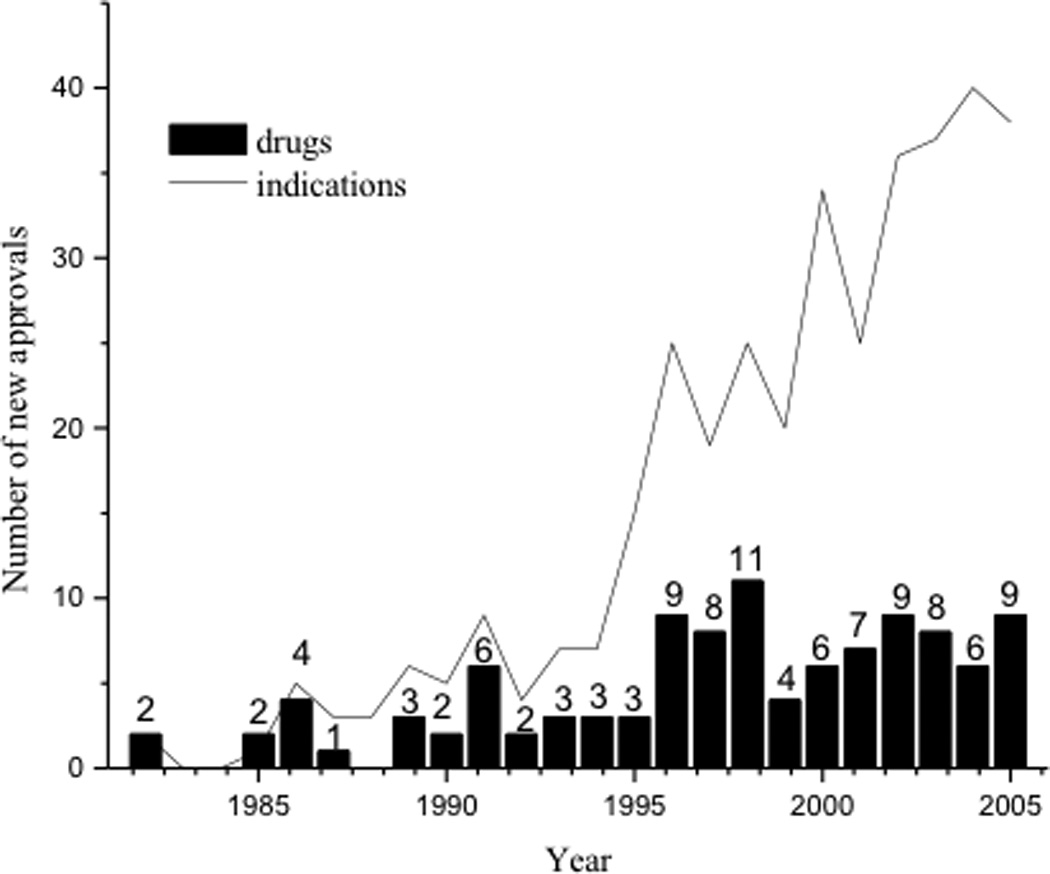
Increasing trend in the use of recombinant proteins and monoclonal antibodies as drugs. The number of new biotech drugs and new indications approved for biotech drugs from 1982 to 2005.
Different types of proteins such as antibodies, receptors, hormones, and enzymes often contain one or more Cys residues, and these may be involved in the formation of intra- and inter-molecular disulfide bond(s) or they may exist as free thiols. In some enzymes, the free Cys residue is part of the catalytic activity site. The presence of a disulfide bond in peptides and proteins has been shown to impose conformational rigidity on a protein. The disulfide bond can have either a left- or a right- handed spiral conformation with a dihedral angle of +90 or −90 degrees, respectively. In a disulfide bond, the most common Chi–1 angle of the Cys residue is −60 degrees with a 5.2–6.1 Å distance between the Cαs of Cys residues.16 During the folding process, the formation of non-native intramolecular disulfide bond(s) may cause protein misfolding, which may lead to aggregation and precipitation.
Native disulfide bonds in proteins are assembled during the oxidative folding process. In eukaryotes, this process occurs in the endoplasmic reticulum (ER) lumen,17 whereas in prokaryotes it takes place in the periplasm.18 Thus, disulfide bonds are only formed in specific cellular compartments. Proteins synthesized by ribosomes are in reduced and unfolded form, and their oxidative refolding in eukaryotes is aided by a series of enzymes such as the endopasmic reticulum oxireductin-1 (Ero1), protein disulfide isomerases (PDI) and Erv2.17,19–21 Many of these enzymes possess C-X-X-C (i.e., PDI) and C-X-C (i.e., Erv2) motifs that are involved in thiol-disulfide redox exchange reactions.22 PDI directly oxidizes thiol groups in a protein via a series of thiol-disulfide exchange reactions by partnering with Ero1 or Erv2. The Ero1 enzyme, which is found inside the ER and is associated with the ER membrane, oxidizes PDI to form an Ero1-PDI mixed disulfide form. If the disulfide bond in the protein substrate is incorrectly formed, PDI catalyzes the reduction of these disulfide bonds and, subsequently, re-oxidizes the protein substrate to form the native disulfide bond. Thus, as a general rule, cytoplasmic proteins contain free thiols, while proteins in other compartments such as the ER can possess disulfide bonds. An exception to this rule is a family of thermophilic organisms which has been discovered to produce intracellular proteins with disulfide bonds. This is presumably due to the presence of protein disulfide oxidoreductase (PDO) in their cytoplasm, which oxidizes the cysteines to disulfides within intracellular proteins.23
In bacterial cells, the oxidative folding machinery parallels the machinery in mammalian cells, and the oxidation reaction is catalyzed by the enzymes DsbA, DsbB, DsbC and DsbD.18 The disulfide bond A (DsbA) enzyme oxidizes thiol groups of the substrate protein to a disulfide bond in the periplasm with the simultaneous reduction of its own disulfide bond. Then, the reduced DsbA is reoxidized by DsbB. The oxidized state of DsbB is maintained by transferring electrons to ubiquinones (benzoquinone derivatives) and menaquinones (naphthoquinone derivatives), components of the respiratory electron transport chain system. When the substrate protein forms a non-native disulfide bond, DsbC enzyme reduces the non-native disulfides. This is followed by the subsequent re-oxidation of the substrate to form the native disulfide by DsbA or DsbC. Finally, DsbD functions to keep DsbC in the reduced state, and the oxidized DsbD can be reduced by cytoplasmic thioredoxin.
2. Disulfide Bond Formation
Prior to the formation of a disulfide bond, the protein may or may not be folded into a native-like structure with the Cys residues in a favorable position to form a disulfide bond.24,25 In the case of bovine pancreatic trypsin inhibitor (BPTI), the formation of all three disulfide bonds in BPTI occurs via stable intermediates with a native conformation containing one or two native disulfide bonds.24,25 No significant amount of non-native disulfide intermediates was observed. However, the formation of only native-like intermediates is not always the case; for example, hirudin, with three disulfide bonds, undergoes oxidative folding via heterogeneous intermediates with two or three non-native disulfide bonds.26 These scrambled disulfide bonds undergo reduction followed by oxidative rearrangement to form the native protein structure. The reductive unfolding of this protein follows a pathway similar to that of the folding process, but reversed. The in vitro oxidative folding of human macrophage colony stimulating factor β (rhm-CSFβ) follows a pathway similar to that of hirudin. The rhm-CSFβ protein is a dimer with three intermolecular disulfide bonds between the two monomers. Each monomer also contains three intramolecular disulfide bonds.27 The oxidative folding of rhm-CSFβ involves the formation of monomeric isomers with native and non-native intramolecular disulfide bonds; the monomer isomerization step is the rate-determining step in this process.27 Upon disulfide shuffling, the protein forms a stable dimeric intermediate that possesses all of the intramolecular disulfides, including a non-native intermolecular disulfide bond. Rearrangement of the non-native disulfide bond creates the native oxidized rhm-CSFβ.
Although the oxidative folding of different proteins could take different pathways, the process normally adheres to some general rules. Disulfide bond formation is usually favored at basic pH, and the presence of an oxidizing agent such as oxidized glutathione increases the rate of oxidative folding, whereas the presence of denaturants such as urea or guanidine hydrochloride (Gdn.HCl) hampers the folding process. These rules do not apply, however, to the oxidative folding of murine prion protein (mPrP 23–231), which has only one intramolecular disulfide bond at Cys179–Cys214.28 The formation of a disulfide bond in reduced mPrP is very slow at pH 8.0 in the absence of denaturant, but mPrP refolds properly in the presence of a denaturant and glutathione at pH 8.0.28 Contrary to intuition, the best pH condition to form a disulfide in mPrP is 4–5. This observation can be explained by the fact that, at alkaline and neutral pH, the reduced and unfolded mPrP is present in a stable intermediate conformation with its Cys residues buried and separated from each other. The addition of glutathione, therefore, does not facilitate oxidation of Cys. The addition of denaturant and a pH of 4.0 facilitate the unfolding of the stable intermediate, bringing the two Cys residues closer and allowing them to form a disulfide bond.
