

Abstract
Activation of the RNA-dependent protein kinase (PKR) has been implicated in the pathogenesis of several neurodegenerative diseases. We find that a compound widely used as a pharmacological inhibitor of this enzyme, referred to as PKR inhibitor (PKRi), {8-(imidazol-4-ylmethylene)-6H-azolidino[5,4-g]benzothiazol-7-one}, protects against the death of cultured cerebellar granule and cortical neurons. PKRi also prevents striatal neurodegeneration and improves behavioral outcomes in a chemically-induced mouse model of Huntington’s disease. Surprisingly, PKRi fails to block the phosphorylation of eIF2α, a downstream target of PKR, and does not reduce the autophosphorylation of PKR enzyme immunoprecipitated from neurons. Furthermore, neurons lacking PKR are fully protected from apoptosis by PKRi demonstrating that neuroprotection by this compound is not mediated by PKR inhibition. Using in vitro kinase assays we investigated whether PKRi affects any other protein kinase. These analyses demonstrated that PKRi has no major inhibitory effect on pro-apoptotic kinases such as the c-Jun N-terminal kinases (JNKs), the p38 MAP kinases and the death-associated protein kinases (DAPKs), or on other kinases including c-Raf, MEK1, MKK6 and MKK7. PKRi does, however, inhibit the activity of certain cyclin-dependent kinases (CDKs) including CDK2 and CDK5 both in vitro and in LK-treated neurons. Consistent with its inhibitory action on mitotic CDKs, the treatment of HT-22 and HEK293T cell lines with PKRi sharply reduces the rate of cell cycle progression. Taken together with the established role of CDK activation in the promotion of neurodegeneration, our results suggest that PKRi exerts its neuroprotective action by inhibiting cyclin-dependent kinases.
Keywords: PKR, eIF2α, Apoptosis, Cerebellar granule neuron, Neuroprotection
Introduction
Neurodegenerative diseases place a tremendous burden on patients and health care resources. A characteristic feature of a variety of different neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD) and prion disease, is protein misfolding and aggregation (reviewed in Kopito, 2000; Hol et al., 2006; Lansbury & Lashuel, 2006). It has been hypothesized that a contributing factor in the formation of the abnormal aggregates in neurodegenerative disorders is sustained endoplasmic reticulum (ER) stress and ER dysfunction (reviewed in Forman et al., 2003; Shastry, 2003; Lindholm et al., 2006; Zhang & Kaufman, 2006). Among the molecules activated by ER stress is the double-stranded RNA-dependent protein kinase (PKR), a kinase that was initially discovered as a key mediator in the cellular response to viral infection. Binding of double-stranded viral RNA to PKR causes its autophosphorylation, an essential step in its activation. A target of PKR is eukaryotic initiation factor 2 (eIF2α), and the phosphorylation of eIF2α leads to the inhibition of protein synthesis. In addition to its anti-viral action, however, PKR is known to regulate other cellular processes such as differentiation, growth, and apoptosis (Chong et al., 1992; Meurs et al., 1993; Lee & Esteban, 1994; Salzberg et al., 2000; Kuyama et al., 2003; Morimoto et al., 2005; Garcia et al., 2007).
A number of recent studies have implicated PKR in the pathogenesis of neurodegenerative diseases. For example, PKR activity is elevated in the brains of patients with AD, PD, HD and ALS (Peel et al., 2001; Chang et al., 2002; Hu et al., 2003; Onuki et al., 2004; Bando et al., 2005). Other laboratories have reported that primary cortical neurons from PKR knockout mice are less susceptible to β-amyloid (Aβ) toxicity, whereas neurons and neuroblastoma cell lines overexpressing a dominant-negative form of PKR display reduced cell death in response to Aβ treatment (Chang et al., 2002).
Recently a small-molecule chemical inhibitor of PKR has become commercially available. In vitro experiments conducted by Jammi et al. (2003) revealed that this compound, {8-(imidazol-4-ylmethylene)-6H-azolidino [5, 4,-g] benzothiazol-7-one}, designated as PKR inhibitor (PKRi), acts in an ATP-competitive manner to block autophosphorylation of PKR and PKR-dependent translation in rabbit reticulocyte lysates. We reported that PKRi prevented the death of cultured cerebellar granule neurons induced by potassium deprivation (Chen et al., 2008). Similar protection was observed by Shimazawa and Hara (2006) in the neuroblastoma SH-SY5Y cell-line following treatment with tunicamycin, an agent that induces ER stress, and more recently by Wang et al. (2007) in cerebellar granule neurons induced to die by treatment with amprolium, a compound that causes thiamine deficiency. These results raise the exciting possibilities that chemical inhibitors of PKR, such as PKRi, may have therapeutic value in the treatment of neurodegenerative diseases.
In this report we analyzed the neuroprotective effects of PKRi in more detail. We find that besides protecting cultured cortical and cerebellar granule neurons from apoptosis, administration of PKRi prevents neurodegeneration and improves behavioral outcome in a chemically-induced mouse model of HD. Surprisingly, neuroprotection by PKRi is not mediated by the inhibition of PKR. Instead, PKRi is a potent inhibitor of cyclin-dependent kinases (CDKs) and glycogen synthase kinase (GSK) in vitro, two kinases that promote neuronal death in a variety of in vitro and in vivo paradigms of neurodegeneration (reviewed in D’Mello & Chin, 2005). Our results indicate that PKRi protects neurons by suppressing the activity of specific cyclin-dependent kinases.
MATERIALS AND METHODS
Materials
All cell culture media and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA, USA). Unless indicated otherwise, all other chemicals were from Sigma-Aldrich (St. Louis, MO, USA). PKRi was purchased from Calbiochem (La Jolla, CA, USA). Antibodies used in this paper were as followed: anti-Phospho-eIF2α (9721S) and anti-active caspase 3 (9661S) were from Cell Signaling Technology (Beverly, MA, USA); anti-PKR(B-10, sc-6282), anti-ATF-3(C-19, sc-188), anti-cyclin A(J-3, sc-6247), anti-CDK5(C-19, sc-596) and anti-CDK2(D-12) (sc-6248) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-Tubulin (T5326) and anti-Brdu (B8434) were from Sigma-Aldrich (St. Louis, MO, USA); Ki67 (RM-9106) was from Lab Vision Corporation (Fremont, CA, USA). Fluorescence conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories, Inc (West Grove, PA, USA). Radioactive materials were from MP Biomedicals (Solon, OH, USA) including [γ-32P] ATP and [32P] orthophosphate.
Cell culture
Animals used in this paper were treated in accordance with the Guidelines of NIH. Cerebellar granule neurons were cultured from 7-day-old Wistar rats which were treated in accordance to the Guidelines of NIH, as described by D’Mello et al (1993) in Basal Minimal Eagle (BME) medium containing 10% FBS, 25mM KCl, 2μM glutamine and 0.2% gentamycin and plated on poly-L-lysine coated dishes (1 X 106 cells/well in 24-well dish and 12 X 106 cells/dish in 60mm dishes). 18–22 hours after plating, arabinofuranosylcytosine (AraC) (10 μM) was added to the culture medium to prevent proliferation of non-neuronal cells. Cortical neurons were cultured from neocortex of embryonic day 17 (E17) Wistar rat embryos (Murphy et al., 1990). Dissociated neurons were cultured in Minimum Essential Medium (MEM) containing GLUTAMAX and supplemented 10% FBS at densities of 8 X 105 cells/well in 24-well dish and 4X 106 cells/dish in 35mm dishes on poly-L-lysine coated dishes. The HT-22 and 293T cell lines were purchased from ATCC (Manassas, VA, USA). HT-22 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/L glucose (without sodium pyruvate) supplemented with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin. 293T cells were cultured with complete DMEM supplemented with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin.
Treatment of neuronal cultures
The treatment of CGNs was performed 6–7 days after plating. Apoptosis was induced by switching the cultures to LK medium (BME containing glutamine and gentamycin with 5mM KCl). PKRi was added at the same time the neuronal medium was switched. Control CGNs were treated in HK medium (BME containing glutamine and gentamycin with 25mM KCl). Viability was quantified 24 hours after treatments as described previously. Apoptosis of cortical neurons was induced one day after plating using 1mM HCA (pH7.5) or 2–3 days after plating using 5μM camptothecin (CPT). PKRi was also added at the same time with either HCA or CPT treatment. Viability was quantified 24 hours after HCA treatment or 6 hours after CPT treatment.
