

Background: γ-Secretase modulators (GSMs) hold potential as disease modifiers in Alzheimer disease; however, their mechanism of action is not completely understood.
Results: Second generation in vivo active GSMs were described and shown to modulate Aβ production via a non-APP targeting mechanism, different from the NSAIDs class of GSMs.
Conclusion: A growing class of second generation GSMs appears to target γ-secretase and displays a different mechanism of action compared with first generation GSMs.
Significance: The identification of in vivo active non-APP targeting second generation GSMs may facilitate the development of novel therapeutics against AD.
Keywords: Alzheimer Disease, Amyloid, Intramembrane Proteolysis, Neurodegenerative Diseases, Pharmacology, Secretases
Abstract
γ-Secretase-mediated cleavage of amyloid precursor protein (APP) results in the production of Alzheimer disease-related amyloid-β (Aβ) peptides. The Aβ42 peptide in particular plays a pivotal role in Alzheimer disease pathogenesis and represents a major drug target. Several γ-secretase modulators (GSMs), such as the nonsteroidal anti-inflammatory drugs (R)-flurbiprofen and sulindac sulfide, have been suggested to modulate the Alzheimer-related Aβ production by targeting the APP. Here, we describe novel GSMs that are selective for Aβ modulation and do not impair processing of Notch, EphB2, or EphA4. The GSMs modulate Aβ both in cell and cell-free systems as well as lower amyloidogenic Aβ42 levels in the mouse brain. Both radioligand binding and cellular cross-competition experiments reveal a competitive relationship between the AstraZeneca (AZ) GSMs and the established second generation GSM, E2012, but a noncompetitive interaction between AZ GSMs and the first generation GSMs (R)-flurbiprofen and sulindac sulfide. The binding of a 3H-labeled AZ GSM analog does not co-localize with APP but overlaps anatomically with a γ-secretase targeting inhibitor in rodent brains. Combined, these data provide compelling evidence of a growing class of in vivo active GSMs, which are selective for Aβ modulation and have a different mechanism of action compared with the original class of GSMs described.
Introduction
The amyloid-β (Aβ)2 peptide plays a pivotal role in Alzheimer disease (AD) pathogenesis. Aβ is a 33–42-amino acid post-proteolytic peptide derived from amyloid precursor protein (APP) as the result of sequential cleavages by β- and γ-secretase, respectively, where the latter activity results in peptides with different C termini and lengths. Genetic and mechanistic data strongly suggest that the amyloidogenic Aβ42 peptide plays a crucial contributing role in Aβ misfolding and AD pathogenesis (1). Accordingly, Aβ and Aβ42 targeting approaches, including β- and γ-secretase directed inhibitors, represent major principles for therapeutic intervention in AD. γ-Secretase is a promiscuous enzyme, with regard to substrate specificity, and catalyzes the proteolysis of more than 50 type 1 membrane proteins, including the Notch and Eph families of receptors (2). γ-Secretase-dependent Notch signaling plays an important role in different contexts of cell signaling, and this feature has complicated the development of γ-secretase inhibitors (GSIs) as a therapeutic strategy for AD (3).
The challenge with γ-secretase inhibition has warranted alternative strategies to combat Aβ generation. Approximately 10 years ago, Koo and co-workers (4) presented the novel concept of γ-secretase modulation (GSM), in which the production of amyloidogenic Aβ42 peptides is lowered whereas the production of shorter Aβ peptides, such as Aβ38, is increased. Several NSAIDs, such as ibuprofen, sulindac sulfide, and indomethacin, exhibit these features, and, most importantly, these drugs appear to be Notch-sparing and thus mitigate a major hurdle associated with γ-secretase inhibition. Importantly, Aβ modulation has been observed in different preclinical animal models, such as in mice and dogs, suggesting that Aβ modulation indeed is a druggable approach (5, 6). Recently, the NSAID-like GSM (R)-flurbiprofen (Tarenflurbil) was tested in phase 3 clinical trials for mild to moderate AD, but the trials did not provide evidence for halting disease progression. It is unclear, however, whether the drug actually lowered CNS Aβ42 levels, and thus questions remain whether the validity of CNS Aβ modulation as a therapeutic approach in AD has been accurately tested (7).
The mechanism by which GSMs of the NSAID class modulate Aβ production is emerging. Experiments with isotope-labeled, nontransition state γ-secretase inhibitors have revealed a noncompetitive mechanism for NSAIDs (8). Pharmacological and biochemical experiments have suggested a direct interaction of certain NSAIDs with the APP-derived immediate substrate for γ-secretase, β-C-terminal fragment, or C99, resulting in altered Aβ production (9, 10). From a CNS drug discovery perspective, the GSMs of the NSAID class exhibit some generally less favorable features however, such as low potency and inefficient blood-brain barrier penetrance.
Lately, non-NSAID second generation GSMs have been described, which are structurally diverse from the NSAIDs and appear to exhibit improved drug-like properties (11). Kounnas et al. (12) reported on the in vivo active second generation GSM compound 4, which was >1000-fold more potent than (R)-flurbiprofen in vitro and did not affect Notch nor E-cadherin signaling in vitro. Moreover, Portelius et al. (6) has demonstrated an Aβ modulatory effect of the second generation GSM E2012 in dog brain, providing additional evidence of CNS efficacy mediated by a second generation GSM.
The mechanism by which the second generation GSMs modulate Aβ is emerging. In a recent publication, the pharmacology of the GSM E2012 and the NSAID class of modulators, respectively, was compared and shown to be differentially effected by familial Alzheimer disease (FAD)-linked PS mutations (13). Inhibitor pulldown experiments with the GSM compound 4 was shown to precipitate both APP and in particular the γ-secretase subunit Pen-2, suggesting that Pen-2 may be the molecular target of GSM compound 4 (12). During the preparation and revision of this study, three reports were published that suggest that second generation GSMs physically interact with PS and not APP (14–16). Collectively, these observations suggest that the first and second generation GSMs may cause Aβ modulation through distinct mechanisms.
In this study, we present the characterization of novel in vivo active GSMs, which do not affect Notch, EphB2, or EphA4 processing, and are much more potent modulators than the NSAID-like class of GSMs. Binding experiments suggest that AstraZeneca (AZ) GSMs interact directly with the γ-secretase complex and not with APP. Displacement binding studies and cellular cross-competition data reveal a noncompetitive relationship between the AZ GSMs and the APP targeting NSAID GSMs (R)-flurbiprofen and sulindac sulfide but a competitive interaction between AZ GSMs and the second generation GSM, E2012. These pharmacological data provide compelling evidence of a growing class of in vivo active GSMs, which most likely modulate Aβ production via a direct γ-secretase targeting mechanism.
MATERIALS AND METHODS
Compounds
Dibenzazepine (DBZ) was obtained from Calbiochem; L685458, (R)-flurbiprofen, and sulindac sulfide were from Sigma, and semagacestat was from Selleck Chemicals. The preparation of AZ1136 and AZ3303 is described in the supplemental material. AZ4800 (WO2010053438), E2012 (US20060004013), and MRK-560 were prepared according to published methods. [3H]AZ8349 and [3H]DBZ were labeled in our laboratories (17).
