

Abstract
Neurological disorders can be modeled in animals so as to recreate specific pathogenic events and behavioral outcomes. Parkinson's Disease (PD) is the second most common neurodegenerative disease of an aging population, and although there have been several significant findings about the PD disease process, much of this process still remains a mystery. Breakthroughs in the last two decades using animal models have offered insights into the understanding of the PD disease process, its etiology, pathology, and molecular mechanisms. Furthermore, while cellular models have helped to identify specific events, animal models, both toxic and genetic, have replicated almost all of the hallmarks of PD and are useful for testing new neuroprotective or neurorestorative strategies. Moreover, significant advances in the modeling of additional PD features have come to light in both classic and newer models. In this review, we try to provide an updated summary of the main characteristics of these models as well as the strengths and weaknesses of what we believe to be the most popular PD animal models. These models include those produced by 6-hydroxydopamine (6-OHDA), 1-methyl-1,2,3,6-tetrahydropiridine (MPTP), rotenone, and paraquat, as well as several genetic models like those related to alpha-synuclein, PINK1, Parkin and LRRK2 alterations.
1. Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease, affecting 1% of the population over 55 years of age [1]. This disease is characterized by the loss of ~50–70% of the dopaminergic neurons in the substantia nigra pars compacta (SNc), a profound loss of dopamine (DA) in the striatum, and the presence of intracytoplasmic inclusions called Lewy bodies (LB), which are composed mainly of α-synuclein and ubiquitin. Although mutations in the α-synuclein gene have thus far been associated only with rare familial cases of PD, α-synuclein is found in all LBs [2]. Therefore, this protein may play an important role in the pathogenesis of this disease. The main features of PD are tremor, rigidity, bradykinesia, and postural instability; however, these motor manifestations can be accompanied by nonmotor symptoms such as olfactory deficits, sleep impairments, and neuropsychiatric disorders [3–5]. Although the complete PD disease process is not yet understood, we have gained a better understanding of its etiology, pathology, and molecular mechanisms, thanks to various animal models [6]. For example, reserpine administration in animals was found to produce a profound depletion of monoamines, including DA, in the brains of injected animals resulting in reserpine syndrome. The symptoms of this syndrome consisted of slowness of movement and rigidity [7] now commonly associated with PD. Interestingly, it was found that L-DOPA was able to reverse many of the symptoms associated with reserpine administration [8], furthering the hypothesis that DA depletion was at the root of PD symptomatology.
For the past several decades, animal models of PD have come in a variety of forms. Typically, they can be divided into those using environmental or synthetic neurotoxins or those utilizing the in vivo expression of PD-related mutations (genetic).
Of the neurotoxic models, compounds that produce both reversible (reserpine) and irreversible (MPTP, 6-OHDA, paraquat, rotenone) effects have been used effectively; however recent studies have focused more on irreversible toxins to produce PD-related pathology and symptomatology. Therefore, the neurotoxins covered in this paper will focus on those that produce an irreversible effect. Neurotoxin-based models produced by 6-hydroxydopamine (6-OHDA) and 1-methyl-1,2,3,6-tetrahydropyridine (MPTP) administration are the most widely used toxic models, while paraquat and rotenone are more recent additions to the stable of toxic agents used to model PD [6, 9]. A common feature of all toxin-induced models is their ability to produce an oxidative stress and to cause cell death in DA neuronal populations that reflect what is seen in PD. Oxidative stress results from increased production of extremely reactive free radicals, including reactive oxidative species (ROS) and peroxynitrite. ROS may be formed during a number of cellular processes, including mitochondrial oxidative respiration and metabolism. There are some drawbacks to the use of these models such as the time factor in these models versus the time factor in the human condition, but these do not negate the value of neurotoxin-based animal models in the study of PD.
Recently, the identification of different genetic mutations (α-synuclein, parkin, LRKK2, PINK1, DJ-1) has led to the development of genetic models of PD [10]. It is important to remember that, at best, only ~10% of PD cases are due to genetic mutations [6], while the vast majority of PD cases arise as sporadic, that is, from unknown origins. Although the above-mentioned genes are mutated in PD and are not overexpressed or knocked out, nonetheless, these animal models are important in that they may reveal specific molecular events that lead to the death of the DA neurons and potential therapeutic targets. In this paper, we try to describe the advantages and disadvantages of all of these animal models and their potential roles in revealing the mechanisms for PD pathogenesis and in testing experimental therapeutics (Table 1).
Table 1.
| Model | Behavioral symptoms | Nigrostriatal damage | Synuclein aggregation/Lewy body formation | Uses of the model | Disadvantages |
|---|---|---|---|---|---|
| 6-OHDA | Rotational behavior after unilateral injection | Loss of DA innervation at injection site (striatum) | No inclusions | Screen therapies that may improve PD symptoms. Study mechanisms of cell death | Requires intracerebral injection, very little synuclein involvement. |
| MPTP | Motor impairments in primates Less obvious motor impairments in acute rodent models |
Loss of DA neurons dependent on dosing regimen, reaching 95% in acute high-dose conditions. Reduced DA levels in striatum concurrent with midbrain DA neuron loss |
Inclusions not prevalent. Few cases of synuclein aggregation in nonhuman primates, as well as increased synuclein immunoreactivity in rodents. | Screen therapies that may improve PD symptoms. Study mechanisms of cell death | Nonprogressive model of cell death. Inclusiones are rare. |
| Rotenone | Reports of decreased motor activity in rodents | Loss of DA neurons accompanied by reduced DA innervation in striatum | Synuclein aggregation in DA neurons. | Test neuroprotective compounds | Substantial morbidity and mortality. Labor and time intensive. |
| Paraquat | No clear motor deficits | Decreased striatal TH immunoreactivity | No inclusions present, but increased synuclein immunoreactivity in DA neurons of the SN | Test neuroprotective strategies | Not extensively tested. Effects in other neurotransmitter systems. |
| α-synuclein | Severe motor deficits in the A53T model, less in the A30P model | Generally no DA neuron degeneration observed | Synuclein aggregation found in DA neurons, generally restricted to A53T model | Study the role of synuclein aggregation in PD, as well as the normal role of synuclein | Generally no DA neuron death observed with synuclein models |
| LRRK2 | Few behavioral deficits seen in Drosophila mutation models | No effect on DA development or maintenance in knockouts, minimal levels of degeneration in mutation models | Generally not observed | Study the role of LRRK2 mutations related to PD | General lack of degeneration and general lack of synuclein aggregation. |
2. Neurotoxic Models
2.1. 6-Hydroxydopamine
6-OHDA is the classic and oft utilized toxin-based animal model of PD [11–13]. A lot of information on the behavioral, biochemical, and physiological effects of dopamine in the CNS has been derived from this model. 6-OHDA was first isolated in the 1950s [14]. Ungersted [15] first used this neurotoxin to lesion the nigro-striatal dopaminergic pathway in the rat nearly 50 years ago, and the use of 6-OHDA remains widespread today for both in vitro and in vivo investigations. Mice, cats, dogs, and monkeys are all sensitive to 6-OHDA; however it is used much more frequently in rats [16–19]. Even though 6-OHDA exhibits a high affinity for several catecholaminergic transporters such as the dopamine transporter (DAT) and norepinephrine transporter (NET) [20], it is often used in conjunction with a selective noradrenaline reuptake inhibitor such as desipramine in order to spare the noradrenergic neurons from damage in animal models of PD [21].