Evaluation of cell viability
Viability of neurons was quantified by diamidino-2-phenylindole hydrochloride (DAPI) staining, which permits the visualization of condensed or fragmented nuclei as previously described (D’Mello et al., 1993). Briefly, cultured neurons were fixed using 4% paraformaldehyde for 15 minutes on ice. Neurons were washed once with phosphate buffered saline (PBS, pH 7.4) after fixation and stained with DAPI (0.1μg/ml in PBS) for 10 minutes. Neurons with nuclei condensation or fragmentation were scored as apoptotic cells.
The viability of HT-22 cells was evaluated by propidium iodide (PI) staining, the Terminal Uridine deoxynucleotidyl transferase dUTP Nick End Labeling (TUNEL) assay and active caspase-3 immunocytochemistry. PI and TUNEL staining were performed 15 hours after PKRi treatment. 1μg/ml PI was added to culture 30 minutes before fixation of the cells with 4% paraformaldehyde. TUNEL assay was performed using DeadEnd™ Fluorometric TUNEL System from Promega (Madison, WI, USA) according to the manufacturer’s instructions. Active caspase 3 immunocytochemistry (1: 200 dilution) was performed 6 hours after PKRi treatment. Cells were fixed and treated with 0.2% Triton for 5 minutes and then blocked with 5% BSA and 5% goat serum in PBS for 30 minutes. The primary antibodies were incubated with the cells overnight at 4°C. After three washes with phosphate-buffered saline (PBS), the cells were incubated with secondary antibodies for 45 minutes at 25°C after which the cells were washed with PBS. To visualize nuclei, cells were stained with DAPI for 15 minutes at 25°C. Cell number of HT22 cells was also quantified using MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay. Briefly, HT-22 cells were plated into 24-well tissue culture dishes at 3X104 cells per well one day before PKRi treatment. After PKRi treatment, cell number was determined 24 hours later by the MTT assay as previously described (Koulich et al., 2001).
Genotyping of PKR−/− mice and culturing of neurons
PKR−/− mice were provided by Dr. John C. Bell from the University of Ottawa, Ottawa, Canada. As previously described (Abraham et al., 1999), the mice are fertile and display no obvious phenotype. Mutant mice were bred and litters genotyped. Cerebellar granule neurons were cultured from the mutant mice as described above for rat cultures.
3-nitropropionic acid (3-NP) injection and behavior test
3-NP administration and analysis of brain sections were performed as previously described (Chin et al., 2004). Briefly, 3-NP was injected to mice intraperioneally at 50mg/kg twice a day and 0.27 mg/kg PKRi once a day for 4 days. After finishing the injection, we performed the behavioral test by using Tru-Scan® activity monitoring system (Coulborn Instruments, PA, USA). The following behavioral parameters were selected: (i) Total movements time (ii) Total movement distance (iii) Average movement per distance and (iv) Mean velocity. Next, the mice were deeply anesthetized. Then, the brains were removed, washed in PBS and fixed in 4% paraformaldehyde overnight. Next day, they were transferred to 20% sucrose over night and rapidly frozen in Cryo-Stat embedding medium (StatLab Medical Products, Lewisville, TX, USA). 40 micron coronal sections were cut on a cryostat and stained with cresyl violet to visulize the brain architecture (Chin et al., 2004).
Protein preparation and Western blot analysis
Cells were lysed with cell lysis buffer (Cell Signaling Technology, Beverly, MA, USA) and proteins were collected by centrifugation at 14,000 rpm for 10 minutes. After the concentration of protein was measured by Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA), protein from each sample was normalized and boiled with 6X SDS loading buffer [375 mM Tris-HCl (pH 6.8 at 25°C), 12% SDS, 60% glycerol, 300 mM dithiothreitol (DTT), 0.012% bromophenol blue] at 95°C for 5 minutes. Next, the proteins were separated by SDS–polyacrylamide gel electrophoresis (PAGE) and electronically transferred to (polyvinylidene difluoride) PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA). After Ponceau S staining to confirm equal protein transfer, the membrane was blocked with blocking buffer [5% milk and 0.1% Tween in Tris-Buffered Saline (TBS)]. The PVDF membrane was then probed with primary antibody diluted in either blocking buffer or bovine serum albumin (BSA) buffer (1% BSA and 0.1% Tween in TBS) at 4°C overnight. The antibody dilutions were 1:1,000, except tubulin which was used at 1:10,000). After 3 washes with TBS-T (0.05% Tween in TBS), the PVDF membrane was probed with horseradish peroxidase-conjugated secondary antibodies with dilution 1:10,000 at 25°C for 1 hour. Western Blot detection was enhanced via chemiluminescence (ECL) kit from GE Health Care Life Science (Piscataway, NJ, USA).
32P-metabolic labeling on endogenous PKR
60mm dishes of 7-day-old neurons were washed twice with warm, phosphate-free BME and incubated in phosphate-free BME containing 25 mM KCl for 4 hours. Next, the cultures were then incubated for 3 hours in HK, LK or LK plus PKRi media containing 250μCi/ml [32P] orthophosphate. After being lysed in ice-cold RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 0.1% SDS, 1 mM Na3VO4,50 mM NaF, 30 mM β-glycerophosphate, 1 mM EDTA, protease inhibitors mixture), the lysates were subjected to immunoprecipitation by using PKR antibody (5 ul) and the products of immunoprecipitation were resolved by SDS–PAGE and transferred electrophoretically to PVDF membrane. After the transfer, labeled proteins were visualized by autoradiography using a Storm860 scanner (Amersham Biosciences, Piscataway, NJ, USA). Data were quantified using ImageQuant software (Amersham Biosciences, Piscataway, NJ, USA) (Liu & D’Mello, 2006).
Kinase profiling
Kinase profiling was performed using the KinaseProfiler Service from Millipore (Billerica, MA, USA) on a fee for service basis. In short, 5–10mU of purified kinase was used along with an appropriate quantity of synthetic substrate in buffer containing optimal amount of [γ-32P] ATP for each kinase with or without 100 nM PKRi. Next the reaction mix was incubated at room temperature for 40 minutes. Then, it was stopped using a 3% phosphoric solution, spotted, washed and dried for scintillation counting.
Immunoprecipitation and CDK kinase assay
Whole cell lysates from HT-22 cells or neurons were incubated with 5 μl of primary CDK2 or CDK5 antibody and 20 μl of Protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight. Immunoprecipitates were collected by centrifugation at 6000 rpm for 30 seconds and washed twice with cell lysis buffer and twice with kinase buffer (40 mM Tris-HCl pH 7.5, 8 mM MgCl2, 50 mMβ-glycerol phosphate, 1 mM DTT). Histone H1 (Millipore, Billerica, MA, USA) was added as a substrate in kinase buffer supplemented with 50 μM ATP and [γ-32P] ATP for 30 minutes at 30°C. The kinase reactions were stopped by addition of 6× SDS sample buffer and boiled at 95°C for 5 minutes. Proteins were resolved by SDS–PAGE and transferred electrophoretically to PVDF membrane. Later, PVDF membrane was subjected to autoradiography. Histone H1 was detected by using Ponceau S staining to confirm equal amount of substrate loaded.
Evaluation of cell proliferation by BrdU and Ki67 staining
3X104 HT-22 cells and 5 X104 293T cells per well were plated one day before PKRi treatment. For BrdU assay, 20μM BrdU was added 24 hours after PKRi treatment. After BrdU incorporation with cells for 2 hours, HT-22 cells were fixed with 4% paraformaldehyde in PBS and washed 3 times using 0.1M PBS containing 0.5% Triton. Then, the cells were incubated with 1N HCl for 10 minutes on ice, 2N HCl for 30 minutes at 37°C, 0.1 M borate buffer (PH 8.5) for 12 minutes at room temperature, and blocked with 0.1M PBS containing 1% BSA and 5% goat serum for 15 minutes. Immunostaining was performed by incubation with a monoclonal Brdu antibody (1:200 dilution) overnight at 4°C. The secondary antibody was incubated for 45 minutes at 25°C. Cell nuclei were stained with DAPI for 15 minutes at 25°C. For Ki67 staining, immunocytochemistry was performed as described above for active caspase 3 staining. The Ki67 antibody was used at 1:200 dilution.