In Vitro Cellular Aβ Assays
HEK/APPSwe, HEK/APP, HEK/PS1, and HEK/PS2 (18) were exposed to compounds for 5 h. Aβ generation was analyzed as described previously (19). Aβ was measured using MSD technology using 6E10 as capture antibody and C-terminally specific antibodies for Aβ42, Aβ40, Aβ39, Aβ38, and Aβ37, respectively.
In Vitro Membrane Aβ Assays
Membranes were prepared from HEK/APPswe cells cultured in Dulbecco's modified Eagle's medium (high glucose) with addition of 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin/streptomycin, nonessential amino acids, and 10 μm Hepes, essentially in accordance with the published method by McLendon et al. (20). In brief, cells grown to 90% confluence were treated with a potent GSI for 20 h, washed three times with ice-cold phosphate-buffered saline (PBS, pH 7.4), and harvested by scraping. Cells were pelleted (centrifugation for 10 min at 1000 × g), resuspended in Lysis buffer (50 mm Tris-HCl, pH 7.4, 1.0 mm EDTA, and complete protease inhibitor (Roche Applied Science)), and incubated for 15 min on ice. Cell suspension was homogenized (Ultra-Turrax T25, two times for 20 s, 11,500 rpm), centrifuged for 10 min at 1000 × g, and post-nuclear supernatant was centrifuged for 45 min at 50,000 × g. Membranes were finally resuspended in assay buffer (50 mm mes, pH 6.4, 1.5 mm MgCl2, 75 mm sodium citrate, and complete protease inhibitor (Roche Applied Science)), homogenized (two times for 10 s, 20,000 rpm), and stored at −80 °C. Carbonate-extracted membranes were prepared by resuspension of the cell pellets in extraction buffer (0.1 m sodium carbonate, pH 11) and incubated on ice for 15 min. Ultracentrifugation of the suspension for 45 min at 50,000 × g resulted in carbonate-extracted membrane pellets that were further resuspended and homogenized (two times for 10 s, 20,000 rpm). The enzymatic reactions were run on membranes with or without sodium carbonate treatment diluted in assay buffer (protein concentration of 1.5 mg/ml). Reaction mixtures were plated in 384-well plates together with test compounds (4% final DMSO concentration). Reaction was initiated by incubation in 37 °C and terminated after 2 h by placing the plate on ice. Aβ peptides were analyzed by ECL (MSD).
In Vitro Cellular NICD Translocation Assay
HEK293 cells stably transfected with a pcDNA3.1hygro vector encoding extracellular truncated human Notch 1 (ΔENotch1) and the N-terminal signal peptide from the full-length Notch 1 were used for analysis of γ-secretase-mediated Notch processing. Cells were expanded in DMEM plus 10% FBS and 300 μg/ml hygromycin before being cryo-preserved in media containing 10% dimethyl sulfoxide. For each experiment, the frozen cells were thawed, washed, and resuspended in fresh media. 10,000 cells/well were plated in 384-well poly-d-lysine-coated cell culture plates and incubated overnight. The following day, fresh cell media containing 3 μm lactacystin was added together with a test compound diluted 1:200 from a prepared compound dilution plate and incubated for 5 h at 37 °C after which cells were washed, fixed with 4% paraformaldehyde in PBS, and immunocytostained using the primary polyclonal anti-NICD (C-20) antibody and Alexa Fluor 594 secondary antibody. Images were captured using the ImageXpressTM scanner (Molecular Devices), and fluorescence automatic measurements were performed using two different analysis algorithms for the average fluorescence in the nucleus as well as the average fluorescence of a 3-μm extra-nuclear ring. The ratio of the nuclear/extra-nuclear fluorescence was calculated/cell, and the mean ratio/well was calculated. % NICD translocation was expressed relative to 0.5% DMSO (100% control) and 500 nm L685458 (21) (0% control). Each concentration was tested in duplicate in at least two separate experiments.
γ-Secretase Substrate Expression Assay
N-terminally truncated versions of EphA4 (amino acids 531–986) (22) and EphB2 (amino acids 529–986) (23), encoding immediate substrates of γ-secretase, were N- and C-terminally fused to the preprotrypsin leader peptide and the V5 and c-Myc immune tags, respectively. HEK293 cells were transiently transfected with either of these constructs or with a construct encoding Myc-tagged dENotch1 (amino acids 1714–2555) and exposed to DBZ (24) and AZ4800 for 15 h prior to harvesting in 2× SDS-PAGE buffer at 95 °C for 15 min. The lysates were subjected to standard SDS-PAGE and Western blotting procedures. Expressed proteins were identified with an anti-Myc antibody (9E10, Invitrogen), and GAPDH immunoreactivity was used to normalize against total protein levels. The experiment was conducted at least three times.
Cellular Cross-competition Assay
To be able to accurately analyze the cross-competition data, it was important to have good data coverage around the pIC50 values of the modulators to be tested. To ensure this, we prepared a 13 concentration 0.5 log serial dilution of AZ4800 in DMSO and mixed this with four or five concentrations of the second GSM to be tested (centered around its pIC50). The compound mixtures were added to HEK/APPswe cells and incubated for 5 h (the final DMSO concentration was 0.5%). GSI L685458 or DMSO was added as controls to measure the Z′ factor for each experiment, and only experiments with Z′ > 0.5 were analyzed. Aβ42 was analyzed in the cell media as described previously (19). For graphical analysis, we only plotted the data from the slope and calculated the velocity (v = molar Aβ42/min) using a standard curve of synthetic Aβ42. For nonlinear global analysis, the complete data set was analyzed.
Aβ Analysis
Aβ peptides were analyzed using MSD technology. Briefly, membrane reactions and conditional cell media were transferred to MSD plates with either 6E10 capture antibody or triplex plates with Aβ40, Aβ42, and Aβ38 capture antibodies. Primary sulfo-tagged detection antibodies specific for either x-37, x-39, or the N terminus of Aβ1-x (6E10), respectively, were added, and the plates were incubated overnight at 4 °C. The following day the plates were processed according to the manufacturer's instructions.
Data Analysis
For IC50 determinations, 10 concentration-response curves were analyzed using GraphPad Prism with the nonlinear regression four-parameter logistic function model. For calculation of % response, the data were normalized to maximum and minimum control responses (0.5% DMSO and 0.5 μm L685458, respectively). The IC50 values reported are the average of at least two independent experiments. Cross-competition modulator experiments were analyzed graphically using a linear regression model (Equation 1). The graphs were interpreted using the reciprocal of Equation 1, which is a linear function of 1/v versus [I1] with a constant value for [I2] for each line; if a change in [I2] shifts the slope of the fitted line and causes the lines of different [I2] to intercept on or to the left of the 1/v axis, simultaneous binding to nonoverlapping sites is occurring. However, if the set of lines of different [I2] are parallel, the binding of the two compounds is suggested to be mutually exclusive, i.e. competitive (25). The cross-competition data were also analyzed using nonlinear global fitting of models for theoretical two inhibition kinetics (Equation 1). Because of the difficulty in determining the cooperativity constant α with high precision, as it is interrelated with the Ki values of the GSMs, we decided to set α to either 1 (noncompetitive) or ∞ (competitive) and determine which of these models fit the data best by using Akaike's information criteria. Each concentration was tested at least in duplicate in two separate experiments.