3. Effects of disulfide bond on protein stability
Intramolecular disulfide bonds are known to contribute to protein thermal stability. Reducing all five disulfide bonds inactivates the Aspergillus niger phytase due to conformational change and/or unfolding.29 Another example of the effect of disulfide bonds on stability is observed in the soybean Bowman-Birk inhibitor (BBI). The crystal structure of BBI has a bow-tie motif, with a trypsin-binding loop at one end and a chymotrypsin-binding loop at the other. In contrast to most globular proteins, it has exposed hydrophobic patches and charged residues containing bound water molecules in its interior.30,31 Interestingly, BBI is remarkably stable against heat and chemical denaturants; this stability can be attributed to the seven intramolecular disulfide bonds within the protein’s structure that “lock” it in its native conformation.30
Introduction of a disulfide bond by Cys mutation has been shown to improve the physical stability of some proteins. In case of Subtilisin E, mutations of Gly61Cys and Ser98Cys followed by the formation of the Cys61–Cys98 disulfide produce an active enzyme with a 4.5 °C increase in melting temperature and a half-life three times longer than that of the native protein.32 In addition, there was no change in its enzymatic activity due to the engineered disulfide bond. Similarly, a Cys39–Cys85 disulfide bond was engineered into dihydrofolate reductase (DHFR) without a significant alteration in its conformation. The disulfide mutant had an improved stability with its ΔG° higher than that of the native protein by 1.8 kcal/mol.33
At this point, it is necessary to discuss the thermodynamics behind the stabilizing effect of disulfide bonds on proteins. This stabilizing effect of disulfides is manifested by an increase in the melting point of the protein. The melting or unfolding transition of a protein is generally regarded as a reversible process between the native and the unfolded states. The differential scanning calorimetry (DSC) is usually employed to determine the reversible thermal unfolding of a protein. Superimposable DSC themorgrams resulted from repeated scans of the same protein imply a two-state reversible unfolding process, which is depicted as N ⇄ U, where N represents the native state and U, the unfolded state of the protein. This is also known as the “all or none” transition in statistical thermodynamics. In such a transition, the protein is either in the N state, with all residues in the n conformation or it is in the U state, where all its residues are in the u conformation. The Gibbs free energy for this process is related to the equilibrium constant of this reaction by the following equation: ΔG = −RT ln Keq. Keq is the ratio of the folding (kf) and unfolding (ku) rate constants. So, Keq = kf/ku. When a disulfide bond is introduced into a protein, there is likely to be a significant increase in free energy of U. The transition state (TS) can either (i) have a large increase in its free energy similar to the unfolded state, in which case, the disulfide bond is lost in the TS or (ii) have a negligible change in its free energy, in this case, TS is more N-like and retains the disulfide bond.34 In (i), the folding rate does not change much, but the unfolding rate decreases significantly. In (ii), the folding rate increases and the unfolding rate remain constant. In any case, the ratio of kf/ku which is equal to Keq decreases, leading to an increase in the free energy (ΔG) of the protein and making it conformationally more stable. The assumption is that there is little or no difference in the enthalpies (ΔH) of the disulfide-containing and reduced protein conformations. It has been proposed that the decrease in the entropy of U can be predicted with the help of the following equation: ΔS = −2.1 − (3/2)R ln n, where n is the number of residues in the loop formed by the intramolecular disulfide bond.35 Similarly, reduction of a disulfide bond or mutation of disulfide-forming Cys residues of a protein can result in increased entropy of U. Consequently, Keq increases and ΔG of the protein decreases. This characteristic was observed in the 110 residue starch-binding domain of Aspergillus Niger glucoamylase. The wild-type protein (SBD) possesses a disulfide bond between C3 and C98, creating a 94 residue loop between its N-terminus and the short loop between its seventh and eighth β-strands. Two different mutants C3G/C98G (GG) and C3S/C98S (SS) were constructed and their stability compared to that of SBD by determining their unfolding temperature and other thermodynamic parameters using DSC and CD techniques.36 The unfolding temperatures were 10 °C lower for the mutants (43 °C) compared to wild type SBD (53 °C). The unfolding processes for SBD, GG and SS were determined to be reversible with GG and SS having about 10 kJ/mol free energy lower than SBD. The contribution of entropic destabilization was much larger compared to that of the enthalpic destabilization.
When multiple disulfide bonds are introduced into a protein, they can act cooperatively to stabilize the protein. The overall increase in the free energy of a protein in that case is higher than the sum of the increases in free energies due to individual disulfide pairs. This phenomenon is seen in the Bacillus circulans xylanase. When disulfide bonds were introduced into it by the following mutations: S110C/N148C (DS1) and A1GC/G187,C188 (cX1), the overall increase in its free energy was 5.4 kcal/mol, 0.8 kcal/ mol more than the sum of the free energy increases (4.6 kcal/mol) due to each mutation separately. Consequently, the melting temperature of this double mutant was 12.4 °C, compared to the 5.0 °C and 3.8 °C increase in melting points in the DS1 and cX1 mutants.37
It is difficult to predict whether increasing conformational rigidity upon addition of a disulfide bond will always improve the stability and/or activity of a protein. A good example that illustrates the effect of the location of a disulfide on structural stability and enzymatic activity was shown in T4 lysozyme. Four mutants of T4 lysozyme with a disulfide bond at Cys9–Cys164, Cys21–Cys142, Cys90–Cys122, and Cys127–Cys154 were designed based on molecular modeling experiments.38 The thermal stabilities of the oxidized mutants were compared to their corresponding reduced mutants as well as to wild type T4 lysozyme. The oxidized Cys9–Cys164 mutant has a melting temperature 6.4 °C higher than that of the wild type while maintaining its enzymatic activity. In contrast, the Cys21–Cys142 mutant has the best thermal stability but has no enzymatic activity. Finally, the Cys90–Cys122 and Cys127–Cys154 mutants have lower thermal stabilities and enzymatic activities compared to that of the wild type enzyme. In another case, Ser52, Ser53, Ser78, Ser79, and Ser80 in alkaline phosphatase (AP) were mutated to the respective Cys residue; a disulfide bond was formed between the new Cys residue and Cys67. All of the disulfide mutants had improved thermal stability but lower enzymatic activities than that of the native AP.39 The decreased enzymatic activity of the mutant appeared to be due to the enhanced rigidity and lower substrate affinity of the active site imposed by the disulfide bond which is located near the residues. Thus, the location of the non-native disulfide bond affects the physicochemical stability and biological properties of the mutant proteins in a manner difficult to predict.
To impose conformational rigidity, a disulfide bond has also been introduced in peptides to form cyclic structures (cyclic peptides), which stabilizes specific secondary structures (i.e., β- and γ-turns). The formation of cyclic peptides in some cases enhances receptor-binding affinity and selectivity, as shown in opioid peptides.40 Several cyclic peptides with a disulfide bond have been marketed as therapeutic agents, including Integrilin®41,42 and oxytocin (Pitocin®).43 Integrilin® is a cyclic Arg-Gly-Asp (RGD) peptide, which is used to treat thrombosis; it binds selectively to gpIIb/IIa receptors on the surface of platelets, thus blocking fibrinogen-mediated platelet aggregation.41,42 Cyclic peptides have also been shown to be chemically and enzymatically more stable than their parent linear analogues. The Asp-mediated chemical degradation of RGD peptides was suppressed by the formation of a cyclic RGD peptide with a disufide bond; thus, the chemical stability of the cyclic RGD peptide is higher than that of the parent linear form.15,44,45
We discussed above the increase in stability of SBD by creating its two mutants: GG and SS. Earlier, a partly similar approach was taken to stabilize SBD. Two mutants were created C3S and C3G. C3G and C3S both showed a 9 C decrease in the melting point of wild type SBD. The explanation for this decreased stability was that the remaining unmutated C98 of SBD was able to form dimers in the native state. Therefore, although addition of disulfide bonds intramolecularly can help increase protein stability, formation of intermolecular disulfide bonds can destabilize a protein.46
4. Analysis of Free Thiol and Disulfide Bond
The presence of a free thiol group in the Cys residues in a protein can be quantitatively determined by different methods, including the use of malemide (i.e., ThioGlo),47 Ellman’s reagent (5,5'-dithiobis-(2-nitrobenzoic acid) or DTNB),48–51 and bimane reagents (Figure 2).52,53 The presence of a disulfide bond can be detected using the crabescein reagent.54 Ellman’s reagent (DTNB) was the earliest reagent widely used for estimating the number of thiol groups. The reaction between DTNB and thiol group(s) produces an equivalent amount of 5-thio-2-nitrobenzoic acid, of which the absorbance can be measured at 412 nm. It was found that the DTNB method could underestimate the concentration of thiol groups in a protein due to its incomplete reaction.47,55 Addition of cytamine as an accelerator of the DNTB-thiol reaction improves the accuracy of thiol determination (Figure 2a).51 As an alternative reagent, 4,4'-dithiodipyridine (DTDP) has a performance similar to that of the DTNB/cystamine in determining thiol concentrations (Figure 2a).51 Upon reaction with the thiol group, DTDP releases 4-thiopyridine (4-TP), which absorbs at 342 nm. One drawback to using DTDP is that some proteins have inherent absorbance at 342 nm, which can interfere with the quantitative measurement of 4-TP. A new derivative of Ellman’s reagent, n-octyl-5-dithio-2-nitrobenzoic acid (ODNB) has been designed; this reagent can react with all thiol groups of a protein, including the buried ones, in a relatively short time (Figure 2a).55
Figure 2.