RESULTS
PKRi is neuroprotective in primary cultured neurons
Primary cultures of cerebellar granule neurons undergo apoptosis when switched from medium containing an elevated level of potassium (HK) to medium containing non-depolarizing levels of potassium (LK) (D’Mello et al., 1993). Treatment with PKRi completely prevents neuronal apoptosis in this paradigm (Chen et al., 2008; also Fig. 1). To examine if this neuroprotection is restricted to cerebellar granule neurons and to LK-induced apoptosis, we evaluated the efficacy of this compound in primary cortical cultures induced to die by treatment with homocysteic acid (HCA), a stimulus that causes oxidative stress (Murphy et al., 1990; Ratan et al., 1994a; 1994b). As shown in Fig. 2A & 2C, substantial neuroprotection was also observed in this paradigm. A similar protection against HCA-induced cell death was observed in the hippocampally derived HT-22 cell line (Supplemental Fig. 1). Camptothecin (CPT) is a DNA damaging agent which induces apoptosis in cultured cortical neurons (Morris & Geller, 1996). As shown in Fig. 2B & 2C, PKRi is protective in this paradigm as well. Thus, PKRi is an effective and versatile neuroprotective agent in tissue culture paradigms.
Figure 1. PKRi is neuroprotective in cerebellar granule neurons.

6 - 7 days after plating, cerebellar granule neurons were switched to HK (control), LK or LK medium containing 10μM PKRi. Viability was quantified by DAPI staining after 24 hours treatments. Percentage of survival was normalized to the control.
(A) Data shown above represent the mean values of viability with their corresponding standard deviations from three independent experiments. * indicates statistical significance between LK and LK+PKRi values (p-value < 0.05). Statistical analysis was performed using One-way ANOVA followed by Bonferroni’s Multiple Comparison Test.
(B) Top row: the cell morphology of cerebellar granule neurons was observed by phase contrast microscope 24 hours after the treatments of HK, LK or LK medium containing 10 μM PKRi. Bottom row: DAPI stained nuclei were observed under fluorescence microscope. Neurons displaying nuclear condensation or fragmentation were quantified as apoptotic.
Figure 2. PKRi is neuroprotective in cortical neurons.

(A) Cultured cortical neurons were untreated (Un, control), treated with 1mM HCA or 1mM HCA containing 10μM PKRi. Viability was quantified by DAPI staining after 24 hours treatment. Percentage of survival was normalized to the untreated control. Data shown above represent the mean values of viability with their corresponding standard deviations from three independent experiments. * indicates statistical significance between HCA and HCA+PKRi values (p-value < 0.05). Statistical analysis was performed using One-way ANOVA followed by Bonferroni’s Multiple Comparison Test.
(B) Cultured cortical neurons were untreated (Un, control), treated with 5μM CPT or 5μM CPT containing 10μM PKRi. Viability was quantified by DAPI staining 6 hours after treatment. Percentage of survival rate was normalized to the control. Data shown above represent the mean values of viability with their corresponding standard deviations from three independent experiments. * indicates statistical significance between CPT and CPT+PKRi values (p-value < 0.05). Statistical analysis was performed using One-way ANOVA followed by Bonferroni’s Multiple Comparison Test.
(C) The morphology of nuclei in cortical neuron cultures was examined 24 hours after HCA treatments or 6 hours after CPT treatments. The proportion of cells with condensed or fragmented nuclei was quantified as apoptosis.
PKRi protects against neurodegeneration and improves behavioral outcome in a mouse model of Huntington’s disease
Administration of mice with 3-nitropropionic acid (3-NP), an irreversible inhibitor of succinate dehydrogenase, results in a Huntington’s disease-like pathology which includes selective striatal neurodegeneration and movement deficits (Beal, 1993; Brouillet et al., 1999). We examined whether PKRi was effective in this in vivo disease model. As shown in Fig. 3A, administration of 3-NP causes a substantial amount of cell loss in the striatum as evidenced by the lack of cresyl violet staining. Co-administration of PKRi at 0.27 mg/kg of body weight protected against 3-NP-induced striatal degeneration. Moreover, mice that were co-administered with 3-NP and PKRi also displayed significantly better locomotor performance as compared with mice receiving 3-NP alone (Fig. 3B).
Figure 3. PKRi prevents striatal lesion and locomotion dysfunction mediated by 3-NP.

(A)Mice were injected with 3-NP, 3-NP + PKRi or saline (Control) twice a day for 4 days. After the last day of injection, whole brains removed and 40 μm coronal sections were stained with cresyl violet. 3-NP injected animals display selective striatal degeneration as evidenced by reduced cresyl violet staining. The extent of 3-NP-induced degeneration is reduced with PKRi.
(B) 3-NP treated mice present significant locomotion dyfunciton, including total movement time, total movement distance, average distance per movement and mean velocity. PKRi injection improves these locomotor activities of mice comparing with 3-NP injection alone. Data shown above represent the mean values of movements with their corresponding standard deviations from seven animals. * indicates statistical significance between 3-NP and 3-NP+PKRi values (p-value < 0.05). Statistical analysis was performed using One-way ANOVA followed by Bonferroni’s Multiple Comparison Test.
PKRi does not inhibit PKR in neuronal cultures
A downstream substrate of PKR is eukaryotic initiation factor eIF2α (Clemens & Elia, 1997). As shown in Fig. 4A, treatment of cerebellar granule neurons with LK resulted in a small increase in eIF2α phosphorylation compared to HK-treated cultures. Unexpectedly, the addition of PKRi to LK-treated cultures does not reduce the increase in eIF2α phosphorylation (Fig. 4A). Given that three other kinases PERK, HRI, and GCN2, can also phosphorylate eIF2α, it is possible the increased phosphorylation in LK is not mediated by PKR, but by one of the other kinases. Arguing against such a possibility, however, is our observation that 2-aminopurine (2-AP), another selective inhibitor of PKR, has been used in several in vitro and in vivo studies (Hu & Conway, 1993; Cheshire et al., 1999; Ruvolo et al., 2001; Ben-Asouli et al., 2002), does inhibit the LK-mediated increase in eIF2α phosphorylation (Fig. 4A).
Figure 4. PKRi does not inhibit PKR efficiently in neurons.

(A) Cerebellar granule neurons were treated with HK, LK, LK medium containing 10mM 2-AP or 10μM PKRi for 3 hours. Lysates from these cultures were subjected to Western blot using a phospho-eIF2α antibody. The same membrane was re-probed with tubulin to ensure equal loading.
(B) Cortical neurons were treated with 1mM HCA or 1mM HCA plus 10μM PKRi for 6 hours and 5μM CPT or, 5μM CPT plus 10μM PKRi for 4 hours. Untreated (Un) and treated lysates were made and subjected to Western blot. eIF2α phosphorylation and ATF-3 expression were determined using phospho-eIF2α and ATF-3 antibodies. The same membrane was reprobed with tubulin to show equal loading.
(C) Cerebellar granule neuron cultures metabolically labeled with [32P] orthophosphate in HK, LK or LK medium containing 10μM PKRi. Lysates were made and PKR was immunoprecipitated. The autophosphorylation of PKR was determined by autoradiography after SDS-PAGE and membrane transferring. The same membrane was also probed with PKR antibody to show equal amount of PKR pull down. Kinase activity was quantified by densitometric analysis of the bands and normalized to HK. The histograms represent the mean of kinase activity with standard deviations from 3 independent experiments. Statistical analysis was performed using one-way ANOVA followed by Bonferroni’s multiple comparison test.
We extended our studies to cortical neurons treated with either homocysteic acid or camptothecin. Although potent inducers of apoptosis, neither homocysteic acid nor camptothecin treatment increases the extent of eIF2α phosphorylation (Fig. 4B). Co-treatment with PKRi actually increases the extent of eIF2α phosphorylation (Fig. 4B), despite its reported inhibitory effect on purified PKR enzyme in vitro (Jammi et al., 2003). The lack of an inhibitory effect on eIF2α phosphorylation in both cerebellar granule neurons and cortical neurons suggests that PKRi is incapable of inhibiting PKR in cultured cells (Fig. 4B).
Because eIF2α phosphorylation is only an indirect indicator of PKR activation, we performed a more direct assay. Activation of PKR requires its autophosphorylation and this phosphorylation of PKR has been used to evaluate its activation (Lasky et al., 1982; Galabru & Hovanessian, 1987). Using 32P-metabolic labeling and immunoprecipitation, we find that PKR autophosphorylation increases in LK-treated cultures (Fig. 4C) consistent with the pattern of eIF2α phosphorylation. Again, PKRi fails to inhibit the increase in PKR autophosphorylation.
Neuroprotection by PKRi does not involve PKR
Biochemical analysis failed to detect an inhibitory effect of PKRi on the activity of PKR, but the possibility our compound protects neurons by blocking some aspect of PKR function could not be completely excluded. To investigate this issue we cultured neurons from mice lacking PKR. Genotyping and expression analysis confirmed mice from which the neuronal cultures were generated lacked a functional PKR gene (Fig. 5A & 5B). As shown in Fig. 5C, LK-treatment of neurons from PKR−/− mice induces apoptosis demonstrating that PKR is not necessary for LK-induced neuronal death. More importantly, treatment with PKRi completely protects PKR−/− neurons from LK-induced death. This finding demonstrates PKRi acts through a PKR-independent mechanism and points to a molecule other than PKR as the target of its neuroprotective action.