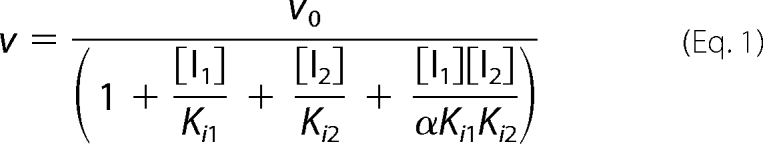 |
Autoradiography
In vitro binding autoradiography on tissue sections was adapted for γ-secretase ligands from previously described protocols (26). Briefly, frozen brains from rats and guinea pigs were sectioned (10 μm) with a cryostat through the sagittal or coronal plane, air-dried, and stored at −80 °C. Adjacent sections were warmed to room temperature, preincubated for 10 min at room temperature in 50 mm Tris buffer, pH 7.4, then transferred to the same buffer containing 1–10 nm of the GSM [3H]AZ8349 or the GSI [3H]DBZ, and incubated for 45 min at RT. The sections were subsequently washed several times and finally air-dried. For competition studies, adjacent tissue sections were incubated in the same buffer together with radiotracers and unlabeled GSMs. Samples and plastic tritium standards (Amersham Biosciences) were exposed to imaging plates (Fuji BAS-TR2040) for 5 days and then processed with a FLA7000 Imaging Reader (Fujifilm). Binding was analyzed with Multigauge software Version 3.0 (Fujifilm) using the relative optical density values generated from co-exposed tritium standards.
Immunohistochemistry
Fresh frozen 10-μm thick tissue sections from adult WT, TG2576 (APPswe), and APPswe/PSdExon9 mice were fixed in 50% acetone for 1 min and 100% acetone for 5 min. The immunohistochemical procedure was carried out using an automated stainer (Ventana Discovery® XT staining module, Ventana, Illkirch, France). A primary rabbit polyclonal anti-APP-directed antibody (Sigma A8717) diluted 1:3000 was manually applied. The Ventana Omni-ultramap kit was used for detection.
In Vivo Efficacy Studies
In vivo drug administration and Aβ(1–42) analysis from 12–18-week-old female C57BL/6 mice (Harlan Laboratory) were given either AZ4800 (75, 150, or 300 μmol/kg), AZ3303 (100 and 300 μmol/kg), or vehicle by oral gavage (10 ml/kg) (8–9 mice/group). The mice were sacrificed under isoflurane anesthesia 1.5 h after administration, and the brains were quickly removed, dissected into hemispheres, snap-frozen on dry ice, and stored at −70 °C. Frozen brain hemisphere was homogenized in 0.2% diethylamine with 50 mm NaCl (18 μl/mg wet weight tissue). Brain homogenates were centrifuged at 133,000 × g for 1 h. Recovered supernatants were neutralized to pH 8.0 with 2 m Tris-HCl, pH 4–5. Analysis of Aβ42 content in brain and plasma was performed with the mouse Aβ(1–42) colorimetric ELISA kit, (Invitrogen) according to the manufacturer's instructions. All animal experiments were conducted in accordance with relevant regulations and guidelines provided by the Swedish Board of Agriculture. The study was approved by an ethical board specialized on animal experiments.
RESULTS
AZ GSMs Modulate Aβ Generation in Cell Culture
We have synthesized three novel chemically distinct compounds, which decrease Aβ42 in tissue culture cells (see chemical structures in Fig. 1 and description of synthesis in supplemental material). To explore their mechanism on APP processing, we measured their effect on Aβ peptide generation in HEK/APPswe cells. Interestingly, all three compounds reduced Aβ40 as well as Aβ42 but appeared 2–15-fold more potent in reducing Aβ42 levels (AZ4800, IC50 = 26 ± 6 nm; AZ3303, IC50 = 74 ± 10 nm; and AZ1136, IC50 = 990 ± 150 nm) versus Aβ40 (AZ4800, IC50 = 60 ± 14 nm; AZ3303, IC50 = 810 ± 70 nm; and AZ1136, IC50 = 1400 ± 100 nm) (Fig. 2A). Although the AZ GSMs had similar inhibitory effects on Aβ40 and Aβ42 secretion, their effect on Aβ(37–39) differed substantially, and a unique Aβ pattern for each of the GSMs was revealed (Fig. 2, B–D). AZ4800 and AZ3303 decreased Aβ39, whereas they increased both Aβ37 and Aβ38, but with different potencies and magnitudes. AZ3303 increased Aβ38 by 550% and Aβ37 by 300%, whereas AZ4800 increased Aβ37 by 750% and Aβ38 by 300%. AZ1136, however, increased both Aβ39 and Aβ37 by 250% but did not affect Aβ38 production. Importantly, total Aβ was not affected by any of the compounds at the concentration range where modulation was observed. The non-NSAID GSM E2012 affected Aβ40/42 production in a similar manner as the AZ GSMs but caused a selective increase in Aβ37 (Fig. 2, A and E). General toxicity of the compounds was tested using the ViaLightTM cell toxicity assay. No toxicity was seen at the concentrations tested (data not shown). We also explored the AZ GSMs in HEK/APP cells and in mouse primary neurons, to make sure that their activity was not dependent on the APPswe mutant nor on overexpressed APP. All compounds retained their modulatory activity in HEK/APP cells and also decreased Aβ42 levels in mouse primary neurons (supplemental Figs. S1 and S2). In addition, we explored the activity of the compounds in HEK293 cells stably expressing either presenilin 1 (PS1) or PS2, i.e. the catalytic subunit of γ-secretase (18). All three AZ GSMs as well as E2012 modulated Aβ production generated by both PS subtypes but displayed a higher potency on Aβ42 inhibition in PS2-overexpressing compared with PS1-overexpressing cells (supplemental Fig. S3). In contrast, the NSAIDs (R)-flurbiprofen and sulindac sulfide modulated Aβ production nonselectively in the same assay (supplemental Fig. S3). These cell culture experiments suggest that AZ4800, AZ1136, and AZ3303 are true Aβ modulators that lower or increase both PS1 and PS2 catalyzed production of some Aβ peptides, without affecting the total amount of Aβ being produced.
FIGURE 1.

Chemical structures of compounds explored in this study. A, AZ4800; B, AZ3303; C, Sulindac Sulfide; D, AZ1136; E, E-2012; F, (R)-flurbiprofen; G, [3H]AZ8349; and H, [3H]DBZ.
FIGURE 2.

Effect of second generation GSMs on APP processing in cellular and cell-free assays. A, Aβ40 and Aβ42 measurement in conditioned media from HEK/APPswe cells. All AZ GSMs and GSM E2012 inhibit Aβ42 (black) and Aβ40 (gray) generation. B–F, Aβ measurement in conditioned media from HEK/APPswe cells. B, AZ1136 increases the levels of Aβ37 and Aβ39. C, AZ4800 increases Aβ37 > Aβ38 and lowers Aβ39. D, AZ3303 increases Aβ38 > Aβ37 and lowers Aβ39. E, E2012 increases Aβ37. AZ1136 (F) and AZ4800 (G) display Aβ modulation in cell membranes derived from HEK/APPswe cells. % Aβ release is set relative to 0.5% DMSO (100%) and 0.5 μm L685458 (0%) controls (mean ± S.E., n = 2).