Although the structure of 6-OHDA is similar to that of dopamine, the presence of an additional hydroxyl group makes it toxic to dopaminergic neurons. This compound does not cross the blood-brain barrier, which necessitates its direct injection into the SNpc, medial forebrain bundle (MFB), or the striatum [22, 23]. It is well known that 6-OHDA destroys catecholaminergic neurons by a combined effect of ROS and quinones [24], and it can induce inflammation in the brain which tends to wane over time. The most common use of 6-OHDA is via unilateral injection into the rat medial forebrain bundle. Injection of 6-OHDA into the SNpc kills approximately 60% of the tyrosine hydroxylase- (TH-) containing neurons in this area of the rodent brain with subsequent loss of TH-positive terminals in the striatum [25] (Figure 1). Several studies have injected this compound directly into the striatum in order to test the hypothesis of retrograde degeneration, explicitly, that TH-positive terminals in the striatum die prior to TH-positive neurons in the SNpc, seemingly a replicate of PD in humans [23, 26, 27]. The magnitude of the lesion depends on the amount of 6-OHDA injected, the site of injection, and the animal species used. This model does not mimic all of the clinical features of PD. Dopamine depletion, nigral dopamine cell loss, and neurobehavioral deficits have been successfully achieved using this model, but it does not seem to affect other brain regions, such as olfactory structures, lower brain stem areas, or locus coeruleus. Although 6-OHDA does not produce or induce proteinaceous aggregates or Lewy-like inclusions like those seen in PD, it has been reported that 6-OHDA does interact with α-synuclein [25]. 6-OHDA is frequently used as a unilateral model because the bilateral injection of this compound into the striatum produces severe adipsia, aphagia, and also death [28, 29] due to the animal's inability to care for itself. One of the most attractive features of the unilateral 6-OHDA model is the fact that each animal can serve as its own control as there is a lesioned and an unlesioned hemisphere. This is particularly useful in behavioral analyses [15] as turning behavior to amphetamine or apomorphine following the unilateral application of 6-OHDA gauges the extent of the induced SNpc or striatal lesion and the efficacy of potential PD therapeutic agents and gene therapies [11, 30].
Figure 1.
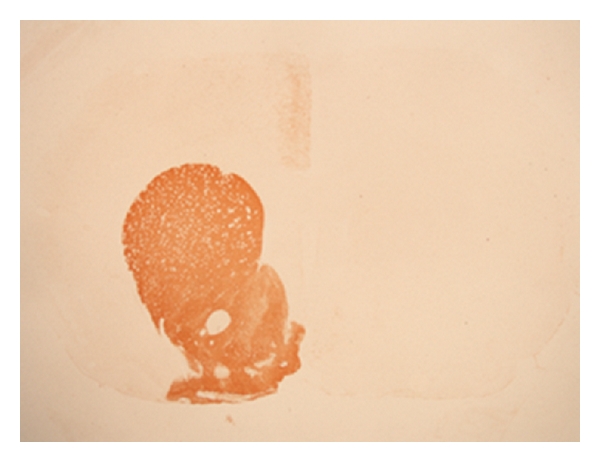
Photomicrograph of a 6-OHDA lesioned rat striatum immunostained for tyrosine hydroxylase (TH). Densities of TH-immunoreactivity striatal fibers are clearly reduced after the 6-OHDA injection (right side) as compared to the densities of striatal TH-immunoreactivity fibers in control rat (left side).
6-OHDA is an attractive candidate as a possible endogenous toxin for the initiation of the PD neurodegenerative process as it is a product of dopamine metabolism [31], and it is the result of hydroxyl radical attack with the presence of excess dopamine; as a neurotoxin, it does produce lesions in the nigrostriatal DA pathway. However, although it has been measured in the brains of levodopa-treated rats subjected to MPTP treatment, 6-OHDA has yet to be recovered from the PD brain. Despite its limitations, this model has contributed enormously to our understanding of PD pathology. 6-OHDA will continue to afford PD researchers a useful animal model for PD research for long time.
2.2. MPTP
In 1982, MPTP was accidentally discovered in a synthesis process gone awry, and, although it may have caused some mayhem in certain circles, today it represents the most important and most frequently used parkinsonian toxin applied in animal models. Young drug addicts developed an idiopathic parkinsonian syndrome after intravenous injection of this compound. After investigating the etiology of their condition, it was found that MPTP was the neurotoxic contaminant responsible for the parkinsonian effect [32]. Oxidative stress, ROS, energy failure, and inflammation have consistently been pointed to as hallmarks of PD. It has been repeatedly demonstrated that MPTP is indeed the gold standard for toxin-based animal models of PD among PD researchers for replicating almost all of these hallmarks [32]. Unfortunately, lacking in this list is the definitive characteristic of PD, LB formation [33, 34]. Interestingly, some studies have demonstrated the production of Lewy body-like inclusions after MPTP administration [35, 36] although these studies have been difficult to replicate. These studies suggest that, under the right circumstances, we may be able to reproduce the majority of hallmarks found in PD.