Figure 5. PKRi protects neurons lacking PKR from LK-induced apoptosis.

(A) Genotyping of PKR−/− and wild-type PKR+/+ mice. Prior to culturing of neurons, the genotypes of the mice were confirmed by PCR.
(B) Western blot analysis of cerebellar granule neurons cultured from 7-day old PKR−/−and wild-type (PKR+/+) mice confirm absence of PKR in the cultures from knockout mice. The same membrane was re-probed with tubulin to show equal loading.
(C) Complete protection by PKRi in PKR−/− neuronal cultures. 7-day old cerebellar granule neurons cultured from mice were treated with HK (Control), LK, or LK medium containing 10 uM PKRi. Cell viability was quantified 24 hours later. Data shown above represent the mean values of viability with their corresponding standard deviations from three independent experiments. * indicates statistical significance between LK and LK + PKRi values (p-value < 0.05). Statistical analysis was performed using one-way ANOVA followed by Bonferroni’s multiple comparison Test. Upper panel: Phase contrast micrograph and fluorescence micrograph of neurons treated with HK, LK, or LK + PKRi (10 uM). Lower panel: Quantification of neuronal viability by DAPI-staining. The data represents the mean values of viability with standard deviation.
Kinase profile of PKRi
PKRi is a 3′ substituted indolone. Crystallographic studies have demonstrated that 3′ substituted indolones are ATP-competitive inhibitors of protein kinases with the selectivity towards a specific kinase depending on the substituents on the indolone core, especially at the C-3 position (Mohammadi et al., 1997; Sun et al., 1998; Sun et al., 2000; Davis et al., 2001). Since PKRi does not activate or act through PKR, its neuroprotective effect likely involves the inhibition of another kinase. It is noteworthy that although Jammi et al. (2003) demonstrated PKRi could inhibit PKR activity in vitro, its selectivity against other kinases was not tested in that study or in any other study since. To investigate this possibility we evaluated the effect of PKRi on different pro-apoptotic kinases including the c-Jun N-terminal kinases (JNKs), stress-activated protein kinases (SAPKs or p38 MAP kinases), GSK3 kinase, and death-associated protein kinases (DAPKs) was evaluated. The effect of PKRi on select CDKs was also examined because inappropriate activation of cyclin-dependent protein kinases (CDKs) has also been shown to induce apoptosis in a variety of neuronal systems (reviewed in Copani et al., 2007; Greene et al., 2007),. As shown in Fig. 6, PKRi highly inhibits CDK1, CDK2 and CDK5. Significant inhibition of GSK3α and GSK3β was also observed. In contrast, the drug inhibited JNK3, CDK6 and MKK6 less effectively and failed to inhibit other pro-apoptotic kinases included in our analysis. Likewise, PKRi had no inhibitory effect on a number of other kinases including c-Raf, MEK1 and MKK7 in these in vitro assays. These results suggest that the neuroprotective action of PKRi could be mediated by inhibition of either GSK3 or cyclin-dependent kinases. Consistent with this notion are reports from a number of different laboratories demonstrating that pharmacological inhibition of either of these kinases is protective against neurodegeneration both in cell culture paradigms as well as in vivo models of neurodegeneration (reviewed in Bhat et al., 2004; D’Mello & Chin, 2005; Copani et al., 2007; Greene et al., 2007).
Figure 6. Kinase profile of PKRi.

The kinase activity of each kinase was performed in vitro with 100nM PKRi. The optimal concentration of ATP was used for each kinase. The data shows the percentage of relative kinase activities compared with control (same kinase but without PKRi treatment). The histogram represents the mean of relative kinase activity with standard deviation. * indicates the p-value < 0.05 and ** indicates p-value< 0.01 using Student’s t-test comparing with untreated kinase reactions.
PKRi is a CDK inhibitor
We and other laboratories have shown that neuronal apoptosis is accompanied by an activation of GSK3α and GSK3β (Hetman et al., 2000; Chen et al., 2004; Chin et al., 2005; Enguita et al., 2005). The activation of GSK3β requires dephosphorylation of its serine residues 9 and 21 (and in the case of GSK3α at homologous residues). As observed previously (Chin et al., 2005; Johnson et al., 2005; Shimazawa et al., 2007; Wang et al., 2007), treatment with LK leads to a reduction in GSK3α/β phosphorylation reflective of the increase in its kinase activity. Not only does PKRi not inhibit the LK-induced reduction of GSK3α/β phosphorylation, but the drug reduces the extent of GSK3α/β phosphorylation even more than seen with LK alone (Fig. 7). The inability of PKRi to inhibit GSK3 activity in neurons indicates that GSK3α/β inhibition is not the mechanism by which PKRi protects neurons.
Figure 7. PKRi treatment activates GSK3 in neurons.
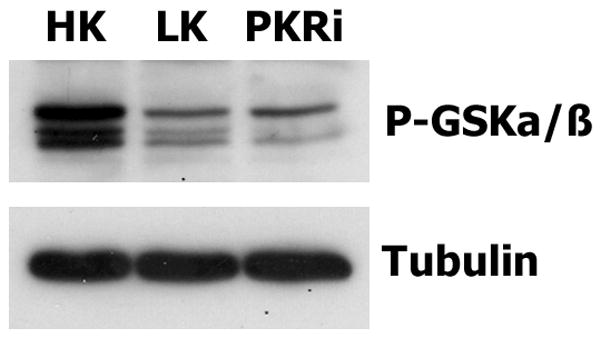
Cerebellar granule neurons were treated with HK, LK or LK medium containing 10μM PKRi for 3 hours and lysates from these cultures subjected to Western blotting analysis using a phospho-GSK3α/β antibody. The same membrane was re-probed with tubulin to ensure equal loading.
We next focused on CDK2 because it was the most potently inhibited of the mitotic CDKs. The ability of PKRi to inhibit the activity of the CDK2 complex immunoprecipitated from cerebellar granule neurons was tested in an in vitro kinase assay. As shown in Fig. 8A, the addition of PKRi to immunoprecipitated neuronal CDK2 inhibits its activity. To examine whether PKRi could inhibit CDK2 activity in intact neurons, we added PKRi to neuronal cultures and evaluated the effect on kinase activity after immunoprecipitation of the enzyme from the cultures. As shown in Fig. 8B, CDK2 activity is slightly elevated in LK-treated neuronal cultures as compared with cultures treated with HK. When added to LK medium, PKRi inhibits CDK2 activity to a level that is even lower than that observed in HK.
Figure 8. PKRi inhibits CDK2 in neurons.

CDK2 was immunoprecipitated from cerebellar granule neurons that were treated as described below. Kinase activity was assayed in an in vitro reaction containing [γ-32P] ATP and using Histone H1 as substrate. Relative activity was visualized by autoradiography. Ponceau S staining of Histone H1 shows that comparable amounts of substrate were used in each reaction. An aliquot of the lysates prior to immunoprecipitation was also analyzed by Western blotting using tubulin antibody to demonstrate that similar quantities of lysate was used for pull-down.
(A) Cerebellar granule neurons were treated with LK for 9 hours. CDK2 was immunoprecipitated and an in vitro kinase assay performed in the presence (+) or absence (−) of 1 μM PKRi.
(B) Cerebellar granule neurons were treated with HK, LK or LK containing 10μM PKRi for 9 hours. Lysates were made and CDK2 was immunoprecipitated and used in an in vitro kinase assay.
To confirm these results in an independent model system, we used HT-22 cells, which as we have described above (Supplemental Figure 1), are protected by PKRi against HCA-toxicity. As shown in Fig. 9A, treatment of this neuroblastoma cell line with PKRi reduces the activity of CDK2. The reduced activity is not because of a decrease in the expression of CDK2 or of associated cyclin A (Fig. 9B & 9C).
Figure 9. PKRi inhibit CDK2 kinase activity in HT-22 cells.

(A) HT-22 cells were untreated (Un) or treated with 10μM PKRi for 3 hours. Lysates were made and CDK2 kinase assay was performed with the Histone H1 as the substrate. The kinase activity was determined by autoradiography. Histone H1 from Ponceau S staining to demonstrate that comparable amounts of substrate were used in each reaction. An aliquot of the lysates prior to immunoprecipitation was also analyzed by Western blotting using tubulin antibody to demonstrate that similar quantities of the lysates were used for pull-down.