AZ GSMs Retain Aβ Modulatory Activity in Cell-free Assays
To further investigate the pharmacological mechanism of the AZ GSMs, we asked whether their impact on Aβ production would require a native cellular context. To address that question, we prepared membranes from HEK/APPswe cells with accumulated APP-derived γ-secretase substrate according to a method previously described by McLendon et al. (20). Part of the nondetergent membrane preparation (denoted TM for total membranes) was further treated with sodium carbonate at pH 11 (denoted CEM for carbonate-extracted membranes) to remove nonmembrane integral proteins. We incubated these membranes with different concentrations of AZ4800 and AZ1136 and analyzed the levels of Aβ37, Aβ38, Aβ40, and Aβ42 in both TM and CEM preparations with the same method as described for the cell culture experiments discussed above. Both GSMs had similar effects on TM and CEM Aβ production. Although the potency for the compounds differed compared with the cellular assay, AZ4800 inhibited both Aβ40 and Aβ42 production, although it increased Aβ37 and Aβ38 levels, which is in accordance with the data obtained using the cellular assay (Fig. 2G). AZ1136 also caused a clear inhibition of Aβ42 and an increase in Aβ37 levels, as observed in the cellular assay (Fig. 2F). For this compound, a slight increase in Aβ38 was also noted, which was not observed in the cellular assay. To verify that the carbonate extraction had worked, we analyzed the specific activity of the membrane batches before and after treatment, and as expected the specific activity increased in the carbonate-treated membranes, indicating that the treatment led to removal of protein content (supplemental Fig. S4). Overall, these experiments suggest that AZ GSMs modulate Aβ directly at the level of APP processing and that they are not dependent on a native cellular context.
AZ GSMs Do Not Affect γ-Secretase-mediated Processing of Notch, EphA4, and EphB2
In the next experiments, we tested the selectivity of GSMs with regard to γ-secretase-dependent Notch, EphA4, and EphB2 processing. To study Notch processing and signaling, we developed a cellular assay using HEK293 cells stably transfected with an N-terminal extracellularly truncated human Notch 1 receptor (ΔENotch1) and measured the distribution of the NICD using immunocytochemistry and an antibody raised against NICD. Under normal conditions, γ-secretase caused the release of NICD, which translocates to the nucleus, whereas in the presence of several established γ-secretase inhibitors such as L685458, DBZ, and MRK-560 (21, 24, 27), NICD is clearly located outside the nuclei (supplemental Fig. S5, A and B). To quantify the level of NICD processing and translocation, we measured the fluorescence intensity inside as well as outside the nuclei and calculated the ratio of nuclear/cytosolic fluorescence for each cell. None of the GSMs showed any inhibition of the NICD translocation in contrast to the potent GSIs L685458, DBZ, MRK-560, and Semagacestat, which all inhibited NICD translocation with high potency (Fig. 3A). Thus, these data suggest that the AZ GSMs display an ∼1000-fold preference for Aβ modulation over NICD formation. We next explored the impact of AZ4800 on the expression of N-terminally truncated EphA4 and EphB2. Similar to αENotch (Fig. 3B), both EphA4 and EphB2 levels were increased in the presence of the GSI DBZ, suggesting that these constructs are bona fide γ-secretase substrates, in which the turnover is regulated by γ-secretase. AZ4800 did not accumulate any of the proteins at a dose that displays full modulation of Aβ (Fig. 3B). These data suggest that AZ4800 and second generation GSMs are selective for Aβ modulation.
FIGURE 3.
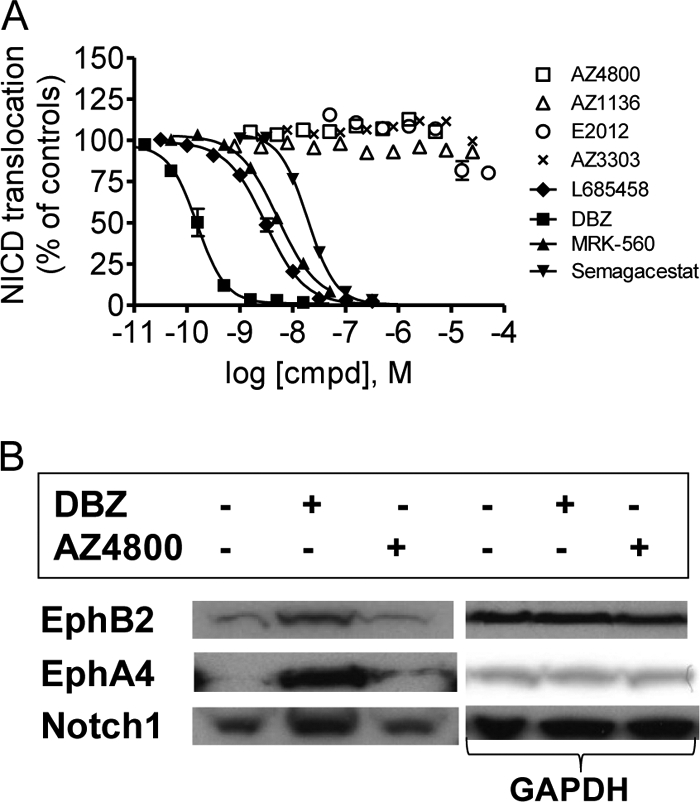
Effect of second generation GSMs on Notch, EphB2, and EphA4 processing. A, none of the AZ GSMs (square, triangle, and ×) or E2012 (○) affect Notch processing, whereas the GSIs L685458, DBZ, MRK-560, and Semagacestat do, as measured by quantifying the amount of nuclearly translocated NICD in HEK293 cells stably transfected with ΔENotch using immunocytochemistry. % NICD translocation is set relative to 0.5% DMSO (100%) and 0.5 μm L685458 (0%) controls (mean ± S.E., n = 2). B, HEK293 cells, transiently transfected with Myc-tagged EphB2, EphA4, and ΔENotch constructs, were exposed to 100 nm DBZ or 1 μm 4800 for 15 h prior to Western blot analysis. DBZ, but not AZ4800, results in accumulation of respective protein, as explored with an anti-Myc antibody (left panel). Each Western blot was probed with an anti-GAPDH antibody to control for loaded protein (right panel). The figure shows one representative blot out of at least three independent experiments.
Second Generation GSMs Modulate Aβ Production via a Mechanism That Is Distinct from the NSAID Class of GSMs
To compare the activity of AZ GSMs with the well characterized NSAIDs (R)-flurbiprofen and sulindac sulfide, we developed a cellular assay based on a biochemical inhibitor cross-competition assay (25). The cross-competition method provides pharmacological mechanistic information as to whether two inhibitors compete for binding to one site on a target molecule or bind to two independent sites of the target enzyme or an associated protein. To evaluate this, we exposed HEK293 cells expressing the Swedish mutation of APP to different compounds and measured the effect of binary combinations of GSMs on Aβ42 levels after 5-h treatments. The results were analyzed graphically using linear regression of reciprocal 1/v plots as well as quantitatively with nonlinear regression. For the graphical analysis, a parallel shift of the regression lines indicates competitive binding, and intercepting lines indicate a degree of noncompetitively between the two compounds tested. We tested all GSMs against AZ4800. AZ4800 in combination with itself, AZ3303, AZ1136, and E2012, respectively, showed a clear parallel shift of the plotted lines indicating AZ4800-competitive binding (Fig. 4, A–D). Interestingly, AZ4800 in combination with (R)-flurbiprofen or sulindac sulfide produced nonparallel intercepting lines indicating noncompetitive interactions (Fig. 4, E and F). Nonlinear global fitting of the data further confirmed the results from the graphical analysis (supplemental Fig. S6).