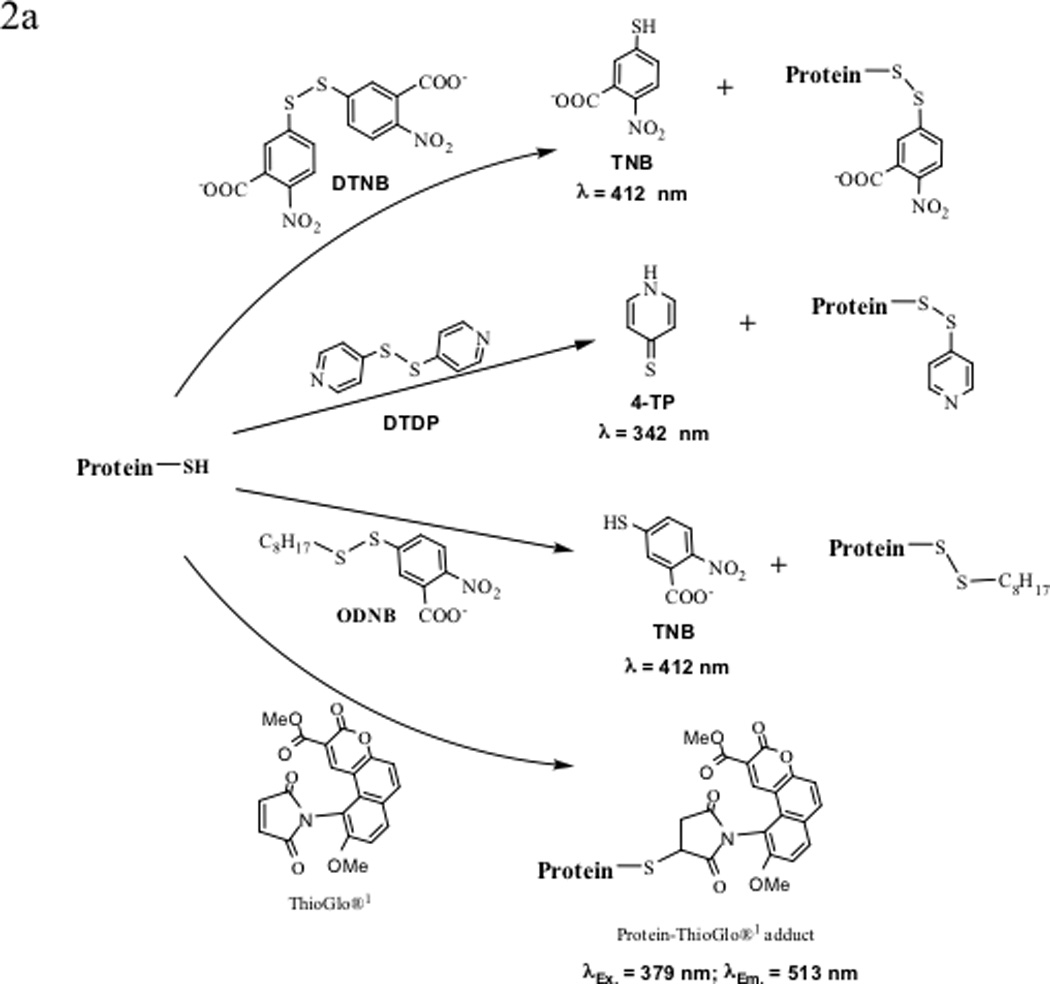
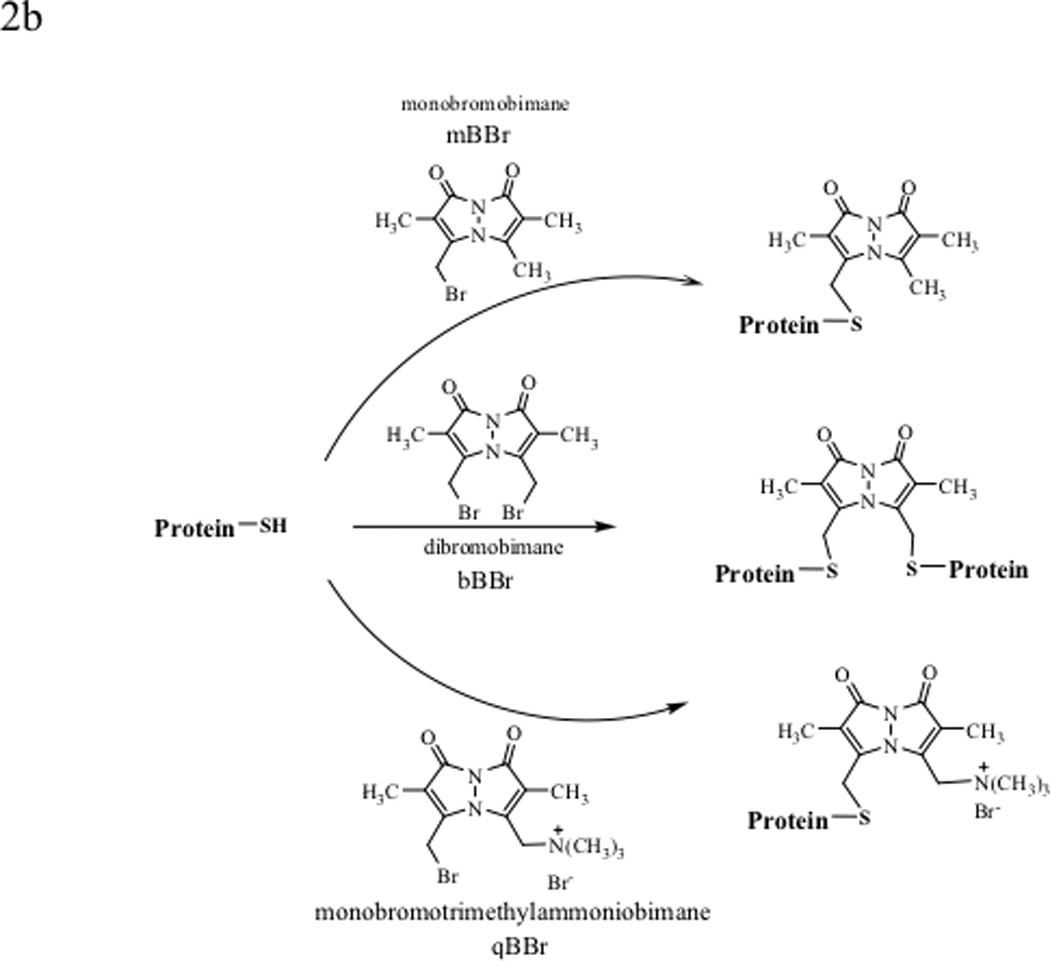
Various labeling reagents for the detection and quantification of protein thiols and disulfides. (a) Labeling protein thiols with 5,5'-dithio-bis(2-nitrobenzoic acid) (DTNB or Ellman’s reagent), 4,4'-dithiodipyridine (DTDP), n-octyl-5-dithio-2-nitrobenzoic acid (ODNB), and ThioGlo®1. (b) Labeling cellular protein thiols with bimanes: monobromobimane (mBBr) and dibromobimane (bBBr) label thiols inside cells as well as on cell surfaces, whereas monobromotrimethylammoniobimane (qBBr) labels only the cell surface protein thiols.
Malemide derivatives are also used to determine the number of thiol groups in a protein. ThioGlo® reagents (ThioGlo®1, λEx= 379 nm and λEm = 513 nm) are maleimide derivatives of naphthopyranone fluorophores that emit fluorescence upon thiol alkylation (Figure 2a). ThioGlo®1 is forty times more sensitive than DTNB and other maleimide reagents. The protein-ThioGlo adduct does not require separation prior to quantitative determination due to the low fluorescence of the starting reagent. One caution is that the reaction should not be carried out at a higher pH (>8) because of the possibility of amine alkylation of the malemide and adduct degradation. Possible disadvantages include the low water solubility of the ThioGlo reagent and the instability of protein-ThioGlo adducts, which can lead to inaccuracy in the quantitative determination of the thiol group.