MPTP is highly lipophilic and after systemic administration rapidly crosses the blood-brain barrier. Once in the brain, MPTP enters astrocytes and is metabolized into MPP+, its active metabolite, by monoamine oxidase-B (MAO-B). Recent findings show that once released from the astrocytes into the extracellular space via the OCT-3 transporter [37], MPP+ is taken up into the neuron by the dopamine transporter (DAT) and can be stored in vesicles via uptake by the vesicular monoamine transporter (VMAT2) [38]. Consequently, mice lacking the DAT are protected from MPTP toxicity [39]. Once inside the neuron, MPP+ is able to inhibit complex 1 of the mitochondrial electron transport chain, resulting in the release of ROS as well as reduced ATP production. Storing into vesicles can decrease MPP+ toxicity [40–42]. Additionally, MPP+ stored in vesicles is thought to expel DA out into the intercellular space where it can be metabolized into a number of compounds, including toxic metabolites, such as DOPAL and where it is can be subjected to superoxide radical (5-cysteinyl-DA) and hydroxyl radical (6-OHDA) attack [43, 44].
MPTP is used mainly in nonhuman primates and mice but has also been used in many other species such as dogs and cats [45]. For unknown reasons, rats are resistant to MPTP and mouse strains vary widely in their sensitivity to the toxin [46]. MPTP can be administered by a variety of regimens, but the most common and reproducible form is still systemic injection (subcutaneous, intravenous) [47]. When MPTP is administered to nonhuman primates, they exhibit behavioral and neuroanatomical similarities to the human condition showing a bilateral parkinsonian syndrome [48] (Figure 2). Another commonly used route is the unilateral intracarotid injection. This causes mostly a unilateral parkinsonism, whose benefits as an animal model were described earlier, but is technically more complicated to perform [49].
Figure 2.
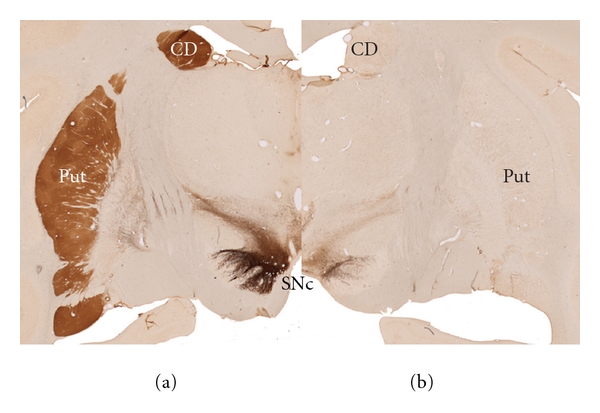
Photomicrographs of nonhuman primate immunostained for tyrosine hydroxylase (TH). Dopaminergic neurons located in the substantia nigra compacta (SNc) project to the caudate (CD) and putamen (PUT). Note the markedly reduced TH immunoreactivity in the substantia nigra and striatum (CD and PUT) in the MPTP-treated monkey (b) compared to control (a).
Usually monkeys are treated with high doses of MPTP for a short time (acute model). Recently, however, new schedules have introduced lower doses of the neurotoxin for longer periods of time (subacute to chronic) to replicate more closely the human pathology [50]. There are recent studies attempting to develop a more progressive model of PD. In addition, models are being developed to study compensatory mechanisms or recovery. These models use low to intermittent doses administered once or twice per week [51–54]. It is well known that monkeys exhibit variability in MPTP susceptibility and that older primates are often more susceptible to MPTP [55]. MPTP-treated monkeys respond well to antiparkinsonian treatments like L-DOPA or apomorphine and, like human pathology, after the treatment develop dyskinesias. Recently, some studies have been taken in order to study and evaluate the nonmotor symptoms of the disease using this model [56–61]. This model has also been used for electrophysiological studies, leading to important findings, including the emergence of deep brain stimulation, which is currently the best surgical method to ameliorate symptoms in PD patients [62, 63].
Currently, the MPTP model is used more in mice than in monkeys. Aside from the obvious financial benefits, the mouse model is employed to test theories about cell death in PD, to work out events in the neuronal death process, and to study other pathological effects of PD. It is also extremely useful as an initial screening tool to test potential treatments for PD. On the other hand, the MPTP monkey model is mainly used to discern behavioral and symptomatic components of PD, as mice do not develop a level of impairment equal to the human condition. Monkeys also represent the last level of PD treatment research prior to any treatment being administered to humans [64]. However, the data generated by mouse models has led to a better understanding of molecular mechanisms involved in PD, and its utility has proven invaluable. One of the most important aspects of the MPTP mouse model is the possibility to work with genetically modified mice [65, 66]. This model can be useful for testing neuroprotective therapies. Currently, MPTP is the standard bearer for toxin-based PD animal models.
3. Pesticide/Herbicide Models
3.1. Paraquat
Paraquat (N,N′-dimethyl-4-4-4′-bypiridinium) (PQ) is a herbicide widely used in agriculture that exhibits a structural resemblance to MPP+, and, because of this structural similarity, it was reasoned that PQ should behave like MPP+. Epidemiological reports suggest that pesticide use increases the risk of developing PD, but, in the case of PQ, there have been only 95 cases of PD linked to its toxicity in humans [67]. PQ exerts its deleterious effects through oxidative stress mediated by redox cycling, which generates ROS. In particular, the superoxide radical, hydrogen peroxide, and hydroxyl radicals lead to the damage of lipids, proteins, DNA and RNA [68, 69]. Recent evidence on the effects of PQ in the nigrostriatal DA system is somewhat ambiguous as some researchers report that, following the systemic application of this herbicide to mice, animals exhibit reduced motor activity and a dose-dependent loss of striatal TH-positive striatal fibers and midbrain SNpc neurons [70, 71]. Other researchers claim that no PQ-induced changes occur in the nigrostriatal DA system [72, 73]. However, in a newer recent study, Rappold et al. [74] demonstrate that PQ, in high doses, employs the organic cationic transporter-3 (OCT-3) and the dopamine transporter (DAT) and is toxic to the DA neurons in the SN. Furthermore, they suggest that the damage done by PQ is caused by radicalized PQ and facilitated by the glial cells. This means that PQ behaves like MPP+ in exerting its toxic effects. Although this study increases our understanding of how PQ may work, it does not end the controversy about PQ and PD.