FIGURE 4.

Pharmacological interaction between first and second generation GSMs. Graphical analysis of modulator cross-competition in a cellular assay for Aβ42. A–F, graphs display AZ4800 versus the following: A, AZ4800; B, AZ3303; C, AZ1136; D, E2012; E, (R)-flurbiprofen; and F, sulindac sulfide. Parallel shift of the lines (A–D) indicates competitive binding to the same or overlapping sites, and intercepting lines (E and F) suggest binding noncompetitive with AZ4800. Lines are fitted to the data by linear regression (mean ± S.E., n = 2). v is amount Aβ42 generated/min analyzed in conditioned media from HEK/APPswe cells.
Distribution of the AZ GSM Molecular Target of Overlaps with γ-Secretase
Next, we conducted a series of autoradiography binding studies to further explore the mode of action by which AZ4800, AZ3303, and AZ1136 modulate γ-secretase-mediated Aβ production. We labeled the GSI DBZ (17) and an analog to the GSM AZ4800 and AZ8349 (see structures in Fig 1), and we incubated them with brain slices from guinea pig in the presence and absence of cold inhibitor and modulator, respectively. The anatomical regions with highest binding of [3H]AZ8349 were associated with the subventricular zone, an area with previously documented high levels of γ-secretase in the adult rodent brain (Fig. 5A). This is also in agreement with the previously shown binding distribution of the GSI [3H]compound D (28). High binding was also observed throughout cortex and the hippocampal formation, most densely in the dentate gyrus but also in striatum, thalamus, and cerebellum. Binding to white matter regions was in general lower. [3H]AZ8349 could be efficiently displaced by its non-3H-labeled precursor, suggesting that the signal was saturable (Fig. 5B). On adjacent sections, [3H]DBZ showed similar binding distribution as [3H]AZ8349, exemplified by the notably strong binding associated to the subventricular zone (Fig. 5C). To address whether APP expression had any impact on the interaction of [3H]AZ8349 to brain slices, we next performed both immunohistochemical studies with anti-APP antibodies and binding studies with [3H]AZ8349 on adjacent brain slices from TG2576 mice and WT littermates, respectively. As expected, a strong APP-like immunoreactivity was obtained in brain slices from TG2576, whereas a much weaker staining was obtained with tissues from WT mice, reflecting the high APP expression levels in the brains of TG2576 (Fig. 5, D–G). [3H]AZ8349, however, bound to both TG2576 and the WT littermates with similar intensity and anatomical distribution, in accordance with what was observed in brain slices from guinea pig (Fig. 5, H–I). Very similar results were also obtained when assessing APPswe/PSdExon9 mice (supplemental Fig. S7). These data clearly suggest that APP does not impact the interaction of [3H]AZ8349 with its target.
FIGURE 5.

Binding distribution of 3H-labeled GSM and GSI to rodent brain sections and comparison with APP immunohistochemistry. 5 nm 3H-labeled GSM AZ8349 (A) and 5 nm 3H-labeled GSI DBZ (C) display excellent anatomical binding overlap to sagittal brain sections from guinea pig. Note very high binding associated to the subventricular zone (SVZ). Nonspecific binding (5 nm 3H-labeled GSM AZ8349 + 5 μm unlabeled AZ8349) is shown (B). D–G, images of immunohistochemistry detecting APP in coronal brain sections from TG2576 mice (E–G) and WT controls (D and E). Note the stronger overall APP-like immunoreactivity in TG2576 mice compared with WT. In contrast, 3H-labeled GSM AZ8349 has no increased binding in TG2576 (I) compared with WT (H). Quantification of binding in the autoradiograms as optical density (photostimulated luminescence (PSL)/mm2) is shown in the bar graphs.
First and Second Generation GSMs Display Different Binding Sites
To further investigate the different mechanism of the first and second generation GSMs, we conducted displacement binding studies on rat brain slices. Approximately 50% of the total [3H]AZ8349 binding was specific, as determined by running the reaction in the presence and absence, respectively, of 10 μm unlabeled AZ8349. In the presence of 10 μm of either AZ4800, AZ8163, and E2012, [3H]AZ8349 (5 nm) was displaced by 32, 51, and 50%, respectively, by approximately the same magnitude as in the presence of excess unlabeled AZ8349 (Fig. 6). However, neither (R)-flurbiprofen nor sulindac sulfide (500 and 100 μm, respectively) showed any displacement of [3H]AZ8349 (Fig. 6, A and B). These data strongly suggest that the AZ GSMs share the same molecular target and that it is different from that of the NSAID class of GSMs.
FIGURE 6.

Displacement of 3H-labeled GSM by different GSMs on rat cryo-cut brain sections. A, rat slices (coronal, 10 μm) were either incubated with 5 nm [3H]AZ8349 alone (panel i) together with 0.5 mm (R)-flurbiprofen (panel ii) or together with 1 μm AZ4800 (panel iii). AZ4800, but not (R)-flurbiprofen, could displace the specific binding of [3H]AZ8349. B, graphical display illustrating the displacement binding studies of 5 nm [3H]AZ8349. Both AZ GSMs (10 μm) and E2012 displace [3H]AZ8349, whereas neither sulindac sulfide 0.1 mm nor (R)-flurbiprofen does (0.5 mm), indicating distinct interaction points. Binding is quantified as PSL/mm2, and data are presented as means ± S.E., n = 3. Tot, total; PSL, photostimulated luminescence.
AZ GSMs Decrease Aβ42 in the Brain of C57BL/6 Mice
Both AZ3303 and AZ4800 exhibit drug-like properties, warranting in vivo testing. In the next series of experiments, we asked whether these GSMs could decrease Aβ42 levels in the brain of wild type (C57BL/6) mice. AZ3303 and AZ4800 were administered as a single dose at different concentrations by oral gavage. The free concentrations of AZ3303 in the brain were 120 ± 30 and 570 ± 210 nm and for AZ4800 130 ± 35, 340 ± 80, and 880 ± 230 nm, respectively, at the doses tested. The brain/plasma ratios for AZ3303 and AZ4800 were 0.73 and 2.4, respectively (average of doses). Aβ42 levels were analyzed in diethylamine-extracted brain homogenates 1.5 h post-drug administration. Both AZ3303 and AZ4800 reduced Aβ42 in a dose-dependent manner (Fig. 7, A and B) by up to 25% for AZ3303 and up to 46% for AZ4800. These data show that both compounds readily reach the brain and exhibit expected CNS Aβ42 lowering activity.
FIGURE 7.