Bimane derivatives (i.e., monobromobimane (mBBr), dibromobimane (bBBr) and monobromotrimethylammoniobimane (qBBr)) have also been developed for quantitative determination of thiol group(s) of proteins in cells (Figure 2b). Bimane derivatives have no intrinsic fluorescence; upon reaction with thiol groups in proteins, they become highly fluorescent.52 Both mBBr and bBBr can be used to measure the total cellular thiol content and qBBr can be used to measure the extracellular and membrane thiol content. Due to the presence of charged group in qBBr, this molecule cannot permeate the cell membranes and it can therefore be used to measure the extracellular thiols. mBBr can penetrate cell membranes and react with thiols in the cells as well as the thiols in the extracellular space. Thus, the difference in the thiol content measured by mBBr and qBBr gives the amount of thiols inside the cells. One of the advantages of using these bimane derivatives is that they are stable during exposure to air and irradiation as well as during general laboratory procedures. Bimane has also been used to determine the intracellular levels of thioredoxin after isolating the modified protein using an antibody affinity Sepahrose column. After elution from the column, the amount of thioredoxin can be determined using a fluorescence spectrometer.56
5. Degradation of Disulfide Bonds
Chemical degradation of disulfide bonds has been studied in both peptides and proteins with most of the disulfide bond degradation observed under neutral and basic conditions (Figure 3).15,57 Degradation of disulfide bonds can be classified into three major pathways: (1) direct attack on the sulfur atom by the hydroxyl anion, opening the disulfide bond to form sulfenic acid/thiolate anion (Fig. 3a); (2) A β-elimination reaction in which the α-proton of the Cys residue is abstracted (which produces dehydroalanine/persulfide (Figure 3b)); and (3) the α-elimination caused by the hydroxyl ion attacking the β-proton of the Cys residue to produce the thiolate/thioaldehyde (Figure 3c).58
Figure 3.
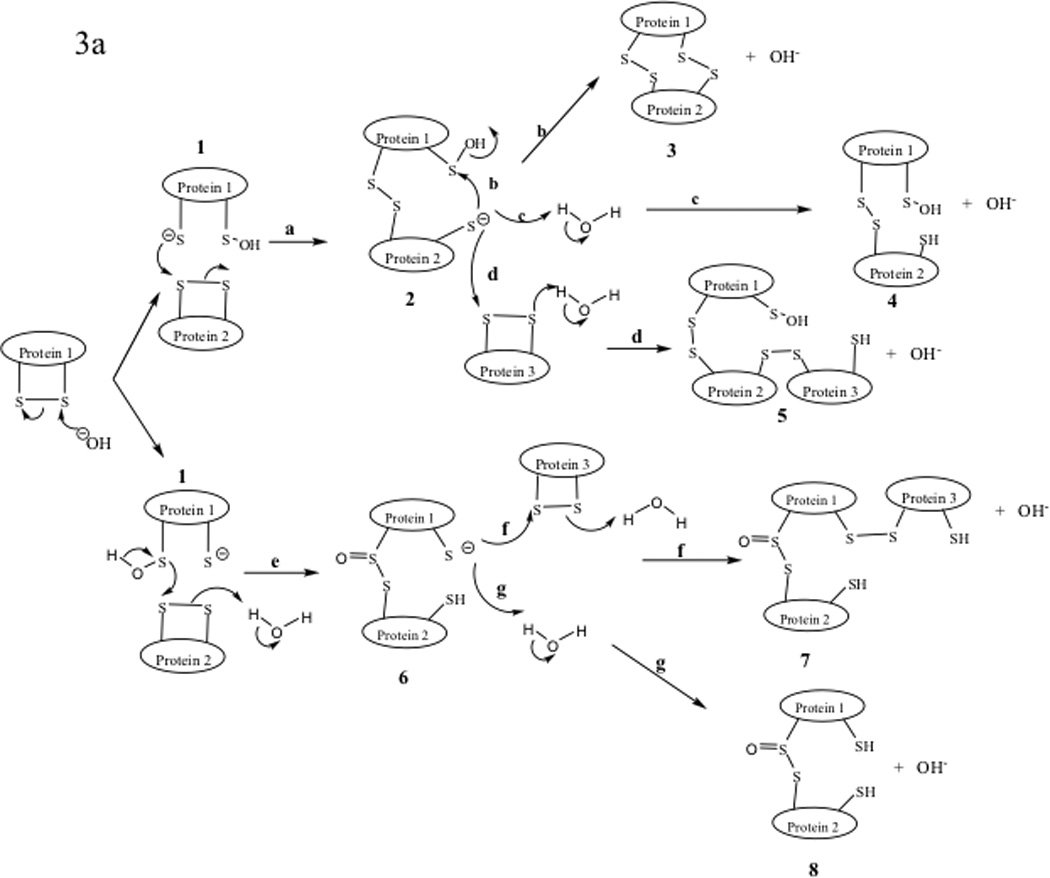

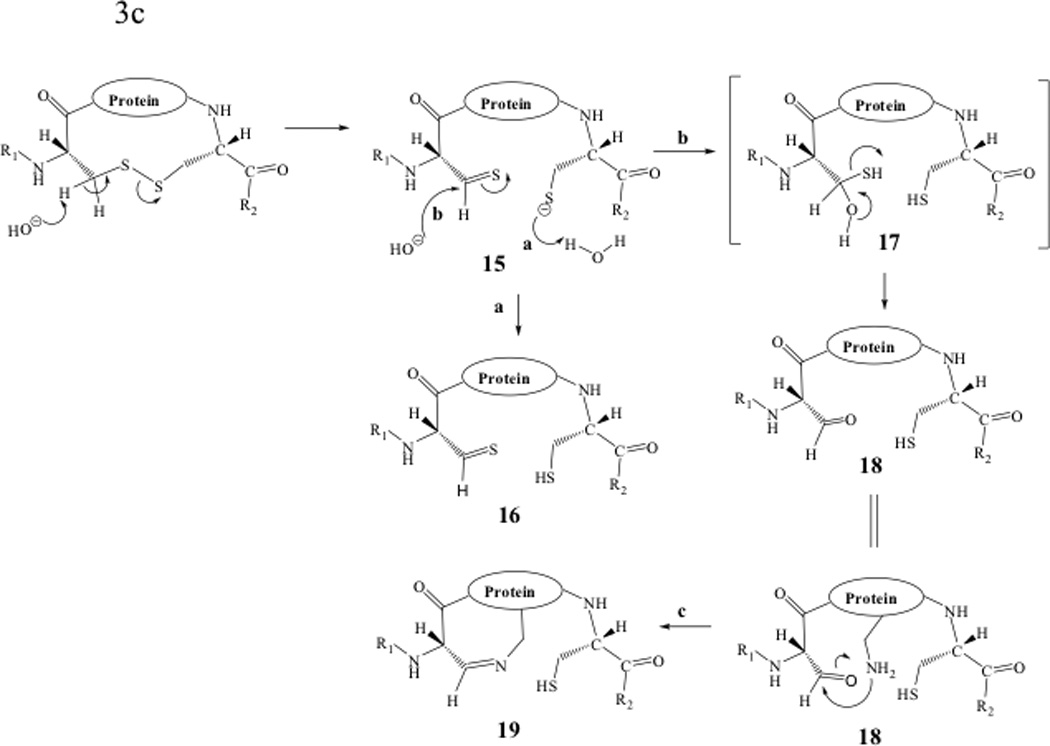
Degradation reactions of protein disulfide bonds in neutral and basic conditions. (a) Direct attack on sulfur atom by hydroxyl anion, (b) β-elimination reaction, and (c) α-elimination reaction.
The hydroxyl attack of the disulfide bond produces sufenic acid and thiolate anion (1, Figure 3a). The thiolate anion 1 can proceed to attack another disulfide bond on another protein molecule to produce a thiolate dimer intermediate 2 (pathway a, Figure 3a). This intermediate (2) undergoes three possible reactions as shown in pathways b, c, and d. In pathway b, intramolecular nucleophilic attack of the sulfenic acid by the thiolate anion produces dimer 3 with two intermolecular disulfide bonds. Intermediate 2 can form sulfenic acid dimer 4 by abstracting a proton from water (pathway c). Higher molecular weight oligomers such as trimer 5 can be produced via intermolecular disulfide bond reactions utilizing pathway d. The thiolate anion 1 can also undergo an intermolecular reaction between the sulfur atom of the sufinic acid group and a disulfide in another molecule to give an intermediate dimer 6 as shown in pathway e. The resulting thiolate 6 oligomerizes by reacting with another protein molecule to generate a trimer 7 (pathway f); further intermolecular reactions lead to higher molecular weight oligomers. Intermediate 6 can also form a dimer molecule with two free thiols (8).