PQ's importance to PD researchers is its ability to induce increases in α-synuclein in individual DA neurons in the SNpc and its ability to induce LB-like structures in DA neurons of the SNpc [75]. The relation of dopaminergic neuron loss with α-synuclein upregulation and aggregation suggests that this model could be valuable for capturing a PD-like pathology. However, the molecular link between oxidative stress and cell death in this model is still unknown. Thus, the significance of PQ in PD research is often limited to the study of the process of LB formation in DA neurons as well as the role of α-synuclein in PD. PQ is only one of the many agricultural chemicals known to cause damage to the dopaminergic system. Maneb (manganese ethylenebisdithiocarbamate) has been shown to decrease locomotor activity and potentiate both the MPTP and the PQ effects [73, 76, 77]. Moreover, the combination of PQ and maneb produced greater effects on the dopaminergic system than either of these chemicals alone. These compounds give credence to the theory that environmental pesticides can cause PD [67, 78–80]. In fact, recent studies have demonstrated that those exposed to PQ or fungicides like maneb or ziram experience a greater risk of developing PD [81, 82]. Further investigations using these models are needed to determine the involvement of environmental exposures in the etiology of PD.
3.2. Rotenone
Unlike PQ, which is a pure herbicide rotenone, is both a herbicide and an insecticide [83]. It is the most potent member of the rotenoid family of neurotoxins found naturally in tropical plants. The half-life of rotenone is 3–5 days depending on its exposure to sunlight, and it is rapidly broken down in soil and in water [83]. For these reasons, rotenone is not considered to be a groundwater pollutant. Rotenone is highly lipophilic and readily crosses the blood-brain barrier. Chronic exposure to low doses of rotenone results in inhibition of the mitochondrial electron transport chain in the rat brain. In animals, rotenone has been administered by different routes. Oral administration appears to cause little neurotoxicity [84, 85]. Chronic systemic administration using osmotic pumps has been the most common delivery regimen, especially in the Lewis rat, which may be more sensitive to rotenone than other strains of rats [86]. Intraperitoneal injections have been reported to elicit behavioral and neurochemical deficits, although mortality is very high [87]. Intravenous administration is able to cause damage to nigrostriatal DA neurons that is accompanied by α-synuclein aggregation, Lewy-like body formation, oxidative stress, and gastrointestinal problems [88]. The apparent beauty of this model is that, like paraquat, it seems to replicate almost all of the hallmarks of PD including causing α-synuclein aggregation and Lewy-like body formation [89, 90]. Interestingly, a subsequent study has found that rotenone is not specific to the DA system and has deleterious effects on other neuronal populations. Likewise, in PD in which neurodegeneration extends beyond the dopaminergic system, rotenone is associated with 35% reduction in serotonin, 26% in noradrenergic, and 29% in cholinergic neurons [89]. However, when rotenone was administered chronically at lower doses to achieve complex I inhibition similar to that observed in patients, it seems to produce a highly selective nigrostriatal degeneration [86] although only about 50% of the treated rats exhibit nigrostriatal lesions. The controversy about the use of rotenone as a model of PD is that although it does augment DA oxidation, evidence is slim on depletion of DA in the nigrostriatal system [91]. Attempts to lesion other animal species such as mice or monkeys have not been successful at all [72, 92]. However, recent studies by two groups have demonstrated that oral administration of rotenone to mice causes nigral degeneration, a decrease of striatal dopamine levels, and motor dysfunction [85, 93, 94]. They also demonstrated α-synuclein aggregation in different areas of the brain [95]. Furthermore, there are no documented cases of rotenone-induced PD in humans. Thus, it is not clear that this model offers any advantage over other toxic models, such as that of 6-OHDA or MPTP.
4. Genetic Models
The underlying principle for studying genetic mutations of a disease is the belief that the clinical similarities between the inherited and sporadic forms of the disease share a common mechanism that can lead to the identification of molecular and biochemical pathways involved in the disease pathogenesis. Genetic mutations in PD are rare and represent only about 10% of all PD cases [6]. And animal models of these mutations (α-synuclein and LRRK2, autosomal dominant PD) and (PINK1/Parkin and DJ-1, autosomal recessive PD) are important as they represent potential therapeutic targets. However, we must first understand the workings of these animal models because it is becoming clearer that there are many facets to PD disease.
Mutations to the α-synuclein gene, which is normally thought to play a role in the synaptic vesicle recycling, were the earliest evidence for genetic link to PD. Two mutations in the α-synuclein gene (A53T, A30P) cause a dominantly inherited form of PD [96] and have been used to create transgenic mice in an effort to recapitulate the pathophysiology of PD. Studies done using α-synuclein transgenic mice have yielded considerable progress, showing that A53T mutations in mice can result in a severe motor phenotype which can eventually lead to paralysis and death [97]. Additionally, mutations to the α-synuclein gene in mice produce inclusions that resemble LBs [98]. However this phenotype is generally restricted to the A53T mutation and not found in A30P transgenic mice. Indeed, it has been shown that knocking out α-synuclein does not affect DA neuron development or maintenance [99, 100] suggesting that the loss of α-synuclein probably plays no role in the degeneration of DA neurons that is seen in PD. Interestingly, studies done in Drosophila expressing mutant α-synuclein show dopaminergic cell loss, reduced TH expression in the SN, filamentous intraneuronal inclusions, and motor deficits [101]. Some of the α-synuclein transgenic mice have olfactory impairments and colonic dysfunction, and it seems that there are other nonmotor abnormalities [102]. Understanding these nonmotor symptoms could offer new model for testing therapies focused on the nonmotor symptoms. However, since the function of α-synuclein has yet to be figured out, the actual role of α-synuclein in PD still remains elusive.
Mutations to the LRRK2 gene have been shown to cause a dominant form of PD [103]. Unlike α-synuclein which is ubiquitous, LRRK2 (leucine rich repeat kinase 2) is localized to membranes. However, similar to α-synuclein transgenic mice, it has been determined that knocking out LRRK2 has no effect on DA neuron development and maintenance [102]. Moreover, Drosophila models are limited in their translation to the human condition, and the LRRK2 mouse model is not particularly a strong model as it shows only minimal levels of neurodegeneration [104].
Mutations to parkin (which accounts for about 50% of the familial cases of PD and 20% of the young onset PD cases), DJ1 (a redox sensitive molecular chaperone and regulator of antioxidant gene expression), and PINK1 (phosphatase and tensin homolog—PTEN-induced novel kinase 1, which is localized to the mitochondrial intermembrane space) cause autosomal recessive forms of PD. Knock-out rodent models of these genes do not demonstrate any nigrostriatal degeneration, present with intranucelar inclusions, or displays any form of DA neuron loss that resembles idiopathic or inherited PD and fail to develope any type of behavioral or pathological phenotype (only PINK1 knock-out mice display reduced DA release in the striatum) [105]. However, recently it has been shown that knocking out parkin in mice at adult age causes neurodegeneration in the SNc [106].