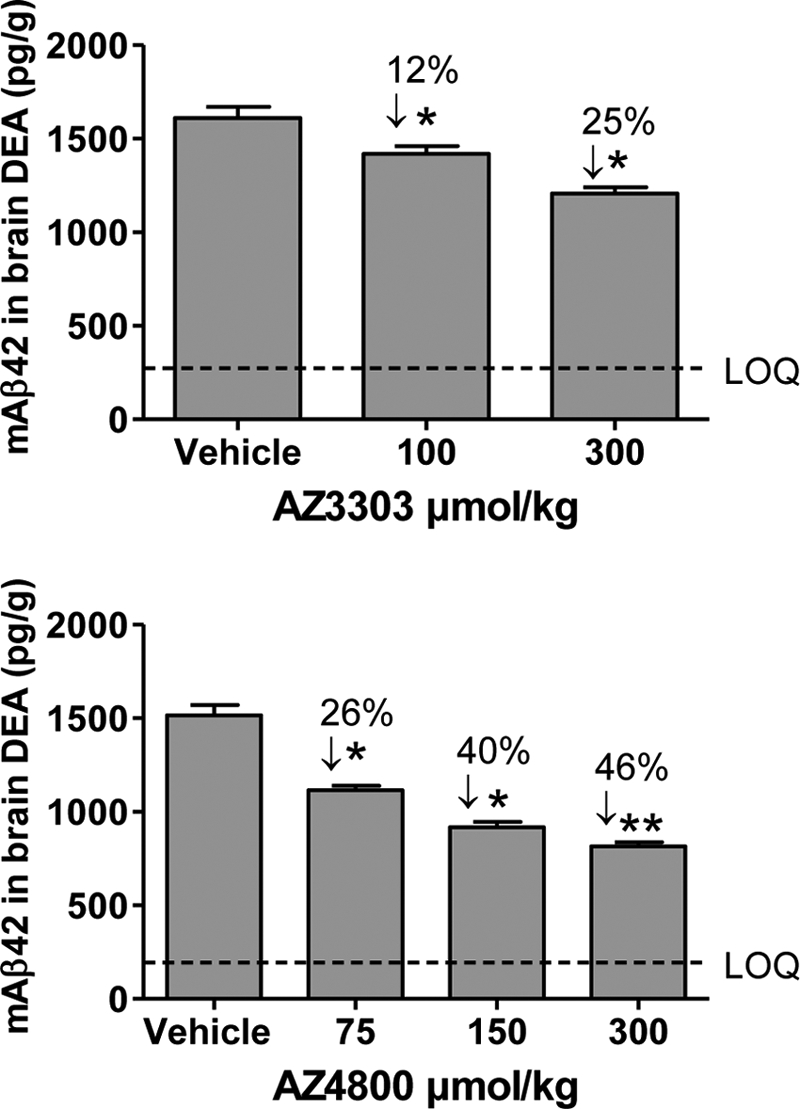
Effect of AZ GSMs on brain Aβ42 levels in C57BL/6 mice. Acute per oral dosing of AZ3303 (top) (100 and 300 μmol/kg) and AZ4800 (bottom) (75, 150, and 300 μmol/kg) causes a statistically significant decrease of Aβ42 levels in diethylamine-extracted brain homogenates at 1.5 h post-drug administration compared with the vehicle group in 12–18-week-old female C57BL/6 mice (n = 8–9 mice/group). Statistical analysis: one-way analysis of variance followed by Dunnett's multiple comparison test (**, p < 0.01; *, p < 0.05). LOQ, limit of quantitation.
DISCUSSION
Recent failures in clinical trials with γ-secretase-directed inhibitors, most likely because of mechanism-based toxicity as a result of impaired Notch signaling, and with the low potency Aβ modulator Tarenflurbil (i.e. (R)-flurbiprofen), highlight the need for alternative and tolerable therapeutic interventions at the level of Aβ production (3, 7). In this report, we describe several novel drug-like molecules (AZ GSMs), which lower Aβ42 levels in vivo and with nanomolar potency in cell culture experiments without affecting total Aβ levels nor the turnover of the γ-secretase substrates Notch, EphA4, or EphB2. Moreover, a 3H-labeled GSM does not co-localize with APP in the mouse brain but rather displays an excellent overlapping binding pattern with a γ-secretase targeting GSI in the rodent brain. Finally, the AZ GSMs exhibit both structural and pharmacological characteristics that are different from the NSAID class of APP-targeting GSMs but similar to the second generation GSM E2012. Combined, our pharmacological data describe a growing class of second generation GSMs, in which the mode of action is different from that of the first generation NSAID class of GSMs and appear to target γ-secretase rather than APP.
The specific mechanism by which these novel GSMs modulate Aβ production remains elusive, but a number of observations suggest that they act at the level of γ-secretase. First, they retain the pharmacology of Aβ modulation in an assay using sodium carbonate-washed membranes, which suggest that the molecular target is restricted to the membrane and membrane integral proteins. Second, binding studies using a 3H-labeled GSM analog revealed particularly strong labeling in the subventricular zone but also throughout the cortex, hippocampus, striatum, thalamus, and cerebellum in mouse, rat, and guinea pig brain sections. This expression pattern is indistinguishable from that obtained with the PS-targeting nontransition state GSI [3H]DBZ and is in line with a number of γ-secretase expression studies and autoradiographic studies using γ-secretase-selective molecular tools such as antibodies and radiolabeled GSIs (28–30). Thus, these observations strongly suggest that this GSM analog interacts either with the γ-secretase complex itself or with a specific γ-secretase-associated protein, such as the recently identified γ-secretase-activating protein GSAP (31). These findings stand in contrast to the mechanism of action described for the most well characterized class of GSMs, the NSAIDs. A growing body of data from a variety of biochemical experiments, such as cross-linking studies, suggests that GSMs of the NSAID class rather interact with APP than with γ-secretase (9, 10). Mechanistically, these molecules have been shown to affect Aβ production by interfering with the dimerization of the transmembrane domain of APP, which may result in an increased efficiency of γ-secretase-mediated processing of the APP transmembrane domain beyond Aβ40 and Aβ42, generating the shorter Aβ37 and Aβ38 peptides (10). Although this is a very elegant and tractable explanation to the GSM mode of action, several observations in our work suggest that GSMs of the NSAID class and the novel GSMs described in this work modulate Aβ production via separate mechanisms. First, (R)-flurbiprofen and sulindac sulfide could not displace the [3H]AZ GSM analog from rat brain tissue sections, suggesting that the binding site of (R)-flurbiprofen and sulindac sulfide is different from that of [3H]AZ GSM. Indeed, binding of [3H]AZ GSM was not affected by APP overexpression in APP TG mouse brain suggesting that the AZ GSMs characterized in this study do not bind to APP. Second, the GSMs of the NSAID class ((R)-flurbiprofen, and sulindac sulfide) and AZ4800 display a noncompetitive relationship in a cell-based cross-competition assay for Aβ42 production. Third, AZ4800 displays a competitive interaction in the same assay versus AZ3303 and AZ1136. Fourth, all AZ GSMs display a preference for PS2-expressing γ-secretases, whereas (R)-flurbiprofen and sulindac sulfide do not. Taken together, these observations suggest that the currently described GSMs of the non-NSAID type represent a class of Aβ modulators with a mode of action distinct from that of the NSAID class of GSMs. Interestingly, we recently learned that the Aβ modulatory activity of E2012 and the NSAIDs class of GSMs, respectively, are differentially affected by a number of FAD-causing PS mutants. Those observations indicate that the E2012 mode of action is different from that of GSMs of the NSAID class (13). In our experiments. E2012 displays a clear competitive relationship with AZ4800 and, similar to the AZ GSMs explored in this study, displays more potent GSM activity toward PS2-overexpressing cells compared with those expressing PS1. These data indicate the presence of several classes of GSMs with distinct modes of action, where E2012 and the novel GSMs reported in this study may share a similar mechanism of Aβ modulation. During the preparation and revision of this report, four separate publications were reported that support that hypothesis by using chemical cross-linking and GSM pulldown experiments, and second generation GSMs were shown to interact directly with the γ-secretase subunit Pen-2 or PS N-terminal fragment (12–16). Thus, a growing number of experiments suggest that second generation GSMs appear to modulate Aβ via a direct γ-secretase targeting mechanism. Moreover, similar to our observations on AZ GSMs and E2012, Ebke et al. (14) found that GSM RO-57-BpB appears more potent on PS2 compared with PS1 secretases. Currently, we do not have an explanation to the preferred modulation of PS2 over PS1 γ-secretases by AZ GSMs. Such information would be of interest and could potentially provide valuable guidance to the generation of GSMs with improved efficacy, because PS1 appears to be the major Aβ-generating enzyme of the brain (19). Although our data strongly suggest that the AZ GSMs modulate Aβ production via targeting γ-secretase, their specific mode of action remains to be determined. The finding that each molecule has a similar effect on Aβ40 and Aβ42 production but that their effect and efficacy on shorter Aβ peptides differ substantially is intriguing. Whether these GSMs affect APP dimerization indirectly via targeting γ-secretase or whether they affect Aβ generation via a distinct mechanism, not involving APP dimerization, remains to be elucidated. Recently, Ihara and co-workers (32) described the concept of Aβ product lines, where the ϵ-cleavage of APP is followed by a γ-secretase-mediated cleavage event every third to fourth amino acid in the APP trans-membrane-spanning helices, which causes the release of Aβ peptides of different lengths. Future studies will clarify how the second generation GSMs affect APP processing according to this model of γ-secretase-mediated Aβ production.