The second degradation pathway is via β-elimination by hydroxyl anion abstraction of the acidic Cα proton of Cys residue to yield a dehydroalanine-persulfide 9 (Figure 3b). The disulfide bond can be reconnected by nucleophilic attack on the dehydroalanine by the persulfide anion (pathway a, Figure 3b). The resulting products (10) form a racemic mixture at the Cα of Cys residue (10, Figure 3b). The persulfide 9 can undergo a sulfur extrusion reaction via pathway b to yield the thiolate anion 11. The thiolate anion in 11 reacts with the alkene functional group of the dehydroalanine to generate the thioether 12; this reaction can also take place in an intermolecular fashion to produce oligomers. A similar reaction has been observed in an IgG1 monoclonal antibody59 and cyclic peptides containing a disulfide bond.15,57 In the IgG1, this thioether bond was observed at C233, which links the heavy chain to the light chain.59 This modification was identified by LC-MS tandem mass spectrometry. Abstraction of a proton from the solvent by the thiolate anion 11 produces dehydroalanine-thiol 13. There is a possibility that the amino group from either the Lys or the N-terminus of the protein may react with the alkene group of the dehydroalanine to produce a secondary amine 14. This reaction has been observed as a degradation product of cyclic peptides.15,57
Finally, the α-elimination reaction proceeds via the abstraction of the α-proton of the Cys residue followed by disulfide bond breakage to produce thiolate /thioaldehyde 15 (Figure 3c). Protonation of the thiolate anion yields thiol-aldehyde 16 (pathway a). In the presence of the hydroxyl anion, the thioaldehyde group can be converted to an aldehyde 18 via intermediate 17 (pathway b). An amino group (i.e., Lys or N-terminus) that is in close proximity to the aldehyde group in 18 may also produce an imine product 19. The imine reaction may produce bimolecular reactions to create oligomers.
Intermolecular covalent bond reactions (disulfide, thioether, amine and imine in Figure 3) producing oligomers which could lead to the association, aggregation and precipitation of proteins. For example, intermolecular disulfide formation causes aggregation of nascent thyroglobulin (Tg) protein. Monomeric Tg is a precursor to the synthesis of thyroid hormone. It contains more than 100 Cys residues and can produce aggregates through the formation of non-native intermolecular disulfide bonds due to the oxidizing environment in the endoplasmic reticulum of thyroid cells.60 During the transport of Tg to the golgi complex, the aggregates of Tg undergo disulfide reduction and dissociation to form monomers with assistance from molecular chaperones. This suggests that the frequency of Cys residues in the protein as well as the redox status of its environment are important factors that contribute to its disulfide-mediated aggregation. Intermolecular disulfide bond reactions in α- and β-crystallin induced by X-ray irradiation were shown to be partly responsible for catarct formation in the rabbit eye lens. It is interesting to note that the reaction between prednisolone and the amino group(s) of crystallin alters crystallin conformation to expose the buried thiol groups.61,62 These exposed thiol groups then undergo oxidation to form intermolecular disulfides, which cause protein aggregation and precipitation.61 It was made clear in a subsequent study that the crystallin that underwent conformation change on reaction with predisolone was α-crystallin.63 In most cases, the disulfide bond can be reversed by reducing agents (i.e., DTT), and its formation can be inhibited by metal complexing agents (i.e., EDTA), or removing metal ions. Intermolecular disulfide bond formation has been suggested to induce the aggregation of BSA. EDTA, citrate, and DTT have been shown to suppress the aggregation of BSA.64
6. Thiol Reaction and Derivatization
The thiol group in Cys can undergo oxidation reactions to form sulfenic (R-S-OH), sulfinic (R-SO2H), and sulfonic (R-SO3H) derivatives as well as the disulfide bond. Thiol groups in proteins are inherently very reactive and can participate in side reactions with reagents used to manipulate other functional groups in a protein and the other solution components. The presence of a free cysteine residue may cause unwanted intramolecular disulfide scrambling, and covalent oligomerization via intermolecular disulfide formation. To prevent these reactions, thiol groups can be derivatized with acetate, acetamide, 1,3-propane sultone65, methyl methanethiosulfonate, methoxycarbonylmethyl disulfide66, maleimide, tetrathionates, and dinitrophenyl alkyl disulfides (DNPSSR)67 (Figure 4). Human serum albumin, [Lys8] vasopressin, bovine insulin, and bovine pancreatic ribonuclease have been alkylated with 1,3-propane sultone to increase their solubility and stability to acid hydrolysis. Alkylating the free thiol of the tetanus toxoid with n-ethylmaleimide inhibits moisture-induced aggregation of the lyophilized toxoid.68
Figure 4.
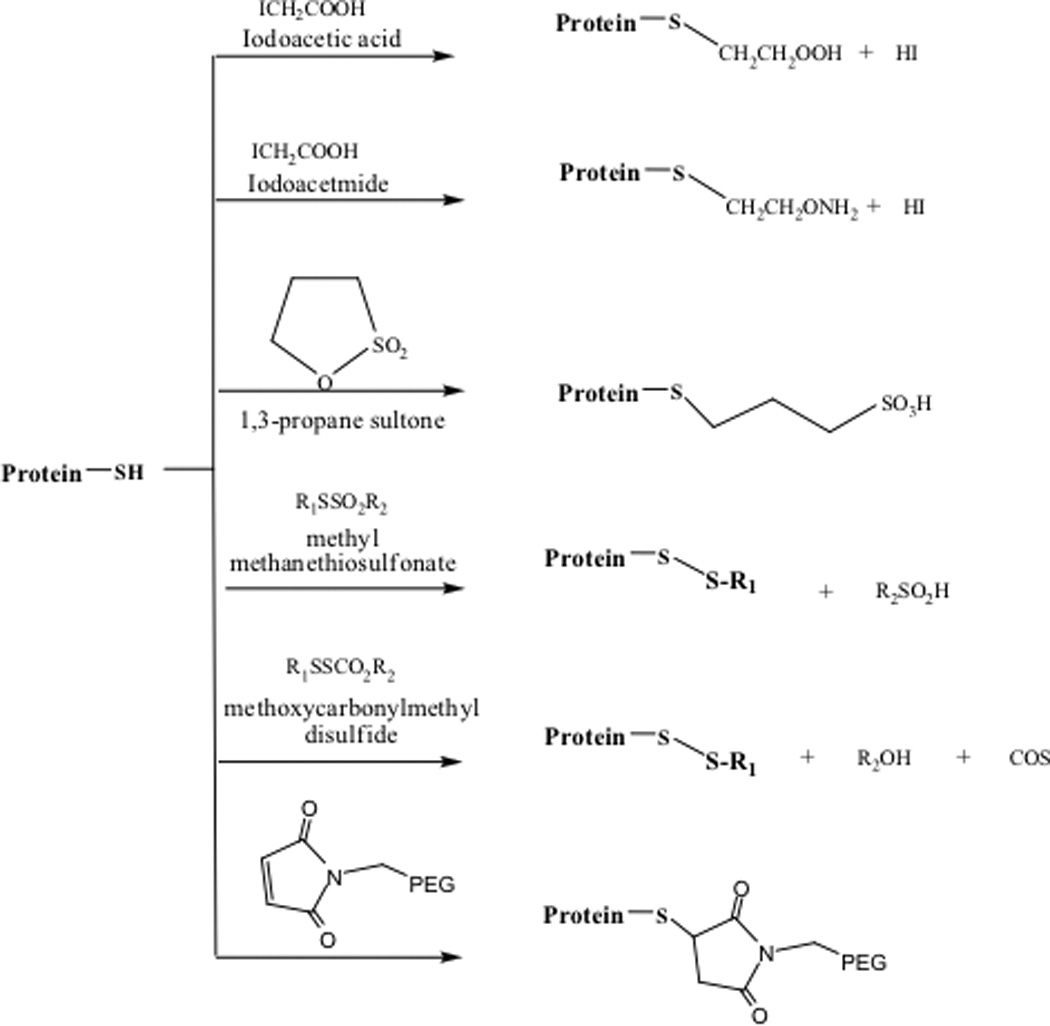
Reagents for derivatization of free thiols in proteins.
To avoid side reactions of the Cys thiol group, Cys has frequently been mutated to Gly, Ala, and Ser residues in proteins. The C6A, C111S and C6S/C111A mutants of human CuZn dismutase (SOD) and the C6A mutant of bovine SOD showed greater resistance to loss of enzymatic activity than did the respective native SODs upon thermal unfolding at 70 °C.69 This corresponds to the fact that the mutants can reversibly refold after denaturation while the unfolded native SOD refolds incorrectly. Cys mutation studies have been used to determine the role of free and disulfide-bonded Cys residues in the function of many proteins. Meprins, cell surface and secreted metalloendopeptidases, are oligomers made up of disulfide-linked multidomain subunits.