Overall, this genetic mouse models are able to recapitulate specific aspects of PD, although none produce the neuronal degeneration associated with PD; therefore these themselves may be defective and may require additional modulations or modifications, like for example the human environment [107].
5. Conclusions
Animal model systems are the closest to humans that we are able to study. A number of animal models of PD have been developed to understand the pathogenesis and test potential therapeutics of this disease. In this paper we have summarized the most prominent aspects characterizing the most popular toxic and genetic models of PD. Each model has advantages and disadvantages as we have discussed in this paper. Toxic models offer some of the hallmarks of PD while genetic models offer others. Meanwhile the toxic models are useful to screen drugs for symptomatic treatment of the disease; transgenic or knockout models are useful for evaluating the role of genetics in PD. The drawback of the toxin models is that most of them resemble PD at late stages, whereas genetic animal models use either overexpression or knock-out technology to model disease from early on. The choice of the model to be used depends on the questions being asked. With toxin models, we are working toward developing a progressive model by tempering the toxic doses used. With genetic models, we are trying to come up with the right balance of contributing components through knock-in or conditional technology. However, there is much progress to be made, because it seems unlikely that a single model, be it toxic or genetic, can fully recapitulate the complexity of human PD. Future models should involve a combination of neurotoxin-induced and genetically induced models ideally taking into account factors of aging and environmental insults.
Acknowledgments
The authors are supported by grants from the National Institutes of Health (NS042269, NS064191, NS38370, NS070276, and NS072182), the US Department of Defense (W81XWH-08-1-0522, W81XWH-08-1-0465, and W81XWH-09-1-0245), the Parkinson Disease Foundation, the Thomas Hartman Foundation For Parkinson's Research, Project A.L.S, the Muscular Dystrophy Association/Wings over Wall Street. J. Blesa is supported by a fellowship from the Spanish Ministry of Education.
References
- 1.Lees AJ, Hardy J, Revesz T. Parkinson’s disease. The Lancet. 2009;373(9680):2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies [8] Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Forno LS. Neuropathology of Parkinson’s disease. Journal of Neuropathology and Experimental Neurology. 1996;55(3):259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell and Tissue Research. 2004;318(1):121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhuri KR, Healy DG, Schapira AHV. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurology. 2006;5(3):235–245. doi: 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- 6.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 7.Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180(4596):p. 1200. doi: 10.1038/1801200a0. [DOI] [PubMed] [Google Scholar]
- 8.Carlsson A. The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacological reviews. 1959;11(2):490–493. [PubMed] [Google Scholar]
- 9.Betarbet R, Sherer TB, Timothy Greenamyre J. Animal models of Parkinson’s disease. BioEssays. 2002;24(4):308–318. doi: 10.1002/bies.10067. [DOI] [PubMed] [Google Scholar]
- 10.Dawson TM, Ko HS, Dawson VL. Genetic Animal Models of Parkinson’s Disease. Neuron. 2010;66(5):646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarting RKW, Huston JP. Unilateral 6-hydroxydopamine lesions of meso-striatal dopamine neurons and their physiological sequelae. Progress in Neurobiology. 1996;49(3):215–266. doi: 10.1016/s0301-0082(96)00015-9. [DOI] [PubMed] [Google Scholar]
- 12.Schwarting RKW, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Progress in Neurobiology. 1996;50(2-3):275–331. doi: 10.1016/s0301-0082(96)00040-8. [DOI] [PubMed] [Google Scholar]
- 13.Schwarting RKW, Huston JP. Behavioral and neurochemical dynamics of neurotoxic meso-striatal dopamine lesions. NeuroToxicology. 1997;18(3):689–708. [PubMed] [Google Scholar]
- 14.Senoh S, Witkop B. Non-enzymatic conversions of dopamine to norepinephrine and trihydroxyphenethylamines. Journal of the American Chemical Society. 1959;81(23):6222–6231. [Google Scholar]
- 15.Ungerstedt U. 6-hydroxy-dopamine induced degeneration of central monoamine neurons. European Journal of Pharmacology. 1968;5(1):107–110. doi: 10.1016/0014-2999(68)90164-7. [DOI] [PubMed] [Google Scholar]
- 16.Roeling TAP, Docter GJ, Voorn P, Melchers BPC, Wolters EC, Groenewegen HJ. Effects of unilateral 6-hydroxydopamine lesions on neuropeptide immunoreactivity in the basal ganglia of the common marmoset, Callithrix jacchus, a quantitative immunohistochemical analysis. Journal of Chemical Neuroanatomy. 1995;9(3):155–164. doi: 10.1016/0891-0618(95)00072-0. [DOI] [PubMed] [Google Scholar]
- 17.Valette H, Deleuze P, Syrota A, et al. Canine myocardial beta-adrenergic, muscarinic receptor densities after denervation: a PET study. Journal of Nuclear Medicine. 1995;36(1):140–146. [PubMed] [Google Scholar]
- 18.Annett LE, Torres EM, Clarke DJ, et al. Survival of nigral grafts within the striatum of marmosets with 6-OHDA lesions depends critically on donor embryo age. Cell Transplantation. 1997;6(6):557–569. doi: 10.1177/096368979700600606. [DOI] [PubMed] [Google Scholar]
- 19.Ruffy R, Leonard M. Chemical cardiac sympathetic denervation hampers defibrillation in the dog. Journal of Cardiovascular Electrophysiology. 1997;8(1):62–67. doi: 10.1111/j.1540-8167.1997.tb00609.x. [DOI] [PubMed] [Google Scholar]
- 20.Sachs C, Jonsson G. Mechanisms of action of 6-hydroxydopamine. Biochemical Pharmacology. 1975;24(1):1–8. doi: 10.1016/0006-2952(75)90304-4. [DOI] [PubMed] [Google Scholar]
- 21.Martin GE, Myers RD, Newberg DC. Catecholamine release by intracerebral perfusion of 6 hydroxydopamine and desipramine. European Journal of Pharmacology. 1976;36(2):299–311. doi: 10.1016/0014-2999(76)90083-2. [DOI] [PubMed] [Google Scholar]
- 22.Perese DA, Ulman J, Viola J, Ewing SE, Bankiewicz KS. A 6-hydroxydopamine-induced selective parkinsonian rat model. Brain Research. 1989;494(2):285–293. doi: 10.1016/0006-8993(89)90597-0. [DOI] [PubMed] [Google Scholar]