A feature of the original discoveries with GSMs of the NSAID type was their Notch sparing capacity (4). This characteristic is also inherited in the AZ GSMs. All compounds display an ∼1000-fold selectivity window when comparing their Aβ42 lowering activity with their effect on the nuclear translocation of the Notch intracellular domain, which is dependent on γ-secretase activity. In addition, AZ4800 did not affect γ-secretase processing of EphB2 and EphA4 (22, 23). This finding is of particular importance given the complex spectra of adverse events identified in clinical trials with GSIs. Thus, these data suggest that γ-secretase-targeting GSMs may provide a much better therapeutic window compared with GSIs. Importantly, we also show that AZ4800 and AZ3303 display good brain penetrating properties and can modulate Aβ42 levels in the brains of C57BL/6 mice after acute oral administration without any overt side effects. These encouraging data show that this novel class of GSMs is more potent than the first generation GSMs and that they could penetrate the blood-brain barrier and rapidly decrease brain Aβ42 levels.
In summary, we have discovered novel potent in vivo-active compounds that could modulate Aβ levels while sparing EphA4, EphB2, and Notch processing, a major challenge for γ-secretase-directed drugs in AD. Our pharmacological data strongly suggest that these compounds act at the level of γ-secretase rather than APP, as proposed for the first generation GSMs, and thus represent molecules exhibiting a novel mode of action for targeting Aβ production in AD therapeutics.
Supplementary Material
Acknowledgments
We are grateful to Drs. Frank Liu and Urban Lendahl for providing cell lines and Dr. Johan Lund for valuable comments to the manuscript.

This article contains supplemental Figs. S1–S7 and Methods.
- Aβ
- amyloid-β
- AD
- Alzheimer disease
- APP
- amyloid precursor protein
- GSM
- γ-secretase modulator
- NSAID
- nonsteroidal anti-inflammatory drug
- AZ
- AstraZeneca
- DBZ
- dibenzazepine
- NICD
- Notch intracellular domain
- GSI
- γ-secretase inhibitor
- total membranes PS
- presenilin
- CEM
- carbonate-extracted membrane
- TM
- total membrane
- MSD
- Meso Scale Discovery.
REFERENCES
- 1. Walsh D. M., Selkoe D. J. (2004) Deciphering the molecular basis of memory failure in Alzheimer disease. Neuron 44, 181–193 [DOI] [PubMed] [Google Scholar]
- 2. Haapasalo A., Kovacs D. M. (2011) The many substrates of presenilin/γ-secretase. J. Alzheimer Dis. 25, 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lundkvist J., Näslund J. (2007) γ-Secretase. A complex target for Alzheimer disease. Curr. Opin. Pharmacol. 7, 112–118 [DOI] [PubMed] [Google Scholar]
- 4. Weggen S., Eriksen J. L., Das P., Sagi S. A., Wang R., Pietrzik C. U., Findlay K. A., Smith T. E., Murphy M. P., Bulter T., Kang D. E., Marquez-Sterling N., Golde T. E., Koo E. H. (2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216 [DOI] [PubMed] [Google Scholar]
- 5. Eriksen J. L., Sagi S. A., Smith T. E., Weggen S., Das P., McLendon D. C., Ozols V. V., Jessing K. W., Zavitz K. H., Koo E. H., Golde T. E. (2003) NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ42 in vivo. J. Clin. Invest. 112, 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Portelius E., Van Broeck B., Andreasson U., Gustavsson M. K., Mercken M., Zetterberg H., Borghys H., Blennow K. (2010) Acute effect on the Aβ isoform pattern in CSF in response to γ-secretase modulator and inhibitor treatment in dogs. J. Alzheimer Dis. 21, 1005–1012 [DOI] [PubMed] [Google Scholar]
- 7. Wan H. I., Jacobsen J. S., Rutkowski J. L., Feuerstein G. Z. (2009) Translational medicine lessons from flurizan's failure in Alzheimer disease (AD) trial. Implication for future drug discovery and development for AD. Clin. Transl. Sci. 2, 242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beher D., Clarke E. E., Wrigley J. D., Martin A. C., Nadin A., Churcher I., Shearman M. S. (2004) Selected nonsteroidal anti-inflammatory drugs and their derivatives target γ-secretase at a novel site. Evidence for an allosteric mechanism. J. Biol. Chem. 279, 43419–43426 [DOI] [PubMed] [Google Scholar]
- 9. Kukar T. L., Ladd T. B., Bann M. A., Fraering P. C., Narlawar R., Maharvi G. M., Healy B., Chapman R., Welzel A. T., Price R. W., Moore B., Rangachari V., Cusack B., Eriksen J., Jansen-West K., Verbeeck C., Yager D., Eckman C., Ye W., Sagi S., Cottrell B. A., Torpey J., Rosenberry T. L., Fauq A., Wolfe M. S., Schmidt B., Walsh D. M., Koo E. H., Golde T. E. (2008) Substrate-targeting γ-secretase modulators. Nature 453, 925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richter L., Munter L. M., Ness J., Hildebrand P. W., Dasari M., Unterreitmeier S., Bulic B., Beyermann M., Gust R., Reif B., Weggen S., Langosch D., Multhaup G. (2010) Amyloid-β42 peptide (Aβ42)-lowering compounds directly bind to Aβ and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc. Natl. Acad. Sci. U.S.A. 107, 14597–14602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oehlrich D., Berthelot D. J., Gijsen H. J. (2011) γ-Secretase modulators as potential disease-modifying anti-Alzheimer drugs. J. Med. Chem. 54, 669–698 [DOI] [PubMed] [Google Scholar]