(B) 3h untreated (Un) or treated 10μM PKRi whole cell lysates were harvested from HT-22 cells. Western blot analysis was performed using cyclin A antibody. The same membrane was re-probed with CDK2. Expression of CDK2 was not changed by PKRi. Western blot using tubulin antibody shows equal amount of protein loading.
(C) HT-22 cells were untreated (Un) or treated with 10μ PKRi for 3 hours. Cyclin A was co-immunoprecipitated from these lysates using CDK2 antibody. IgG was used as control (Ctrl). Western blot was performed using Cyclin A antibody to show the amounts of cyclin A pulled down. The same membrane was re-probed with CDK2 to show equal amounts of CDK2 were immunoprecipitated. Western blot using tubulin antibody show equal amount of input protein was administrated before immunoprecipitation.
Our results demonstrate that PKRi inhibits CDK2 activity but do not establish that this action effectively inhibits cell cycle progression. To investigate this issue, we examined the effect of PKRi on the proliferation of HT-22 cells. Treatment of HT-22 cultures with PKRi results in a decrease in total cell number when examined 24 hours after addition of the drug (Fig. 10A). To rule out the possibility that the reduced cell number is due to toxicity we performed propidium-iodide staining. As shown in Fig. 10B (left panel), no increase in propidium iodide staining was observed in the presence of PKRi. We used TUNEL-staining (Fig. 10B, middle panel) and active-caspase-3 immunocytochemistry to detect cell death (Fig. 10B, right panel). No increase in the extent of cell death was observed in the presence of PKRi. The results of the three cell death assays eliminate toxicity as being responsible for the reduced cell numbers.
Figure 10. Reduced HT-22 cell proliferation by PKRi is not associated with drug toxicity.

(A) HT-22 cells were treated with PKRi at the indicated doses and the MTT assay utilized to evaluate the number of cells in the dish. Data was normalized to untreated cells (Control). Data shown above represent the mean values of proliferation rates with their corresponding standard deviations from three independent experiments. * indicates statistical significance between control and PKRi treatment values (p-value < 0.05). Statistical analysis was performed using one-way ANOVA followed by Bonferroni’s multiple comparison test.
(B) HT-22 cells were untreated (Un), treated with 10μM PKRi or 1mM HCA. HCA treatment was used as a positive control for cell death. Left: PI staining was performed to detect cell death 15 hours after treatments. PI positive cells show red flourescence. DAPI staining allows for determination of apoptotic condensed and/or fragmented nuclei. Middle: TUNEL assay was performed 15 hours after treatments. DAPI staning provides the morphology of nuclei. Right: After 6 hours treatments, active caspase 3 immunoreactivity is shown in red. DAPI staning provides the morphology of nuclei.
To more directly examine the effects of PKRi on cell proliferation we measured BrdU-incorporation, a commonly-used assay of cell proliferation. As shown in Fig. 11A & 11B, the rate of cell cycle progression was substantially reduced in HT-22 cells treated with PKRi. Similar results were obtained with roscovitine, a well-characterized CDK inhibitor (Meijer, 1996; Rudolph et al., 1996; De Azevedo et al., 1997; Meijer et al., 1997). The inhibition of cell cycle progression by PKRi was confirmed by the almost complete absence of Ki-67 immunoreactivity (Fig. 11B). Ki-67 is a protein that is expressed during all active phases of the cell cycle and is a widely used proliferation marker (Scholzen & Gerdes, 2000). Similar results were obtained using HEK293T cells (Fig. 11B) and in the striatal N1E115 cell-line (data not shown).
Figure 11. PKRi inhibits HT-22 cell proliferation and DNA synthesis.

(A) HT-22 cells were treated with increasing concentrations of PKRi as indicated. BrdU incorporation assay was performed 24 hours after PKRi treatment to evaluate the rate of cell cycle progression. Data shown above represent the mean values and standard deviations of BrdU positive rates with standard deviations from three independent experiments. * indicates statistical significance between control and PKRi treatment values (p-value < 0.05). Statistical analysis was performed using one-way ANOVA followed by Bonferroni’s multiple comparison test.
(B) HT-22 and 293T cells were untreated (Un), treated with 10μM PKRi or with 50μM Roscovitine (Ros) for 24 hours. The BrdU or Ki67 positive cells were visualized by performing immunocytochemistry with antibodies against BrdU or Ki67. Texas Red-conjugated secondary antibody was used to detect the immunoreactivity. DAPI staining was performed to visualize the morphology of cell nuclei.
DISCUSSION
Our laboratory has previously reported that a number of different chemical compounds containing a 3′ substituted indolone core can protect neurons from apoptosis (Johnson et al., 2005). Among the 3′ indolones we tested more recently was PKRi, a compound sold as an inhibitor of PKR and used by many as such (Shimazawa & Hara, 2006; Eley et al., 2007; Eley & Tisdale, 2007; Ito et al., 2007; Russell et al., 2007; Wang et al., 2007). PKRi is impressive in its ability to prevent LK-induced apoptosis of cerebellar granule neurons (Chen et al., 2008). In contrast to many other 3′ substituted indolones which display toxicity at doses that are 5 – 10 times higher that their neuroprotective concentrations, PKRi is not toxic even when used at very high doses (Chen et al., 2008). We report that this compound can also prevent the death of primary cortical neurons caused by homocysteic acid, a compound that produces oxidative stress. Additionally, PKRi inhibits apoptosis caused by the DNA damaging agent, camptothecin, in cortical neuron cultures. Most importantly, PKRi is beneficial in a chemically-induced mouse model of Huntington’s disease. Our previous (Chen et al., 2008) and current findings along with the findings from other groups published recently (Shimazawa & Hara, 2006; Shimazawa et al., 2007; Wang et al., 2007) demonstrate that PKRi is an effective and versatile neuroprotective agent that could have value in the treatment of neurodegenerative diseases. It may be noted however, that although capable of reducing 3-NP induced striatal degeneration rather substantially, PKRi administration only partially improved locomotor activity in 3-NP treated mice. The reason for this is not clear but likely due to non-selective effects of PKRi on cells and organ systems outside the nervous system. Lethal actions of this compound when administered at high doses to rats has been reported recently (Ingrand et al., 2007). We have also noticed that when administered to adult mice at doses of 0.7 mg/kg body weight or higher, PKRi is generally lethal. Given these serious side effects, structural modifications of PKR are necessary before it can be considered for pre-clinical testing.
PKRi was identified by Jammi et al (2003) in a screen of a small library of 26 different ATP-binding site directed chemical inhibitors. The screen was performed in vitro using a kinase domain-containing fragment of human PKR expressed and purified from bacteria as a GST-fusion protein. Since its discovery, PKRi has been used in a number of studies to inhibit PKR (Shimazawa & Hara, 2006; Eley et al., 2007; Eley & Tisdale, 2007; Ito et al., 2007; Russell et al., 2007; Wang et al., 2007). A surprising finding of our study is that in cultured cerebellar and cortical neurons, PKRi does not inhibit PKR discernibly as judged by its inability to reduce eIF2α phosphorylation and PKR autophosphorylation. On the contrary, in cortical neurons PKRi treatment stimulates eIF2α phosphorylation. A similar stimulatory effect of PKRi on eIF2α phosphorylation was observed in cerebellar granule neurons and Hela cells treated with tunicamycin, an agent that induces endoplasmic reticulum stress (data not shown). To conclusively rule out the possibility that PKRi acted by inhibiting PKR, we utilized neuronal cultures generated from mice lacking PKR. We discovered that PKRi was completely effective in protecting these cultures from cell death indicating that a molecule other than PKR was the target of PKRi at least in the context of neuroprotection.
The selectivity of PKRi against kinases other than PKR has not been reported previously. Because it did not inhibit PKR in cultured neurons and because PKR was not necessary for the neuroprotective effect of PKRi, we tested whether this compound inhibited other pro-apoptotic kinases. We discovered that PKRi inhibits CDK1, CDK2, and CDK5 potently when tested against purified enzyme. We confirmed that PKR inhibits CDK2 in intact neurons and when added to CDK2 complex immunoprecipitated from neuronal cultures. Furthermore, the treatment of different cell lines with PKRi reduces their ability to proliferate. Increased expression of cyclins and activation of CDKs has been reported in a number of different paradigms in vivo and in vitro of neurodegeneration (Herrup & Busser, 1995; Yang et al., 2001). Neuroprotection through the inhibition of specific CDKs using molecular approaches as well as pharmacological agents has been amply demonstrated (D’Mello & Chin, 2005; Copani et al., 2007; Greene et al., 2007). It is therefore likely that PKRi exerts its neuroprotective action through its anti-CDK activity. Although the efficacy of PKRi can be explained entirely on the basis of its anti-proliferative action, it is possible that its inhibitory effect on other kinases also contribute to neuroprotection. For example, PKRi inhibits the non-mitotic CDK5 kinase strongly when tested in vitro. We have confirmed that the inhibition of CDK5 by PKRi also occurs in intact cerebellar granule neurons (Supplemental figure 2). Activation of CDK5 has previously been implicated in promoting neuronal death in several experimental paradigms, including cerebellar granule neurons (Jorda et al., 2005; Verdaguer et al., 2005a; Verdaguer et al., 2005b). Further studies are necessary to examine whether inhibition of CDK5 contributes to PKRi-mediated neuroprotection, and if so, to what extent.