- 23.Przedborski S, Levivier M, Jiang H, et al. Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience. 1995;67(3):631–647. doi: 10.1016/0306-4522(95)00066-r. [DOI] [PubMed] [Google Scholar]
- 24.Cohen G. Oxy-radical toxicity in catecholamine neurons. NeuroToxicology. 1984;5(1):77–82. [PubMed] [Google Scholar]
- 25.Blandini F, Armentero MT, Martignoni E. The 6-hydroxydopamine model: news from the past. Parkinsonism and Related Disorders. 2008;14(2):S124–S129. doi: 10.1016/j.parkreldis.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 26.Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59(2):401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- 27.Lee CS, Sauer H, Björklund A. Dopaminergic neuronal degeneration and motor impairments following axon terminal lesion by intrastriatal 6-hydroxydopamine in the rat. Neuroscience. 1996;72(3):641–653. doi: 10.1016/0306-4522(95)00571-4. [DOI] [PubMed] [Google Scholar]
- 28.Ungerstedt U. Adipsia and aphagia after 6-hydroxydopamine induced degeneration of the nigro-striatal dopamine system. Acta Physiologica Scandinavica, Supplement. 1971;367:95–122. doi: 10.1111/j.1365-201x.1971.tb11001.x. [DOI] [PubMed] [Google Scholar]
- 29.Bourn WM, Chin L, Picchioni AL. Enhancement of audiogenic seizure by 6-hydroxydopamine. Journal of Pharmacy and Pharmacology. 1972;24(11):913–914. doi: 10.1111/j.2042-7158.1972.tb08916.x. [DOI] [PubMed] [Google Scholar]
- 30.Knoll J. The pharmacology of (-)deprenyl. Journal of Neural Transmission. Supplementum. 1986;22:75–89. [PubMed] [Google Scholar]
- 31.Graham JD, Lewis MJ, Williams J. Proceedings: The effect of delta-1-tetrahydrocannabinol on the noradrenaline and dopamine content of the brain and heart of the rat. British Journal of Pharmacology. 1974;52(3):p. 446. [PMC free article] [PubMed] [Google Scholar]
- 32.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219(4587):979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 33.Forno LS, DeLanney LE, Irwin I, Langston JW. Similarities and differences between MPTP-induced parkinsonsim and Parkinson’s disease. Neuropathologic considerations. Advances in neurology. 1993;60:600–608. [PubMed] [Google Scholar]
- 34.Halliday G, Herrero MT, Murphy K, et al. No Lewy pathology in monkeys with over 10 years of severe MPTP parkinsonism. Movement Disorders. 2009;24(10):1519–1523. doi: 10.1002/mds.22481. [DOI] [PubMed] [Google Scholar]
- 35.Kowall NW, Hantraye P, Brouillet E, Beal MF, McKee AC, Ferrante RJ. MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. NeuroReport. 2000;11(1):211–213. doi: 10.1097/00001756-200001170-00041. [DOI] [PubMed] [Google Scholar]
- 36.Fornai F, Schlüter OM, Lenzi P, et al. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and α-synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(9):3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui M, Aras R, Christian WV, et al. The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(19):8043–8048. doi: 10.1073/pnas.0900358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(7):2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bezard E, Gross CE, Fournier MC, Dovero S, Bloch B, Jaber M. Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter. Experimental Neurology. 1999;155(2):268–273. doi: 10.1006/exnr.1998.6995. [DOI] [PubMed] [Google Scholar]
- 40.Gainetdinov RR, Fumagalli F, Wang YM, et al. Increased MPTP neurotoxicity in vesicular monoamine transporter 2 heterozygote knockout mice. Journal of Neurochemistry. 1998;70(5):1973–1978. doi: 10.1046/j.1471-4159.1998.70051973.x. [DOI] [PubMed] [Google Scholar]
- 41.Guillot TS, Shepherd KR, Richardson JR, et al. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. Journal of Neurochemistry. 2008;106(5):2205–2217. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guillot TS, Miller GW. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Molecular Neurobiology. 2009;39(2):149–170. doi: 10.1007/s12035-009-8059-y. [DOI] [PubMed] [Google Scholar]
- 43.Burke WJ, Kumar VB, Pandey N, et al. Aggregation of α-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathologica. 2008;115(2):193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- 44.Panneton WM, Kumar VB, Gan Q, Burke WJ, Galvin JE. The neurotoxicity of DOPAL: behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS One. 2010;5(12) doi: 10.1371/journal.pone.0015251. Article ID e15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zigmond MJ, Berger TW, Grace AA, Stricker EM. Compensatory responses to nigrostriatal bundle injury. Studies with 6-hydroxydopamine in an animal model of parkinsonism. Molecular and Chemical Neuropathology. 1989;10(3):185–200. doi: 10.1007/BF03159728. [DOI] [PubMed] [Google Scholar]
- 46.Chiueh CC, Markey SP, Burns RS. Neurochemical and behavioral effects of systemic and intranigral administration of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the rat. European Journal of Pharmacology. 1984;100(2):189–194. doi: 10.1016/0014-2999(84)90221-8. [DOI] [PubMed] [Google Scholar]
- 47.Przedborski S, Jackson-Lewis V, Naini AB, et al. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. Journal of Neurochemistry. 2001;76(5):1265–1274. doi: 10.1046/j.1471-4159.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- 48.Langston JW, Langston EB, Irwin I. MPTP-induced parkinsonism in human and non-human primates—clinical and experimental aspects. Acta Neurologica Scandinavica. 1984;70(supplement 100):49–54. [PubMed] [Google Scholar]
- 49.Bankiewicz KS, Oldfield EH, Chiueh CC. Hemiparkinsonism in monkeys after unilateral internal carotid artery infusion of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Life Sciences. 1986;39(1):7–16. doi: 10.1016/0024-3205(86)90431-5. [DOI] [PubMed] [Google Scholar]
- 50.Iravani MM, Syed E, Jackson MJ, Johnston LC, Smith LA, Jenner P. A modified MPTP treatment regime produces reproducible partial nigrostriatal lesions in common marmosets. European Journal of Neuroscience. 2005;21(4):841–854. doi: 10.1111/j.1460-9568.2005.03915.x. [DOI] [PubMed] [Google Scholar]