- 12. Kounnas M. Z., Danks A. M., Cheng S., Tyree C., Ackerman E., Zhang X., Ahn K., Nguyen P., Comer D., Mao L., Yu C., Pleynet D., Digregorio P. J., Velicelebi G., Stauderman K. A., Comer W. T., Mobley W. C., Li Y. M., Sisodia S. S., Tanzi R. E., Wagner S. L. (2010) Modulation of γ-secretase reduces β-amyloid deposition in a transgenic mouse model of Alzheimer disease. Neuron 67, 769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kretner B., Fukumori A., Gutsmiedl A., Page R. M., Luebbers T., Galley G., Baumann K., Haass C., Steiner H. (2011) Attenuated Aβ42 responses to low potency γ-secretase modulators can be overcome for many pathogenic presenilin mutants by second-generation compounds. J. Biol. Chem. 286, 15240–15251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ebke A., Luebbers T., Fukumori A., Shirotani K., Haass C., Baumann K., Steiner H. (2011) Novel γ-secretase enzyme modulators directly target presenilin protein. J. Biol. Chem. 286, 37181–37186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ohki Y., Higo T., Uemura K., Shimada N., Osawa S., Berezovska O., Yokoshima S., Fukuyama T., Tomita T., Iwatsubo T. (2011) Phenylpiperidine-type γ-secretase modulators target the transmembrane domain 1 of presenilin 1. EMBO J. 30, 4815–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crump C. J., Fish B. A., Castro S. V., Chau D. M., Gertsik N., Ahn K., Stiff C., Pozdnyakov N., Bales K. R., Johnson D. S., Li Y. M. (2011) Piperidine acetic acid-based γ-secretase modulators directly bind to Presenilin-1. ACS Chem. Neurosci. 2, 705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malmquist J., Bernlind A., Sandell J., Ström P., Waldman M. (2012) J. Label. Comp. Pharm., 55, 80–83 [Google Scholar]
- 18. Strömberg K., Hansson E. M., Laudon H., Bergstedt S., Näslund J., Lundkvist J., Lendahl U. (2005) γ-Secretase complexes containing N- and C-terminal fragments of different presenilin origin retain normal γ-secretase activity. J. Neurochem. 95, 880–890 [DOI] [PubMed] [Google Scholar]
- 19. Borgegård T., Minidis A., Juréus A., Malmborg J., Rosqvist S., Gruber S., Almqvist H., Yan H., Bogstedt A., Olsson F., Dahlström J., Ray C., Närhi K., Malinowsky D., Hagström E., Jin S., Malmberg A., Lendahl U., Lundkvist J. (2011) In vivo analysis using a presenilin-1-specific inhibitor. Presenilin-1-containing γ-secretase complexes mediate the majority of CNS Aβ production in the mouse. Alzheimer Dis. Res. J. 3, 29–45 [Google Scholar]
- 20. McLendon C., Xin T., Ziani-Cherif C., Murphy M. P., Findlay K. A., Lewis P. A., Pinnix I., Sambamurti K., Wang R., Fauq A., Golde T. E. (2000) Cell-free assays for γ-secretase activity. FASEB J. 14, 2383–2386 [DOI] [PubMed] [Google Scholar]
- 21. Shearman M. S., Beher D., Clarke E. E., Lewis H. D., Harrison T., Hunt P., Nadin A., Smith A. L., Stevenson G., Castro J. L. (2000) L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid-β protein precursor γ-secretase activity. Biochemistry 39, 8698–8704 [DOI] [PubMed] [Google Scholar]
- 22. Inoue E., Deguchi-Tawarada M., Togawa A., Matsui C., Arita K., Katahira-Tayama S., Sato T., Yamauchi E., Oda Y., Takai Y. (2009) Synaptic activity prompts γ-secretase-mediated cleavage of EphA4 and dendritic spine formation. J. Cell Biol. 185, 551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Litterst C., Georgakopoulos A., Shioi J., Ghersi E., Wisniewski T., Wang R., Ludwig A., Robakis N. K. (2007) Ligand binding and calcium influx induce distinct ectodomain/γ-secretase-processing pathways of EphB2 receptor. J. Biol. Chem. 282, 16155–16163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Milano J., McKay J., Dagenais C., Foster-Brown L., Pognan F., Gadient R., Jacobs R. T., Zacco A., Greenberg B., Ciaccio P. J. (2004) Modulation of notch processing by γ-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol. Sci. 82, 341–358 [DOI] [PubMed] [Google Scholar]
- 25. Tian G., Ghanekar S. V., Aharony D., Shenvi A. B., Jacobs R. T., Liu X., Greenberg B. D. (2003) The mechanism of γ-secretase. Multiple inhibitor binding sites for transition state analogs and small molecule inhibitors. J. Biol. Chem. 278, 28968–28975 [DOI] [PubMed] [Google Scholar]
- 26. Juréus A., Swahn B. M., Sandell J., Jeppsson F., Johnson A. E., Johnström P., Neelissen J. A., Sunnemark D., Farde L., Svensson S. P. (2010) Characterization of AZD4694, a novel fluorinated Aβ plaque neuroimaging PET radioligand. J. Neurochem. 114, 784–794 [DOI] [PubMed] [Google Scholar]
- 27. Churcher I., Beher D., Best J. D., Castro J. L., Clarke E. E., Gentry A., Harrison T., Hitzel L., Kay E., Kerrad S., Lewis H. D., Morentin-Gutierrez P., Mortishire-Smith R., Oakley P. J., Reilly M., Shaw D. E., Shearman M. S., Teall M. R., Williams S., Wrigley J. D. (2006) 4-substituted cyclohexyl sulfones as potent, orally active γ-secretase inhibitors. Bioorg. Med. Chem. Lett. 16, 280–284 [DOI] [PubMed] [Google Scholar]
- 28. Yan X. X., Li T., Rominger C. M., Prakash S. R., Wong P. C., Olson R. E., Zaczek R., Li Y. W. (2004) Binding sites of γ-secretase inhibitors in rodent brain. Distribution, postnatal development, and effect of deafferentiation. J. Neurosci. 24, 2942–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Elder G. A., Tezapsidis N., Carter J., Shioi J., Bouras C., Li H. C., Johnston J. M., Efthimiopoulos S., Friedrich V. L., Jr., Robakis N. K. (1996) Identification and neuron-specific expression of the S182/presenilin I protein in human and rodent brains. J. Neurosci. Res. 45, 308–320 [DOI] [PubMed] [Google Scholar]
- 30. Blanchard V., Czech C., Bonici B., Clavel N., Gohin M., Dalet K., Revah F., Pradier L., Imperato A., Moussaoui S. (1997) Immunohistochemical analysis of presenilin 2 expression in the mouse brain. Distribution pattern and co-localization with presenilin 1 protein. Brain Res. 758, 209–217 [DOI] [PubMed] [Google Scholar]
- 31. He G., Luo W., Li P., Remmers C., Netzer W. J., Hendrick J., Bettayeb K., Flajolet M., Gorelick F., Wennogle L. P., Greengard P. (2010) γ-Secretase activating protein is a therapeutic target for Alzheimer disease. Nature 467, 95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takami M., Nagashima Y., Sano Y., Ishihara S., Morishima-Kawashima M., Funamoto S., Ihara Y. (2009) γ-Secretase. Successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl-terminal fragment. J. Neurosci. 29, 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.