It is well established that the activation of c-Jun is a central event in the promotion of neuronal death (Estus et al., 1994; Ham et al., 1995; Herdegen et al., 1998; Watson et al., 1998; Crocker et al., 2001). More recently we and others have reported that ATF-3 induction is also a common feature of neuronal death both in vivo and in vitro (Tsujino et al., 2000; Averill et al., 2004; Ohba et al., 2004; Holtz et al., 2006; Huang et al., 2006; Chen et al., 2008). In cerebellar granule neurons, siRNA-mediated suppression of ATF-3 has a protective effect (Tsujino et al., 2000; Averill et al., 2004; Ohba et al., 2004; Holtz et al., 2006; Huang et al., 2006; Chen et al., 2008). We find that PKRi completely inhibits the apoptosis-regulated increase of ATF-3 expression in cerebellar granule neurons as well as cortical neurons. A similar inhibition of c-Jun activation by this compound was previously reported (Chen et al., 2008). Our results suggest that the induction of c-jun and ATF-3 expression in neuronal apoptosis is connected to the activation of cell cycle components and attempted reentry into the cell cycle. Although not much is known about the relationship between ATF-3 and the cell cycle, studies using proliferating cell types have shown that c-Jun stimulates cell cycle progression by inducing expression of cyclin D, E2F, and cyclin A (Vanhara et al., 2007; Shen et al., 2008). Inhibition of the CDK inhibitor p21WAF1/CIP1 by c-Jun has also been recently reported (Kolomeichuk et al., 2008; Stepniak et al., 2008). These findings place c-Jun activation upstream of CDK activation. Our study indicates that PKRi inhibits CDK2 directly. At least in neurons, therefore, inhibition of CDK2 activity leads to the suppression of apoptosis-associated c-Jun activation. The molecular mechanism by which CDK inhibition leads to reduced c-Jun or ATF-3 expression is unclear. It is also conceivable, however, that the inhibition of c-Jun and ATF-3 by PKRi is a distinct effect that is unconnected to CDK2 inhibition.
In summary, our study provides three important conclusions - (1) PKRi is an effective and versatile neuroprotective agent, a modified form of which (given its toxicity in mice at high doses) may have value in the treatment of human neurodegenerative disorders; (2) The neuroprotective action of this compound is not mediated by the inhibition of PKR, and (3) PKRi inhibits CDKs 1, 2, and 5 and blocks proliferation of cells in culture. In view of the established efficacy of a number of other structurally diverse CDK inhibitors to prevent neuronal death, the neuroprotective effect of PKRi is likely to be due to its action against CDKs. Our findings suggest that caution must be exercised when using PKRi as an inhibitor of PKR in cultured cells and in vivo.
Supplementary Material
(A) HT-22 cells were treated with 1 mM HCA or 1 mM HCA plus 10μM PKRi. After 24-hour treatment, MTT assay was performed. Survival is represented as percentage of control cultures. Control cultures for HCA-treated cells received no HCA (untreated cultures). In the case of HCA + PKRi treated cells, the control cultures received PKRi but not HCA. The need for separate controls is necessitated by the fact that PKRi treatment stops cell proliferation and therefore cannot be directly compared to untreated cultures which continue to proliferate. Data shown represents the mean values of viability with their corresponding standard deviations from 3 independent experiments. ** indicates the p-value < 0.001 using Student’s t-test.
(B) HT-22 cells were untreated (Un), treated with 1mM HCA or 1mM HCA plus 10μM PKRi. After 15 hours of treatment, TUNEL assay was performed. DAPI staining shows the morphology of nuclei.
Cerebellar granule neurons were treated with HK, LK or LK containing 10μM PKRi for 3 hours. Lysates were made and CDK5 was immunoprecipitated to perform an in vitro kinase assay containing [γ-32P] ATP and using Histone H1 as substrate. Relative activity was visualized by autoradiography. Ponceau S staining of Histone H1 shows that comparable amounts of substrate were used in each reaction. An aliquot of the lysates prior to immunoprecipitation was also analyzed by Western blotting using tubulin antibody to demonstrate that similar quantities of lysate was used for pull-down.
Acknowledgments
This research was funded by grants from the National Institutes of Health (NS40408 and NS047201) to S.R.D. We are grateful to Dr. John C. Bell, Ottawa Health Research Institute (University of Ottawa), Canada for providing the PKR knockout mice.
Abbreviations
- 3-NP
3-nitropropionic acid
- BrdU
Bromodeoxyuridine
- CPT
Camptothecin
- CDK
Cyclin-dependent kinase
- DAPI
Diamidino-2-phenylindole hydrochloride
- ER
Endoplasmic reticulum
- eIF2α
Eukaryotic initiation factor 2
- GSK
Glyocogen synthase kinase
- HCA
Homocysteic acid
- HK
High potassium
- LK
Low potassium
- PKR
Double-stranded RNA-dependent protein kinase
- PKRi
PKR inhibitor
- PI
Propidium iodide
- TUNEL
Terminal uridine deoxynucleotidyl transferase dUTP nick end labeling
References
- Abraham N, Stojdl DF, Duncan PI, Methot N, Ishii T, Dube M, Vanderhyden BC, Atkins HL, Gray DA, McBurney MW, Koromilas AE, Brown EG, Sonenberg N, Bell JC. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J Biol Chem. 1999;274:5953–5962. doi: 10.1074/jbc.274.9.5953. [DOI] [PubMed] [Google Scholar]
- Averill S, Michael GJ, Shortland PJ, Leavesley RC, King VR, Bradbury EJ, McMahon SB, Priestley JV. NGF and GDNF ameliorate the increase in ATF3 expression which occurs in dorsal root ganglion cells in response to peripheral nerve injury. Eur J Neurosci. 2004;19:1437–1445. doi: 10.1111/j.1460-9568.2004.03241.x. [DOI] [PubMed] [Google Scholar]
- Bando Y, Onuki R, Katayama T, Manabe T, Kudo T, Taira K, Tohyama M. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson’s disease and Huntington’s disease. Neurochem Int. 2005;46:11–18. doi: 10.1016/j.neuint.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Beal JA. Some risk management considerations. Implant Soc. 1993;3(7):9. [PubMed] [Google Scholar]
- Ben-Asouli Y, Banai Y, Pel-Or Y, Shir A, Kaempfer R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell. 2002;108:221–232. doi: 10.1016/s0092-8674(02)00616-5. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem. 2004;89:1313–1317. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Conde F, Beal MF, Hantraye P. Replicating Huntington’s disease phenotype in experimental animals. Prog Neurobiol. 1999;59:427–468. doi: 10.1016/s0301-0082(99)00005-2. [DOI] [PubMed] [Google Scholar]
- Chang RC, Suen KC, Ma CH, Elyaman W, Ng HK, Hugon J. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J Neurochem. 2002;83:1215–1225. doi: 10.1046/j.1471-4159.2002.01237.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. Faseb J. 2004;18:1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- Chen HM, Wang L, D’Mello SR. Inhibition of ATF-3 expression by B-Raf mediates the neuroprotective action of GW5074. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05226.x. [DOI] [PubMed] [Google Scholar]
- Cheshire JL, Williams BR, Baldwin AS., Jr Involvement of double-stranded RNA-activated protein kinase in the synergistic activation of nuclear factor-kappaB by tumor necrosis factor-alpha and gamma-interferon in preneuronal cells. J Biol Chem. 1999;274:4801–4806. doi: 10.1074/jbc.274.8.4801. [DOI] [PubMed] [Google Scholar]
- Chin PC, Liu L, Morrison BE, Siddiq A, Ratan RR, Bottiglieri T, D’Mello SR. The c-Raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and Akt-independent mechanism. J Neurochem. 2004;90:595–608. doi: 10.1111/j.1471-4159.2004.02530.x. [DOI] [PubMed] [Google Scholar]