- 51.Hantraye P, Varastet M, Peschanski M, et al. Stable parkinsonian syndrome and uneven loss of striatal dopamine fibres following chronic MPTP administration in baboons. Neuroscience. 1993;53(1):169–178. doi: 10.1016/0306-4522(93)90295-q. [DOI] [PubMed] [Google Scholar]
- 52.Bezard E, Imbert C, Deloire X, Bioulac B, Gross CE. A chronic MPTP model reproducing the slow evolution of Parkinson’s disease: evolution of motor symptoms in the monkey. Brain Research. 1997;766(1-2):107–112. doi: 10.1016/s0006-8993(97)00531-3. [DOI] [PubMed] [Google Scholar]
- 53.Mounayar S, Boulet S, Tandé D, et al. A new model to study compensatory mechanisms in MPTP-treated monkeys exhibiting recovery. Brain. 2007;130(11):2898–2914. doi: 10.1093/brain/awm208. [DOI] [PubMed] [Google Scholar]
- 54.Blesa J, Juri C, Collantes M, et al. Progression of dopaminergic depletion in a model of MPTP-induced Parkinsonism in non-human primates. An 18F-DOPA and 11C-DTBZ PET study. Neurobiology of Disease. 2010;38(3):456–463. doi: 10.1016/j.nbd.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 55.Ovadia A, Zhang Z, Gash DM. Increased susceptibility to MPTP toxicity in middle-aged rhesus monkeys. Neurobiology of Aging. 1995;16(6):931–937. doi: 10.1016/0197-4580(95)02012-8. [DOI] [PubMed] [Google Scholar]
- 56.Pessiglione M, Guehl D, Hirsch EC, Féger J, Tremblay L. Disruption of self-organized actions in monkeys with progressive MPTP-induced parkinsonism. I. Effects of task complexity. European Journal of Neuroscience. 2004;19(2):426–436. doi: 10.1111/j.0953-816x.2003.03088.x. [DOI] [PubMed] [Google Scholar]
- 57.Schneider JS. Modeling Cognitive Deficits Associated with Parkinsonism in the Chronic-Low-Dose MPTP-Treated Monkey. Boca Raton, Fla, USA: CRC Press; 2006. [PubMed] [Google Scholar]
- 58.Anderson G, Noorian AR, Taylor G, et al. Loss of enteric dopaminergic neurons and associated changes in colon motility in an MPTP mouse model of Parkinson’s disease. Experimental Neurology. 2007;207(1):4–12. doi: 10.1016/j.expneurol.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fox SH, Chuang R, Brotchie JM. Serotonin and Parkinson’s disease: on movement, mood, and madness. Movement Disorders. 2009;24(9):1255–1266. doi: 10.1002/mds.22473. [DOI] [PubMed] [Google Scholar]
- 60.Barraud Q, Obeid I, Aubert I, et al. Neuroanatomical study of the A11 diencephalospinal pathway in the non-human primate. PLoS One. 2010;5(10) doi: 10.1371/journal.pone.0013306. Article ID e13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vezoli J, Fifel K, Leviel V, et al. Early presymptomatic and long-term changes of rest activity cycles and cognitive behavior in a MPTP-monkey model of Parkinson's disease. PLoS One. 2011;6(8) doi: 10.1371/journal.pone.0023952. Article ID e23952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergman H, Wichmann T, DeLong MR. Reversal of experimental Parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249(4975):1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- 63.Guridi J, Herrero MT, Luquin MR, et al. Subthalamotomy in parkinsonian monkeys. Behavioural and biochemical analysis. Brain. 1996;119(5):1717–1727. doi: 10.1093/brain/119.5.1717. [DOI] [PubMed] [Google Scholar]
- 64.Bezard E, Przedborski S. A tale on animal models of Parkinson's Disease. Movement Disorders. 2011;26(6):993–1002. doi: 10.1002/mds.23696. [DOI] [PubMed] [Google Scholar]
- 65.Vila M, Jackson-Lewis V, Vukosavic S, et al. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2837–2842. doi: 10.1073/pnas.051633998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dauer W, Kholodilov N, Vila M, et al. Resistance of α-synuclein null mice to the parkinsonian neurotoxin MPTP. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(22):14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berry C, La Vecchia C, Nicotera P. Paraquat and parkinson’s disease. Cell Death and Differentiation. 2010;17(7):1115–1125. doi: 10.1038/cdd.2009.217. [DOI] [PubMed] [Google Scholar]
- 68.Day BJ, Patel M, Calavetta L, Chang LY, Stamler JS. A mechanism of paraquat toxicity involving nitric oxide synthase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(22):12760–12765. doi: 10.1073/pnas.96.22.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Przedborski S, Ischiropoulos H. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxidants and Redox Signaling. 2005;7(5-6):685–693. doi: 10.1089/ars.2005.7.685. [DOI] [PubMed] [Google Scholar]
- 70.Brooks AI, Chadwick CA, Gelbard HA, Cory-Slechta DA, Federoff HJ. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Research. 1999;823(1-2):1–10. doi: 10.1016/s0006-8993(98)01192-5. [DOI] [PubMed] [Google Scholar]
- 71.McCormack AL, Thiruchelvam M, Manning-Bog AB, et al. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiology of Disease. 2002;10(2):119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- 72.Thiffault C, Langston JW, Di Monte DA. Increased striatal dopamine turnover following acute administration of rotenone to mice. Brain Research. 2000;885(2):283–288. doi: 10.1016/s0006-8993(00)02960-7. [DOI] [PubMed] [Google Scholar]
- 73.Thiruchelvam M, Brockel BJ, Richfield EK, Baggs RB, Cory-Slechta DA. Potentiated and preferential effects of combined paraquat and maneb on nigrostriatal dopamine systems: environmental risk factors for Parkinson’s disease? Brain Research. 2000;873(2):225–234. doi: 10.1016/s0006-8993(00)02496-3. [DOI] [PubMed] [Google Scholar]
- 74.Rappold PM, Cui M, Chesser AS, et al. Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter-3. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(51):20766–20771. doi: 10.1073/pnas.1115141108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of α-synuclein in mice: paraquat and α-synuclein. Journal of Biological Chemistry. 2002;277(3):1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- 76.Takahashi RN, Rogerio R, Zanin M. Maneb enhances MPTP neurotoxicity in mice. Research Communications in Chemical Pathology and Pharmacology. 1989;66(1):167–170. [PubMed] [Google Scholar]
- 77.Kachroo A, Irizarry MC, Schwarzschild MA. Caffeine protects against combined paraquat and maneb-induced dopaminergic neuron degeneration. Experimental Neurology. 2010;223(2):657–661. doi: 10.1016/j.expneurol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Butterfield PG, Valanis BG, Spencer PS, Lindeman CA, Nutt JG. Environmental antecedents of young-onset Parkinson’s disease. Neurology. 1993;43(6):1150–1158. doi: 10.1212/wnl.43.6.1150. [DOI] [PubMed] [Google Scholar]