Mutations of C320A and C289A of mouse meprin-α both resulted in an increased susceptibility to proteolytic degradation and heat deactivation. Although the mutants retained their catalytic activity toward a bradykinin peptide, they displayed decreased activity toward the protein substrate azocasein. It was found that the C320A mutant only formed monomers, while the C289A mutant formed dimers and oligomers, suggesting that Cys 320 but not C289 is involved in inter-subunit disulfide formation.70 Mutation of either Cys residue resulted in a change in protein structure causing it to lose its enzymatic activity toward a protein substrate. Thus, Cys mutation is a technique that can not only shed light on the importance of the state of Cys residues (paired or reduced, intra- or interprotein pairing) in a particular protein but can also help to distinguish the different roles of each Cys in the protein.
The Cys residue of therapeutic proteins has also been PEGylated to improve their physicochemical stability, pharmacokinetics and pharmacodynamics properties. PEGylated proteins usually have lower glomerular filtration and immunogenicity. The molecular weight of these PEG reagents can range from 2 to 50 kDa. The PEGylation of a Cys residue is more selective than PEGylation of Lys residue. PEGylation of Lys residues gives a heterogenous mixture of products with a distribution of conjugated products, which is difficult to control. The site-specific PEGylation the Cys residue is normally carried out with PEG-Maleimide, PEG-acrylamide, PEG-acrylate, PEG-vinyl sulfone, PEG-epoxide or PEG-iodoacetamide. In a protein without a free thiol, a surface residue away from the active site can be mutated to a Cys residue followed by PEGylation. This method has been successfully used to improve the half-life of granulocyte colony stimulating factor (G-CSF)71 and the Q5C analog of interferon-α (IFN-α)72, immunotoxin anti-Tac(Fv)-PE38 (LMB2),73 and Eryhtropoietin (EPO).74 When LMB2 was stabilized by PEGylation using 5– or 20– kDa PEG-maleimide, there was an increase in plasma stability (five- to eight- fold) and antitumor activity (three- to four- fold).73 In this case, LMB2’s animal toxicity and immunogenecity were significantly lower than that of the parent toxin. Native disulfide bond can also be conjugated with PEG by reacting with PEG-monosulfone; the product forms a three carbon bridge with the two sulfur atoms of the native disulfide bond. Interferon α-2b, anti-CD4+ antibody, and L-asparaginase have also been PEGylated in this manner. In the case of interferon α-2b, the PEGylated product maintains its biological activity as well as its tertiary structure, which was confirmed by NMR and X-ray crystallography.
7. Conclusions
Thiol groups and disulfides have important roles in the stability and solubility of proteins. Thiols and disulfides are inherently very reactive. Increased research is being devoted to studying the role of thiols and disulfides in problems associated with protein molecules. Studies have shown some disulfide bonds in proteins to be essential for either protein stability and/ or activity. On the other hand, some non-native disulfide bonds in proteins are a result of altered in vitro or in vivo conditions and are detrimental to protein stability and/ or activity. Various reagents have been designed to detect and quantify free thiols and disulfides in proteins. The choice of the appropriate labeling agent should be made after careful consideration since they all have advantages and disadvantages. Changes in native thiol and/or disulfide content in proteins can be blocked by either the addition of appropriate redox reagents or irreversible conjugation with various agents as described above. Mutation of the Cys residue to another amino acid (i.e., Ser, Ala) can be an alternative strategy when the residue is not essential to protein activity. Protein molecules are highly complex systems. Our knowledge of the role of Cys residue in these macromolecular systems is continuously expanding. It is hoped that the growing appreciation for the role of thiols and disulfides in protein structure may lead to new methods to improve the success in formulating proteins as therapeutic agents.
Preliminary work in Dr. Siahaan’s laboratory has shown that the protein EC1 forms intermolecular disulfide bonds, piquing our interest into studying the effect of this disulfide bond on EC1 stability. EC1 is the N-terminal domain of E-cadherin, a transmembrane protein found in the adherens junctions in epithelial and endothelial cells. E-cadherin functions as a homophilic cell adhesion molecule75 and the EC1 domain has been suggested to have a role in the intercellular interactions of E-cadherin.76 EC1 contains a single Cys residue near its N-terminus. Across the cadherin family, the N-terminus of the first domain is implicated to form domain-swapped dimers. The first domain of only E-cadherin in the cadherin family contains a Cys residue. The oxidation of this Cys residue leads to a covalent dimer formation and subsequent physical oligomerization of EC1. We, therefore, propose to study the effect of EC1 dimerization and oligomerization on its structure and stability.
References
- 1.Guide to Biotechnology, editor. Bio. Approved Biotechnology Drugs. 2007. [Google Scholar]
- 2.Cyran R. The market for bioengineered protein drugs BCC Research Report and Reviews. ed. 2004. [Google Scholar]
- 3.Nonoyama A, Laurence JS, Garriques L, Qi H, Le T, Middaugh CR. A biophysical characterization of the peptide drug pramlintide (AC137) using empirical phase diagrams. Journal of Pharmaceutical Sciences. 2007 doi: 10.1002/jps.21197. [DOI] [PubMed] [Google Scholar]
- 4.Fan H, Li H, Zhang M, Middaugh CR. Effects of solutes on empirical phase diagrams of human fibroblast growth factor 1. J Pharm Sci. 2007;96:1490–1503. doi: 10.1002/jps.20796. [DOI] [PubMed] [Google Scholar]
- 5.Brandau DT, Joshi SB, Smalter AM, Kim S, Steadman B, Middaugh CR. Stability of the Clostridium botulinum type A neurotoxin complex: an empirical phase diagram based approach. Mol Pharm. 2007;4:571–582. doi: 10.1021/mp0601244. [DOI] [PubMed] [Google Scholar]
- 6.Goolcharran C, Stauffer LL, Cleland JL, Borchardt RT. The effects of a histidine residue on the C-terminal side of an asparaginyl residue on the rate of deamidation using model pentapeptides. J Pharm Sci. 2000;89:818–825. doi: 10.1002/(SICI)1520-6017(200006)89:6<818::AID-JPS14>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 7.Goolcharran C, Cleland JL, Keck R, Jones AJ, Borchardt RT. Comparison of the rates of deamidation, diketopiperazine formation and oxidation in recombinant human vascular endothelial growth factor and model peptides. AAPS PharmSci. 2000;2:E5. doi: 10.1208/ps020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai MC, Hageman MJ, Schowen RL, Borchardt RT, Topp EM. Chemical stability of peptides in polymers. 1. Effect of water on peptide deamidation in poly(vinyl alcohol) and poly(vinyl pyrrolidone) matrixes. J Pharm Sci. 1999;88:1073–1080. doi: 10.1021/js980227g. [DOI] [PubMed] [Google Scholar]
- 9.Zhao F, Yang J, Schoneich C. Effects of polyaminocarboxylate metal chelators on iron-thiolate induced oxidation of methionine- and histidine-containing peptides. Pharm Res. 1996;13:931–938. doi: 10.1023/a:1016021716274. [DOI] [PubMed] [Google Scholar]
- 10.Yao Y, Yin D, Jas GS, Kuczer K, Williams TD, Schoneich C, Squier TC. Oxidative modification of a carboxyl-terminal vicinal methionine in calmodulin by hydrogen peroxide inhibits calmodulin-dependent activation of the plasma membrane Ca-ATPase. Biochemistry. 1996;35:2767–2787. doi: 10.1021/bi951712i. [DOI] [PubMed] [Google Scholar]
- 11.Li S, Nguyen TH, Schoneich C, Borchardt RT. Aggregation and precipitation of human relaxin induced by metal-catalyzed oxidation. Biochemistry. 1995;34:5762–5772. doi: 10.1021/bi00017a008. [DOI] [PubMed] [Google Scholar]