- Chin PC, Majdzadeh N, D’Mello SR. Inhibition of GSK3beta is a common event in neuroprotection by different survival factors. Brain Res Mol Brain Res. 2005;137:193–201. doi: 10.1016/j.molbrainres.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Chong KL, Feng L, Schappert K, Meurs E, Donahue TF, Friesen JD, Hovanessian AG, Williams BR. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. Embo J. 1992;11:1553–1562. doi: 10.1002/j.1460-2075.1992.tb05200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens MJ, Elia A. The double-stranded RNA-dependent protein kinase PKR: structure and function. J Interferon Cytokine Res. 1997;17:503–524. doi: 10.1089/jir.1997.17.503. [DOI] [PubMed] [Google Scholar]
- Copani A, Caraci F, Hoozemans JJ, Calafiore M, Sortino MA, Nicoletti F. The nature of the cell cycle in neurons: focus on a “non-canonical” pathway of DNA replication causally related to death. Biochim Biophys Acta. 2007;1772:409–412. doi: 10.1016/j.bbadis.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Lamba WR, Smith PD, Callaghan SM, Slack RS, Anisman H, Park DS. c-Jun mediates axotomy-induced dopamine neuron death in vivo. Proc Natl Acad Sci U S A. 2001;98:13385–13390. doi: 10.1073/pnas.231177098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello SR, Chin PC. Treating neurodegenerative conditions through the understanding of neuronal apoptosis. Curr Drug Targets CNS Neurol Disord. 2005;4:3–23. doi: 10.2174/1568007053005118. [DOI] [PubMed] [Google Scholar]
- D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis ST, Benson BG, Bramson HN, Chapman DE, Dickerson SH, Dold KM, Eberwein DJ, Edelstein M, Frye SV, Gampe RT, Jr, Griffin RJ, Harris PA, Hassell AM, Holmes WD, Hunter RN, Knick VB, Lackey K, Lovejoy B, Luzzio MJ, Murray D, Parker P, Rocque WJ, Shewchuk L, Veal JM, Walker DH, Kuyper LF. Prevention of chemotherapy-induced alopecia in rats by CDK inhibitors. Science. 2001;291:134–137. doi: 10.1126/science.291.5501.134. [DOI] [PubMed] [Google Scholar]
- De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem. 1997;243:518–526. doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- Eley HL, Russell ST, Tisdale MJ. Attenuation of muscle atrophy in a murine model of cachexia by inhibition of the dsRNA-dependent protein kinase. Br J Cancer. 2007;96:1216–1222. doi: 10.1038/sj.bjc.6603704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eley HL, Tisdale MJ. Skeletal muscle atrophy, a link between depression of protein synthesis and increase in degradation. J Biol Chem. 2007;282:7087–7097. doi: 10.1074/jbc.M610378200. [DOI] [PubMed] [Google Scholar]
- Enguita M, DeGregorio-Rocasolano N, Abad A, Trullas R. Glycogen synthase kinase 3 activity mediates neuronal pentraxin 1 expression and cell death induced by potassium deprivation in cerebellar granule cells. Mol Pharmacol. 2005;67:1237–1246. doi: 10.1124/mol.104.007062. [DOI] [PubMed] [Google Scholar]
- Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Lee VM, Trojanowski JQ. ‘Unfolding’ pathways in neurodegenerative disease. Trends Neurosci. 2003;26:407–410. doi: 10.1016/S0166-2236(03)00197-8. [DOI] [PubMed] [Google Scholar]
- Galabru J, Hovanessian A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J Biol Chem. 1987;262:15538–15544. [PubMed] [Google Scholar]
- Garcia MA, Meurs EF, Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Greene LA, Liu DX, Troy CM, Biswas SC. Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim Biophys Acta. 2007;1772:392–401. doi: 10.1016/j.bbadis.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N- terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–2395. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci. 2000;20:2567–2574. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hol EM, Fischer DF, Ovaa H, Scheper W. Ubiquitin proteasome system as a pharmacological target in neurodegeneration. Expert Rev Neurother. 2006;6:1337–1347. doi: 10.1586/14737175.6.9.1337. [DOI] [PubMed] [Google Scholar]
- Holtz WA, Turetzky JM, Jong YJ, O’Malley KL. Oxidative stress-triggered unfolded protein response is upstream of intrinsic cell death evoked by parkinsonian mimetics. J Neurochem. 2006;99:54–69. doi: 10.1111/j.1471-4159.2006.04025.x. [DOI] [PubMed] [Google Scholar]
- Hu JH, Zhang H, Wagey R, Krieger C, Pelech SL. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J Neurochem. 2003;85:432–442. doi: 10.1046/j.1471-4159.2003.01670.x. [DOI] [PubMed] [Google Scholar]
- Hu Y, Conway TW. 2-Aminopurine inhibits the double-stranded RNA-dependent protein kinase both in vitro and in vivo. J Interferon Res. 1993;13:323–328. doi: 10.1089/jir.1993.13.323. [DOI] [PubMed] [Google Scholar]
- Huang WL, Robson D, Liu MC, King VR, Averill S, Shortland PJ, Priestley JV. Spinal cord compression and dorsal root injury cause up-regulation of activating transcription factor-3 in large-diameter dorsal root ganglion neurons. Eur J Neurosci. 2006;23:273–278. doi: 10.1111/j.1460-9568.2005.04530.x. [DOI] [PubMed] [Google Scholar]
- Ingrand S, Barrier L, Lafay-Chebassier C, Fauconneau B, Page G, Hugon J. The oxindole/imidazole derivative C16 reduces in vivo brain PKR activation. FEBS Lett. 2007;581:4473–4478. doi: 10.1016/j.febslet.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Ito M, Onuki R, Bando Y, Tohyama M, Sugiyama Y. Phosphorylated PKR contributes the induction of GRP94 under ER stress. Biochem Biophys Res Commun. 2007;360:615–620. doi: 10.1016/j.bbrc.2007.06.087. [DOI] [PubMed] [Google Scholar]
- Jammi NV, Whitby LR, Beal PA. Small molecule inhibitors of the RNA- dependent protein kinase. Biochem Biophys Res Commun. 2003;308:50–57. doi: 10.1016/s0006-291x(03)01318-4. [DOI] [PubMed] [Google Scholar]
- Johnson K, Liu L, Majdzadeh N, Chavez C, Chin PC, Morrison B, Wang L, Park J, Chugh P, Chen HM, D’Mello SR. Inhibition of neuronal apoptosis by the cyclin-dependent kinase inhibitor GW8510: identification of 3′ substituted indolones as a scaffold for the development of neuroprotective drugs. J Neurochem. 2005;93:538–548. doi: 10.1111/j.1471-4159.2004.03004.x. [DOI] [PubMed] [Google Scholar]
- Jorda EG, Verdaguer E, Canudas AM, Jimenez A, Garcia de Arriba S, Allgaier C, Pallas M, Camins A. Implication of cyclin-dependent kinase 5 in the neuroprotective properties of lithium. Neuroscience. 2005;134:1001–1011. doi: 10.1016/j.neuroscience.2005.04.061. [DOI] [PubMed] [Google Scholar]
- Kolomeichuk SN, Bene A, Upreti M, Dennis RA, Lyle CS, Rajasekaran M, Chambers TC. Induction of apoptosis by vinblastine via c-Jun autoamplification and p53-independent down-regulation of p21WAF1/CIP1. Mol Pharmacol. 2008;73:128–136. doi: 10.1124/mol.108.039750. [DOI] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Koulich E, Nguyen T, Johnson K, Giardina C, D’Mello S. NF-kappaB is involved in the survival of cerebellar granule neurons: association of IkappaBbeta [correction of Ikappabeta] phosphorylation with cell survival. J Neurochem. 2001;76:1188–1198. doi: 10.1046/j.1471-4159.2001.00134.x. [DOI] [PubMed] [Google Scholar]
- Kuyama M, Nakanishi G, Arata J, Iwatsuki K, Fujimoto W. Expression of double-stranded RNA-activated protein kinase in keratinocytes and keratinocytic neoplasia. J Dermatol. 2003;30:579–589. doi: 10.1111/j.1346-8138.2003.tb00439.x. [DOI] [PubMed] [Google Scholar]
- Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- Lasky SR, Jacobs BL, Samuel CE. Mechanism of interferon action. Characterization of sites of phosphorylation in the interferon-induced phosphoprotein P1 from mouse fibroblasts: evidence for two forms of P1. J Biol Chem. 1982;257:11087–11093. [PubMed] [Google Scholar]
- Lee SB, Esteban M. The interferon-induced double-stranded RNA-activated protein kinase induces apoptosis. Virology. 1994;199:491–496. doi: 10.1006/viro.1994.1151. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Liu L, D’Mello SR. Phosphorylation of IkappaB-beta is necessary for neuronal survival. J Biol Chem. 2006;281:1506–1515. doi: 10.1074/jbc.M510402200. [DOI] [PubMed] [Google Scholar]