- 79.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50(5):1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- 80.Barlow BK, Richfield EK, Cory-Slechta DA, Thiruchelvam M. A fetal risk factor for Parkinson’s disease. Developmental Neuroscience. 2004;26(1):11–23. doi: 10.1159/000080707. [DOI] [PubMed] [Google Scholar]
- 81.Tang CC, Poston KL, Dhawan V, Eidelberg D. Abnormalities in metabolic network activity precede the onset of motor symptoms in Parkinson’s disease. Journal of Neuroscience. 2010;30(3):1049–1056. doi: 10.1523/JNEUROSCI.4188-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tanner CM, Kame F, Ross GW, et al. Rotenone, paraquat, and Parkinson's disease. Environmental Health Perspectives. 2011;119(6):866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hisata J. Lake and stream rehabilitation: rotenone use and health risks. Final supplemental environmental impact statement. Washington Satet Deparment of Fish and Wildlife; 2002. [Google Scholar]
- 84.Inden M, Kitamura Y, Takeuchi H, et al. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. Journal of Neurochemistry. 2007;101(6):1491–1504. doi: 10.1111/j.1471-4159.2006.04440.x. [DOI] [PubMed] [Google Scholar]
- 85.Inden M, Kitamura Y, Abe M, Tamaki A, Takata K, Taniguchi T. Parkinsonian rotenone mouse model: reevaluation of long-term administration of rotenone in C57BL/6 mice. Biological and Pharmaceutical Bulletin. 2011;34(1):92–96. doi: 10.1248/bpb.34.92. [DOI] [PubMed] [Google Scholar]
- 86.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nature Neuroscience. 2000;3(12):1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 87.Alam M, Mayerhofer A, Schmidt WJ. The neurobehavioral changes induced by bilateral rotenone lesion in medial forebrain bundle of rats are reversed by L-DOPA. Behavioural Brain Research. 2004;151(1-2):117–124. doi: 10.1016/j.bbr.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 88.Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT. A highly reproducible rotenone model of Parkinson’s disease. Neurobiology of Disease. 2009;34(2):279–290. doi: 10.1016/j.nbd.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Höglinger GU, Féger J, Prigent A, et al. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. Journal of Neurochemistry. 2003;84(3):491–502. doi: 10.1046/j.1471-4159.2003.01533.x. [DOI] [PubMed] [Google Scholar]
- 90.Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and α-synuclein aggregation. Experimental Neurology. 2003;179(1):9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- 91.Wu Y-N, Johnson SW. Dopamine oxidation facilitates rotenone-dependent potentiation of N-methyl-d-aspartate currents in rat substantia nigra dopamine neurons. Neuroscience. 2011;195:138–144. doi: 10.1016/j.neuroscience.2011.08.041. [DOI] [PubMed] [Google Scholar]
- 92.Ferrante RJ, Schulz JB, Kowall NW, Beal MF. Systemic administration of rotenone produces selective damage in the striatum and globus pallidus, but not in the substantia nigra. Brain Research. 1997;753(1):157–162. doi: 10.1016/s0006-8993(97)00008-5. [DOI] [PubMed] [Google Scholar]
- 93.Takeuchi H, Yanagida T, Inden M, et al. Nicotinic receptor stimulation protects nigral dopaminergic neurons in rotenone-induced Parkinson’s disease models. Journal of Neuroscience Research. 2009;87(2):576–585. doi: 10.1002/jnr.21869. [DOI] [PubMed] [Google Scholar]
- 94.Pan-Montojo F, Anichtchik O, Dening Y, et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One. 2010;5(1) doi: 10.1371/journal.pone.0008762. Article ID e8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pan-Montojo FJ, Funk RHW. Oral administration of rotenone using a gavage and image analysis of alpha-synuclein inclusions in the enteric nervous system. Journal of Visualized Experiments. 2010;(44) doi: 10.3791/2123. Article ID e2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krüger R, Kuhn W, Müller T, et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nature Genetics. 1998;18(2):106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 97.Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VMY. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron. 2002;34(4):521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 98.Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in α-synuclein mice: Implications for neurodegenerative disorders. Science. 2000;287(5456):1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 99.Abeliovich A, Schmitz Y, Fariñas I, et al. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25(1):239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 100.Thomas B, Mandir AS, West N, et al. Resistance to MPTP-Neurotoxicity in α-synuclein knockout mice is complemented by human α-synuclein and associated with increased β-synuclein and Akt activation. PLoS One. 2011;6(1) doi: 10.1371/journal.pone.0016706. Article ID e16706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404(6776):394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 102.Wang D, Tang B, Zhao G, et al. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Molecular Neurodegeneration. 2008;3(1, article no. 3) doi: 10.1186/1750-1326-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 104.Li Y, Liu W, Oo TF, et al. Mutant LRRK2R1441G BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nature Neuroscience. 2009;12(7):826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moore DJ, Dawson TM. Value of genetic models in understanding the cause and mechanisms of Parkinson’s disease. Current Neurology and Neuroscience Reports. 2008;8(4):288–296. doi: 10.1007/s11910-008-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shin J-H, Ko HS, Kang H, et al. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in parkinson's disease. Cell. 2011;144(5):689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Peng J, Oo ML, Andersen JK. Synergistic effects of environmental risk factors and gene mutations in Parkinson’s disease accelerate age-related neurodegeneration. Journal of Neurochemistry. 2010;115(6):1363–1373. doi: 10.1111/j.1471-4159.2010.07036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]