- 12.Wakankar AA, Borchardt RT. Formulation considerations for proteins susceptible to asparagine deamidation and aspartate isomerization. J Pharm Sci. 2006;95:2321–2336. doi: 10.1002/jps.20740. [DOI] [PubMed] [Google Scholar]
- 13.Manning MC, Patel K, Borchardt RT. Stability of protein pharmaceuticals. Pharm Res. 1989;6:903–918. doi: 10.1023/a:1015929109894. [DOI] [PubMed] [Google Scholar]
- 14.Xie M, Shahrokh Z, Kadkhodayan M, Henzel WJ, Powell MF, Borchardt RT, Schowen RL. Asparagine deamidation in recombinant human lymphotoxin: hindrance by three-dimensional structures. J Pharm Sci. 2003;92:869–880. doi: 10.1002/jps.10342. [DOI] [PubMed] [Google Scholar]
- 15.Bogdanowich-Knipp SJ, Chakrabarti S, Williams TD, Dillman RK, Siahaan TJ. Solution stability of linear vs. cyclic RGD peptides. J Pept Res. 1999;53:530–541. doi: 10.1034/j.1399-3011.1999.00052.x. [DOI] [PubMed] [Google Scholar]
- 16.Richardson JS. In: The anatomy and taxonomy of protein structure. Advances In Protein Chemistry, editor. New York: Academic Press; 1981. pp. 223–231. [DOI] [PubMed] [Google Scholar]
- 17.Gruber CW, Cemazar M, Heras B, Martin JL, Craik DJ. Protein disulfide isomerase: the structure of oxidative folding. Trends Biochem Sci. 2006;31:455–464. doi: 10.1016/j.tibs.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Kadokura H, Katzen F, Beckwith J. Protein disulfide bond formation in prokaryotes. Annual Review of Biochemistry. 2003;72:111–135. doi: 10.1146/annurev.biochem.72.121801.161459. [DOI] [PubMed] [Google Scholar]
- 19.Frand AR, Cuozzo JW, Kaiser CA. Pathways for protein disulphide bond formation. Trends in Cell Biology. 2000;10:203–210. doi: 10.1016/s0962-8924(00)01745-1. [DOI] [PubMed] [Google Scholar]
- 20.Sevier CS, Kaiser CA. Conservation and diversity of the cellular disulfide bond formation pathways. Antioxid Redox Signal. 2006;8:797–811. doi: 10.1089/ars.2006.8.797. [DOI] [PubMed] [Google Scholar]
- 21.Kulp MS, Frickel EM, Ellgaard L, Weissman JS. Domain architecture of protein-disulfide isomerase facilitates its dual role as an oxidase and an isomerase in Ero1p-mediated disulfide formation. J Biol Chem. 2006;281:876–884. doi: 10.1074/jbc.M511764200. [DOI] [PubMed] [Google Scholar]
- 22.Fomenko DE, Gladyshev VN. Genomics perspective on disulfide bond formation. Antioxid Redox Signal. 2003;5:397–402. doi: 10.1089/152308603768295131. [DOI] [PubMed] [Google Scholar]
- 23.Beeby M, O'Connor BD, Ryttersgaard C, Boutz DR, Perry LJ, Yeates TO. The genomics of disulfide bonding and protein stabilization in thermophiles. PLoS biology. 2005;3:e309. doi: 10.1371/journal.pbio.0030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weissman JS, Kim PS. A kinetic explanation for the rearrangement pathway of BPTI folding. Nat Struct Biol. 1995;2:1123–1130. doi: 10.1038/nsb1295-1123. [DOI] [PubMed] [Google Scholar]
- 25.Dadlez M, Kim PS. A third native one-disulphide intermediate in the folding of bovine pancreatic trypsin inhibitor. Nat Struct Biol. 1995;2:674–679. doi: 10.1038/nsb0895-674. [DOI] [PubMed] [Google Scholar]
- 26.Arolasa JL, Aviles FX, Chang JY, Ventura S. Folding of small disulfide-rich proteins: clarifying the puzzle. Trends Biochem Sci. 2006;31:292–301. doi: 10.1016/j.tibs.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Zhang YH, Yan X, Maier CS, Schimerlik MI, Deinzer ML. Conformational analysis of intermediates involved in the in vitro folding pathways of recombinant human macrophage colony stimulating factor beta by sulfhydryl group trapping and hydrogen/deuterium pulsed labeling. Biochemistry. 2002;41:15495–15504. doi: 10.1021/bi0203841. [DOI] [PubMed] [Google Scholar]
- 28.Lu B-Y, Beck PJ, Chang J-Y. Oxidative folding of murine prion mPrP(23–231) European Journal of Biochemistry. 2001;268:3767–3773. doi: 10.1046/j.1432-1327.2001.02283.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang X-Y, Meng F-G, Zhou H-M. The role of disulfide bonds in the conformational stability and catalytic activity of phytase. Biochemistry and cell biology. 2004;82:329–334. doi: 10.1139/o03-082. [DOI] [PubMed] [Google Scholar]
- 30.Singh RR, Appu Rao AG. Reductive unfolding and oxidative refolding of a Bowman-Birk inhibitor from horsegram seeds (Dolichos biflorus): evidence for 'hyperreactive' disulfide bonds and rate-limiting nature of disulfide isomerization in folding. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology. 2002;1597:280–291. doi: 10.1016/s0167-4838(02)00301-1. [DOI] [PubMed] [Google Scholar]
- 31.Voss RH, Ermler U, Essen L-O, Wenzl G, Kim Y-M, Flecker P. Crystal Structure of the Bifunctional Soybean Bowman-Birk Inhibitor at 0.28-nm Resolution. Structural Peculiarities in a Folded Protein Conformation. European Journal of Biochemistry. 1996;242:122–131. doi: 10.1111/j.1432-1033.1996.0122r.x. [DOI] [PubMed] [Google Scholar]
- 32.Takagi H, Takahashi T, Momose H, Inouye M, Maeda Y, Matsuzawa H, Ohta T. Enhancement of the thermostability of subtilisin E by introduction of a disulfide bond engineered on the basis of structural comparison with a thermophilic serine protease. J Biol Chem. 1990;265:6874–6878. [PubMed] [Google Scholar]
- 33.Villafranca JE, Howell EE, Oatley SJ, Nguyen Huu X, Kraut J. An engineered disulfide bond in dihydrofolate reductase. Biochemistry. 1987;26:2182–2189. doi: 10.1021/bi00382a017. [DOI] [PubMed] [Google Scholar]
- 34.Wedemeyer WJ, Welker E, Narayan M, Scheraga HA. Disulfide Bonds and Protein Folding. Biochemistry. 2000;39:7032–7032. doi: 10.1021/bi005111p. [DOI] [PubMed] [Google Scholar]
- 35.Pace CN, Grimsley GR, Thomson JA, Barnett BJ. Conformational stability and activity of ribonuclease T1 with zero, one, and two intact disulfide bonds. J Biol Chem. 1988;263:11820–11825. [PubMed] [Google Scholar]
- 36.Sugimoto H, Nakaura M, Kosuge Y, Imai K, Miyake H, Karita S, Tanaka A. Thermodynamic Effects of Disulfide Bond on Thermal Unfolding of the Starch-Binding Domain of Aspergillus niger Glucoamylase. Bioscience, Biotechnology, and Biochemistry. 2007;71:1535–1541. doi: 10.1271/bbb.70098. [DOI] [PubMed] [Google Scholar]
- 37.Davoodi J, Wakarchuk WW, Carey PR, Surewicz WK. Mechanism of stabilization of Bacillus circulans xylanase upon the introduction of disulfide bonds. Biophysical Chemistry. 2007;125:453–461. doi: 10.1016/j.bpc.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Matsumura M, Becktel WJ, Levitt M, Matthews BW. Stabilization of Phage T4 Lysozyme by Engineered Disulfide Bonds. PNAS. 1989;86:6562–6566. doi: 10.1073/pnas.86.17.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asgeirsson B, Adalbjornsson BV, Gylfason GA. Engineered disulfide bonds increase active-site local stability and reduce catalytic activity of a cold-adapted alkaline phosphatase. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics. 2007;1774:679–687. doi: 10.1016/j.bbapap.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 40.Berezowska I, Chung NN, Lemieux C, Wilkes BC, Schiller PW. Cyclic dermorphin tetrapeptide analogues obtained via ring-closing metathesis. Acta Biochim Pol. 2006;53:73–76. [PubMed] [Google Scholar]
- 41.Thibault G, Tardif P, Lapalme G. Comparative specificity of platelet alpha(IIb)beta(3) integrin antagonists. J Pharmacol Exp Ther. 2001;296:690–696. [PubMed] [Google Scholar]
- 42.Rossi ML, Zavalloni D. Inhibitors of platelets glycoprotein IIb/IIIa (GP IIb/IIIa) receptor: rationale for their use in clinical cardiology. Mini Rev Med Chem. 2004;4:703–709. doi: 10.2174/1389557043403666. [DOI] [PubMed] [Google Scholar]
- 43.Gimpl G, Burger K, Politowska E, Ciarkowski J, Fahrenholz F. Oxytocin receptors and cholesterol: interaction and regulation. Exp Physiol. 2000;85(Spec No):41S–49S. doi: 10.1111/j.1469-445x.2000.tb00006.x. [DOI] [PubMed] [Google Scholar]
- 44.Bogdanowich-Knipp SJ, Jois DS, Siahaan TJ. The effect of conformation on the solution stability of linear vs. cyclic RGD peptides. J Pept Res. 1999;53:523–529. doi: 10.1034/j.1399-3011.1999.00055.x. [DOI] [PubMed] [Google Scholar]