- Meijer L. Chemical inhibitors of cyclin-dependent kinases. Trends Cell Biol. 1996;6:393–397. doi: 10.1016/0962-8924(96)10034-9. [DOI] [PubMed] [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1993;90:232–236. doi: 10.1073/pnas.90.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, Hubbard SR, Schlessinger J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276:955–960. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- Morimoto H, Ozaki A, Okamura H, Yoshida K, Kitamura S, Haneji T. Okadaic acid induces tyrosine phosphorylation of IkappaBalpha that mediated by PKR pathway in human osteoblastic MG63 cells. Mol Cell Biochem. 2005;276:211–217. doi: 10.1007/s11010-005-4440-y. [DOI] [PubMed] [Google Scholar]
- Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol. 1996;134:757–770. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. Faseb J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- Ohba N, Kiryu-Seo S, Maeda M, Muraoka M, Ishii M, Kiyama H. Expression of damage-induced neuronal endopeptidase (DINE) mRNA in peri-infarct cortical and thalamic neurons following middle cerebral artery occlusion. J Neurochem. 2004;91:956–964. doi: 10.1111/j.1471-4159.2004.02784.x. [DOI] [PubMed] [Google Scholar]
- Onuki R, Bando Y, Suyama E, Katayama T, Kawasaki H, Baba T, Tohyama M, Taira K. An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer’s disease. Embo J. 2004;23:959–968. doi: 10.1038/sj.emboj.7600049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL, Rao RV, Cottrell BA, Hayden MR, Ellerby LM, Bredesen DE. Double-stranded RNA-dependent protein kinase, PKR, binds preferentially to Huntington’s disease (HD) transcripts and is activated in HD tissue. Hum Mol Genet. 2001;10:1531–1538. doi: 10.1093/hmg/10.15.1531. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994a;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994b;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- Rudolph B, Saffrich R, Zwicker J, Henglein B, Muller R, Ansorge W, Eilers M. Activation of cyclin-dependent kinases by Myc mediates induction of cyclin A, but not apoptosis. Embo J. 1996;15:3065–3076. [PMC free article] [PubMed] [Google Scholar]
- Russell ST, Eley H, Tisdale MJ. Mechanism of attenuation of angiotensin-II-induced protein degradation by insulin-like growth factor-I (IGF-I) Cell Signal. 2007;19:1583–1595. doi: 10.1016/j.cellsig.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Ruvolo PP, Gao F, Blalock WL, Deng X, May WS. Ceramide regulates protein synthesis by a novel mechanism involving the cellular PKR activator RAX. J Biol Chem. 2001;276:11754–11758. doi: 10.1074/jbc.M011400200. [DOI] [PubMed] [Google Scholar]
- Salzberg S, Vilchik S, Cohen S, Heller A, Kronfeld-Kinar Y. Expression of a PKR dominant-negative mutant in myogenic cells interferes with the myogenic process. Exp Cell Res. 2000;254:45–54. doi: 10.1006/excr.1999.4721. [DOI] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Shastry BS. Neurodegenerative disorders of protein aggregation. Neurochem Int. 2003;43:1–7. doi: 10.1016/s0197-0186(02)00196-1. [DOI] [PubMed] [Google Scholar]
- Shen Q, Uray IP, Li Y, Krisko TI, Strecker TE, Kim HT, Brown PH. The AP-1 transcription factor regulates breast cancer cell growth via cyclins and E2F factors. Oncogene. 2008;27:366–377. doi: 10.1038/sj.onc.1210643. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Hara H. Inhibitor of double stranded RNA-dependent protein kinase protects against cell damage induced by ER stress. Neurosci Lett. 2006;409:192–195. doi: 10.1016/j.neulet.2006.09.074. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Ito Y, Inokuchi Y, Hara H. Involvement of double-stranded RNA-dependent protein kinase in ER stress-induced retinal neuron damage. Invest Ophthalmol Vis Sci. 2007;48:3729–3736. doi: 10.1167/iovs.06-1122. [DOI] [PubMed] [Google Scholar]
- Stepniak D, Wiesner M, de Ru AH, Moustakas AK, Drijfhout JW, Papadopoulos GK, van Veelen PA, Koning F. Large-scale characterization of natural ligands explains the unique gluten-binding properties of HLA-DQ2. J Immunol. 2008;180:3268–3278. doi: 10.4049/jimmunol.180.5.3268. [DOI] [PubMed] [Google Scholar]
- Sun L, Tran N, Liang C, Hubbard S, Tang F, Lipson K, Schreck R, Zhou Y, McMahon G, Tang C. Identification of substituted 3-[(4,5,6, 7-tetrahydro-1H-indol-2-yl)methylene]-1,3-dihydroindol-2-ones as growth factor receptor inhibitors for VEGF-R2 (Flk-1/KDR), FGF-R1, and PDGF-Rbeta tyrosine kinases. J Med Chem. 2000;43:2655–2663. doi: 10.1021/jm9906116. [DOI] [PubMed] [Google Scholar]
- Sun L, Tran N, Tang F, App H, Hirth P, McMahon G, Tang C. Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J Med Chem. 1998;41:2588–2603. doi: 10.1021/jm980123i. [DOI] [PubMed] [Google Scholar]
- Tsujino H, Kondo E, Fukuoka T, Dai Y, Tokunaga A, Miki K, Yonenobu K, Ochi T, Noguchi K. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: A novel neuronal marker of nerve injury. Mol Cell Neurosci. 2000;15:170–182. doi: 10.1006/mcne.1999.0814. [DOI] [PubMed] [Google Scholar]
- Vanhara P, Bryja V, Horvath V, Kozubik A, Hampl A, Smarda J. c-Jun induces apoptosis of starved BM2 monoblasts by activating cyclin A-CDK2. Biochem Biophys Res Commun. 2007;353:92–97. doi: 10.1016/j.bbrc.2006.11.124. [DOI] [PubMed] [Google Scholar]
- Verdaguer E, Alvira D, Jimenez A, Rimbau V, Camins A, Pallas M. Inhibition of the cdk5/MEF2 pathway is involved in the antiapoptotic properties of calpain inhibitors in cerebellar neurons. Br J Pharmacol. 2005a;145:1103–1111. doi: 10.1038/sj.bjp.0706280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdaguer E, Jorda EG, Alvira D, Jimenez A, Canudas AM, Folch J, Rimbau V, Pallas M, Camins A. Inhibition of multiple pathways accounts for the antiapoptotic effects of flavopiridol on potassium withdrawal-induced apoptosis in neurons. J Mol Neurosci. 2005b;26:71–84. doi: 10.1385/JMN:26:1:071. [DOI] [PubMed] [Google Scholar]
- Wang X, Fan Z, Wang B, Luo J, Ke ZJ. Activation of double-stranded RNA-activated protein kinase by mild impairment of oxidative metabolism in neurons. J Neurochem. 2007;103:2380–2390. doi: 10.1111/j.1471-4159.2007.04978.x. [DOI] [PubMed] [Google Scholar]
- Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb Exp Pharmacol. 2006:69–91. doi: 10.1007/3-540-29717-0_3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) HT-22 cells were treated with 1 mM HCA or 1 mM HCA plus 10μM PKRi. After 24-hour treatment, MTT assay was performed. Survival is represented as percentage of control cultures. Control cultures for HCA-treated cells received no HCA (untreated cultures). In the case of HCA + PKRi treated cells, the control cultures received PKRi but not HCA. The need for separate controls is necessitated by the fact that PKRi treatment stops cell proliferation and therefore cannot be directly compared to untreated cultures which continue to proliferate. Data shown represents the mean values of viability with their corresponding standard deviations from 3 independent experiments. ** indicates the p-value < 0.001 using Student’s t-test.
(B) HT-22 cells were untreated (Un), treated with 1mM HCA or 1mM HCA plus 10μM PKRi. After 15 hours of treatment, TUNEL assay was performed. DAPI staining shows the morphology of nuclei.
Cerebellar granule neurons were treated with HK, LK or LK containing 10μM PKRi for 3 hours. Lysates were made and CDK5 was immunoprecipitated to perform an in vitro kinase assay containing [γ-32P] ATP and using Histone H1 as substrate. Relative activity was visualized by autoradiography. Ponceau S staining of Histone H1 shows that comparable amounts of substrate were used in each reaction. An aliquot of the lysates prior to immunoprecipitation was also analyzed by Western blotting using tubulin antibody to demonstrate that similar quantities of lysate was used for pull-down.