- 45.Bogdanowich-Knipp SJ, Jois SD, Siahaan TJ. Effect of conformation on the conversion of cyclo-(1,7)-Gly-Arg-Gly-Asp-Ser-Pro-Asp-Gly-OH to its cyclic imide degradation product. J Pept Res. 1999;54:43–53. doi: 10.1034/j.1399-3011.1999.00091.x. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka A, Karita S, Kosuge Y, Senoo K, Obata H. Thermal unfolding of mutant forms C509G and C509S of starch binding domain-fragment of Aspergillus niger glucoamylase. Netsusokutei. 1999;26:136. [Google Scholar]
- 47.Wright SK, Viola RE. Evaluation of methods for the quantitation of cysteines in proteins. Analytical Biochemistry. 1998;265:8–14. doi: 10.1006/abio.1998.2858. [DOI] [PubMed] [Google Scholar]
- 48.Ellman GL. Tissue sulfhydryl groups. Archives of Biochemistry and Biophysics. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 49.Robyt JF, Ackerman RJ, Chittenden CG. Reaction of protein disulfide groups with Ellman's reagent: a case study of the number of sulfhydryl and disulfide 27 groups in Aspergillus oryzae -amylase, papain, and lysozyme. Arch Biochem Biophys. 1971;147:262–269. doi: 10.1016/0003-9861(71)90334-1. [DOI] [PubMed] [Google Scholar]
- 50.Sedlak J, Lindsay RH. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman's reagent. Anal Biochem. 1968;25:192–205. doi: 10.1016/0003-2697(68)90092-4. [DOI] [PubMed] [Google Scholar]
- 51.Riener CK, Kada G, Gruber HJ. Quick measurement of protein sulfhydryls with Ellman's reagent and with 4,4'-dithiodipyridine. Anal Bioanal Chem. 2002;373:266–276. doi: 10.1007/s00216-002-1347-2. [DOI] [PubMed] [Google Scholar]
- 52.Kosower NS, Kosower EM, Newton GL, Ranney HM. Bimane fluorescent labels: labeling of normal human red cells under physiological conditions. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:3382–3386. doi: 10.1073/pnas.76.7.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kosower NS, Newton GL, Kosower EM, Ranney HM. Bimane fluorescent labels. Characterization of the bimane labeling of human hemoglobin. Biochimica et Biophysica Acta. 1980;622:201–209. [PubMed] [Google Scholar]
- 54.Packard B, Edidin M, Komoriya A. Site-directed labeling of a monoclonal antibody: targeting to a disulfide bond. Biochemistry. 1986;25:3548–3552. doi: 10.1021/bi00360a012. [DOI] [PubMed] [Google Scholar]
- 55.Faulstich H, Tews P, Heintz D. Determination and derivatization of protein thiols by n-octyldithionitrobenzoic acid. Analytical Biochemistry. 1993;208:357–362. doi: 10.1006/abio.1993.1061. [DOI] [PubMed] [Google Scholar]
- 56.Chinn PC, Pigiet V, Fahey RC. Determination of thiol proteins using monobromobimane labeling and high-performance liquid chromatographic analysis: application to Escherichia coli thioredoxin. Analytical Biochemistry. 1986;159:143–149. doi: 10.1016/0003-2697(86)90319-2. [DOI] [PubMed] [Google Scholar]
- 57.He HT, Gürsoy RN, Kupczyk-Subotkowska L, Tian J, Williams T, Siahaan TJ. Synthesis and chemical stability of a disulfide bond in a model cyclic pentapeptide: Cyclo(1,4)-Cys-Gly-Phe-Cys-Gly-OH. Journal of Pharmaceutical Sciences. 2006;95:2222–2234. doi: 10.1002/jps.20701. [DOI] [PubMed] [Google Scholar]
- 58.Florence MT. Degradation of protein disulphide bonds in dilute alkali. Biochem J. 1980;189:507–520. doi: 10.1042/bj1890507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tous GI, Wei Z, Feng J, Bilbulian S, Bowen S, Smith J, Strouse R, McGeehan P, Casas-Finet J, Schenerman MA. Characterization of a novel modification to monoclonal antibodies: Thioether cross-link of heavy and light Chains. Anal Chem. 2005;77:2675–2682. doi: 10.1021/ac0500582. [DOI] [PubMed] [Google Scholar]
- 60.Kim PS, Kim KR, Arvan P. Disulfide-linked aggregation of thyroglobulin normally occurs during nascent protein folding. Am J Physiol. 1993;265:C704–C711. doi: 10.1152/ajpcell.1993.265.3.C704. [DOI] [PubMed] [Google Scholar]
- 61.Bucala R, Manabe S, Urban RC, Cerami A. Nonenzymatic modification of lens crystallins by prednisolone induces sulfhydryl oxidation and aggregate formation: In vitro and in vivo studies. Experimental Eye Research. 1985;41:353–363. doi: 10.1016/s0014-4835(85)80026-9. [DOI] [PubMed] [Google Scholar]
- 62.Manabe S, Bucala R, Cerami A. Nonenzymatic addition of glucocorticoids to lens proteins in steroid-induced cataracts. The Journal of Clinical Investigation. 1984;74:1803–1810. doi: 10.1172/JCI111599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hook DWA, Harding JJ. The effect of modification of alpha-crystallin by prednisolone-21-hemisuccinate and fructose 6-phosphate on chaperone activity. Developments in Ophthalmology. 2002;35:150–160. doi: 10.1159/000060819. [DOI] [PubMed] [Google Scholar]
- 64.Sean T, Kelly ALZ. Effects of intermolecular thiol-disulfide interchange reactions on BSA fouling during microfiltration. Biotechnology and Bioengineering. 1994;44:972–982. doi: 10.1002/bit.260440814. [DOI] [PubMed] [Google Scholar]
- 65.Ruegg UT, Rudinger J. Alkylation of cysteine thiols with 1,3-propane sultone. Methods in enzymology. 47:116–122. doi: 10.1016/0076-6879(77)47013-7. [DOI] [PubMed] [Google Scholar]
- 66.Smith DJ, Miggio ET, Kenyon GL. Simple alkanethiol groups for temporary blocking of sulfhydryl groups of enzymes. Biochemistry. 1975;14:766–771. doi: 10.1021/bi00675a019. [DOI] [PubMed] [Google Scholar]
- 67.Koizumi S, Suzuki T, Takahashi S, Satake K, Takeuchi T, Umezawa H, Nagatsu T. Sulfhydryl modification and activation of phenylalanine hydroxylase by dinitrophenyl alkyl disulfide. Biochemistry. 1987;26:6461–6465. doi: 10.1021/bi00394a025. [DOI] [PubMed] [Google Scholar]
- 68.Inhibition of moisture-induced aggregation of tetanus toxoid by protecting thiol groups. Proceedings of the International Symposium on Controlled Release of Bioactive Materials. 1994;21ST:54. [Google Scholar]
- 69.Hallewell RA, Imlay KC, Lee P, Fong NM, Gallegos C, Getzoff ED, Tainer JA, Cabelli DE, Tekamp-Olson P, Mullenbach GT, Cousens LS. Thermostabilization of recombinant human and bovine CuZn superoxide dismutases by replacement of free cysteines. Biochemical and Biophysical Research Communications. 1991;181:474–480. doi: 10.1016/s0006-291x(05)81443-3. [DOI] [PubMed] [Google Scholar]
- 70.Marchand P, Volkmann M, Bond JS. Cysteine Mutations in the MAM Domain Result in Monomeric Meprin and Alter Stability and Activity of the Proteinase. J Biol Chem. 1996;271:24236–24241. doi: 10.1074/jbc.271.39.24236. [DOI] [PubMed] [Google Scholar]
- 71.Rosendahl MS, Doherty DH, Smith DJ, Bendale AM, Cox GN. Site specific protein PEGylation: Application to Cysteine Analogs of Recombinant Human Granulocyte Colony-Stimulating factor. Bioprocess international. 2005;3:52–62. [PMC free article] [PubMed] [Google Scholar]
- 72.Rosendahl MS, Doherty DH, Smith DJ, Carlson SJ, Chlipala EA, Cox GN. A long acting, highly potent interferon α-2 conjugate created using site-specific PEGylation. Bioconjugate Chemistry. 2005;16:200–207. doi: 10.1021/bc049713n. [DOI] [PubMed] [Google Scholar]
- 73.Tsutsumi Y, Onda M, Nagata S, Lee B, Kreitman RJ, Pastan I. Site-specific chemical modification with polyethylene glycol of recombinant immuntoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. 2000;97:8548–8553. doi: 10.1073/pnas.140210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Long DL, Doherty DH, Eisenberg SP, Smith DJ, Rosendahl MS, Christensen KR, Edwards DP, Chlipala EA, Cox GN. Design of homogeneous, monopegylated erythropoietin analogs with preserved in vitro bioactivity. Exp Hematol. 2006;34:697–704. doi: 10.1016/j.exphem.2006.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;v251:1451(1455). doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 76.Nose A, Tsuji K, Takeichi M. Localization of specificity determining sites in cadherin cell adhesion molecules. Cell. 1990;61:147–155. doi: 10.1016/0092-8674(90)90222-z. [DOI] [PubMed] [Google Scholar]